

Summary
Objective
The DBA/1 mouse is a relevant animal model of sudden unexpected death in epilepsy (SUDEP), as it exhibits seizure-induced respiratory arrest (S-IRA) evoked by acoustic stimulation, followed by cardiac arrhythmia and death. Defects in serotonergic neurotransmission may contribute to S-IRA. The tryptophan hydroxylase-2 (TPH2) enzyme converts L-tryptophan to 5-hydroxytryptophan (5-HTP), a precursor for central nervous system (CNS) serotonin (5-HT) synthesis; and DBA/1 mice have a polymorphism that decreases TPH2 activity. We, therefore, hypothesized that supplementation with 5-HTP may bypass TPH2 and suppress S-IRA in DBA/1 mice.
Methods
TPH2 expression was examined by Western blot in the brainstem of DBA/1 and C57BL/6J mice both with and without acoustic stimulation. Changes in breathing and cardiac electrical activity in DBA/1 and C57BL/6J mice that incurred sudden death during generalized seizures evoked by pentylenetetrazole (PTZ) were studied by plethysmography and electrocardiography. The effect of 5-HTP administration on seizure-induced mortality evoked by acoustic stimulation or by PTZ was investigated in DBA/1 mice.
Results
Repetitive acoustic stimulation resulted in reduced TPH2 protein in the brainstem of DBA/1 mice as compared with C57BL/6J mice. S-IRA evoked by acoustic stimulation in DBA/1 mice was significantly reduced by 5-HTP. Following S-IRA, cardiac electrical activity could be detected for minutes before terminal asystole and death in both DBA/1 and C57BL/6J mice after PTZ treatment. The incidence of S-IRA by PTZ administration was greater in DBA/1 than in C57BL/6J mice, and administration of 5-HTP also significantly reduced S-IRA by PTZ in DBA/1 mice.
Significance
Our data suggest that S-IRA is the primary event leading to death incurred in most DBA/1 and some C57BL/6J mice during PTZ-evoked seizures. Suppression of S-IRA by 5-HTP suggests that 5-HT transmission contributes to the pathophysiology of S-IRA, and that 5-HTP, an over-the-counter supplement available for human consumption, may be clinically useful in preventing SUDEP.
Keywords: SUDEP, Generalizedseizures, Pentylenetetrazole, 5-HT, Therapeutics
Sudden unexpected death in epilepsy (SUDEP) is the major cause of death in patients with epilepsy, accounting for up to 17% of deaths in this population.1–4 SUDEP ranks second to stroke in public health burden when years of potential life lost from SUDEP are compared with those from other common neurologic diseases.5 Studies in humans and animal models suggest that respiratory dysfunction and cardiac arrhythmia may contribute to SUDEP,3,4,6–8 although several other pathophysiologic mechanisms are also proposed.9–13 It has been observed that most witnessed SUDEP patients display difficulties in breathing after generalized tonic–clonic seizures,14–18 and analysis of video electroencephalography (EEG) and electrocardiography (ECG) data found that most patients with SUDEP or near-SUDEP initially exhibit respiratory dysfunction, leading to apnea, followed by terminal asystole.19 This sequential pattern of terminal events is also observed in DBA/1 mice; these mice are subjected to seizure-induced respiratory arrest (S-IRA) after generalized audiogenic seizures (AGSz), with subsequent cardiac arrest and death, validating the DBA/1 mouse as a relevant model of SUDEP.3
Several selective serotonin (5-hydroxytryptamine or 5-HT) reuptake inhibitors (SSRIs), which elevate 5-HT levels in the synaptic cleft, prevent S-IRA in DBA/1 mice evoked by AGSz.20–22 An SSRI is also found effective in reducing S-IRA evoked by maximal electroshock in Lmx1b(f/f) mice on a primarily C57BL/6J background,23 a strain that is resistant to AGSz. These previous studies suggest that a deficiency of 5-HT neurotransmission may play an important role in the pathogenesis of S-IRA. However, in addition to 5-HT, SSRIs are known to affect other neurotransmission systems.3,24 These off-target effects of SSRIs may contribute to or mask their actions on S-IRA, as it has been shown that not all SSRIs are effective in preventing S-IRA in DBA/1 mice.25 Abnormalities of 5-HT neurotransmission have been observed in both DBA/1 and DBA/2 mice, which may contribute to their tendency for S-IRA.3 L-Tryptophan from dietary sources is converted to 5-hydroxytryptophan (5-HTP) by the enzyme tryptophan hydroxylase-2 (TPH2) in the brain, and 5-HTP is then converted to 5-HT by aromatic L-amino acid decarboxylase. TPH2 is the rate-limiting enzyme in 5-HT synthesis,26 and a polymorphism of TPH2 in DBA/1 mice results in reduced activity of this enzyme,26–28 which may contribute to S-IRA.
In the current study, we further address the role of 5-HT neurotransmission in the pathogenesis of S-IRA in DBA/1 mice. We first quantified TPH2 enzyme protein in the brainstem of both DBA/1 and S-IRA-resistant C57BL/6J mice, and studied the effect of priming on its expression. We then attempted to enhance central 5-HT synthesis in DBA/1 mice through acute and repeated peripheral supplementation with its chemical precursor, 5-HTP; and, finally, we studied the effect of 5-HTP administration on S-IRA in DBA/1 mice evoked by either acoustic stimulation or pentylenetetrazole (PTZ), a proconvulsant widely used to mimic human generalized tonic–clonic seizures.29
Materials and Methods
Animals
All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Massachusetts General Hospital. DBA/1 mice were purchased from Harlan Laboratories (Indianapolis, IN, U.S.A.), and C57BL/6J mice were purchased from Jackson Laboratory (Bar Harbor, ME). Both strains were housed and bred in the Massachusetts General Hospital Center for Comparative Medicine animal facility and provided with rodent food and water ad libitum. Unless otherwise mentioned, DBA/1 mice were “primed” starting from postnatal day 26–28 by subjecting to acoustic stimulation daily for 3–4 days to establish consistent susceptibility to S-IRA. Primed DBA/1 mice at approximately 1 month of age (10–15 g) were used in experiments with acoustic stimulation, and separate groups of DBA/1 (13–22 g) and C57BL/6J mice (17–25 g) at 2–3 months of age were used in PTZ experiments.
Induction of seizures and seizure-induced sudden death
Seizures and seizure-induced sudden death were evoked either by acoustic stimulation or by the proconvulsant PTZ, as described previously.22,30 To induce AGSz in DBA/1 mice, each mouse was placed in a cylindrical acrylic glass chamber in a sound-isolated room. AGSz were induced by acoustic stimulation using an electric bell (96 dB SPL) (UC4-150, Zhejiang People’s Electronics, China). The acoustic stimulus was given for a maximum duration of 60 s or until the mouse exhibited tonic seizures, which culminated in tonic hindlimb extension and S-IRA. Mice with S-IRA were resuscitated within 5 s after the final respiratory gasp using a rodent respirator (Harvard Apparatus 680, Holliston, MA, U.S.A.).22 The susceptibility to S-IRA in primed DBA/1 mice was always reconfirmed 24 h before drug or vehicle administration. In the acute treatment protocol, 5-HTP (100–150 mg/kg) or vehicle was administered intraperitoneally 1 h before induction of S-IRA. In the repeated treatment protocol, 5-HTP (50–100 mg/kg, i.p.) or vehicle was administered once a day for 2 days, and induction of S-IRA was performed 1 h after the second administration. The volume injected was 0.1–0.3 ml for each mouse. S-IRA and AGSz behaviors were videotaped for offline analysis. Recovery of S-IRA susceptibility in DBA/1 mice was tested 24 h after drug administration or at 24-h intervals thereafter until the S-IRA susceptibility returned.
Generalized clonic and/or tonic–clonic seizures were also induced in separate groups of DBA/1 and C57BL/6J mice by systemic administration of PTZ (75 mg/kg, i.p.). 5-HTP (100–200 mg/kg) or vehicle was administered once a day for 2 days, and i.p. PTZ was administered 1 h after the second treatment.
5-HTP (Cat # 107751) and PTZ (Cat # P6500) were purchased from Sigma-Aldrich (St. Louis, MO, U.S.A.) and dissolved in saline for systemic administration.
Expression of TPH2 in the brainstem
The expression of TPH2 was studied by Western blot in nonprimed DBA/1 mice, primed DBA/1 mice, C57BL/6J mice exposed to the acoustic stimulation, and C57BL/6J mice without exposure to the acoustic stimulation, as described previously.31,32 In primed DBA/1 mice, only those mice that exhibited S-IRA were used for Western blot analysis. The C57BL/6J mice exposed to the acoustic stimulation were subjected to the mean duration of acoustic stimulus that the primed DBA/1 mice received. Mice were decapitated under isoflurane anesthesia, and the brains were removed and stored at −80°C. The whole brainstems were isolated for TPH2 protein analysis by one rostral cut that corresponds to bregma −3 mm and one caudal cut that corresponds to bregma −9 mm.33 To ensure consistency in isolating the brainstem, we used experimental DBA/1 mice and control C57BL/6J mice at similar ages and isolated the brainstems of mice in the experimental and control groups at the same time. Each brainstem tissue was homogenized, and total protein was quantified using the bicinchoninic acid (BCA) protein assay kit (Pierce Technology, Iselin, NJ, U.S.A.). Sixty micrograms of total protein from each animal was resolved by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Following SDS-PAGE, the protein was transferred to nitrocellulose, which was then blocked with blocking buffer (Thermo Fisher Scientific, Waltham, MA, U.S.A.) and probed with antibodies. The primary antibody for TPH2 (T0678, Sigma-Aldrich) was used at a dilution of 1:1,000 to detect TPH2 (55 kDa), and the primary antibody for β-actin (1:10,000, Sigma-Aldrich) was used to detect β-actin (42 kDa). The secondary antibody (A21424, Life Technologies, Grand Island, NY, U.S.A.) was diluted at 1:5,000. Bands were visualized by enhanced chemiluminescence (GE Healthcare Bio-Science, Pittsburgh, PA, U.S.A.), and optical density was quantified using ImageJ Software. The optical density of TPH2 band was normalized to that of β-actin protein in the same tissue sample. The resulting relative optical density in C57BL/6J mice exposed to the acoustic stimulation, primed DBA/1 mice, and C57BL/6J mice without exposure to the acoustic stimulation and nonprimed DBA/1 mice was normalized to that of C57BL/6J mice exposed to the acoustic stimulation.
Plethysmography and ECG recordings
The terminal responses of respiratory and cardiac systems during seizure-induced sudden death evoked by PTZ were examined using whole-body plethysmography and ECG recordings in DBA/1 and C57BL/6J mice. Both primed and nonprimed DBA/1 mice were used in this experiment to determine if priming affects the incidence of seizure-induced sudden death evoked by PTZ. After injected with PTZ (75 mg/kg, i.p.), a mouse of either strain was placed in a clear, custom-built whole-body plethysmography chamber constructed from polyvinyl chloride pipe with a volume of ~20 cubic inches.3,22 The closed chamber was continuously flushed with room air (0.3–1 liter per minute) using mass flow controllers (Model GE50A; MKS Instruments Inc, Andover, MA, U.S.A.). CO2 levels were monitored by analyzing gas exiting the chamber with a Capnomac Ultima medical gas analyzer (GE Healthcare, Buckinghamshire, United Kingdom). Changes in air pressure in the chamber were monitored using a pressure transducer and demodulator (Models CD15 and MP45-14-871; Validyne Engineering, Northridge, CA, U.S.A.). If a mouse displayed S-IRA, as indicated by a flat plethysmography breathing signal, it was quickly removed from the plethysmography chamber (or continuously monitored for up to 10 min prior to removal from the chamber), and three subdermal needle electrodes (Model F-E2, Grass Instruments, Warwick, RI, U.S.A.) were inserted into skin across its thorax (two electrodes were placed at the root of each forelimb, and the ground electrode was placed on the abdomen) for ECG analysis (gain 20,000; bandpass filter 0.3–3,000 Hz; P511 AC amplifier, Grass Technologies, Warwick, RI, U.S.A.). Cardiac electrical activity was continuously monitored for up to 10 min after placement of the needle electrodes. Custom software written using LabView 2013 (National Instruments, Austin, TX, U.S.A.) interfaced with several USB-6009 data acquisition boards (National Instruments) was used for gas flow control, data acquisition (at 128 Hz), and data analysis.
Statistical analysis
Data are reported as mean ± standard deviation (SD). Comparison of TPH2 expression among C57BL/6J and DBA/1 mice was performed using one-way analysis of variance (ANOVA) with Tukey’s test as post hoc analysis. The incidence of S-IRA evoked by PTZ among primed DBA/1, nonprimed DBA/1 and C57BL/6J mice was analyzed using Mann-Whitney U test, and the incidence of S-IRA between drug and control groups was compared using Wilcoxon signed rank test. Statistical significance was inferred if p < 0.05.
Results
Repetitive acoustic stimulation reduces the TPH2 protein in DBA/1 mice
We examined the expression of TPH2 in the brainstem of primed DBA/1 mice, nonprimed DBA/1 mice, C57BL/6J mice exposed to the acoustic stimulation, and C57BL/6J mice without exposure to the acoustic stimulation using Western blot. As compared with the relative optical density of TPH2 in C57BL/6J mice exposed to the acoustic stimulation (100%, n = 5), to which that of all four groups was normalized, the relative optical density of TPH2 was significantly reduced in the brainstem of primed DBA/1 mice (66.3 ± 11.9%, n = 5) (p < 0.05). The TPH2 protein relative expression levels were not significantly different between C57BL/6J mice without exposure to the acoustic stimulation (85.8 ± 15.8%, n = 5) and nonprimed DBA/1 mice (83.3 ± 28.4%, n = 5), and the TPH2 relative expression levels in both groups were not significantly different from those in C57BL/6J mice exposed to the acoustic stimulation and in primed DBA/1 mice (Fig. 1).
Figure 1.

Repetitive acoustic stimulation reduces TPH2 protein in DBA/1 mice. Western blot analysis was performed to compare TPH2 protein in the brainstem of primed DBA/1 mice (P-DBA/1), nonprimed DBA/1 mice (NP-DBA/1), C57BL/6J mice with exposure to the acoustic stimulation (S-C57), and C57BL/6J mice without exposure to the acoustic stimulation (NS-C57). The error bars denote SDs. *p < 0.05: significantly different from the C57BL/6J mice exposed to the acoustic stimulation.
5-HTP suppresses S-IRA evoked by acoustic stimulation in primed DBA/1 mice
Initial acoustic stimulation evokes S-IRA only in a small fraction of DBA/1 mice (10–30%) after generalized AGSz,3 which are characterized by wild running and tonic–clonic seizures culminating in tonic hindlimb extension, followed by S-IRA, and primed DBA/1 mice exhibit consistent S-IRA after tonic hindlimb extension. As compared with the vehicle control, the incidence of S-IRA was significantly reduced 1 h after acute systemic administration of 5-HTP (125 and 150 mg/kg, i.p.) (p < 0.01 and p < 0.05, respectively). 5-HTP at 100 mg/kg did not alter S-IRA as compared with the vehicle control (Fig. 2A). Although tonic seizures were blocked in 55–60% of DBA/1 mice at high doses of 5-HTP (125 and 150 mg/kg), these animals still exhibited wild running and/or clonic seizures. Thus, 5-HTP at the doses tested did not block susceptibility to AGSz in primed DBA/1 mice.
Figure 2.

5-HTP reduces S-IRA evoked by acoustic stimulation in primed DBA/1 mice. Effects of acute (A) and repeated (once daily for 2 days; B) systemic administration of 5-HTP on the incidence of S-IRA. 5-HTP was delivered by intraperitoneal injection. *p < 0.05; **p < 0.01: significantly different from the vehicle (saline) control; N, number of mice.
The effect of repeated treatment with 5-HTP on S-IRA was also examined. Repeated treatment with 5-HTP (100 mg/kg, i.p.) once daily for 2 days significantly reduced S-IRA in primed DBA/1 mice as compared with the vehicle control (p < 0.01). Repeated treatment with 5-HTP at 75 mg/kg reduced S-IRA, but this reduction did not reach statistical significance when compared with the control. Repeated treatment with 5-HTP at 50 mg/kg exerted no effect on S-IRA in primed DBA/1 mice (Fig. 2B). Like acute administration of 5-HTP, repeated treatment with 5-HTP for 2 days did not block the susceptibility of DBA/1 mice to AGSz, although tonic seizures were blocked in 56% of DBA/1 mice at 100 mg/kg 5-HTP.
DBA/1 and C57BL/6J mice display S-IRA during sudden death evoked by PTZ
To further test the relevance of the DBA/1 mouse as an animal model of SUDEP, we monitored changes in breathing and cardiac electrical activity during sudden death evoked by PTZ using whole-body plethysmography and ECG recordings. To determine if priming process exerts effect on the incidence of S-IRA by PTZ, we included both primed and nonprimed DBA/1 mice in this experiment. PTZ (75 mg/kg, i.p.) evoked generalized clonic and/or tonic–clonic seizures in primed (n = 6) and nonprimed DBA/1 mice (n = 5). Most DBA/1 mice incurred sudden death immediately after generalized tonic–clonic seizures. When a DBA/1 mouse exhibited generalized tonic–clonic seizures evoked by PTZ, S-IRA always follows as indicated by a flat plethysmography signal (Fig. 3A, C). Following apnea, each mouse was quickly removed from plethysmography chamber, and ECG recordings were obtained (Fig. 3B, D). In both primed DBA/1 and nonprimed DBA/1 mice, following S-IRA, cardiac electrical activity could be detected for several minutes prior to terminal asystole (Fig. 3A, B). These changes in breathing and cardiac electrical activity were also invariantly observed in two of the three C57BL/6J mice that died from generalized seizures evoked by PTZ (an ECG signal could not be obtained for technical reasons in one C57BL/6J mouse that died from S-IRA) (Fig. 3C, D). In a separate group of primed DBA/1 mice (n = 5), breathing was continuously monitored up to 10 min in the plethysmography chamber after S-IRA evoked by PTZ, showing that breathing did not return in these animals. Subsequent ECG recordings outside the chamber demonstrated that all mice still displayed cardiac activity prior to terminal asystole. Of all the primed DBA/1 (n = 11), nonprimed DBA/1 (n = 5) and C57BL/6J mice (n = 16) tested, two mice (one nonprimed DBA/1 and one C57BL/6J mouse) exhibited S-IRA after generalized tonic–clonic seizures, but breathing spontaneously returned during ECG recordings. These two mice died from S-IRA after the occurrence of the second generalized tonic–clonic seizures with initial PTZ injection.
Figure 3.

DBA/1 and C57BL/6J mice exhibit S-IRA during seizure-induced sudden death evoked by PTZ. Examples of sequential breathing signal and ECG recordings in a nonprimed DBA/1 mouse (A, B) and a C57BL/6J mouse (C, D) during seizure-induced sudden death evoked by PTZ (75 mg/kg, i.p.), demonstrating that S-IRA occurred after generalized seizures, and that cardiac activity could be observed for minutes prior to terminal asystole. In panels from A to D, the breathing and ECG signals inside the dashed-line squares were expanded to show the details of signal changes around generalized tonic–clonic seizures and following seizures. The arrows between A and B and between C and D indicate the time delay in transferring the apneic animal from the plethysmography chamber to the bench for ECG analysis.
We compared the incidence of S-IRA evoked by PTZ in DBA/1 and C57BL/6J mice. PTZ administration caused 90.9% of S-IRA in primed DBA/1 mice (n = 11), and 100% in nonprimed DBA/1 mice (n = 5). The incidence of S-IRA evoked by PTZ was not significantly different between primed and nonprimed DBA/1 mice. However, PTZ at the same dose evoked only 18.8% of S-IRA in C57BL/6J mice (n = 16). The incidence of S-IRA evoked by PTZ was significantly greater in primed DBA/1 or nonprimed DBA/1 mice as compared with that in C57BL/6J mice (p < 0.01) (Fig. 4).
Figure 4.
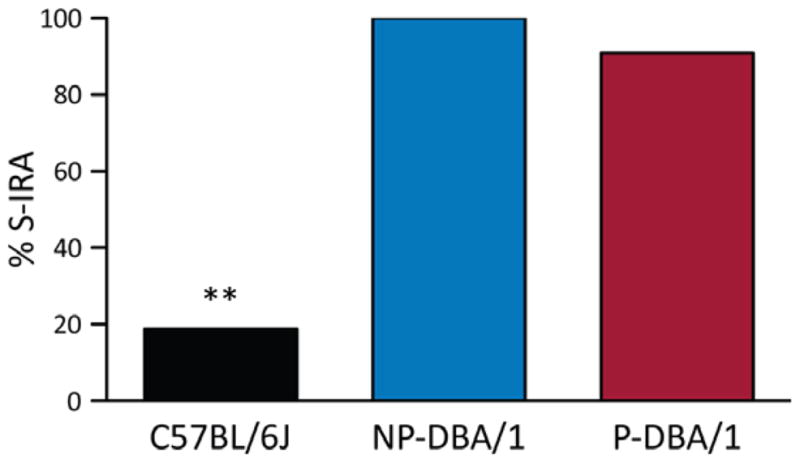
PTZ evokes a greater incidence of S-IRA in DBA/1 mice than C57BL/6J mice. Comparison of S-IRA evoked by PTZ (75 mg/kg, i.p.) among primed DBA/1 (P-DBA/1), nonprimed DBA/1 (NP-DBA/1), and C57BL/6J mice. **p < 0.01: significantly different from either primed or nonprimed DBA/1 mice.
5-HTP reduces S-IRA evoked by PTZ in DBA/1 mice
To determine if S-IRA suppressing effect of 5-HTP is dependent on the methods by which seizures are induced, we also examined the effect of 5-HTP on S-IRA evoked by PTZ in nonprimed DBA/1 mice, never exposed to the acoustic stimulation. S-IRA was significantly reduced in DBA/1 mice with repeated 5-HTP (200 mg/kg, i.p.) treatment over 2 days (16.7%, n = 6) (p < 0.05) compared to control mice (85.7%, n = 7). Treatment using 100 mg/kg 5-HTP once daily for 2 days did not reach statistical significance (33.3%, n = 6; p = 0.07). The incidence of S-IRA was not significantly different between 100 mg/kg and 200 mg/kg 5-HTP treatment (Fig. 5).
Figure 5.
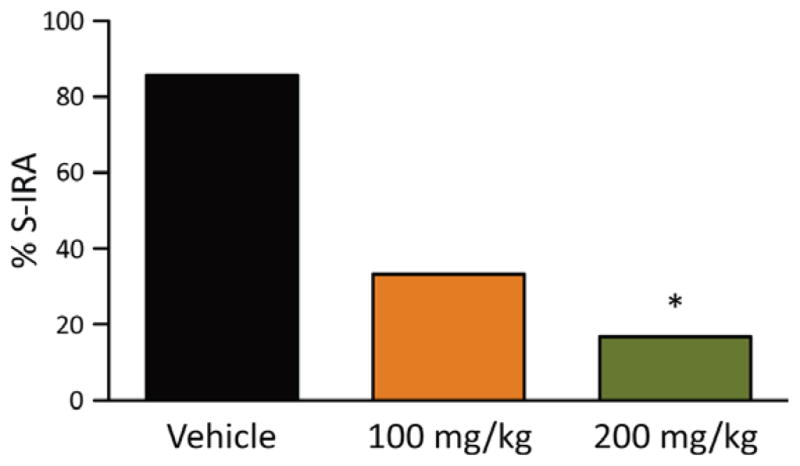
5-HTP reduces S-IRA evoked by PTZ in DBA/1 mice. Incidence of S-IRA in vehicle or 5-HTP-treated mice following generalized seizures evoked by PTZ. 5-HTP or vehicle (saline) was administered by i.p. injection once daily for 2 days in nonprimed DBA/1 mice. *p < 0.05: significantly different from the vehicle control.
Discussion
In the current study, we observed that the TPH2 expression is reduced in the brainstem of primed DBA/1 mice as compared with that of C57BL/6J mice exposed to acoustic stimulation. Systemic administration of 5-HTP, which is well documented to enhance brain 5-HT synthesis34–37 by bypassing TPH2, not only reduces S-IRA evoked by acoustic stimulation in primed DBA/1 mice, but also decreases that evoked by PTZ in DBA/1 mice that have never been exposed to the acoustic stimulation. DBA/1 and C57BL/6J mice always exhibit S-IRA immediately after generalized tonic–clonic seizures evoked by PTZ, and persistent cardiac activity is observed for minutes prior to terminal asystole.
S-IRA is the common initiating event during seizure-induced sudden death
SUDEP or near-SUDEP patients exhibit breathing difficulties after generalized tonic–clonic seizures,15,16,19 followed by apnea and subsequent death with terminal asystole.19 These changes in breathing and cardiac electrical activity were also observed during seizure-induced sudden death evoked by AGSz in DBA/1 mice,3 by maximal electroshock in Lmx1b(f/f) mice,23 or by hyperthermia in a Dravet syndrome mouse model.38 In the current work, we further tested the relevance of the DBA/1 mouse as an animal model of SUDEP by evoking generalized clonic and/or tonic–clonic seizures using PTZ, a chemoconvulsant that is widely used to model human generalized seizures.29 PTZ produced a greater incidence of S-IRA in both primed and nonprimed DBA/1 relative to C57BL/6J mice. This difference in PTZ-evoked S-IRA that we observed between DBA/1 and C57BL/6J mice parallels that of earlier work demonstrating susceptibility of DBA/1 mice to S-IRA during AGSz, unlike C57BL/6J mice, which are resistant.3 Both primed and nonprimed DBA/1 mice exhibited S-IRA after generalized tonic–clonic seizures evoked by PTZ, and cardiac activity could be detected for minutes prior to terminal asystole. Of interest, S-IRA and subsequent terminal asystole were also observed in the limited number of C57BL/6J mice that incurred seizure-induced sudden death following PTZ administration. Our data and previous studies suggest that S-IRA is a common terminal event leading to SUDEP.
It should be noted that in our current study, two (~6%) mice exhibited S-IRA evoked by PTZ after initial generalized seizures and did not die. However, both mice did subsequently die from S-IRA after second generalized seizures. Other studies, as well, have found that a small proportion of DBA/2 mice (15%) display tonic hindlimb extension evoked by acoustic stimulation without S-IRA.39 These studies suggest that S-IRA in some rare cases does not always lead to sudden death in these animal models.
Patients with Dravet syndrome have a high rate of SUDEP, and most cases of Dravet syndrome are linked to mutations of sodium channels, which are usually expressed in both heart and brain.40–42 Although S-IRA and subsequent terminal asystole occur in a mouse model of Dravet syndrome,38 a combination of S-IRA and cardiac arrest is most likely involved in sudden death in Dravet syndrome.38,40
5-HT neurotransmission is involved in S-IRA in DBA/1 mice
Deficits in 5-HT neurotransmission may predispose to S-IRA. 5-HT2C receptor knockout mice exhibit seizures that often end up with S-IRA.43 The SSRI fluoxetine suppresses S-IRA in DBA/1 and DBA/2 mice evoked by the acoustic stimulation,20,22,39 and S-IRA is reduced in DBA/1 mice by the SSRIs, sertraline and fluvoxamine.21,25 The SSRI citalopram inhibits S-IRA in Lmx1b(f/f) mice evoked by maximal electroshock.23 Consistent with these studies in animal models of SUDEP, SSRIs are also reported to reduce respiratory dysfunction in patients with partial seizures in a retrospective study.44 However, the SSRI paroxetine is less effective in preventing S-IRA than several other SSRIs in DBA/1 mice.25 In addition to 5-HT neurotransmission, SSRIs also exert effects on other neurotransmitters and receptors. For example, SSRIs inhibit norepinephrine and dopamine transporters45 and bind to 5-HT, adrenergic, and muscarinic cholinergic receptors.24,45 In our current study, we demonstrate that 5-HTP supplementation reduces S-IRA in DBA/1 mice evoked by either acoustic stimulation or PTZ. The preventive effect of 5-HTP against S-IRA is in agreement with elevation of brain 5-HT levels by 5-HTP.34,37 These data support the hypothesis that deficits in 5-HT neurotransmission contribute to S-IRA in DBA/1 mice, and are consistent with the theory that SSRIs prevent S-IRA by promoting 5-HT neurotransmission, and not through off-target effects. It is of note that 5-HTP also increases the brain levels of melatonin,46 a hormone regulating circadian rhythm.47 Further studies are needed to examine if melatonin exerts effect on S-IRA.
Our data showed that, as compared with corresponding C57BL/6J control mice, the amount of TPH2 protein is reduced in the brainstem of primed DBA/1 mice but is not altered in that of nonprimed DBA/1 mice, indicating that a decrease in TPH2 expression in primed DBA/1 mice may be related to the priming process. 5-HTP administration does not block susceptibility to AGSz in primed DBA/1 mice, suggesting that reduced TPH2 expression is unlikely to cause increased AGSz susceptibility associated with priming in these mice. Given that S-IRA evoked by PTZ was not different between primed and nonprime DBA/1 mice, it is unlikely that the small reduction in TPH2 expression contributes substantially to S-IRA in primed DBA/1 mice. It is of note that the antibody we used does not discriminate between TPH2 and TPH1. Although TPH2 is expressed predominantly in the CNS, controversy exists regarding whether TPH1 is expressed in the brain.48 Further studies using TPH2-specific antibody will be useful to confirm our results. We recognize that enzyme protein expression levels may not reflect enzyme activity, and further studies are needed to investigate if a reduction in TPH2 enzymatic activity promotes S-IRA.
Like SSRIs, 5-HTP is clinically used as a medication to treat a variety of psychiatric conditions, including depression,46 and is widely available as an over-the-counter oral supplement with minimal side effects. 5-HTP may be advantageous to SSRIs in enhancing 5-HT availability, as it is more “specific” to augment 5-HT levels in the brain, whereas SSRIs may produce nonspecific off-target effects and clearly cause a number of undesired clinical side effects.
In summary, we demonstrated that treatment with 5-HTP, as used to bypass a putative deficiency in TPH2 activity, reduces S-IRA in DBA/1 mice following generalized seizures evoked by PTZ or acoustic stimulation. These data suggest that 5-HT neurotransmission plays an important role in the pathogenesis of S-IRA, regardless of seizure induction method. Our study also suggests that 5-HTP may have merit as a potential therapy for the prevention of SUDEP. Finally, we determined that regardless of the mouse strain, DBA/1 or C57BL/6J, S-IRA always occurs after PTZ-evoked generalized seizures with persistence of cardiac activity for minutes before terminal asystole, implying S-IRA is a primary instigator in SUDEP.
Key Points.
TPH2 protein is reduced in primed DBA/1 mice as compared with C57BL/6J mice exposed to the acoustic stimulation
DBA/1 and C57BL/6J mice exhibit S-IRA after generalized tonic–clonic seizures evoked by PTZ
Following S-IRA, cardiac electrical activity can be detected for minutes prior to terminal asystole
5-HTP reduces S-IRA evoked by either acoustic stimulation or PTZ
Acknowledgments
We thank Dr. Han Lu (in Dr. Zhongcong Xie’s lab) for help with Western blot experiments. This work was supported by R03NS078591 and CURE (Citizens United for Research in Epilepsy) foundation to HJF, and R01HL117871 to JFC. We thank the support from the Massachusetts General Hospital Department of Anesthesia, Critical Care and Pain Medicine. Haiting Zhao is a recipient of fellowship from China Scholarship Council (CSC 201506370075). Xiaoxuan Yang is partly supported by Chinese Medical Association (CMA).
Biography

Dr. Honghai Zhang is a postdoctoral research fellow at Massachusetts General Hospital.
Footnotes
Disclosure
None of the authors has any conflict of interest to disclose. We confirm that we have read the journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- 1.Tomson T, Nashef L, Ryvlin P. Sudden unexpected death in epilepsy: current knowledge and future directions. Lancet Neurol. 2008;7:1021–1031. doi: 10.1016/S1474-4422(08)70202-3. [DOI] [PubMed] [Google Scholar]
- 2.Hughes JR. A review of sudden unexpected death in epilepsy: prediction of patients at risk. Epilepsy Behav. 2009;14:280–287. doi: 10.1016/j.yebeh.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 3.Feng HJ, Faingold CL. Abnormalities of serotonergic neurotransmission in animal models of SUDEP. Epilepsy Behav. 2015 Aug 10; doi: 10.1016/j.yebeh.2015.06.008. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lhatoo S, Noebels J, Whittemore V, et al. Sudden unexpected death in epilepsy: identifying risk and preventing mortality. Epilepsia. 2015;56:1700–1706. doi: 10.1111/epi.13134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thurman DJ, Hesdorffer DC, French JA. Sudden unexpected death in epilepsy: assessing the public health burden. Epilepsia. 2014;55:1479–1485. doi: 10.1111/epi.12666. [DOI] [PubMed] [Google Scholar]
- 6.Opeskin K, Berkovic SF. Risk factors for sudden unexpected death in epilepsy: a controlled prospective study based on coroners cases. Seizure. 2003;12:456–464. doi: 10.1016/s1059-1311(02)00352-7. [DOI] [PubMed] [Google Scholar]
- 7.Massey CA, Sowers LP, Dlouhy BJ, et al. Mechanisms of sudden unexpected death in epilepsy: the pathway to prevention. Nat Rev Neurol. 2014;10:271–282. doi: 10.1038/nrneurol.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aiba I, Noebels JL. Spreading depolarization in the brainstem mediates sudden cardiorespiratory arrest in mouse SUDEP models. Sci Transl Med. 2015;7:282ra246. doi: 10.1126/scitranslmed.aaa4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stollberger C, Finsterer J. Cardiorespiratory findings in sudden unexplained/unexpected death in epilepsy (SUDEP) Epilepsy Res. 2004;59:51–60. doi: 10.1016/j.eplepsyres.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 10.Hirsch LJ. Is sudden unexpected death in epilepsy due to postictal brain shutdown? Ann Neurol. 2010;68:773–775. doi: 10.1002/ana.22242. [DOI] [PubMed] [Google Scholar]
- 11.Shen HY, Li T, Boison D. A novel mouse model for sudden unexpected death in epilepsy (SUDEP): role of impaired adenosine clearance. Epilepsia. 2010;51:465–468. doi: 10.1111/j.1528-1167.2009.02248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klassen TL, Bomben VC, Patel A, et al. High-resolution molecular genomic autopsy reveals complex sudden unexpected death in epilepsy risk profile. Epilepsia. 2014;55:e6–e12. doi: 10.1111/epi.12489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sarkis RA, Thome-Souza S, Poh MZ, et al. Autonomic changes following generalized tonic clonic seizures: an analysis of adult and pediatric patients with epilepsy. Epilepsy Res. 2015;115:113–118. doi: 10.1016/j.eplepsyres.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 14.Langan Y, Nashef L, Sander JW. Sudden unexpected death in epilepsy: a series of witnessed deaths. J Neurol Neurosurg Psychiatry. 2000;68:211–213. doi: 10.1136/jnnp.68.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.So EL, Sam MC, Lagerlund TL. Postictal central apnea as a cause of SUDEP: evidence from near-SUDEP incident. Epilepsia. 2000;41:1494–1497. doi: 10.1111/j.1528-1157.2000.tb00128.x. [DOI] [PubMed] [Google Scholar]
- 16.Bateman LM, Li CS, Seyal M. Ictal hypoxemia in localization-related epilepsy: analysis of incidence, severity and risk factors. Brain. 2008;131:3239–3245. doi: 10.1093/brain/awn277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blum AS. Respiratory physiology of seizures. J Clin Neurophysiol. 2009;26:309–315. doi: 10.1097/WNP.0b013e3181b7f14d. [DOI] [PubMed] [Google Scholar]
- 18.Pezzella M, Striano P, Ciampa C, et al. Severe pulmonary congestion in a near miss at the first seizure: further evidence for respiratory dysfunction in sudden unexpected death in epilepsy. Epilepsy Behav. 2009;14:701–702. doi: 10.1016/j.yebeh.2009.02.012. [DOI] [PubMed] [Google Scholar]
- 19.Ryvlin P, Nashef L, Lhatoo SD, et al. Incidence and mechanisms of cardiorespiratory arrests in epilepsy monitoring units (MORTEMUS): a retrospective study. Lancet Neurol. 2013;12:966–977. doi: 10.1016/S1474-4422(13)70214-X. [DOI] [PubMed] [Google Scholar]
- 20.Faingold CL, Tupal S, Randall M. Prevention of seizure-induced sudden death in a chronic SUDEP model by semichronic administration of a selective serotonin reuptake inhibitor. Epilepsy Behav. 2011;22:186–190. doi: 10.1016/j.yebeh.2011.06.015. [DOI] [PubMed] [Google Scholar]
- 21.Faingold CL, Randall M. Effects of age, sex, and sertraline administration on seizure-induced respiratory arrest in the DBA/1 mouse model of sudden unexpected death in epilepsy (SUDEP) Epilepsy Behav. 2013;28:78–82. doi: 10.1016/j.yebeh.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 22.Zeng C, Long X, Cotten JF, et al. Fluoxetine prevents respiratory arrest without enhancing ventilation in DBA/1 mice. Epilepsy Behav. 2015;45:1–7. doi: 10.1016/j.yebeh.2015.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buchanan GF, Murray NM, Hajek MA, et al. Serotonin neurones have anti-convulsant effects and reduce seizure-induced mortality. J Physiol. 2014;592:4395–4410. doi: 10.1113/jphysiol.2014.277574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dale E, Bang-Andersen B, Sanchez C. Emerging mechanisms and treatments for depression beyond SSRIs and SNRIs. Biochem Pharmacol. 2015;95:81–97. doi: 10.1016/j.bcp.2015.03.011. [DOI] [PubMed] [Google Scholar]
- 25.Faingold CL, Kommajosyula SP, Long X, et al. Serotonin and sudden death: differential effects of serotonergic drugs on seizure-induced respiratory arrest in DBA/1 mice. Epilepsy Behav. 2014;37:198–203. doi: 10.1016/j.yebeh.2014.06.028. [DOI] [PubMed] [Google Scholar]
- 26.Zhang X, Beaulieu JM, Sotnikova TD, et al. Tryptophan hydroxylase-2 controls brain serotonin synthesis. Science. 2004;305:217. doi: 10.1126/science.1097540. [DOI] [PubMed] [Google Scholar]
- 27.Kulikov AV, Osipova DV, Naumenko VS, et al. Association between Tph2 gene polymorphism, brain tryptophan hydroxylase activity and aggressiveness in mouse strains. Genes Brain Behav. 2005;4:482–485. doi: 10.1111/j.1601-183X.2005.00145.x. [DOI] [PubMed] [Google Scholar]
- 28.Osipova DV, Kulikov AV, Mekada K, et al. Distribution of the C1473G polymorphism in tryptophan hydroxylase 2 gene in laboratory and wild mice. Genes Brain Behav. 2010;9:537–543. doi: 10.1111/j.1601-183X.2010.00586.x. [DOI] [PubMed] [Google Scholar]
- 29.White HS. Clinical significance of animal seizure models and mechanism of action studies of potential antiepileptic drugs. Epilepsia. 1997;38(Suppl 1):S9–S17. doi: 10.1111/j.1528-1157.1997.tb04523.x. [DOI] [PubMed] [Google Scholar]
- 30.Lopez-Meraz ML, Gonzalez-Trujano ME, Neri-Bazan L, et al. 5-HT1A receptor agonists modify epileptic seizures in three experimental models in rats. Neuropharmacology. 2005;49:367–375. doi: 10.1016/j.neuropharm.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 31.Uteshev VV, Tupal S, Mhaskar Y, et al. Abnormal serotonin receptor expression in DBA/2 mice associated with susceptibility to sudden death due to respiratory arrest. Epilepsy Res. 2010;88:183–188. doi: 10.1016/j.eplepsyres.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 32.Faingold CL, Randall M, Mhaskar Y, et al. Differences in serotonin receptor expression in the brainstem may explain the differential ability of a serotonin agonist to block seizure-induced sudden death in DBA/2 vs. DBA/1 mice. Brain Res. 2011;1418:104–110. doi: 10.1016/j.brainres.2011.08.043. [DOI] [PubMed] [Google Scholar]
- 33.Paxinos G, Franklin K. Paxinos and Franklin’s the Mouse Brain in Stereotaxic Coordinates. New York: Academic Press; 2013. [Google Scholar]
- 34.Gartside SE, Cowen PJ, Sharp T. Effect of 5-hydroxy-L-tryptophan on the release of 5-HT in rat hypothalamus in vivo as measured by microdialysis. Neuropharmacology. 1992;31:9–14. doi: 10.1016/0028-3908(92)90154-h. [DOI] [PubMed] [Google Scholar]
- 35.Lin MT, Tsay HJ, Su WH, et al. Changes in extracellular serotonin in rat hypothalamus affect thermoregulatory function. Am J Physiol. 1998;274:R1260–R1267. doi: 10.1152/ajpregu.1998.274.5.R1260. [DOI] [PubMed] [Google Scholar]
- 36.Halladay AK, Wagner GC, Sekowski A, et al. Alterations in alcohol consumption, withdrawal seizures, and monoamine transmission in rats treated with phentermine and 5-hydroxy-L-tryptophan. Synapse. 2006;59:277–289. doi: 10.1002/syn.20239. [DOI] [PubMed] [Google Scholar]
- 37.Pascucci T, Andolina D, Mela IL, et al. 5-Hydroxytryptophan rescues serotonin response to stress in prefrontal cortex of hyperphenylalaninaemic mice. Int J Neuropsychopharmacol. 2009;12:1067–1079. doi: 10.1017/S1461145709990381. [DOI] [PubMed] [Google Scholar]
- 38.Bravo E, Kim Y, Richerson GB. Ventilatory arrest is the primary initiating event that leads to sudden death after heat-induced seizures in a Dravet mouse model. AES Annual Meeting. 2015;69:3.364. Abstract. [Google Scholar]
- 39.Tupal S, Faingold CL. Evidence supporting a role of serotonin in modulation of sudden death induced by seizures in DBA/2 mice. Epilepsia. 2006;47:21–26. doi: 10.1111/j.1528-1167.2006.00365.x. [DOI] [PubMed] [Google Scholar]
- 40.Kalume F. Sudden unexpected death in Dravet syndrome: respiratory and other physiological dysfunctions. Respir Physiol Neurobiol. 2013;189:324–328. doi: 10.1016/j.resp.2013.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Catterall WA. Sodium channels, inherited epilepsy, and antiepileptic drugs. Annu Rev Pharmacol Toxicol. 2014;54:317–338. doi: 10.1146/annurev-pharmtox-011112-140232. [DOI] [PubMed] [Google Scholar]
- 42.Chopra R, Isom LL. Untangling the dravet syndrome seizure network: the changing face of a rare genetic epilepsy. Epilepsy Curr. 2014;14:86–89. doi: 10.5698/1535-7597-14.2.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brennan TJ, Seeley WW, Kilgard M, et al. Sound-induced seizures in serotonin 5-HT2c receptor mutant mice. Nat Genet. 1997;16:387–390. doi: 10.1038/ng0897-387. [DOI] [PubMed] [Google Scholar]
- 44.Bateman LM, Li CS, Lin TC, et al. Serotonin reuptake inhibitors are associated with reduced severity of ictal hypoxemia in medically refractory partial epilepsy. Epilepsia. 2010;51:2211–2214. doi: 10.1111/j.1528-1167.2010.02594.x. [DOI] [PubMed] [Google Scholar]
- 45.Shank RP, Vaught JL, Pelley KA, et al. McN-5652: a highly potent inhibitor of serotonin uptake. J Pharmacol Exp Ther. 1988;247:1032–1038. [PubMed] [Google Scholar]
- 46.Birdsall TC. 5-Hydroxytryptophan: a clinically-effective serotonin precursor. Altern Med Rev. 1998;3:271–280. [PubMed] [Google Scholar]
- 47.Reiter RJ. Pineal melatonin: cell biology of its synthesis and of its physiological interactions. Endocr Rev. 1991;12:151–180. doi: 10.1210/edrv-12-2-151. [DOI] [PubMed] [Google Scholar]
- 48.Cote F, Thevenot E, Fligny C, et al. Disruption of the nonneuronal tph1 gene demonstrates the importance of peripheral serotonin in cardiac function. Proc Natl Acad Sci U S A. 2003;100:13525–13530. doi: 10.1073/pnas.2233056100. [DOI] [PMC free article] [PubMed] [Google Scholar]