

Abstract
The emergence of antibiotic-resistant bacterial species, such as vancomycin-resistant enterococci (VRE), necessitates the development of new antimicrobials. Here, we investigate the spectrum of antibacterial activity of three phenylthiazole-substituted aminoguanidines. These compounds possess potent activity against VRE, inhibiting growth of clinical isolates at concentrations as low as 0.5 μg/mL. The compounds exerted a rapid bactericidal effect, targeting cell wall synthesis. Transposon mutagenesis suggested three possible targets: YubA, YubB (undecaprenyl diphosphate phosphatase (UPPP)) and YubD. Both UPPP as well as undecaprenyl diphosphate synthase were inhibited by compound 1. YubA and YubD are annotated as transporters and may also be targets since 1 collapsed the proton motive force in membrane vesicles. Using Caenorhabditis elegans, we demonstrate that two compounds (1, 3, at 20 μg/mL) retain potent activity in vivo, significantly reducing the burden of VRE in infected worms. Taken altogether, the results indicate that compounds 1 and 3 warrant further investigation as novel antibacterial agents against drug-resistant enterococci.
Keywords: Enterococci, cytological profiling, cell wall, peptidoglycan, Caenorhabditis elegans, undecaprenyl diphosphate phosphatase, uncoupler
INTRODUCTION
According to the US CDC, there are approximately 1.7 million hospital-acquired infections (HAIs) in the US each year resulting in nearly 100,000 deaths and an estimated $20 billion in healthcare costs1. Many of the organisms responsible have become resistant to most antibiotics, contributing to prolonged illness, high treatment costs, increased treatment failure, and death2, 3. Bacterial pathogens such as vancomycin-resistant enterococci (VRE), in particular isolates of Enterococcus faecalis and Enterococcus faecium, are of particular concern and have been identified as leading sources of nosocomial infections4. These range from skin infections to intra-abdominal infections to urinary tract infections, with immunocompromised individuals—in particular the elderly and patients undergoing organ transplants and cancer chemotherapy—being at particular risk4.
Enterococcal infections were initially susceptible to many therapeutic agents including β-lactams (in particular ampicillin), glycopeptides (vancomycin), fluoroquinolones, and aminoglycosides5. However, the ability of enterococci to colonize the gastrointestinal tract of patients hospitalized for long periods has permitted these organisms to acquire resistance, particularly after repeated drug exposure4, severely limiting the number of effective therapeutic options available. Moreover, VRE strains have been isolated that exhibit resistance to newer antibacterial agents, including linezolid and daptomycin6, 7. The problem of antibiotic resistance is compounded by the diminishing number of new antibiotics being approved. From 1980–1984, 19 new antibiotics were approved by the US FDA but this number plummeted to one new approval from 2010–20128. The approval of three new antibiotics in 2014 indicates regulatory agencies understand the urgent need for new treatment options9 although the ever-present nature of resistance development necessitates the continuous search for new drugs, and new drug leads.
Our group recently explored the antimicrobial activity of a broad range of phenylthiazole aminoguanidines against drug-resistant staphylococci10–14, three of which (1–3, Figure 1) we find have potent activity against both E. faecalis and E. faecium. We examine here the activity of these compounds against clinical isolates of VRE; their mechanisms of action; potential synergies with other antibiotics; toxicity, and activity in an in vivo model of VRE infection in Caenorhabditis elegans.
Figure 1.
Chemical structures of phenylthiazole compounds investigated.
RESULTS
Antibacterial Activity of Compounds 1–3 Against the ESKAPE Pathogens (Enterococcus faecium, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp.)
We first investigated the antibacterial activity of phenylthiazole compounds 1–3 against a panel of ESKAPE pathogens using the broth micro-dilution method. Examination of the spectrum of activity of 2 and 3 revealed that they were inactive against most Gram-negative pathogens (minimum inhibitory concentration, MIC > 128 μg/mL), Table 1, the exception being with A. baumannii where the MIC was 8 μg/mL, similar to that found with erythromycin. Compound 1 also exhibited limited activity against the same pathogens (MIC ranges from 8–64 μg/mL), indicating that all three compounds are generally ineffective against Gram-negative bacteria. These results are in contrast to our earlier results on S. aureus, differences that could be due to the presence of the outer membrane (OM) in the Gram-negative bacteria, and/or efflux pumps. We thus next investigated whether the presence of the OM and/or efflux pumps did in fact contribute to the lack of antibacterial activity observed for 1–3 against Gram-negative bacteria.
Table 1.
Minimum inhibitory concentration (MIC) of phenylthiazole compounds 1–3 and control antibiotics against the ESKAPE pathogens (excluding S. aureus). For Gram-negative bacteria, the MIC of each compound in the presence of ¼ × MIC of colistin (COL) was used to examine the impact of the outer membrane on negating the antibacterial activity of the compounds.
| 1 | 2 | 3 | Erythromycin | Linezolid | |||||
|---|---|---|---|---|---|---|---|---|---|
| Bacterial strain | (−) COL |
(+) COL |
(−) COL |
(+) COL |
(−) COL |
(+) COL |
(−) COL |
(+) COL |
|
|
Acinetobacter baumanii ATCC 19606 |
8 | 1 | 8 | 1 | 8 | 1 | 8 | 0.5 | – |
|
Escherichia coli O157:H7 ATCC 35150 |
16 | 4 | >128 | 4 | >128 | 1 | 64 | 1 | – |
|
Enterobacter cloacae BAA-1154 |
64 | 1 | >128 | 1 | >128 | 1 | 128 | 1 | – |
|
Klebsiella pneumoniae BAA-1706 |
16 | 1 | >128 | 2 | >128 | 1 | 128 | 0.5 | – |
|
Pseudomonas aeruginosa ATCC 15442 |
64 | 1 | >128 | 1 | >128 | 1 | 128 | 1 | – |
| Escherichia coli 1411 | 64 | – | >128 | – | >128 | – | – | – | >128 |
| Escherichia coli 1411 ΔacrAB | 4 | – | 4 | – | 8 | – | – | – | 8 |
|
Enterococcus faecium (VRE) ATCC 700221 |
0.5 | – | 0.5 | – | 0.5 | – | – | – | 0.5 |
In the presence of a sub-inhibitory concentration of the membrane-disrupting antibiotic colistin, which permeabilizes the outer membrane, the MIC of compounds 1–3 against Gram-negative bacteria decreased dramatically. For example, 2 and 3 were inactive against K. pneumoniae, P. aeruginosa and E. coli when tested alone (MIC > 128 μg/mL), but in the presence of sub-inhibitory levels of colistin, both compounds exhibited potent antibacterial activity (MIC from 1 to 4 μg/mL). This behavior is similar to that seen with the antibiotic erythromycin, whose activity is known to be impeded by the presence of the OM. For example, with K. pneumoniae, P. aeruginosa, and E. coli, we find that erythromycin alone lacks activity (MIC > 128 μg/mL), but in the presence of colistin, erythromycin has potent activity against each organism (MIC from 0.5 to 1 μg/mL). It thus appears likely that the OM impedes the entry of both the phenylthiazole compounds as well as erythromycin into Gram-negative bacteria.
When the antibacterial activity of compounds 1–3 was examined against E. coli 1411 and a mutant strain containing a deletion of the gene encoding the AcrAB efflux pump, there was a major decrease in the MIC in the mutant. Against E. coli 1411, compound 1 inhibited growth at 64 μg/mL, and compounds 2 and 3 were inactive (MIC > 128 μg/mL). However, against the mutant strain (Escherichia coli 1411 ΔacrAB), all three compounds were potent inhibitors of bacterial growth (MIC of 4 μg/mL). This behavior is similar to that observed with linezolid, a known substrate of the AcrAB efflux pump15. It thus appears that the lack of activity of the phenylthiazoles against Gram-negative bacterial pathogens is due both to the presence of the OM as well as drug efflux pumps. Fortunately, in earlier work we found that compounds 1–3 exhibited potent antibacterial activity against the Gram-positive pathogen, methicillin-resistant S. aureus (MIC values ranged from 1.3 to 5.6 μg/mL)10, and as shown in Table 1, all three compounds are potent inhibitors of the growth of another important Gram-positive pathogen, vancomycin resistant E. faecium (MIC = 0.5 μg/mL). Thus, the phenylthiazole compounds appear to be potent leads against clinically-relevant Gram-positive pathogens, including MRSA and VRE. Plus, the fact that they synergize with cell wall biosynthesis inhibitors in S. aureus suggests, perhaps, a similar target area in enterococci.
Phenylthiazole Compounds Retain Their Potent Activity Against Clinical Isolates of Drug-Resistant Enterococci
To further evaluate the antibacterial activity of compounds 1–3 against enterococci, we determined MIC values against 24 strains of E. faecalis and E. faecium (including 16 strains resistant to vancomycin), isolated from diverse sources including blood, urine, peritoneal fluid, sputum, and feces (Table S1), from infected patients. All three compounds exhibited potent antibacterial activity against all isolates tested (Table 2). Interestingly, 1–3 were most active against isolates of vancomycin-resistant E. faecium, inhibiting growth at concentrations ranging from 0.5 to 4 μg/mL (Table S2), with MIC50 values of 1 (for compounds 2 and 3) to 4 μg/mL (for compound 1). These values are similar to those we find with the antibiotic linezolid (MIC range from 0.5 to 2 μg/mL). Against E. faecalis, 1 and 3 retained their potent antibacterial activity (MIC range from 1–8 μg/mL), but there was an increase in the MIC of compound 2 (MIC = 16 μg/mL, against multiple strains). However, all compounds proved quite effective at inhibiting the growth of VRE. The compounds also retained activity against isolates exhibiting resistance to ampicillin, ciprofloxacin, gentamicin, streptomycin, erythromycin, and doxycycline. This indicates that cross-resistance between these antibiotics and 1–3, against enterococci, is unlikely to occur.
Table 2.
Minimum inhibitory concentration (MIC50) (in μg/mL) and minimum bactericidal concentration (MBC50) of phenylthiazole compounds 1–3, vancomycin, and linezolid against 50% of vancomycin-sensitive (VSE) and vancomycin-resistant Enterococcus faecalis or E. faecium (VRE) strains.
| Bacterial species (number of isolates) | 1 | 2 | 3 | Vancomycin | Linezolid | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| MIC50 | MBC50 | MIC50 | MBC50 | MIC50 | MBC50 | MIC50 | MBC50 | MIC50 | MBC50 | |
| Enterococcus faecium (3) | 4 | 4 | 8 | 16 | 4 | 4 | 1 | – | 2 | 16 |
| Vancomycin-resistant Enterococcus faecium (10) | 2 | 4 | 1 | 16 | 1 | 4 | >64 | >64 | 1 | 128 |
| Enterococcus faecalis (5) | 4 | 4 | 8 | 16 | 8 | 8 | 1 | – | 1 | 64 |
| Vancomycin-resistant Enterococcus faecalis (6) | 4 | 4 | 2 | 4 | 4 | 4 | >64 | >64 | 1 | 64 |
We next investigated whether the antibacterial effects were bacteriostatic or bactericidal. To address this, we determined the minimum bactericidal concentrations, MBCs. Against most strains of E. faecalis and E. faecium, the MBC values were equal to or one-fold higher than the MIC values for compounds 1 and 3, indicating that the two compounds are bactericidal. A similar trend was observed for compound 2 against E. faecalis, however, against E. faecium, the MBC for 2 was more than four-fold higher than the MIC, indicating 2 may be bacteriostatic, particularly against vancomycin-resistant isolates. As expected, ampicillin exhibited bactericidal activity against enterococci strains sensitive to this antibiotic, while linezolid exhibited bacteriostatic activity against both E. faecalis and E. faecium (MBC was more than four-fold higher than the MIC).
Compounds 1 and 3 Rapidly Eradicate Vancomycin-Resistant Enterococci as Determined by Time-kill Analysis
In order to confirm the bactericidal activity of the phenylthiazole compounds against VRE, we carried out time-kill assays. As shown in Figure 2, compounds 1 and 3 exhibited rapid bactericidal activity against both vancomycin-resistant E. faecium as well as vancomycin-resistant E. faecalis. Compound 1 (at 4 × MIC) was capable of completely eradicating both strains of VRE within two hours, and no bacterial re-growth was observed over the following 22 hours. Compound 3 matched compound 1 in completely eliminating vancomycin-resistant E. faecalis within two hours (Figure 2B), but required four hours to achieve the same effect against vancomycin-resistant E. faecium (Figure 2A). Compound 2 also exhibited rapid bactericidal activity against E. faecalis, completely eradicating bacterial growth within two hours. However, although 2 produced a gradual reduction in CFU against vancomycin-resistant E. faecium after 24 hours, the compound was not capable of completely eradicating the bacteria. This supports the MBC results for 2 against vancomycin-resistant E. faecium, indicating that 2 is bacteriostatic. The bacteriostatic activity of linezolid against both E. faecium and E. faecalis is also confirmed since the antibiotic was not able to generate a 103 × reduction in VRE CFU over 24 hours.
Figure 2.
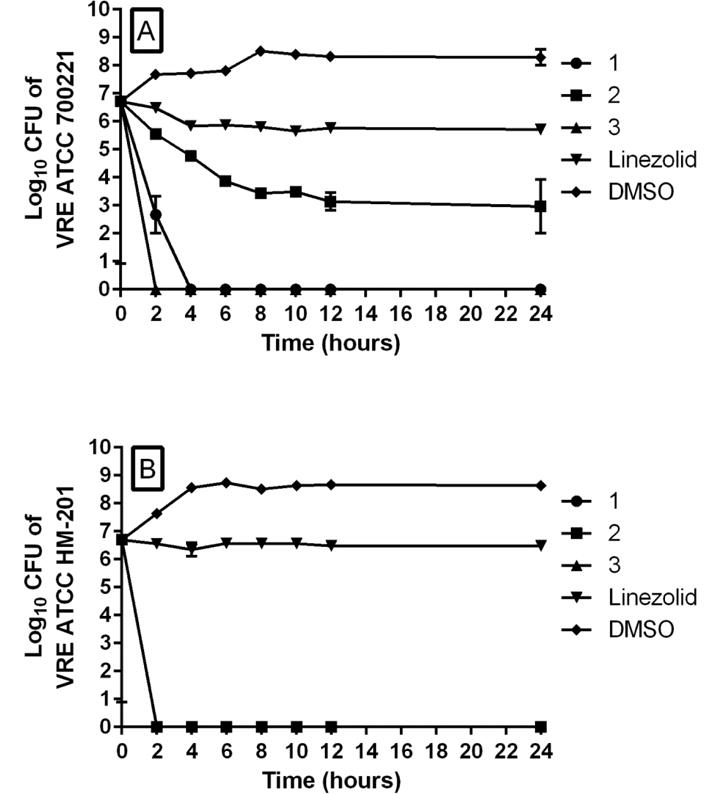
Time-kill analysis of phenylthiazole compounds 1, 2, 3, and linezolid (all tested at 4 × MIC) over a 24 hour incubation period at 37 °C against A) vancomycin-resistant Enterococcus faecium ATCC 700221 and B) vancomycin-resistant Enterococcus faecalis HM-201. DMSO served as a negative control. The error bars represent standard deviation values obtained from triplicate samples used for each compound/antibiotic studied.
Compounds 1–3 Exhibit Limited Toxicity to Human Colorectal Cells
Enterococci are commensal organisms of the human gastrointestinal tract. We thus next examined the toxicity of all three compounds against a human colorectal (HRT-18) cell line. When the compounds were incubated with cells for a short period (two hours), compound 1 was non-toxic up to 20 μg/mL (Figure 3A). Compounds 2 and 3 exhibited an improved toxicity profile since both were not toxic up to 40 μg/mL. When compounds were incubated with HRT-18 cells for 24 hours (Figure 3B), the toxicity profile of 1 remained the same (not toxic up to 20 μg/mL), while 2 and 3 were toxic at 40 μg/mL, but non-toxic at 20 μg/mL.
Figure 3.
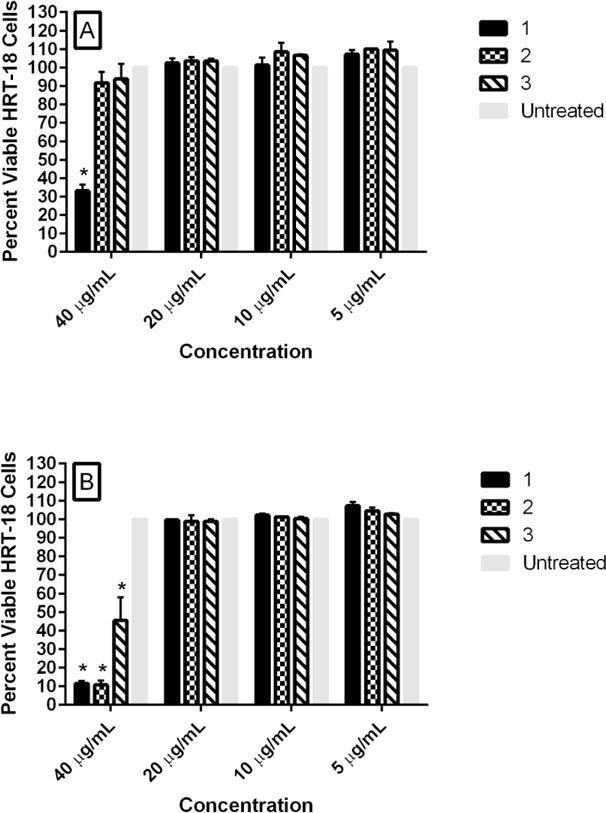
Percent viable mammalian cells (measured as average absorbance ratio (test agent relative to untreated cells)) for cytotoxicity analysis of phenylthiazole compounds 1, 2, and 3 at 5, 10, 20, and 40 μg/mL against human colorectal (HRT-18) cells using the MTS 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assay. Untreated cells served as a negative control to determine a baseline measurement for the cytotoxic impact of each compound. Test agents were incubated with cells for either A) two hours or B) 24 hours. The absorbance values represent an average of a minimum of three samples analyzed for each compound. Error bars represent standard deviation values for the absorbance values. A one-way ANOVA (with post-hoc Dunnett’s multiple comparisons test), P ≤ 0.05, demonstrated statistical difference between the values obtained for compounds 1, 2, and 3 relative to untreated cells at 40 μg/mL (denoted by an asterisk, *).
Single-Step and Multi-Step Resistance Selection of Enterococci to Compounds 1–3
Given the promising results described above, we next sought to examine the likelihood that enterococci will develop resistance to 1–3, using two different methods. First, we attempted to isolate spontaneous mutants exhibiting resistance using a single-step resistance selection experiment. In the presence of a high inoculum of E. faecalis NR-31975 (5 × 108 CFU/mL), no resistant mutants to 1–3 were isolated at concentrations of 2 ×, 4 ×, or 8 × MIC. When the inoculum size was increased (to 5 × 1010 CFU/mL), the same result was obtained, indicating a frequency of resistance > 2 × 10−11. We next attempted to isolate resistant mutants to 1–3 via a multi-step resistance selection experiment using two strains of VRE: E. faecalis ATCC 51299 and E. faecium ATCC HM-968. A four-fold increase in MIC was categorized as resistance. Against E. faecium (Figure 4A), there was no shift in the MIC observed for compounds 1 and 3 over 14 serial passages (similar to results obtained with linezolid). A one-fold increase (that is, the MIC increases from x → 2x) in MIC was observed for linezolid after the third passage and for compound 2, after the eleventh passage. However, no additional increase in the MIC of either agent was observed thereafter. Against E. faecalis (Figure 4B), a one-fold increase in MIC for compounds 1 and 3 was observed after the sixth passage; no further increase in MIC was observed until the last passage. For compound 2, a one-fold increase in MIC was observed after the sixth passage and an additional two-fold increase was observed after the eleventh passage. No additional increase was observed thereafter. No increase in MIC was observed for linezolid over the 14 serial passages. These results indicate a low likelihood for enterococci to develop rapid resistance to, in particular, 1 and 3, consistent with the single-step results.
Figure 4.
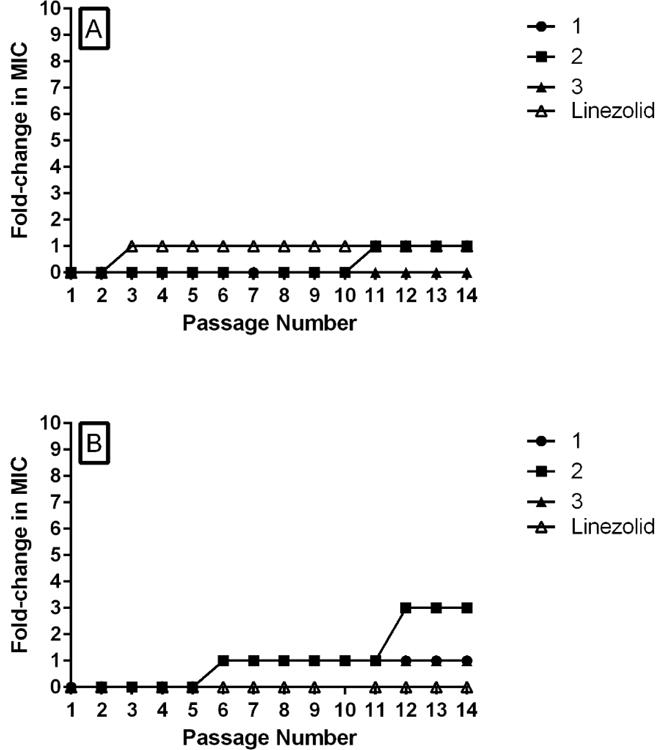
Multi-step resistance selection of compounds 1, 2, 3, and linezolid against A) vancomycin-resistant Enterococcus faecium HM-968 and B) vancomycin-resistant Enterococcus faecalis ATCC 51299. Bacteria were serially passaged over a fourteen-day period and the broth microdilution assay was used to determine the minimum inhibitory concentration of each compound against VRE after each successive passage. A four-fold shift in the MIC is defined as resistance to the test agent.
Compound 1 Exerts its Antibacterial Activity by Inhibiting Cell Wall Synthesis
In order to investigate the mechanism of action of the phenylthiazoles, compound 1 was subjected to Bacterial Cytological Profiling (BCP) in representative Gram-positive (Bacillus subtilis) and Gram-negative (E. coli) bacteria. BCP identifies the likely pathway targeted by novel antibiotics by comparing their cytological effects with those found using a library of cytological profiles generated by using antibacterials with known mechanisms of action (MOAs), or by the rapid proteolytic depletion of essential proteins16–18. When E. coli ΔtolC (which lacks an effective efflux pump) was treated with compound 1, cells lysed and formed spheroplasts after two hours (Figure 5). Spheroplasts and misshapen cells were very prevalent in the presence of 0.5 M sucrose, which osmotically stabilizes cells lacking a functional cell wall. Lysis and cell shape defects were observed as early as 30 minutes after addition of compound 1 to the medium. In contrast, cells incubated with the cell wall biosynthesis inhibitor ᴅ-cycloserine formed misshapen cells and spheroplasts after 30 minutes (Figure 5), and cells were completely lysed after two hours (data not shown). These results suggest that compound 1 inhibits cell wall biosynthesis in E. coli ΔtolC.
Figure 5.
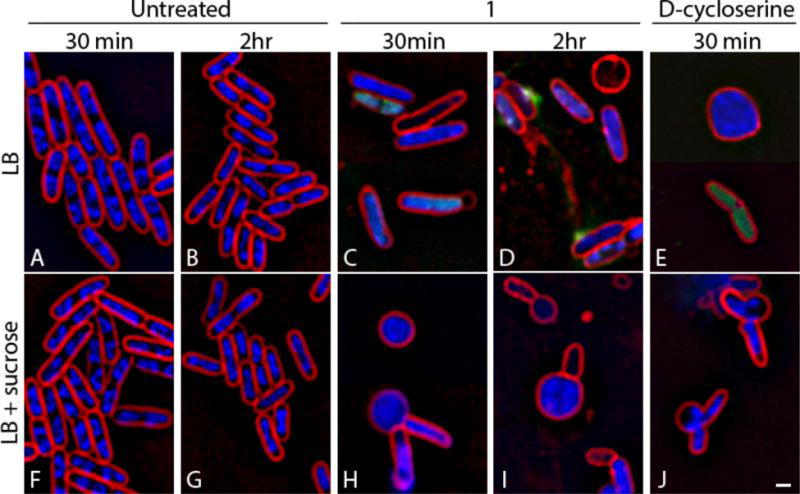
Effects of compound 1 on cell wall biosynthesis in E. coli ΔtolC. (A, B, F, G) Untreated cells. (C, D, H, I) Cells treated with compound 1 for either 30 minutes or two hours at 5 × MIC (25 μg/mL). (E, J) Cells treated with ᴅ-cycloserine at 5 × MIC (125 μg/mL) for 30 minutes. Cells (F–J) were treated in the presence of 0.5 M sucrose to facilitate visualization of cell shape defects. Cells were stained with FM 4–64 (red), DAPI (blue), and SYTOX Green (green). Scale bar is 1 μm.
To determine whether 1 had the same effect in a Gram-positive bacterium, we examined the effects of 1 in B. subtilis, again using BCP. B. subtilis incubated with 1 at 5 × MIC rapidly lysed, with 95% (n = 131) of cells being permeable to Sytox Green (a nucleic acid stain that is impermeable to live cells) within 30 minutes of treatment. Since disruption of either the cell wall or the cell membrane can result in permeabilized cells (Figure S1), we next investigated the effects of 1 with control agents in the presence of dimethylsulfone (a.k.a. methylsulfonyl methane, MSM), which osmotically stabilizes cells for better observation of cell shape defects. Cells treated with 1 for two hours in the presence of MSM were slightly misshapen or bent and contained pools of membrane (Figures S2 and Figure 6). Figure 6 shows four examples of cells containing a small bulge at the site of the bend that could be visualized with phase contrast microscopy. These cells appeared very similar to vancomycin-treated cells, which also show subtle cell shape-defects, forming bends, bulges, and pools of membrane (Figure S2 and Figure 6A). Unlike compound 1 and vancomycin, ᴅ-cycloserine generated very obvious cell shape defects (Figure S2), and Triton X-100 detergent-treated cells were lysed without affecting overall cell shape. MSM suppresses cell lysis and permeability defects for cell wall active antibiotics, but not for membrane active compounds (Figure S2 and Figure 6)17. We found MSM dramatically suppressed the permeability phenotypes of compound 1, as well as vancomycin and ᴅ-cycloserine, but had no effect on Triton X-100 treated cells, suggesting that 1 inhibits cell wall/peptidoglycan synthesis (Figure 6) in both B. subtilis and E. coli ΔtolC, implicating broadly conserved target(s) and MOA. But what are the actual targets of 1?
Figure 6.
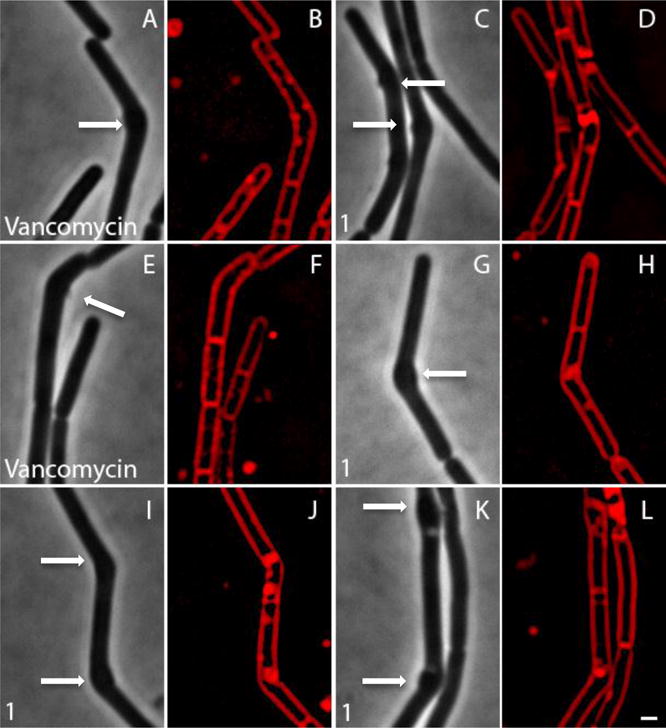
Comparison of cell shape defects in Bacillus subtilis treated with compound 1 or vancomycin. All cells were grown in LB in the presence of MSM at 37°C and are shown at two hours. Both compound 1 and vancomycin lead to slight bending of the cells and bulges, as indicated by the arrows.
Target Identification
Peptidoglycan biosynthesis involves numerous enzymes and a simplified metabolic pathway is shown in Figure 7. The first step involves the sequential addition of two molecules of isopentenyl diphosphate (IPP, 4) to dimethylallyl diphosphate (DMAPP, 5) to form the (C15) isoprenoid farnesyl diphosphate (FPP, 6) in a reaction catalyzed by farnesyl diphosphate synthase (FPPS), with IPP/DMAPP being produced by the mevalonate pathway in S. aureus and the non-mevalonate (methylerythritol phosphate, MEP) pathway in B. subtilis and E. coli. FPP then reacts with 8 additional IPP molecules to form the (C55) isoprenoid undecaprenyl diphosphate (UPP, 7) in a reaction catalyzed by undecaprenyl diphosphate synthase (UPPS). UPP is converted to undecaprenyl monophosphate (UP, 8) by undecaprenyl diphosphate phosphatase (UPPP), then UP reacts with UDP-N-acetylmuramyl pentapeptide (9) to form Lipid I (10) in a reaction catalyzed by MraY, followed by conversion to Lipid II, and after several more steps, peptidoglycan (11) is formed. Drugs such as ampicillin and vancomycin inhibit at some of these later stages in cell wall synthesis (transpeptidation), as they interfere with peptidoglycan crosslinking, resulting in defects in cell wall structure.
Figure 7.
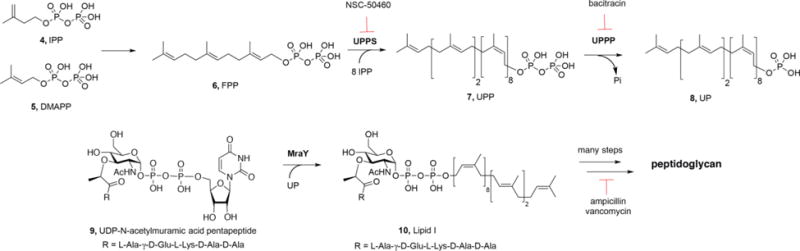
Schematic illustration of some key steps in cell wall biosynthesis in many Gram-positive bacteria, and sites of action of known drugs and inhibitors discussed in the text.
We first sought to see if 1 resulted in changes in the concentration of UDP-N-acetylmuramyl pentapeptide (compound 9), the final soluble cell wall precursor in peptidoglycan biosynthesis, in E. faecalis NR-31975 cells. We found that treatment of cells with compound 1 resulted in a similar LC-MS result to treatment of cells with vancomycin, a known inhibitor of bacterial cell wall synthesis. This can be seen in the results shown in Figure S4 in which there are large increases in UDP-N-acetylmuramyl pentapeptide accumulation with 1 or vancomycin treatment, implicating inhibition of peptidoglycan biosynthesis. A peak was present in the chromatograms at the same retention time (~8 minutes) for both 1 and vancomycin-treated samples, and had the correct m/z for the pentapeptide, m/z = 1150.3588, a <1 ppm error. These results support inhibition of a target in the peptidoglycan biosynthesis pathway, but they do not suggest a specific molecular target.
We next tried to identify the molecular target of the phenylthiazole 1 by using a target overexpression experiment, in Bacillus subtilis. The metabolic function(s) in B. subtilis inhibited by the thiazole should in principle be restored by over-expression of the targeted protein(s) via genomic insertion of a transposon with a strong outward-oriented promoter. That is, resistance to compound 1 should be achieved by over-expression of the drug-resistance gene—which could be a molecular target in peptidoglycan biosynthesis—but also perhaps an efflux pump19. In the presence of a high concentration of compound 1, only bacterial colonies where the transposon successfully inserts adjacent to the biological target/resistance mechanism survive, due to overexpression of the target/resistance mechanism by the bacterium. Using this approach we identified three possible targets: 1) yubA, locus tag BSU31160, a putative inner membrane AI-2E (autoinducer-2-exporter) family protein; 2) yubB, locus tag BSU31150, undecaprenyl diphosphate phosphatase (UPPP) and 3) yubD, locus tag BSU31130, a putative major facilitator superfamily transporter. Clearly, these results strongly suggest UPPP as a likely target since UPPP is in the peptidoglycan biosynthesis pathway, Figure 7. If UPPP (YubB) is inhibited by 1, there would be a decrease in UP levels and this would be expected to lead to accumulation of the MraY substrate 9 (since it would not have a substrate to react with).
To test this hypothesis we used the E. coli UPPP expression system described previously20, 21 and determined the IC50 for inhibition by 1. We found a 6 μM IC50 (corresponding to 2 μg/mL), consistent with a UPPP target, Figure 8A. We also tested for human FPPS (HsFPPS) and E. coli UPPS (EcUPPS) inhibition. There was no inhibition of FPPS (data not shown), but UPPS was inhibited with a 19 μM IC50 (corresponding to 6.3 μg/mL), Figure 8B. What is interesting here is that UPPS as well as UPPP are both inhibited at low μM levels and since these two enzymes are adjacent to each other in the biosynthetic pathway, this multi-target inhibition is expected to contribute to their activity, in cells, and is very similar to the dual UPPS/UPPP inhibition we have reported with other inhibitors21.
Figure 8.
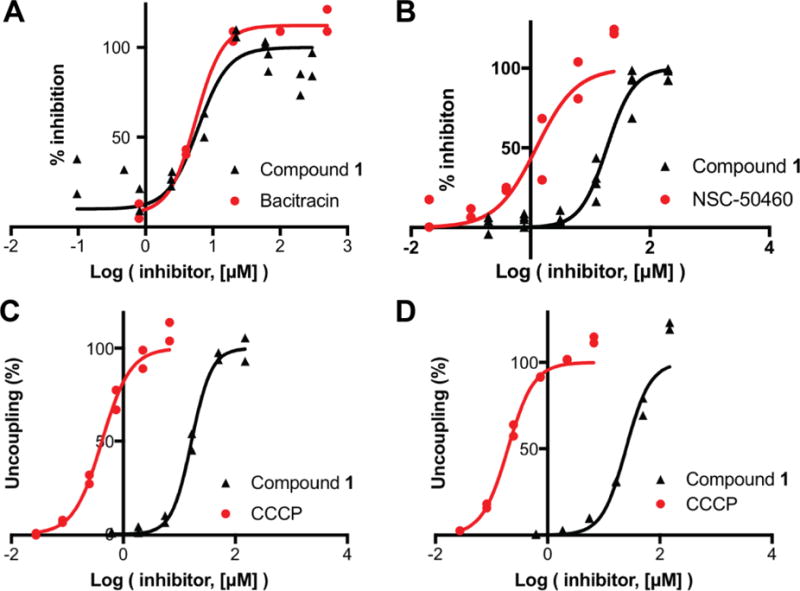
Dose response curves for enzyme inhibition by 1, and its effects on the PMF. A, UPPP (YubB) inhibition; bacitracin control. B, UPPS inhibition, bisamidine NSC-50460 control. C, PMF collapse in E. coli IMVs, ATP driven PMF, CCCP control. D, As C but succinate/O2-driven PMF generation.
What, then—if anything—is the involvement of YubA and YubD in the activity of 1 in cells? At present, these proteins have not been characterized in detail but both are annotated as transporters, raising the question: could the phenythiazoles also target membrane transporters? Upon inspection of the structure of 1 (as well as 2 and 3) it is clear that each compound has a polar aminoguanidine “headgroup” (pKa ~7) and a lipophilic “tail”. In other work, we and others have shown that many such compounds—lipophilic bases—can act as protonophore uncouplers, collapsing the proton motive force (PMF) in cells, as determined by using fluorescence probes as well as by 31P NMR spectroscopy22–24. That work led to a re-appraisal of the mechanism of action (MOA) of the tuberculosis (TB) drug lead SQ109, as well as the MOA of TB drugs in clinical use such as bedaquiline and clofazimine23. More importantly, many TB drug leads that had been thought to target the trehalose monomycolate transporter MmpL3 (mycobacterial membrane protein large 3) in a direct fashion are now thought to actually function by collapsing the PMF, inhibiting the function of PMF-driven transporters. If 1 were to also collapse the PMF, this could indirectly inhibit the transporters YubA and YubD, identified in the transposon mutagenesis experiments.
To determine whether 1 is a protonophore uncoupler, we used the E. coli inverted membrane vesicle (IMV) system we used previously24. Results with 1 and the potent, known uncoupler CCCP (m-chlorophenyl carbonyl cyanide phenylhydrazone) are shown in Figure 8C and Figure 8D with both ATP-powered PMF generation as well as succinate/O2-powered PMF generation. The IMVs have their ATPase on the outside of the vesicle so ATP hydrolysis through the ATPase, or succinate/O2, drives H+ into the vesicles, the fluorophore ACMA (9-amino-6-chloro-2-methoxyacridine) accumulates and its fluorescence is self-quenched (the signal goes down). Addition of CCCP or 1 collapses the PMF and fluorescence increases (back to normal). For CCCP, the EC50 is 0.4 μM in ATP and 0.2 μM in succinate; for 1, the EC50 for PMF collapse is 12 μM in ATP and 25 μM in succinate, Figure 8C and Figure 8D. This is relatively weak uncoupling but could lead to inhibition of some transporters, including drug efflux pumps.
At present, the three-dimensional structures of YubA, YubB (UPPP) and YubD have not been reported. However, a structural model for UPPP (together with site-directed mutagenesis results) for UPPP has been reported20 and is shown in Figure 9, together with RaptorX predicted structural models for YubA and YubD, and an X-ray structure for UPPS25 (PDB ID code 1X0626). YubA and YubD are both membrane proteins and the proteins with known structures that were used to create these homology models are all transporters, including PMF-driven multi-drug efflux pumps. We thus propose that 1 inhibits both UPPS and UPPP by directly binding to these proteins, in addition to potentially affecting YubA and/or YubD function, by acting as a protonophore uncoupler, reducing ΔpH, with such multi-targeting contributing to the very low rate of resistance that we observe.
Figure 9.
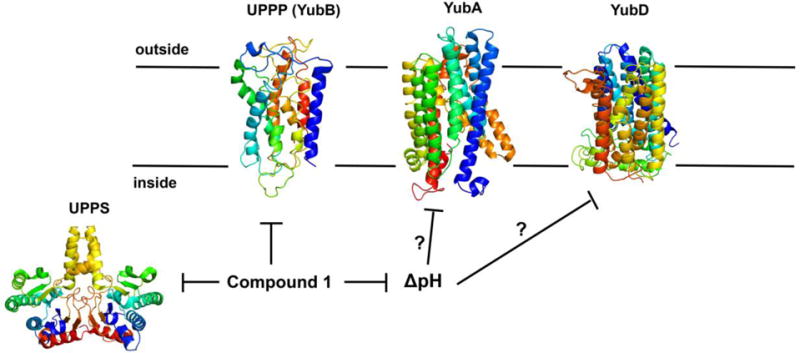
Structures of proposed targets of compound 1. The UPPS structure is the X-ray structure of EcUPPS, PDB ID code 1X06. The membrane protein structures are models for UPPP (YubB), YubA and YubD. Compound 1 inhibits UPPS and UPPP at low μM levels and collapses the PMF (in EcIMVs), suggested to affect the activity of YubA or YubD, putative transporters identified as targets in the transposon mutagenesis experiment (together with UPPP/YubB).
Resensitization of Enterococci to the Effects of Other Antibiotics
The discovery that the phenylthiazole 1 inhibits peptidoglycan synthesis led us to investigate its ability to resensitize VRE to the effects of known antibiotics. Compounds 1–3 have thus far been shown to be potent single-agent inhibitors of the growth of drug-resistant strains of S. aureus, E. faecium and E. faecalis. However, combination therapy using two or more antibiotics has multiple potential advantages over monotherapy including reducing the size and frequency of doses needed to resolve infection, and mitigating toxicity issues associated with single agents (such as vancomycin). Additionally, pairing a bactericidal agent (such as a cell wall synthesis inhibitor) with an aminoglycoside is already known to be necessary for treating certain enterococcal infections, such as endocarditis.
Previously, we demonstrated that 1 resensitizes vancomycin-resistant S. aureus (VRSA) to vancomycin11. With aminoglycoside antibiotics, resistance is due to an inability to cross the cell wall to reach the bacterial ribosome5, and increased accumulation of such antibiotics has been observed in enterococci in the presence of a cell wall synthesis inhibitor5. We therefore next examined whether VRE exposed to a sub-inhibitory concentration (½ × MIC) of compound 1 were resensitized to the effects of vancomycin and gentamicin. Phenylthiazole 1 was able to resensitize E. faecium ATCC 700221 to the effect of vancomycin —a 256-fold decrease in the MIC being observed (Table 3). However, against E. faecium HM-968 and two strains of E. faecalis (HM-201 and HM-934), no resensitization to vancomycin was observed. Likewise, a sub-inhibitory concentration of 1 was unable to resensitize either E. faecium (ATCC 700221) or E. faecalis (ATCC 51299) to the effect of gentamicin (data not shown). Nevertheless, the results found with vancomycin were of interest, so we next examined whether the thiazole compounds would exhibit synergistic relationships with antibiotics frequently used to treat enterococcal infections. Using checkerboard assays, we first tested compound 1 in combination with ampicillin, ciprofloxacin, doxycycline, or linezolid against E. faecalis ATCC 51299. While ampicillin (FIC index = 3.00), doxycycline (FIC index = 2.00), and linezolid (FIC index = 2.00) exhibited indifferent or additive relationships, ciprofloxacin demonstrated synergy with compound 1 (FIC index = 0.50). We then examined if synergisms between compounds 1–3 and ciprofloxacin would be observed when tested against additional strains of E. faecalis (ATCC 49532 and ATCC 49533), but all three compounds exhibited only an additive relationship (FIC index ranging from 0.63 to 1.00, Table S3).
Table 3.
Minimum inhibitory concentration (MIC) (μg/mL) of vancomycin alone and in the presence of a subinhibitory concentration of compound 1 against vancomycin-resistant enterococci.
| Strain | Vancomycin | Vancomycin + ½ × MIC Compound 1 | Fold-improvement in MIC of vancomycin |
|---|---|---|---|
| E. faecium ATCC 700221 | 256 | 1 | 256-fold |
| E. faecium HM-968 | 1024 | >128 | No change |
| E. faecalis HM-201 | 512 | >128 | No change |
| E. faecalis HM-934 | >512 | >128 | No change |
Compounds 1 and 3 Retain Their Potent Antibacterial Activity In Vivo Against VRE
The finding that the thiazole compounds possess good in vitro activity against many VRE strains and exert their antibacterial effect by targeting cell wall synthesis prompted us to investigate the efficacy of these compounds in the C. elegans animal model. Vancomycin-resistant E. faecalis HM-201, a highly pathogenic strain, was examined with compounds 1 and 3 since these compounds exhibited rapid bactericidal activity, in vitro. Based upon the results from the HRT-18 cell growth inhibition experiments, we chose a dose of 20 μg/mL. To verify that this concentration was not toxic to C. elegans, worms were exposed to compounds 1, 3, and a control antibiotic (linezolid), and viability was observed over a 24-hour period. All worms survived when exposed to compound 1 (Figure 10A) as well as linezolid, and 90% survived after exposure to compound 3. C. elegans AU37 worms were then infected with VRE HM-201 and subsequently treated for 18 hours with 20 μg/mL 1, 3, linezolid, or PBS. After treatment, worms were lysed and the VRE burden determined. Compounds 1 (89% decrease) and 3 (94% decrease) produced significant decreases in VRE burden in infected C. elegans (Figure 10B). In contrast, the bacteriostatic antibiotic linezolid was unable to reduce the burden of VRE in infected worms at this concentration.
Figure 10.
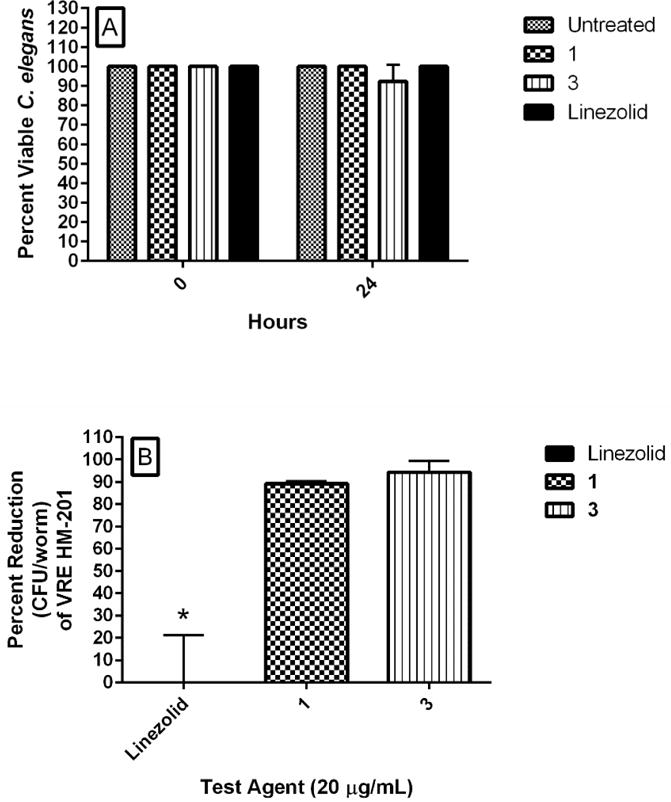
In vivo examination of toxicity and antibacterial activity of phenylthiazole compounds 1 and 3 (tested at 20 μg/mL) in C. elegans AU37 infected with vancomycin-resistant Enterococcus faecalis HM-201. Linezolid served as a positive control. A) Worms (in L4 stage of growth) were treated with 20 μg/mL of compounds 1, 3, or linezolid and percent viable worms was determined after 24 hours of exposure. B) Worms (in L4 stage of growth) infected with vancomycin-resistant E. faecalis HM-201 for two hours before transferring 20–25 worms to wells of a 96-well plate. Test agents were added and incubated with worms for 18 hours. Worms were sacrificed and the number of viable colony-forming units of E. faecalis HM-201 in infected worms was determined for each treatment regimen. The figure shows the percent reduction of E. faecalis HM-201 (relative to untreated worms). Asterisks (*) denote statistical significance (P < 0.05) in bacterial reduction relative to the positive control (linezolid) using a two-tailed Student’s t-test (with post-hoc Holm-Sidak test for multiple comparisons).
In silico Examination of the Pharmacokinetic Profile of Compounds 1 and 3
The results obtained in the C. elegans infection experiment led us to examine the pharmacokinetic profiles of compounds 1 and 3 in order to identify appropriate routes of administration. Utilizing computer modeling, the pharmacokinetic profiles of both compounds, in addition to linezolid, were simulated utilizing a dose of 600 mg (as is commonly administered for linezolid in human patients for the treatment of enterococcal infections). As shown in Table 4, the results indicate that neither compound 1 nor compound 3 would be suitable for oral use for the treatment of systemic enterococcal infections since neither is predicted to reach a concentration in plasma/blood sufficient to inhibit bacterial growth. The maximum plasma concentration (Cmax) predicted for compound 1 is 1.02 μg/mL, whereas the MIC50 ranges from 2 to 4 μg/mL. Similarly for compound 3, the Cmax is predicted to be 1.83 μg/mL, while the MIC50 ranges from 1 to 8 μg/mL. Intravenous administration of compounds 1 and 3 is predicted to result in slow rates of clearance (8.22 mL/min-kg and 8.71 mL/min-kg, respectively) and moderate half-lives (6.42 and 8.38 hours, respectively) which could alleviate the need for multiple daily dosing. The low values obtained for the volume of distribution at steady-state (2.10 L/kg for compound 1 and 2.55 L/kg for compound 3) are similar to the value obtained for linezolid (1.12 L/kg), indicating that 1 and 3 are not expected to distribute extensively into tissues. These pharmacokinetic simulations, combined with previous in vitro studies with human liver microsomes12, indicate compound 1 has a high affinity to hepatic microsomal enzymes. To address this issue, intravenous infusion of 1 and 3 may be considered in order to treat systemic enterococcal infections.
Table 4.
In silico pharmacokinetic analysis for compounds 1, 3, and linezolid (simulated at 600 mg).
| Oral | Intravenous | |||||
|---|---|---|---|---|---|---|
| 1 | 3 | Linezolid | 1 | 3 | Linezolid | |
| Cmax1 (μg/mL) | 1.02 | 0.82 | 5.33 | – | – | – |
| tmax2 (hours) | 1.67 | 1.83 | 2.75 | – | – | – |
| AUClast3 (μg -hour/mL) | 7.32 | 6.31 | 104.18 | 17.33 | 16.29 | 122.21 |
| Fraction absorbed (FAlast) | 0.86 | 0.84 | 0.92 | – | – | – |
| Bioavailability, F (%) | 42.1 | 38.48 | 79.77 | – | – | – |
| CL4 (mL/min-kg) | – | – | – | 8.22 | 8.71 | 1.09 |
| t1/25 (hours) | – | – | – | 6.42 | 8.38 | 12.31 |
| MRT6 (hours) | – | – | – | 4.25 | 4.87 | 17.04 |
| Vd7 (L/kg) | – | – | – | 4.57 | 6.32 | 1.11 |
| Vss8 (L/kg) | – | – | – | 2.10 | 2.55 | 1.12 |
| AUC (μg-hour/L) | – | – | – | 17.38 | 16.40 | 130.60 |
Cmax = maximum concentration of drug in plasma/blood
tmax = time required to reach Cmax
AUC = area under the curve
CL = rate of clearance
t1/2 = half-life
MRT = mean residence time
Vd = volume of distribution
Vss = volume of distribution at steady-state
DISCUSSION
The burden of resistance to currently available antibiotics necessitates the development of new antibacterial agents, targeting in particular the ESKAPE microorganisms, a significant threat given their ability to evade many antibiotics and the host immune response. One member of this group are the vancomycin-resistant Enterococci, commensal microorganisms of the human gastrointestinal tract. Their intrinsic resistance (or reduced susceptibility) towards multiple antibiotics (including penicillin-based antibiotics, cephalosporins, aminoglycosides, fluoroquinolones, and trimethoprim-sulfamethoxazole) limits the number of therapeutic agents available27. In addition, some species exhibit differing levels of sensitivity to specific antibiotics, which further complicates treatment options. For example, although E. faecium is typically susceptible to clindamycin and quinupristin-dalfopristin, some strains of E. faecalis are resistant to both agents28.
The present study identifies three phenylthiazole compounds that exhibit potent activity against both drug-resistant E. faecalis and E. faecium. When examined against a clinically-relevant panel of E. faecalis, the MIC50 of compounds 1–3 ranged from 4 to 8 μg/mL. Against a panel of E. faecium, the MIC50 of compounds 1–3 ranged from 1 to 8 μg/mL. The compounds maintain their activity against strains exhibiting resistance to numerous antibiotics including ampicillin, linezolid, and vancomycin, an important finding given the emergence of ampicillin-resistant strains of E. faecium. Although vancomycin has been frequently used to treat infections caused by these strains, more than 80% of ampicillin-resistant E. faecium in the United States now exhibit resistance to glycopeptide antibiotics like vancomycin and in addition, these strains exhibit high-level resistance to aminoglycoside antibiotics such as gentamicin and streptomycin28. Given that compounds 1–3 exhibit potent activity against both ampicillin-resistant and vancomycin-resistant enterococci, they represent potentially important leads for the treatment of drug-resistant infections caused by both E. faecium and E. faecalis.
As noted above, enterococci are intrinsically resistant to many antibiotics. However, enterococci also have the ability to acquire genetic material via horizontal gene transfer. Indeed, more than 25% of the genome of E. faecalis is composed of DNA acquired externally29. This ability to acquire exogenous genomic material has contributed in part to the rapid development of resistance to newer antibacterial agents, such as daptomycin and tigecycline28. We thus examined if we could isolate enterococcal mutants exhibiting resistance to the phenylthiazoles. Initially, a single-step resistance selection experiment using E. faecalis NR-39175 was conducted in order to isolate mutants exhibiting resistance to compounds 1–3. Even at an inoculum size > 1010 CFU/mL, no resistant mutants were isolated, which corresponds to a low frequency of mutation, > 2 × 10−11. Next, we serially passaged compounds 1–3 against two vancomycin-resistant strains (one E. faecalis and one E. faecium) over a 14-day period. Once again, no resistant mutants (categorized as a >four-fold increase in MIC) were isolated, indicating that rapid resistance to these thiazole agents is unlikely to occur.
To further understand the antibacterial activity of these phenylthiazoles, Bacterial Cytological Profiling was employed in order to narrow down the possibilities of the compounds’ mechanism of action. BCP suggests the mechanism of action of an antibiotic by comparing changes in cytological parameters for new compounds to those observed using a library of drugs with known mechanisms of action. Since BCP has not yet been developed for E. faecalis or E. faecium, we examined the effect of compound 1 in B. subtilis, as well as in an efflux pump-defective mutant of E. coli that is sensitive to many antibiotics that typically are ineffective against Gram-negative bacteria. In both organisms, we obtained results that suggested inhibition of cell wall synthesis was a likely target. In E. coli, cells lysed or formed spheroplasts as early as 30 minutes after exposure to compound 1. In B. subtilis, notable cell shape defects were observed, similar to the effects seen with vancomycin in osmotically buffered media. We then found an increased accumulation of the final soluble precursor of peptidoglycan synthesis in the bacterial cytoplasm (9, UDP-N-acetylmuramyl pentapeptide) both in the presence of compound 1, and vancomycin, again implicating inhibition of peptidoglycan (cell wall) biosynthesis.
We then used a transposon mutagenesis assay that suggested three possible targets: the isoprenoid biosynthesis protein undecaprenyl diphosphate phosphatase (UPPP, a.k.a. YubB) as well as two putative transporters, YubA and YubD. We found that 1 inhibited an expressed UPPP (YubB) at low μg/mL concentrations, in addition to inhibiting the previous enzyme in the pathway, UPPS (at 6.3 μg/mL). The roles of YubA and YubD are currently unknown, but based on bioinformatics and computational modeling they are both predicted to be transporters (with some template models being PMF-driven multi-drug efflux pumps). This suggested the possibility that 1 might be a protonophore uncoupler, which (using E. coli inverted membrane vesicles) was found to be the case.
Taken together, these results suggested that the phenylthiazoles might resensitize VRE to the effects of vancomycin and the aminoglycoside antibiotic gentamicin since in earlier work we found that compounds 1 and 2 were capable of resensitizing vancomycin-resistant S. aureus to the effects of vancomycin. When similar experiments were conducted with compound 1 against VRE, only one strain of vancomycin-resistant E. faecium (ATCC 700221) was resensitized, with a large improvement in the MIC being observed in the presence of a sub-inhibitory concentration of 1. Sensitivity to vancomycin was not restored in the remaining VRE strains. Closer inspection of the susceptibility data of these strains to vancomycin suggested one reason for this behavior: the MIC of vancomycin against E. faecium ATCC 700221 was 256 μg/mL, but in the remaining strains, the MIC of vancomycin was ≥ 512 μg/mL or higher. Thus, strains of VRE that exhibit high-level resistance to vancomycin may not be amenable to resensitization, a similar effect having been observed with aminoglycoside antibiotics30.
As noted above, recent reports have demonstrated that antibacterial agents that inhibit peptidoglycan biosynthesis can lead to increased uptake of aminoglycoside antibiotics, by enterococci5, leading to increased sensitivity to the aminoglycosides. However, we did not observe this when we examined our compounds, at sub-inhibitory concentrations, with gentamicin. This is likely due to the fact that the isolates tested exhibited high level resistance to aminoglycosides (MIC > 128 mg/L), which has been shown to nullify the effectiveness of pairing a cell wall biosynthesis inhibitor with an aminoglycoside28. High level resistance to aminoglycosides occurs through enterococci acquiring mutations in the target (ribosome) or enzymes (such as aminoglycoside acetyltransferases, AACs) that transfer acetyl groups to the amino groups of aminoglycoside antibiotics, rendering them ineffective31. Thus, the ability of the phenylthiazole compounds to resensitize VRE to the effects of vancomycin (and potentially, aminoglycosides) may be strain-specific, and be limited to isolates exhibiting low-to-moderate resistance.
The final step in our study was to determine if the phenylthiazole compounds had antibacterial activity against VRE in an animal model of infection. We used C. elegans, a well-established in vivo model for early-stage drug discovery32–34. Both compounds 1 and 3 (at 20 μg/mL) proved superior to the bacteriostatic antibiotic, linezolid, in reducing the burden of VRE in infected worms. However, in silico pharmacokinetic analysis revealed that oral administration of these compounds (simulating a dose of 600 mg) would not achieve plasma concentrations sufficient to inhibit VRE growth, due to limited permeability across the gastrointestinal tract. One possible reason for the predicted limited oral bioavailability is that the aminoguanidine is partially charged at physiological pH and might be a substrate for the P-gp efflux system35. A second reason is that a Caco-2 bidirectional permeability analysis of compounds 1 and 3 revealed limited ability to cross from the apical to basolateral surface of a polarized cell monolayer. While these effects would limit the utility of these compounds for oral treatment of systemic enterococcal infections, they may open the door for intravenous use for treating systemic infections, or for use as decolonizing agents. That is, an alternative approach to quelling infection is to reduce or eliminate VRE from the gastrointestinal tract of colonized patients at high risk of infection4, 36.
For example, VRE colonization in liver transplant patients has been linked to an increased risk of infection and death37. However, decolonization of VRE is very difficult given that enterococci in the gastrointestinal tract can range from 1–10 million CFU/g of stool37. Bacteriostatic antibiotics such as linezolid are unable to significantly reduce this burden. Other antibacterial agents have been examined for use as decolonizing agents, including bacitracin and gentamicin, but many patients are unable to tolerate their side-effects37. The ability of the phenylthiazole compounds to rapidly eradicate a high inoculum of VRE within two hours (as shown with the time-kill assay) suggests that they warrant further investigation as decolonizing agents. Specifically, their inability to cross the GI tract and reach the bloodstream may prove beneficial. Ramoplanin, an orally administered lipoglycodepsipeptide antibiotic that inhibits cell wall synthesis was recently approved for the treatment of Clostridium difficile infection. As with our compounds, ramoplanin is not absorbed systemically, but when administered orally, was shown to suppress gastrointestinal colonization of VRE in up to 90% of patients in a phase II clinical trial, and a phase III clinical trial validated the efficacy of ramoplanin as a decolonizing agent to prevent bloodstream infections caused by VRE37.
An alternative application for the phenylthiazoles would of course be to examine their ability to treat skin infections caused by enterococci. Though not as frequent as infections caused by S. aureus, enterococci have been associated with 8% of all complicated skin and soft tissue infections, particularly in the lower extremities, and in polymicrobial infections38, 39. Moreover, in one study39, polymicrobial skin infections caused by S. aureus, enterococci, and other bacterial species were present in nearly one half of all affected patients. We previously demonstrated that compounds 1 and 3 were potent topical antibacterial agents in a murine model of MRSA skin infection with both compounds reducing the burden of MRSA in infected skin wounds by 96%10. Future studies with these phenylthiazole compounds will aim to address their potential application as decolonizing agents, as well as topical antibacterial agents for the treatment of enterococcal skin infections in appropriate animal models. Additionally, designing new analogues focused on improving the limitations of compounds 1–3 (namely improving permeability and metabolic stability) while maintaining their potent anti-VRE activity will be pursued. This approach was successful in discovering second-generation phenylthiazoles with an enhanced pharmacokinetic profile that were potent inhibitors of MRSA growth40. However, most of the second-generation phenylthiazoles were not active against VRE (MIC > 64 μg/mL) or were significantly less potent than the first-generation compounds. This indicates different structural optimization strategies have to be adopted to balance the compounds’ potent anti-VRE activity while enhancing their pharmacokinetic profile.
CONCLUSIONS
In this study, we demonstrate that three phenylthiazole compounds exhibit potent antimicrobial activity in vitro against clinically-relevant strains of vancomycin-resistant enterococci. These agents are rapidly bactericidal against both E. faecalis and E. faecium and are not toxic to mammalian tissues at 20 μg/mL, both in cell culture (HRT-18) and in an animal model (C. elegans). We were not able to isolate VRE mutants exhibiting resistance to these compounds. The phenylthiazoles exert their antibacterial effect at least in part by inhibiting isoprenoid biosynthesis, targeting undecaprenyl diphosphate phosphatase (UPPP) and undecaprenyl diphosphate synthase (UPPS). The results of transposon mutagenesis identified UPPP (YubB) as well as YubA and YubD as possible targets. The latter two are putative transporters and we find that in addition to inhibiting UPPP and UPPS, 1 collapses the PMF (in membrane vesicles), suggesting it may also block YubA and/or YubD function. The antimicrobial activity of 1 and 3 against VRE was confirmed in vivo in a C. elegans animal model. The compounds reduced the bacterial load in infected worms by ~90%. Collectively, these results provide valuable information to be used in the development of this class of compounds as antimicrobial and decolonizing agents for infections caused by vancomycin-resistant enterococci.
EXPERIMENTAL SECTION
Synthesis of Thiazole Compounds 1–3
Synthetic schemes, spectral data, and purity (>95%, determined by HPLC) of compounds 1–3 (Figure 1), in addition to all intermediates, have been reported elsewhere12, 14.
Bacterial Strains and Reagents Used
Clinical isolates of E. faecalis and E. faecium were obtained through BEI Resources (Table S1). Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter cloacae were obtained from the American Type Culture Collection (Manassas, VA, USA). Escherichia coli OP50, E. coli 1411 and E. coli ΔAcrAB were described before41, 42. The human colorectal cell line (HRT-18) was obtained from the American Type Culture Collection (Manassas, VA, USA). Ampicillin (IBI Scientific, Peosta, IA), chloramphenicol (Sigma-Aldrich, St. Louis, MO, USA), ciprofloxacin (Enzo Life Sciences, Farmingdale, NY, USA), doxycycline monohydrate (Alfa Aesar, Ward Hill, MO, USA), linezolid (Chem-Impex International Inc., Wood Dale, IL, USA), and vancomycin hydrochloride (Gold Biotechnology Inc., St. Louis, MO, USA) were from the vendors noted above. Compounds were then dissolved in dimethyl sulfoxide (DMSO) (for ampicillin, doxycycline, linezolid, and vancomycin), ethanol (for chloramphenicol), or 0.1 N HCl (for ciprofloxacin), in order to prepare stock solutions (10 mg/mL). Tryptic soy broth (TSB), tryptic soy agar (TSA), and brain heart infusion broth (BHI) were purchased from Becton, Dickinson and Company (Franklin Lakes, NJ, USA). Phosphate-buffered saline (PBS) (Sigma-Aldrich, St. Louis, MO, USA), RPMI-1640 medium (American Type Culture Collection, Manassas, VA, USA), fetal horse serum (American Type Culture Collection, Manassas, VA, USA), and 96-well plates (CellTreat Scientific Products, Shirley, MA, USA) were all purchased from the vendors listed above. Nematode growth medium (NGM) and M9 medium were prepared as described in the literature43.
Determination of Minimum Inhibitory Concentration (MIC) and Minimum Bactericidal Concentration (MBC)
The MICs of thiazole compounds 1–3 and control antibiotics (linezolid, vancomycin) were determined against all bacterial strains tested using a broth microdilution method, following the guidelines outlined by the Clinical and Laboratory Standards Institute, as described elsewhere14, 44. Strains of E. faecalis, A. baumannii, E. cloacae, E. coli and P. aeruginosa were cultured in TSB, while E. faecium strains were cultured in BHI. For Gram-negative bacterial strains, the MIC was also tested in the presence of a sub-inhibitory concentration (¼ × MIC) of colistin (to permeabilize the outer membrane). Plates containing test agents and bacteria were incubated at 37 °C for 18–22 hours prior to determining the MIC. The MIC was taken to be the lowest concentration of each test agent where bacterial growth could not be visualized.
The MBC for each test agent against E. faecalis and E. faecium was determined using methods described previously14, with the following modifications. Aliquots (5 μL) of E. faecalis were transferred to TSA plates while aliquots of E. faecium were transferred onto BHI agar plates. Plates were incubated at incubated at 37 °C for 18–22 hours before the MBC, the concentration where >99% reduction in bacterial cell count was observed, was determined.
Time-Kill Analysis of Compounds 1–3 and Linezolid against VRE
Vancomycin-resistant E. faecalis ATCC HM-201 and vancomycin-resistant E. faecium ATCC 700221 cells in late logarithmic growth phase were diluted to ~5 × 106 colony-forming units (CFU)/mL and (in triplicate) exposed to concentrations equivalent to 4 × MIC of 1–3 or linezolid in either TSB (for E. faecalis) or BHI (for E. faecium). 100 μL samples were collected after 0, 2, 4, 6, 8, 10, 12, and 24 hours of incubation at 37 °C, and subsequently serially diluted in PBS. Bacteria were then transferred to either TSA (for E. faecalis) or BHI agar plates (for E. faecium) and were incubated at 37 °C for 18–22 hours before viable CFU/mL values were determined.
Cytotoxicity Analysis of Phenylthiazole Compounds in Cell Culture
Compounds 1–3 were assayed at concentrations of 5 μg/mL, 10 μg/mL, 20 μg/mL, and 40 μg/mL against a human colorectal (HRT-18) cell line to determine their effects to mammalian cells in vitro, as described elsewhere45. Cells were cultured in RPMI-1640 medium with 10% fetal horse serum at 37 °C with 5% CO2. Cells were incubated with compounds in 96-well plates at 37 °C and 5% CO2 for either 2 or 24 hours prior to addition of the assay reagent MTS 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (Promega, Madison, WI, USA). Absorbance readings (at OD490) were taken using a kinetic microplate reader (Molecular Devices, Sunnyvale, CA, USA). The quantity of viable cells after treatment with each compound is expressed as a percentage of the viability of untreated cells.
Single-Step Resistance Selection
The frequency of spontaneous single-step resistance of E. faecalis ATCC 31975 to the phenylthiazole compounds was carried out as described elsewhere46, 47. Briefly, bacterial cultures (ranging from 5 × 108 CFU/mL to 5 × 1010 CFU/mL) were spread onto TSA plates (10-mm diameter) containing compound 1, 2, or 3 at either 2 ×, 4 ×, or 8 × MIC. Plates were incubated aerobically at 37°C for 48 hours. The MIC of colonies present on each plate was checked (to determine any shift relative to the wild-type strain) using the broth microdilution method outlined above. The frequency of resistance was calculated as the number of resistant colonies per inoculum48.
Multi-Step Resistance Selection of VRE to Phenylthiazole Compounds
To assess the ability of VRE to develop resistance to the phenylthiazole compounds after repeated exposure, a multi-step resistance selection experiment was performed, as described elsewhere49. The broth microdilution method for MIC determination against a clinical isolate of vancomycin-resistant E. faecalis (ATCC 51299) and an isolate of vancomycin-resistant E. faecium (ATCC HM-968) was repeated for 14 passages over a period of two weeks. The initial inoculum was prepared to a McFarland standard of 0.5. The solution was subsequently diluted 1:300 in either TSB (for E. faecalis) or BHI (for E. faecium) to reach a starting inoculum of 1 × 105 CFU/mL. For each subsequent passage, the inoculum for the MIC determination was adjusted to a final density of approximately 5 × 105 CFU/mL using the contents of a well containing a sub-inhibitory concentration of the compound (where bacterial growth was observed from the previous passage). Bacteria were then transferred to a new 96-well microtiter plate. Compounds 1–3 and linezolid were added (in triplicate) to wells in the first row of the microtiter plate, and then serially diluted along the vertical axis. The plate was incubated at 37 °C for a minimum of 18 hours before the MIC was determined, by visual inspection. Resistance was classified as a greater than four-fold increase in the initial MIC, as reported elsewhere49.
Bacterial Cytological Profiling of Phenylthiazole Compounds against Bacillus subtilis and E. coli
Cells were grown in Luria Bertani (LB) medium at 30 °C (E. coli) or 37 °C (B. subtilis) until the optical density at 600 nm (OD600) was ~0.2. Cells were then left untreated, treated with compounds, or treated with compounds in the presence of 0.5 M sucrose (E. coli) or MSM (B. subtilis), as described previously16–18. After 30 minutes or two hours, cells were stained with FM 4–64 (1 μg/mL) to visualize the membranes; DAPI (2 μg/mL E. coli, 1 μg/ml B. subtilis) to visualize the DNA, and SYTOX Green (1 μg/mL), a vital stain which is normally excluded from cells with an intact membrane but brightly stains cells that are lysed18. Images were collected using a Delta Vision Spectris Deconvolution microscope, as described previously18.
Inhibition of Cell Wall Synthesis in Enterococci by Compound 1 via UDP-N-acetylmuramyl-pentapeptide Accumulation
To investigate whether the phenylthiazole compounds exhibit their effect on enterococci by inhibiting cell wall synthesis, we determined the accumulation of the final soluble cell wall precursor (UDP-N-acetylmuramyl-pentapeptide) inside bacterial cells. We used the procedure described previously50, with the following modifications: E. faecalis NR-31975, in early logarithmic growth stage (OD600 ~ 0.5), was incubated with 130 μg/mL chloramphenicol for 15 minutes at 37 °C. Bacteria were subsequently incubated with either 10 × MIC of compound 1 or vancomycin (positive control) for 30 minutes at 37 °C. Untreated samples served as a negative control. Samples were then centrifuged at 12,000 × g, the supernatant discarded, and the pellet re-suspended in 1 mL of sterile deionized water. The cell pellet was boiled at 100 °C for 30 minutes before samples were chilled on ice for 10 minutes. UDP-N-acetylmuramyl-pentapeptide levels were measured using an Agilent High Performance Liquid Chromatograph coupled to a time-of-flight mass spectrometer (HPLC-MS). A Waters XBridge Phenyl (2.1 × 100 mm, 3.5 um) column was used, with mobile phases of water, 0.1% formic acid (Buffer A) and acetonitrile, 0.1% formic acid (Buffer B). A gradient of 5–20% Buffer B over 14 minutes was then employed at a flow rate of 0.3 mL/min, with an electrospray source in positive ionization mode. Extracted ion chromatograms (EICs) were generated at a m/z of 1150.3588 (20 ppm window). The mass error for UDP-N-acetylmuramyl-pentapeptide was less than 1 ppm.
Molecular Target Identification Using Genomic Insertion of a Transposon with a Strong Outward-Oriented Promoter
Overexpression of the target/resistance mechanism was carried out using a transposon with a strong outward-oriented promoter for the random overexpression of neighboring genes, in Bacillus subtilis. The pEP26 delivery vector carrying the transposon with the promoter (TnHyJump) was transformed into B. subtilis, as described elsewhere19. For transposon integration into bacterial DNA, cells were grown for 10 hours at 25 °C, serially diluted, sub-cultured in dual-selection TSA plates containing 5 μg/ml chloramphenicol (for transposon selection) and 3 × MIC compound 1 (for selection of compound resistance), then incubated overnight at 42 °C. Growth at 42 °C is non-permissive for the maintenance of the delivery vector, so chloramphenicol/compound 1 resistance arises mainly from the chromosomal insertion of the transposon. 12 colonies out of 142 colonies on 3 × MIC compound 1 were screened for MIC shift (resistance) against compound 1 using the broth microdilution method to confirm resistance. Genomic DNA was extracted from resistant colonies (recombinants that were capable of growth at concentrations that were inhibitory to the control) and were sent to the Purdue Genomics Core Facility for sequencing. Insertion sites were identified by sequencing. Transposon location within the resistant B. subtilis genome, orientation, and flanking genes were determined by performing a BLASTN search on the NCBI public BLAST server.
HsFPPS, EcUPPS and EcUPPP Inhibition Assays
Human FPPS, E. coli UPPS and UPPP (denoted EcUPPS and EcUPPP) were expressed, purified, and assayed, as described previously20, 51, 52. Briefly, compound 1 was prepared as a 20 mM stock solution in DMSO and then serially diluted from 200 μM to 0.2 μM. EcUPPS (0.8 mg) was incubated with compound 1 at room temperature for 30 minutes in HEPES buffer (100 mM HEPES, 50 mM NaCl, 0.5 mM MgCl2, 0.02% DDM (w/v), pH 7.5) before adding the reaction mixture with 10 μM IPP and FPP, 0.375 U/mL inorganic phosphatase. The 100 μL reaction was quenched by the same volume of malachite green mixture from a malachite green phosphate assay kit (BioAssay Systems). For the EcUPPP inhibition assay, the 20 mM stock solution of compound 1 was serially diluted from 300 μM to 0.8 μM. Compound 1 was incubated with 0.125 μM EcUPPP at room temperature for 15 minutes in HEPES buffer (50 mM HEPES, 150 mM NaCl, 10 mM MgCl2, 0.02% DDM (w/v), pH 7.5), followed by the addition of 35 μM FPP and incubation at 37 °C for 20 minutes. The 100 μL reaction was then mixed with the same volume of the malachite green mixture. The released phosphate in EcUPPS and EcUPPP assays was monitored by absorbance at 620 nm after 30 minutes development. Dose response curves were constructed using GraphPad Prism (Graphpad Software, San Diego, CA).
Uncoupler Assays
Proton translocation out of EcIMVs was measured by the fluorescence increase of ACMA. The excitation and emission wavelengths were 410 and 480 nm, respectively. IMVs (0.1mg/mL membrane protein), 2 μM ACMA and 0.5 mM ATP/succinate were added in HEPES buffer (10 mM HEPES-KOH, 5 mM MgSO4, 100 mM KCl, pH 7.5). The uncoupler CCCP and compound 1 were serially diluted in the reaction mixture. Dose response curves were constructed using GraphPad PRISM (Graphpad Software, San Diego, CA).
Re-sensitization of VRE to Vancomycin and Aminoglycoside Antibiotics
TSB (for E. faecalis ATCC HM-201 and E. faecalis ATCC 51299) or BHI (for E. faecium ATCC 700221 and E. faecium HM-968) were inoculated with VRE (5×105 CFU/mL), as described previously13, with the following modifications. Aliquots (5 mL) of the bacterial suspensions were divided into micro-centrifuge tubes and compounds 1, 2, or 3 (at ½ × MIC) were introduced into each tube. After sitting at room temperature for 30 minutes, 1 mL samples from each tube were transferred to a new micro-centrifuge tube, prior to addition of the antibiotic (either vancomycin or gentamicin at concentrations equivalent to their MIC). Plates containing the test agents and bacteria were then incubated at 37 °C for 18–21 hours after which the MIC values were measured. A fold-reduction was calculated by comparing the MIC of the antibiotic alone compared to the MIC of the antibiotic given in combination with the thiazole compound.
Combination Therapy of Thiazole Compounds With Conventional Antibiotics
Possible synergistic interactions between the thiazole compounds and ampicillin, ciprofloxacin, doxycycline, and linezolid were determined via checkerboard assay53. Initially, compound 1 was examined in combination with the four antibiotics against a single strain of E. faecalis (ATCC 51299) prior to investigating compounds 1–3 in combination with ciprofloxacin against E. faecalis ATCC 49532 and E. faecalis ATCC 49533. Bacteria equivalent to a McFarland standard of 0.5 were prepared in PBS. The bacteria were then diluted in TSB to achieve a starting cell density of 1 × 105 CFU/mL. TSB was transferred to all wells of a 96-well micro-titer plate. The phenylthiazole compounds and antibiotics were diluted in TSB to achieve a starting concentration equivalent to 2 × or 4 × the MIC. Compounds were serially diluted along the horizontal axis of the microtiter plate while the antibiotics were serially diluted along the vertical axis. Plates were incubated for 20 hours at 37 °C. The MIC of the test compound in combination with each antibiotic studied was taken to be the lowest concentration of each compound/antibiotic where no visible growth of bacteria was observed. The fractional inhibitory concentration index (ΣFIC) was calculated for each combination, as described previously11. A synergistic relationship was classified as an FIC index less than or equal to 0.5. FIC values above 0.5 but less than 2.0 were characterized as additive, values between 2.0 and 4.0 characterized as indifference, while FIC values above 4.0 were classified as antagonistic.
In Vivo Analysis of Toxicity and Efficacy of Thiazole Compounds
To examine the toxicity of the thiazole compounds in vivo and to examine their efficacy in treating a VRE infection in vivo, we used the C. elegans animal model. The temperature-sensitive sterile mutant strain C. elegans AU37 [sek-1(km4); glp-4(bn2) I] was used because this strain is sterile at room temperature and capable of laying eggs only at 15 °C. Additionally, the strain is quite susceptible to infection, due to a mutation in the sek-1 gene of the p38 mitogen-activated protein kinase pathway34, 54. Briefly, worms were grown for five days at 15 °C (permitting worms to lay eggs) on NGM agar plates seeded with a lawn of E. coli OP50. Eggs were harvested by bleaching55 and maintained for 24 hours at room temperature with gentle agitation, for hatching. Hatched larvae were transferred to a new NGM plate seeded with E. coli OP50 and were kept at room temperature for 4–5 days until the worms reached their adult growth stage. Adult worms were then collected and washed three times with PBS in a 1:10 (worm:PBS) ratio to remove E. coli.
In order to examine the toxicity of the thiazole compounds to C. elegans, 15–20 adult worms were transferred to wells of a 96-well microtiter plate. Worms were incubated with either 10 or 20 μg/mL of compounds 1, 3, linezolid (positive control), or sterile water (negative control) (in triplicate). After 24, 48, 72, 96, and 120 hours, worms were examined microscopically to examine viability. The number of worms that survived each treatment regimen were counted and results are presented as percent viable worms (after 24 hour exposure to each test agent).
To test the antibacterial activity of the thiazole compounds against VRE in vivo, adult worms were transferred to TSA agar plates seeded with a lawn of E. faecalis HM-201, for infection42. After two hours of infection, worms were collected and washed with M9 buffer, three times, before transferring 20–25 worms to wells in a 96-well microtiter plate. Worms were incubated with 20 μg/mL of compounds 1, 3, linezolid (positive control), or sterile water (negative control) (in triplicate). After treatment for 20 hours, worms were washed three times with M9 buffer and then examined microscopically to examine morphological changes, and viability. They were subsequently lysed in micro-centrifuge tubes containing 200 mg of 1.0-mm silicon carbide particles (Biospec Products, Bartlesville, OK) that were vortexed for one minute. Samples were serially diluted and plated onto TSA plates containing 50 μg/mL gentamicin to select for VRE. Plates were incubated at 37 °C for 18 hours before viable CFUs were determined.
In Silico Pharmacokinetic Analysis
The pharmacokinetic profile (oral and intravenous dose of 600 mg) was simulated using chemPK version 2.0 (Cyprotex Inc., Cheshire, United Kingdom), for compounds 1, 3, and linezolid.
Supplementary Material
Acknowledgments
The authors would like to thank BEI Resources, NIAID, and NIH for the clinical isolates of enterococci. We would also like to thank Dr. Daniel Kearns for sharing the Bacillus subtilis DS8137 strain containing the pEP26 plasmid used for the target overexpression experiment; Professor Rey-Ting Guo for providing EcUPPP, and Xinxin Feng for help with the uncoupler experiments.
This work was supported by US-Egypt Joint Grant, Grant ID# USC17:241 from the US National Academy of Sciences (NAS) and the Science & Technology Development Funds (STDF), Egypt, and in part by the United States Public Health Service (NIH grants CA158191 and GM06537).
ABBREVIATIONS
- BCP
Bacterial Cytological Profiling
- EcIMV
E. coli inverted membrane vesicles
- FPPS
farnesyl diphosphate synthase
- UPPS
undecaprenyl diphosphate synthase
- UPPP
undecaprenyl diphosphate phosphatase
Footnotes
SUPPORTING INFORMATION
Supporting information including Supplemental Figures and Tables are provided free of charge on the ACS publications website.
References
- 1.Centers for Disease Control and Prevention. Preventing healthcare-associated infections. 2016 Dec 5; http://www.cdc.gov/washington/~cdcatWork/pdf/infections.pdf.
- 2.World Health Organization. Antimicrobial resistance global report on surveillance 2014. WHO Press; France: 2014. pp. 1–232. [Google Scholar]
- 3.Centers for Disease Control and Prevention. Antibiotic resistance threats in the United States, 2013. Atlanta: CDC; 2013. pp. 1–114. [Google Scholar]
- 4.Patel R. Clinical impact of vancomycin-resistant enterococci. J Antimicrob Chemother. 2003;51(Suppl 3):iii13–21. doi: 10.1093/jac/dkg272. [DOI] [PubMed] [Google Scholar]
- 5.Arias CA, Contreras GA, Murray B. E. Management of multidrug-resistant enterococcal infections. Clin Microbiol Infect. 2010;16:555–562. doi: 10.1111/j.1469-0691.2010.03214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kelesidis T, Humphries R, Uslan DZ, Pegues DA. Daptomycin nonsusceptible enterococci: an emerging challenge for clinicians. Clin Infect Dis. 2011;52:228–234. doi: 10.1093/cid/ciq113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonora MG, Solbiati M, Stepan E, Zorzi A, Luzzani A, Catania MR, Fontana R. Emergence of linezolid resistance in the vancomycin-resistant Enterococcus faecium multilocus sequence typing C1 epidemic lineage. J Clin Microbiol. 2006;44:1153–1155. doi: 10.1128/JCM.44.3.1153-1155.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hede K. Antibiotic resistance: An infectious arms race. Nature. 2014;509:S2–3. doi: 10.1038/509S2a. [DOI] [PubMed] [Google Scholar]
- 9.Thangamani S, Mohammad H, Younis W, Seleem MN. Drug repurposing for the treatment of staphylococcal infections. Curr Pharm Des. 2015;21:2089–2100. doi: 10.2174/1381612821666150310104416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mohammad H, Cushman M, Seleem MN. Antibacterial evaluation of synthetic thiazole compounds in vitro and in vivo in a methicillin-resistant Staphylococcus aureus (MRSA) skin infection mouse model. PLoS One. 2015;10:e0142321. doi: 10.1371/journal.pone.0142321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mohammad H, Mayhoub AS, Cushman M, Seleem MN. Anti-biofilm activity and synergism of novel thiazole compounds with glycopeptide antibiotics against multidrug-resistant staphylococci. J Antibiot (Tokyo) 2015;68:259–266. doi: 10.1038/ja.2014.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohammad H, Mayhoub AS, Ghafoor A, Soofi M, Alajlouni RA, Cushman M, Seleem MN. Discovery and characterization of potent thiazoles versus methicillin- and vancomycin-resistant Staphylococcus aureus. J Med Chem. 2014;57:1609–1615. doi: 10.1021/jm401905m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mohammad H, Reddy PV, Monteleone D, Mayhoub AS, Cushman M, Hammac GK, Seleem MN. Antibacterial characterization of novel synthetic thiazole compounds against methicillin-resistant Staphylococcus pseudintermedius. PLoS One. 2015;10:e0130385. doi: 10.1371/journal.pone.0130385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mohammad H, Reddy PV, Monteleone D, Mayhoub AS, Cushman M, Seleem MN. Synthesis and antibacterial evaluation of a novel series of synthetic phenylthiazole compounds against methicillin-resistant Staphylococcus aureus (MRSA) Eur J Med Chem. 2015;94:306–316. doi: 10.1016/j.ejmech.2015.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bohnert JA, Kern WV. Selected arylpiperazines are capable of reversing multidrug resistance in Escherichia coli overexpressing RND efflux pumps. Antimicrob Agents Chemother. 2005;49:849–852. doi: 10.1128/AAC.49.2.849-852.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lamsa A, Liu WT, Dorrestein PC, Pogliano K. The Bacillus subtilis cannibalism toxin SDP collapses the proton motive force and induces autolysis. Mol Microbiol. 2012;84:486–500. doi: 10.1111/j.1365-2958.2012.08038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lamsa A, Lopez-Garrido J, Quach D, Riley EP, Pogliano J, Pogliano K. Rapid inhibition profiling in Bacillus subtilis to identify the mechanism of action of new antimicrobials. ACS Chem Biol. 2016;11:2222–2231. doi: 10.1021/acschembio.5b01050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nonejuie P, Burkart M, Pogliano K, Pogliano J. Bacterial cytological profiling rapidly identifies the cellular pathways targeted by antibacterial molecules. Proc Natl Acad Sci U S A. 2013;110:16169–16174. doi: 10.1073/pnas.1311066110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pozsgai ER, Blair KM, Kearns DB. Modified mariner transposons for random inducible-expression insertions and transcriptional reporter fusion insertions in Bacillus subtilis. Appl Environ Microbiol. 2012;78:778–785. doi: 10.1128/AEM.07098-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang HY, Chou CC, Hsu MF, Wang AH. Proposed carrier lipid-binding site of undecaprenyl pyrophosphate phosphatase from Escherichia coli. J Biol Chem. 2014;289:18719–18735. doi: 10.1074/jbc.M114.575076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Desai J, Zhang Y, Malwal SR, Shin CJ, Feng X, Sun H, Liu G, Guo RT, Oldfield E. Bacterial cell growth inhibitors targeting undecaprenyl diphosphate synthase and undecaprenyl diphosphate phosphatase. ChemMedChem. 2016;11:2311–2319. doi: 10.1002/cmdc.201600342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feng X, Zhu W, Schurig-Briccio LA, Lindert S, Shoen C, Hitchings R, Li J, Wang Y, Baig N, Zhou T, Kim BK, Crick DC, Cynamon M, McCammon JA, Gennis RB, Oldfield E. Antiinfectives targeting enzymes and the proton motive force. Proc Natl Acad Sci U S A. 2015;112:E7073–82. doi: 10.1073/pnas.1521988112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li W, Upadhyay A, Fontes FL, North EJ, Wang YH, Crans DC, Grzegorzewicz AE, Jones V, Franzblau SG, Lee RE, Crick DC, Jackson M. Novel insights into the mechanism of inhibition of MmpL3, a target of multiple pharmacophores in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2014;58:6413–6423. doi: 10.1128/AAC.03229-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li K, Schurig-Briccio LA, Feng X, Upadhyay A, Pujari V, Lechartier B, Fontes FL, Yang H, Rao G, Zhu W, Gulati A, No JH, Cintra G, Bogue S, Liu YL, Molohon K, Orlean P, Mitchell DA, Freitas-Junior L, Ren F, Sun H, Jiang T, Li Y, Guo RT, Cole ST, Gennis RB, Crick DC, Oldfield E. Multitarget drug discovery for tuberculosis and other infectious diseases. J Med Chem. 2014;57:3126–3139. doi: 10.1021/jm500131s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kallberg M, Wang HP, Wang S, Peng J, Wang ZY, Lu H, Xu JB. Template-based protein structure modeling using the RaptorX web server. Nat Protoc. 2012;7:1511–1522. doi: 10.1038/nprot.2012.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo RT, Ko TP, Chen APC, Kuo CJ, Wang AHJ, Liang PH. Crystal structures of undecaprenyl pyrophosphate synthase in complex with magnesium, isopentenyl pyrophosphate, and farnesyl thiopyrophosphate - roles of the metal ion and conserved residues in catalysis. J Biol Chem. 2005;280:20762–20774. doi: 10.1074/jbc.M502121200. [DOI] [PubMed] [Google Scholar]
- 27.Hollenbeck BL, Rice LB. Intrinsic and acquired resistance mechanisms in enterococcus. Virulence. 2012;3:421–433. doi: 10.4161/viru.21282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kristich CJ, Rice LB, Arias CA. Enterococcal infection-treatment and antibiotic resistance. In: Gilmore MS, Clewell DB, Ike Y, Shankar N, editors. Enterococci: From Commensals to Leading Causes of Drug Resistant Infection. Massachusetts Eye and Ear Infirmary; Boston: 2014. pp. 123–185. [PubMed] [Google Scholar]
- 29.Manson JM, Hancock LE, Gilmore MS. Mechanism of chromosomal transfer of Enterococcus faecalis pathogenicity island, capsule, antimicrobial resistance, and other traits. Proc Natl Acad Sci U S A. 2010;107:12269–12274. doi: 10.1073/pnas.1000139107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lorian V. Antibiotics in Laboratory Medicine. 4th. Williams & Wilkins; Baltimore: 1996. [Google Scholar]
- 31.Arias CA, Murray BE. Emergence and management of drug-resistant enterococcal infections. Expert Rev Anti-Infect Ther. 2008;6:637–655. doi: 10.1586/14787210.6.5.637. [DOI] [PubMed] [Google Scholar]
- 32.Squiban B, Kurz CL. C. elegans: An all in one model for antimicrobial drug discovery. Curr Drug Targets. 2011;12:967–977. doi: 10.2174/138945011795677854. [DOI] [PubMed] [Google Scholar]
- 33.Ewbank JJ, Zugasti O. C. elegans: model host and tool for antimicrobial drug discovery. Dis Models & Mech. 2011;4:300–304. doi: 10.1242/dmm.006684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moy TI, Ball AR, Anklesaria Z, Casadei G, Lewis K, Ausubel FM. Identification of novel antimicrobials using a live-animal infection model. Proc Natl Acad Sci U S A. 2006;103:10414–10419. doi: 10.1073/pnas.0604055103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seelig A. A general pattern for substrate recognition by P-glycoprotein. Eur J Biochem. 1998;251:252–261. doi: 10.1046/j.1432-1327.1998.2510252.x. [DOI] [PubMed] [Google Scholar]
- 36.Tran TT, Palmer HR, Weimar MR, Arias CA, Cook GM, Murray BE. Oral bacitracin: A consideration for suppression of intestinal vancomycin-resistant enterococci (VRE) and for VRE bacteremia from an apparent gastrointestinal tract source. Clin Infect Dis. 2015;60:1726–1728. doi: 10.1093/cid/civ130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng VC, Chen JH, Tai JW, Wong SC, Poon RW, Hung IF, To KK, Chan JF, Ho PL, Lo CM, Yuen KY. Decolonization of gastrointestinal carriage of vancomycin-resistant Enterococcus faecium: case series and review of literature. BMC Infect Dis. 2014;14:514. doi: 10.1186/1471-2334-14-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.File TM, Jr, Tan JS. Treatment of bacterial skin and soft tissue infections. Surg Gynecol Obstet. 1991;172(Suppl):17–24. [PubMed] [Google Scholar]
- 39.Giordano P, Weber K, Gesin G, Kubert J. Skin and skin structure infections: treatment with newer generation fluoroquinolones. Ther Clin Risk Manage. 2007;3:309–317. doi: 10.2147/tcrm.2007.3.2.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seleem MA, Disouky AM, Mohammad H, Abdelghany TM, Mancy AM, Bayoumi SA, Elshafeey A, El-Morsy A, Seleem MN, Mayhoub AS. Second-generation phenylthiazole antibiotics with enhanced pharmacokinetic properties. J Med Chem. 2016;59:4900–4912. doi: 10.1021/acs.jmedchem.6b00233. [DOI] [PubMed] [Google Scholar]
- 41.Abushahba MF, Mohammad H, Seleem MN. Targeting Multidrug-resistant staphylococci with an anti-rpoA peptide nucleic acid conjugated to the HIV-1 TAT cell penetrating peptide. Mol Ther Nucleic Acids. 2016;5:e339. doi: 10.1038/mtna.2016.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thangamani S, Mohammad H, Abushahba MF, Sobreira TJ, Hedrick VE, Paul LN, Seleem MN. Antibacterial activity and mechanism of action of auranofin against multi-drug resistant bacterial pathogens. Sci Rep. 2016;6:22571. doi: 10.1038/srep22571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stiernagle T. Maintenance of C. elegans. WormBook [Online] 2006:1–11. doi: 10.1895/wormbook.1.101.1. http://www.wormbook.org/ (accessed June 9, 2015). [DOI] [PMC free article] [PubMed]
- 44.Clinical and Laboratory Standards Institute, CLSI. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically—Ninth Edition: Approved Standard M07-A9. Wayne, PA: 2012. [Google Scholar]
- 45.Allen CN, Harpur ES, Gray TJB, Hirst BH. Toxic effects of nonsteroidal antiinflammatory drugs in a human intestinal epithelial-cell line (Hct-8), as assessed by the MTT and neutral red assays. Toxicol In Vitro. 1991;5:183–191. doi: 10.1016/0887-2333(91)90016-7. [DOI] [PubMed] [Google Scholar]
- 46.Baldoni D, Haschke M, Rajacic Z, Zimmerli W, Trampuz A. Linezolid alone or combined with rifampin against methicillin-resistant Staphylococcus aureus in experimental foreign-body infection. Antimicrob Agents Chemother. 2009;53:1142–1148. doi: 10.1128/AAC.00775-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nagai K, Davies TA, Pankuch GA, Dewasse BE, Jacobs MR, Appelbaum PC. In vitro selection of resistance to clinafloxacin, ciprofloxacin, and trovafloxacin in Streptococcus pneumoniae. Antimicrob Agents Chemother. 2000;44:2740–2746. doi: 10.1128/aac.44.10.2740-2746.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singh MP, Petersen PJ, Weiss WJ, Janso JE, Luckman SW, Lenoy EB, Bradford PA, Testa RT, Greenstein M. Mannopeptimycins, new cyclic glycopeptide antibiotics produced by Streptomyces hygroscopicus LL-AC98: antibacterial and mechanistic activities. Antimicrob Agents Chemother. 2003;47:62–69. doi: 10.1128/AAC.47.1.62-69.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Farrell DJ, Robbins M, Rhys-Williams W, Love WG. Investigation of the potential for mutational resistance to XF-73, retapamulin, mupirocin, fusidic acid, daptomycin, and vancomycin in methicillin-resistant Staphylococcus aureus isolates during a 55-passage study. Antimicrob Agents Chemother. 2011;55:1177–1181. doi: 10.1128/AAC.01285-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmitt P, Wilmes M, Pugniere M, Aumelas A, Bachere E, Sahl HG, Schneider T, Destoumieux-Garzon D. Insight into invertebrate defensin mechanism of action: oyster defensins inhibit peptidoglycan biosynthesis by binding to lipid II. J Biol Chem. 2010;285:29208–29216. doi: 10.1074/jbc.M110.143388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baykov AA, Evtushenko OA, Avaeva SM. A malachite green procedure for ortho-phosphate determination and its use in alkaline phosphatase-based enzyme-immunoassay. Anal Biochem. 1988;171:266–270. doi: 10.1016/0003-2697(88)90484-8. [DOI] [PubMed] [Google Scholar]
- 52.Durrant JD, Cao R, Gorfe AA, Zhu W, Li J, Sankovsky A, Oldfield E, McCammon JA. Non-bisphosphonate inhibitors of isoprenoid biosynthesis identified via computer-aided drug design. Chem Biol Drug Des. 2011;78:323–332. doi: 10.1111/j.1747-0285.2011.01164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Orhan G, Bayram A, Zer Y, Balci I. Synergy tests by E test and checkerboard methods of antimicrobial combinations against Brucella melitensis. J Clin Microbiol. 2005;43:140–143. doi: 10.1128/JCM.43.1.140-143.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jakobsen H, Bojer MS, Marinus MG, Xu T, Struve C, Krogfelt KA, Lobner-Olesen A. The alkaloid compound harmane increases the lifespan of Caenorhabditis elegans during bacterial infection, by modulating the nematode’s innate immune response. PLoS One. 2013;8:e60519. doi: 10.1371/journal.pone.0060519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Powell JR, Ausubel FM. Models of Caenorhabditis elegans infection by bacterial and fungal pathogens. Methods Mol Biol. 2008;415:403–427. doi: 10.1007/978-1-59745-570-1_24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.