

Abstract
The Japanese sea cucumber (Apostichopus japonicus Selenka 1867) is an economically important species as a source of seafood and ingredient in traditional medicine. It is mainly found off the coasts of northeast Asia. Recently, substantial exploitation and widespread biotic diseases in A. japonicus have generated increasing conservation concern. However, the genomic knowledge base and resources available for researchers to use in managing this natural resource and to establish genetically based breeding systems for sea cucumber aquaculture are still in a nascent stage. A total of 312 Gb of raw sequences were generated using the Illumina HiSeq 2000 platform and assembled to a final size of 0.66 Gb, which is about 80.5% of the estimated genome size (0.82 Gb). We observed nucleotide-level heterozygosity within the assembled genome to be 0.986%. The resulting draft genome assembly comprising 132 607 scaffolds with an N50 value of 10.5 kb contains a total of 21 771 predicted protein-coding genes. We identified 6.6–14.5 million heterozygous single nucleotide polymorphisms in the assembled genome of the three natural color variants (green, red, and black), resulting in an estimated nucleotide diversity of 0.00146. We report the first draft genome of A. japonicus and provide a general overview of the genetic variation in the three major color variants of A. japonicus. These data will help provide a comprehensive view of the genetic, physiological, and evolutionary relationships among color variants in A. japonicus, and will be invaluable resources for sea cucumber genomic research.
Keywords: Sea cucumber genome, Apostichopus japonicas, Color variants, Genetic variation, Population genomics
Data description
Background information on Apostichopus japonicus
The class Holothuroidea (also known as sea cucumbers) belongs to the phylum Echinodermata and comprises approximately 1250 recorded species worldwide, including some species that are of commercial and medical value [1, 2]. Apostichopus japonicus Selenka 1867 is one of the well-known, commercially important sea cucumber species and occurs in the northwestern Pacific coast including China, Japan, Korea, and the Far Eastern seas. This species exhibits a wide array of dorsal/ventral color variants (in particular green, red, and black; Fig. 1), which differ in their biological and morphological attributes (e.g., shape of ossicle, habitat preference, spawning period, and polian vesicles) [1, 3]. The red variant is found on rock pebbles and gravel substrate and has higher salinity and temperature tolerance than the other color variants [4, 5]. Green and black variants are found on sandy and muddy bottoms at shallower depths, and the green variant has greater plasticity in thermotolerance than the red variant [6, 7].
Figure 1.

Three color variants of A. japonicus (green, red, and black).
Recently, overexploitation and the prevalence of biotic diseases (viral infections) in sea cucumber aquaculture have generated increasing conservation concern [8, 9]. However, the genomic knowledge base and resources available for researchers to use in managing this natural resource or establishing genetically based breeding systems are still in a nascent stage [10].
Sample collection and genomic DNA extraction
Specimens of the three color A. japonicus variants (green, red, and black) were collected from same geographical location (GPS data: 34.1 N, 127.18 E, Geomun-do, Yeosu, Republic of Korea). Genomic DNA of each color variant was extracted manually from body wall tissues of single male specimens. Briefly, we ground the tissues to fine powder using mortar and pestle with liquid nitrogen freezing. Tissue powders were digested for 1 hour at 65°C in CTAB buffer (2% cetyltrimethylammonium bromide, 1.4 M NaCl, 20 mM EDTA, 100 mM Tris-HCl, and pH 8.0), followed by Phenol/Chloroform extraction and ethanol precipitation.
Sequencing and quality control
Using the standard protocol provided by Illumina (San Diego, USA), we constructed both short-insert (180 and 400 bp) and long-insert (2 kb) libraries for 2 × 101 bp paired-end reads, which were sequenced using a HiSeq 2000 instrument. For the green color variant, a total of 225 Gb of raw data was generated from all three libraries. In the case of the red and black color variants, 40 and 47 Gb of raw reads, respectively, were produced by 400 bp short-insert library. The raw reads were preprocessed using Trimmomatic v0.33 [11] and Trim Galore [12], in which reads containing adapter sequences, poly-N sequences, or low-quality bases (below a mean Phred score of 20) were removed. To correct errors in the raw sequences, we used ALLPATHS-LG v52488 [13]. Approximately 208, 39, and 42 billion clean reads were obtained for green, red, and black color variant samples, respectively (Table 1). The A. japonicus genome size was estimated to be approximately 0.9 Gb based on k-mer measurement (Fig. 2), which is fully consistent with genome size measured by flow cytometry (∼0.82 Gb) [14]. Based on this estimation, the clean sequence reads correspond to about 356-fold coverage of the A. japonicus genome.
Table 1.
Statistics on total reads of the A. japonicus genome
| Variants | Insertion size (bp) | Total readsa (raw data) | Total readsa (w/o adaptor) | Total readsa (error corrected) | % error corrected |
|---|---|---|---|---|---|
| Green | 180 | 498 608 646 | 474 117 288 | 466 062 920 | 1.70 |
| 400 | 897 432 174 | 842 766 704 | 831 964 242 | 1.28 | |
| 2000 (v1) | 293 701 464 | 270 513 434 | 268 573 812 | 0.72 | |
| 2000 (v2) | 538 359 438 | 496 446 984 | 493 387 418 | 0.62 | |
| Total | 2228 101 722 | 2083 844 410 | 2059 988 392 | 1.14 | |
| Red | 400 | 397 799 042 | 394 984 810 | 383 734 440 | 2.85 |
| Black | 400 | 460 597 940 | 423 543 558 | 416 007 614 | 1.78 |
aThe length of each read is 101 bp.
Figure 2.
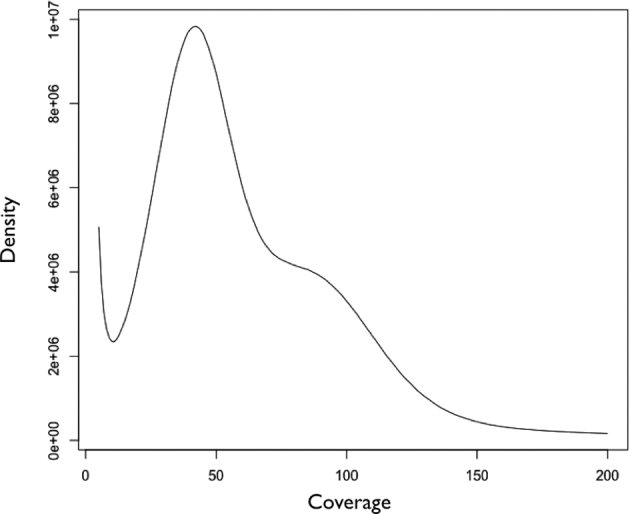
K-mer distribution of the A. japonicus genome.
Assembly
For whole-genome assembly, we used reads only from green color variant libraries and employed Platanus v1.2.4 [15], which is well suited for high-throughput short reads and heterozygous diploid genomes. Briefly, error corrected paired-end (insert size: 180 bp and 400 bp) reads were input for contig assembly. Next, all cleaned paired-end (insert size: 180 bp and 400 bp) and mate-paired (insert size: two 2 kb samples) reads were mapped onto the contigs for scaffold building and were utilized for gap filling (any nucleotide represented by “N” in scaffolds). After gap filling by Platanus, the gaps that still remained in the resulting scaffolds were closed using GapCloser (a module of SOAPdenovo2 [16]). The final genome assembly was 0.66 Gb in total length, which is about 80.5% of the estimated genome size by flow cytometry (0.82 Gb) [14], and is composed of 132 607 scaffolds and unscaffolded contigs (that are longer than or equal to 1 kb) with an N50 value of 10.5 kb (Table 2). We assessed the completeness of the assembly using CEGMA v2.4.010312 [17] and BUSCO v1.22 [18]. Then 73.4% of the core eukaryotic genes (based on the 248 core essential genes) and 60.7% of the metazoan single-copy orthologs (based on the 843 genes) were identifiable in the genome. Because assembling highly heterozygous genomes is a major challenge in de novo genome sequencing, we further sought to explore whether there are other assemblers that could produce better genome assembly statistics. We applied two popular genome assemblers, SOAPdenovo2 2.04-r240 [16] and ALLPATHS-LG v52488 [13], and as expected [15], the Platanus assembler was superior to the others (Table S1).
Table 2.
Statistics on Apostichopus japonicus genome assembly
| Statistics | Values |
|---|---|
| Total assembled bases (bp) | 664 375 288 |
| Average length of scaffolds (bp) | 5010 |
| Number of scaffolds | 132 607 |
| Number of contigs | 197 146 |
| Length of longest scaffold (bp) | 131 537 |
| GC content (%) | 35.92 |
| Scaffold N50 (bp) | 10 488 |
| Contig N50 (bp) | 5525 |
| Number of genes | 21 771 |
| Number of exons per gene | 4.67 |
| Average exon length (bp) | 209 |
| Number of introns per gene | 4.21 |
| Average intron length (bp) | 1048 |
Annotation
To identify genomic repeat elements in the A. japonicus genome assembly, we ran RepeatMasker (version 4.0.6) [19] using the Repbase transposable element library (release 20150807) [20] and the de novo repeat library constructed by RepeatModeler (version 1.0.8) [21]. Approximately 27.2% of the A. japonicus genome was identified as interspersed repeats.
Protein-coding genes were predicted using four steps. First, ab initio gene prediction was performed with trained AUGUSTUS v3.2.1 [22] using hints from splicing alignment of transcripts to the repeat-masked assembled genome with BLAT [23] and PASA v2.0.2 [24]. To obtain high-quality spliced alignments of expressed transcript sequences for the AUGUSTUS training set, we collected high-throughput messenger RNA sequencing (RNA-seq) data from our previous [25] (from body wall tissue of adult stage specimens) and other transcriptome (from embryo, larva, and juvenile stages [developmental-stage specific]; from gonads, intestines, respiratory trees, and coelomic fluid of adults [tissue-specific]) [26] studies, and assembled reads from the RNA-seq dataset using Trinity v2.1.1 [27]. Second, for homology-based gene prediction, homologous proteins in other species (from UniProt [28]) were mapped to the repeat-masked assembled genome using tBLASTn [29] with an E-value ≤ 1 × 10−5. The aligned sequences were predicted using GeneWise v2.4.0 [30] to search for precise spliced alignment and gene structures. Third, for homology-based gene prediction with transcriptome evidence, existing RNA-seq reads [23, 25] were mapped to the repeat-masked assembled genome using TopHat v2.1.0 [31], and gene models were built using Cufflinks v2.2.1 [32]. Finally, the resulting gene sets from each approach were integrated into a comprehensive and non-redundant consensus gene set. We predicted a total of 21 771 (≥ 50 amino acids) genes in the assembled A. japonicus genome, including 101 776 exons (average 4.67 exons per gene), and an average gene size of 5402 nucleotides (average transcript size of 982 nucleotides) (Table. 2).
Genetic polymorphism among natural color variants
To provide a general overview of the total genetic variation in the species, we realigned reads from the green color variant to the assembled genome using BWA v0.7.13 [33]. Picard v1.141 (http://broadinstitute.github.io/picard/) was used to mark and remove duplicates. Before single nucleotide polymorphism (SNP) and small insertion and deletion (indel) calling, we realigned reads with indels using GATK RealignerTargetCreator and IndelRealigner v3.5 [34] to avoid misalignment around indels. Next, GATK Haplotypecaller was used to call SNPs and indels from the resulting sequences. In this study, we observed nucleotide-level heterozygosity within the assembled genome to be 0.986%; namely, we identified a total of 6 550 122 SNPs at the assembled genome, for a heterozygous SNP rate of 0.00986 per site. This high rate of nucleotide polymorphism is not uncommon in marine invertebrates and also has been found in the sea urchin genome (∼1%; at least one SNP per 100 bases) [35], which belongs to the same phylum.
To measure nucleotide diversity in A. japonicus, the aforementioned analyses were repeated for red and black color variants separately, and VCFtools v0.1.14 [36] with sliding window analysis (bin 10 kb, step 1 kb) was used to calculate nucleotide diversity. We identified 6.6–14.5 million heterozygous SNPs (1.7–3.7 million small indels) in the assembled genome from the three natural color variants (Table 3), resulting in an estimated nucleotide diversity of 0.00146.
Table 3.
SNP and small indel statistics among three color variants
| Variants | Percent heterozygous SNP loci | Percent small indel loci |
|---|---|---|
| Green | 6 550 122 | 1 662 708 |
| Red | 14 509 713 | 3 681 007 |
| Black | 12 627 560 | 3 198 584 |
In summary, we report the first draft genome of A. japonicus Fig. 3 and provide a general overview of the genetic variation in its three color variants (green, red, and black). These data will help elucidate the genetic, physiological, and evolutionary relationships among different color variants in A. japonicus and will be invaluable resources for sea cucumber genomic research.
Figure 3.

Schematic workflow of A. japonicus genome assembly and annotation. The left side represents the genome assembly and the right side represents the transcriptome assembly that was performed in previous publications. To achieve suitable gene prediction, we integrated these two assembly results.
Availability of supporting data
The raw dataset of all A. japonicus genome libraries and the assembly were deposited in the NCBI database with BioProject accession number PRJNA335936, SRA accession number SRP082485, and GenBank accession number MODV00000000. The additional dataset associated with genome annotation along with further supporting data are available in the GigaScience Database, GigaDB [37]. The RNA-seq datasets used in this study were downloaded from the ENA database with accession number PRJEB12167 and the NCBI database with SRA accession number SRA046386.
Abbreviations
Indel: insertion and deletion; RNA-seq: high-throughput messenger RNA sequencing; SNP: single nucleotide polymorphism.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CP designed the study. CP, JKP, and SJC contributed to the project coordination. JJ, HGL, HHH, and SJ collected the samples and extracted the genomic DNA. CP, JO, SGL, and SC conducted the genome analyses. CP, JKP, JJ, and EK wrote the paper. All authors read and approved the final manuscript.
Supplementary Material
Acknowledgements
This work was supported by research grants from the Marine Biotechnology Program (PJT200620, Genome Analysis of Marine Organisms and Development of Functional Applications) funded by the Ministry of Oceans and Fisheries of the Republic of Korea to CP, JKP, and SJC and from the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (NRF-2015R1C1A1A02036896) to CP. This work was also supported by National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (NRF-2015R1A4A1041997) to JKP. This work was carried out with the support of “Cooperative Research Program for Agriculture Science & Technology Development (Signaling regulations and disease mechanisms research on exposure to biological, chemical and environmental hazard substance, PJ01052301)” Rural Development Administration, Republic of Korea to SJC.
References
- 1. Choe S, Oshima Y. On the morphological and ecological differences between two commercial forms, “Green” and “Red”, of the Japanese common sea cucumber, Stichopus japonicus Selenka. Nippon Suisan Gakkaishi. 1961;27:97–105. [Google Scholar]
- 2. Kanno M, Kijima A. Quantitative and qualitative evaluation on the color variation of the Japanese sea cucumber Stichopus japonicus. Suisanzoshoku. 2002;50:63–9. [Google Scholar]
- 3. Hongsheng Yang J-FH, Mercier A. The Sea Cucumber Apostichopus japonicus: History, Biology and Aquaculture. Academic Press, 2015. [Google Scholar]
- 4. Yamamoto K, Handa T, Fujimoto K. Differences in tolerance to low-salinity among red, blue and black (color pattern) of the Japanese common sea cucumber, Apostichopus japonicus from ventilation in the respiratory tree. Suisan Zoshoku. 2003;51:321–26. [Google Scholar]
- 5. Yamamoto K, Handa T, Fujimoto K. Effects of water temperature on ventilation of the Japanese common sea cucumber, Apostichopus japonicus of different color pattern. Aquaculture Science. 2005;53(1):67–74. [Google Scholar]
- 6. Choe S. Biology of the Japanese Common Sea Cucumber Stichopus japonicus Selenka. Pussan [sic]: Pusan National University, 1963. [Google Scholar]
- 7. Dong Y-W, Ji T-T, Meng X-L, Dong S-L, Sun W-M. Difference in thermotolerance between green and red color variants of the Japanese sea cucumber, Apostichopus japonicus Selenka: Hsp70 and heat-hardening effect. Biol Bull. 2010;218(1):87–94. [DOI] [PubMed] [Google Scholar]
- 8. Bordbar S, Anwar F, Saari N. High-value components and bioactives from sea cucumbers for functional foods–a review. Mar Drugs. 2011;9(10):1761–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Purcell SW. Value, market preferences and trade of Beche-de-mer from Pacific Island sea cucumbers. PLoS One. 2014;9(4):e95075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Long KA, Nossa CW, Sewell MA, Putnam NH, Ryan JF. Low coverage sequencing of three echinoderm genomes: The brittle star Ophionereis fasciata, the sea star Patiriella regularis, and the sea cucumber Australostichopus mollis. Gigascience. 2016;5:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bolger AM, Lohse M, Usadel B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Krueger F. Trim Galore!: A Wrapper Tool Around Cutadapt and FastQC to Consistently Apply Quality and Adapter Trimming to FastQ files. http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (5 June 2015, date last accessed). [Google Scholar]
- 13. Gnerre S, Maccallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ et al. . High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci U S A. 2011;108(4):1513–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu Jin ZX-j, Su Lin, Liu Shi-lin, Ru Shao-guo, Yang Hong-sheng. Genome size determination of sea cucumber (Apostichopus japonicus). J Fish China. 2012;36:(5). doi:10.3724/SP.J.1231.2012.27753. [Google Scholar]
- 15. Kajitani R, Toshimoto K, Noguchi H, Toyoda A, Ogura Y, Okuno M et al. . Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 2014;24(8):1384–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J et al. . SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 2012;1(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Parra G, Bradnam K, Korf I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23(9):1061–7. [DOI] [PubMed] [Google Scholar]
- 18. Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–2. [DOI] [PubMed] [Google Scholar]
- 19. Smit AFA, Hubley R, Green P. RepeatMasker 4.0.6. http://www.repeatmasker.org/ (29 October 2015, date last accessed). [Google Scholar]
- 20. Bao W, Kojima KK, Kohany O. Repbase update, a database of repetitive elements in eukaryotic genomes. Mob DNA. 2015;6:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Smit A, Hubley R. RepeatModeler Open-1.0. http://www.repeatmasker.org/ (29 May 2014, date last accessed). [Google Scholar]
- 22. Stanke M, Diekhans M, Baertsch R, Haussler D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 2008;24(5):637–44. [DOI] [PubMed] [Google Scholar]
- 23. Kent WJ. BLAT–the BLAST-like alignment tool. Genome Res. 2002;12(4):656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Haas BJ, Delcher AL, Mount SM, Wortman JR, Smith RK Jr, Hannick LI et al. . Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003;31(19):5654–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jo J, Park J, Lee HG, Kern EM, Cheon S, Jin S et al. . Comparative transcriptome analysis of three color variants of the sea cucumber Apostichopus japonicus. Mar Geonomics. 2016. doi:10.1016/j.margen.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 26. Du H, Bao Z, Hou R, Wang S, Su H, Yan J et al. . Transcriptome sequencing and characterization for the sea cucumber Apostichopus japonicus (Selenka, 1867). PloS One. 2012;7(3):e33311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I et al. . Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. UniProt C UniProt: a hub for protein information. Nucleic Acids Res. 2015;43(Database issue):D204–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K et al. . BLAST+: Architecture and applications. BMC Bioinformatics. 2009;10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li W, Cowley A, Uludag M, Gur T, McWilliam H, Squizzato S et al. . The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 2015;43(W1):W580–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR et al. . Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A et al. . The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research. 2010;20(9):1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sea Urchin Genome Sequencing C Sodergren E, Weinstock GM, Davidson EH, Cameron RA, Gibbs RA et al. . The genome of the sea urchin Strongylocentrotus purpuratus. Science. 2006;314(5801):941–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA et al. . The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jo J, Oh J, Lee H-G, Hong H-H, Lee S-G, Cheon S, Kern EMA, Jin S, Cho S-J, Park J-K, Park C. Supporting data for the “Draft genome of the sea cucumber Apostichopus japonicus and genetic polymorphism among color variants”. GigaScience Database. 2016. http://dx.doi.org/10.5524/100257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.