

Abstract
Background
Immunotherapy is an emerging paradigm for the treatment of cancer, but the potential efficacy of many drugs cannot be sufficiently tested in the mouse. We sought to develop a rational combination of motolimod—a novel Toll-like receptor 8 (TLR8) agonist that stimulates robust innate immune responses in humans but diminished responses in mice—with pegylated liposomal doxorubicin (PLD), a chemotherapeutic that induces immunogenic cell death.
Methods
We followed an integrative pharmacologic approach including healthy human volunteers, non-human primates, NSG-HIS (“humanized immune system”) mice reconstituted with human CD34+ cells, and cancer patients to test the effects of motolimod and to assess the combination of motolimod with PLD for the treatment of ovarian cancer.
Results
The pharmacodynamic effects of motolimod monotherapy in NSG-HIS mice closely mimicked those in non-human primates and healthy human subjects, while the effects of the motolimod/PLD combination in tumor-bearing NSG-HIS mice closely mimicked those in patients with ovarian cancer treated in a phase 1b trial (NCT01294293). The NSG-HIS mouse helped elucidate the mechanism of action of the combination, and revealed a positive interaction between the two drugs in vivo. The combination produced no dose-limiting toxicities in ovarian cancer patients. Two subjects (15%) had complete responses and 7 subjects (53%) had disease stabilization. A phase 2 study was consequently initiated.
Conclusions
These results are the first to demonstrate the value of pharmacologic approaches integrating the NSG-HIS mouse, non-human primates, and cancer patients for the development of novel immunomodulatory anticancer agents with human specificity.
Introduction
Most chemotherapeutic agents are believed to work independently of the host's immune system, and some even appear to exert immunosuppressive functions. However, select chemotherapeutic agents such as anthracyclines may promote tumor cell death through mechanisms that activate antitumor immune response (1), thereby converting the tumor microenvironment (TME) from an immunologic sanctuary to a setting characterized by immunogenic tumor cell death. This transition is associated with the surface expression of stress-associated markers such as calreticulin (CRT) on injured cells, which induce active phagocytosis by myeloid dendritic cells (mDCs) and promote antigen processing and cross-presentation (2). Furthermore, the release into the TME of high-motility group box 1 (HMGB1) protein, a danger-associated molecular pattern (DAMP) molecule, triggers the innate immune response (3, 4). Despite these immune-activating processes, tumor-infiltrating mDCs are likely to be suppressed by factors within the TME (5), and phagocytosis of necrotic cells may not be a sufficient stimulus for optimal cross-presentation of tumor antigens by tumor DCs to trigger protective cytotoxic T cell responses.
Toll-like receptors (TLRs) comprise a family of 13 surface or endosomal receptors that recognize ligands that are broadly shared by pathogens, collectively referred to as pathogen-associated molecular pattern (PAMP) molecules. PAMPs activate DCs and stimulate the release of cytokines and chemokines, thus facilitating the development of adaptive immune responses (6, 7). In humans, TLR8 is distinguished from other endosomally-expressed TLRs by its unique appearance on mDCs, monocytes, and natural killer (NK) cells. The natural ligand of TLR8 is foreign single-stranded RNA; engagement triggers cellular activation and maturation, including the release of Th1-polarizing cytokines. Based on a small genetic deletion in the murine TLR8 gene (8), the activity of TLR8 agonists in mice is significantly attenuated compared to that in humans and other species. Consequently, the in vivo biological activity of TLR8 agonists has not been extensively studied to date. Nonetheless, the specific expression of TLR8 within human myeloid cells offers unique opportunities for anticancer therapy.
We have developed a synthetic, small molecule agonist of TLR8 (motolimod; previously identified as VTX-2337) comprising a 2-aminobenzazepine core (9). Motolimod is selective for TLR8 and potently stimulates CD14+ monocytes and HLA-DR+CD11c+ mDCs, but not HLADR+CD11c-CD123+ plasmacytoid DCs (9). In mDCs, motolimod activated NF-κB and induced expression of IL-12 and TNFα. A completed phase 1 clinical study has demonstrated the tolerability and biological activity of motolimod monotherapy, and revealed predictable pharmacokinetic and pharmacodynamic profiles (10).
Given the stimulatory effects of motolimod on mDCs and monocytes, we hypothesized that motolimod would be an optimal partner for immunomodulatory chemotherapies such as anthracyclines, since simultaneous activation of PAMP and DAMP pathways during tumor antigen exposure could produce positive interactions. To test this hypothesis, we chose pegylated liposomal doxorubicin (PLD) because of favorable pharmacokinetics and pharmacodynamics, limited hematopoietic toxicity, and preferential accumulation in tumors (11). We chose ovarian cancer because of the implicated role of immune mechanisms (12-14) and because PLD is standard of care in relapsed disease (15).
Because the activity of TLR8 agonists in mice varies from that in humans (16), we pursued an integrative pharmacologic approach to study the combination of PLD and motolimod in parallel models including in vitro human, in vivo non-human primate, and in vivo humanized mouse, to acquire knowledge on the mechanism of action of the combination, which was then evaluated in a phase 1b clinical study in women with recurrent ovarian cancer. Motolimod plus PLD induced potent immune activation, which was similar to that observed in the NSG-HIS mouse and in healthy human volunteers, and was well tolerated without unexpected or synergistic toxicities.
Materials and Methods
Drugs
Motolimod (VTX-2337) was supplied by VentiRx Pharmaceuticals, Inc. (Seattle, WA). For preclinical studies, PLD (Ben Venue Laboratories Inc., Bedford, OH) was purchased from the University of Pennsylvania Hospital pharmacy. For the GOG-9925 study, PLD was supplied by each clinical site's pharmacy.
Human volunteers
Five men and 5 women healthy volunteers enrolled in a randomized, open-label, phase 1 study of motolimod (Study VRXP-A105) received two doses of motolimod 2.5 mg/m2 SC separated by one week (17). Plasma samples were collected pre-dose and 6 hours after dosing, and analyzed for mediator levels as described above. This clinical study was conducted in accordance with Good Clinical Practice guidelines and the ethical principles based on the Declaration of Helsinki. Approval for study procedures was obtained from the institutional review boards of each study site and all subjects provided written informed consent upon study enrollment.
Primate studies
Studies in cynomolgus monkeys were conducted at Charles River Laboratories, Preclinical Services, (Shrewsbury MA) in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Study protocols were reviewed and approved by the Charles River Institutional Animal Care and Use Committee. Three male Cynomolgus monkeys were administered a single dose of motolimod 3.6 mg/m2. Plasma samples were collected pre-dose and 6 hours post-dose, and analyzed as described above. For in vitro studies, 7 mL of peripheral blood was collected from 3 males and 3 females and added to TruCulture tubes (Myriad-RBM) containing 2 mL of culture media plus 300 nM motolimod. Tubes were incubated for 24 hours; supernatants were stored at -35°C until analysis for mediator levels as described above.
NSG-HIS mice and immune analyses
We used NOD-scid IL2rγnull (NSG) mice reconstituted with human immune system (NSG-HIS) to test the biological effects of motolimod and the combination with PLD. To generate human ovarian tumors, we injected mice with OVCAR5 cells. Details of mouse experiments as well as flow cytometry analysis of human leukocytes harvested from NSG-HIS mice, and testing of reactive human TILs from mouse tumors, are provided in supplemental material.
Measurement of cytokines in human, primate, and murine blood
Human and non-human primate cytokines in biological samples (TruCulture supernatants and plasma samples) were measured using Luminex-based technology (HumanMAP panel; Myriad RBM; Austin, TX). Plasma samples from CD-1 mice were pooled at each time point, and cytokines were measured using the Myriad-RBM RodentMAP® panel.
In vitro tumor cell viability assessment and Western blot analysis
OVCAR5 cells were treated with PLD (0.1, 1 and 10 μg/ml) and/or recombinant human TNFα (20 ng/ml). Cell viability was assessed by AnnexinV staining (Pharmingen #559763). Protein extraction and western blot analysis were performed as described elsewhere (18). Primary antibody against c-FLIP (Cell Signaling #3210 rabbit) and secondary antibody (BioRad 172-1019) were used at room temperature. Protein bands were visualized using ECL (Amersham #RPN2132) with X-films (Bioexpress # F-9023).
Clinical Study Design
GOG-9925 was a phase 1b, open-label, dose-escalation study of motolimod in combination with either PLD or paclitaxel in subjects with advanced ovarian cancer. The trial utilized a traditional 3+3 dose-escalation design to identify the maximum tolerated dose (MTD) of each combination (19). Primary objectives were to assess safety and tolerability, secondary objectives were to evaluate pharmacokinetics and pharmacodynamics, and exploratory objectives included assessment of antitumor activity. The study was conducted at four Gynecologic Oncology Group centers in the US (June 2011 to September 2013) in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki. Approval was obtained from each site's Institutional Review Board. All subjects provided written informed consent. For each subject in cohort 1, PLD 40 mg/m2 was administered on day 1, followed by 2.5 mg/m2 motolimod on day 3; each subject was assessed over ≥ 24 hours for safety. In the absence of overt toxicity, the next subject in the cohort was enrolled. Enrollment of subsequent cohorts proceeded without such staggering. All enrolled subjects were assessed for dose-limiting toxicities (DLTs), defined as treatment-related adverse events (AEs) that occurred during the first cycle of treatment and met standard pre-defined toxicity criteria. DLTs and other toxicities were the basis for decisions regarding dose escalation, de-escalation, and cohort expansion. Details on eligibility criteria and study assessment are provided in supplemental material.
Statistical analyses
Details on statistical analyses are provided in supplemental material.
Results
Motolimod induces immune activation in NSG-HIS mice similar to non-human primates and humans
To screen for suitable preclinical models, we compared the response to motolimod in vitro of PBMCs obtained from human healthy volunteers, non-human primates and wild-type CD1 mice. Confirming the reported phylogenetic differences in TLR8, leukocytes from wild-type mice were minimally responsive to motolimod relative to non-human primate and human leukocytes (Supplementary Figure S1). We thus sought an immunocompetent murine model that would allow us to explore the therapeutic potential of motolimod for tumor immunotherapy in vivo at clinically relevant doses. We selected the NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NOD-scid-IL2rγnull NSG) mouse which, when reconstituted with human donor hematopoietic stem cells, develops all lineages of the human immune system (NSG-HIS) (20). NSG mice were given 2.75 Gy whole-body irradiation and then intravenously injected with ∼1×105 human donor cord blood CD34+ cells. Consistent with published data (21), a high level of human hematopoietic cell engraftment was seen in NSG-HIS mice within 12 weeks, giving rise to human immune T and B-lymphocytes as well as monocytes and dendritic cells repopulating peripheral blood, spleen, and lymph nodes (Supplementary Figure S2).
We next tested whether human immune cells in NSG-HIS mice were sensitive to the effects of motolimod. Compared to untreated controls, mice treated with motolimod at 1.5 or 15 mg/m2 showed a dose-dependent increase in plasma levels of human cytokines and chemokines, including IL-6, IL-12p70, TNFα, MCP-1, and MIP-1β 6 hours following treatment that declined to baseline by 24 hours (Figure 1A). Although no individual cytokines were significantly increased (due to the small sample size), a mixed-effect linear model combining all analytes showed a significant treatment effect at the highest dose (p=0.01). Consistent with activation of human monocytes in vivo, motolimod upregulated the co-stimulatory markers CD86 in human CD14+ and CD11c+ cells harvested from NSG-HIS spleens (Figure 1B and C).
Figure 1.
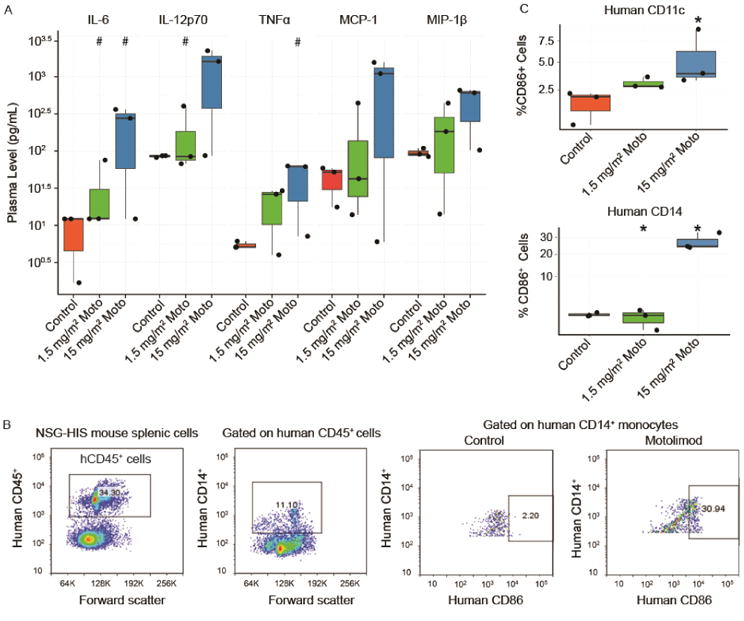
Motolimod induces human immune cell activation in the NSG-HIS mouse. (A) NSG-HIS mice (n=3 mice/group) were administered vehicle (control) or motolimod at 1.5 or 15 mg/m2. Plasma samples were obtained 6 hours after dosing and analyzed by HumanMAP® assay. A mixed-effect linear model combining all analytes showed a significant treatment effect at the highest dose (p≤0.01). Representative cytokines and chemokines are displayed. Increase(s) that approached but did not achieve statistical significance with respect to the control (p≤0.1; one-sided Wilcoxon rank-sum test) are identified with (#). (B) Representative images of flow cytometry analysis of human immune cells collected from NSG-HIS a spleen 6 hours after administration of vehicle (control) or motolimod 1.5 or 15 mg/m2 shows upregulation of the costimulatory marker hCD86 in hCD14+ spleen cells following treatment with motolimod. (C) Significant upregulation of hCD86 in hCD14+ and hCD11c+ cells in NSG-HIS mouse spleens 6 hours after dosing with motolimod 15 mg/m2 (n=3 mice/group). Significant increase(s) with respect to the control (p≤0.05; one-sided Wilcoxon rank-sum test) are identified with an asterisk (*).
To further assess the suitability of the NSG-HIS mouse model, we conducted comparative pharmacodynamic studies assessing the in vivo plasma biomarker response to motolimod in NSG-HIS mice, non-human primates and healthy human volunteers. Doses that were proven to be safe and biologically active for each species were used (10, 17). A dose-dependent increase in cytokines and chemokines indicative of TLR8-induced monocyte activation— including IL-6, MCP-1, and MIP-1β—were observed in all species (Figure 2). Collectively, these data demonstrate that NSG-HIS mice exhibit sensitivity to TLR8 stimulation similar to that of non-human primates and humans.
Figure 2.
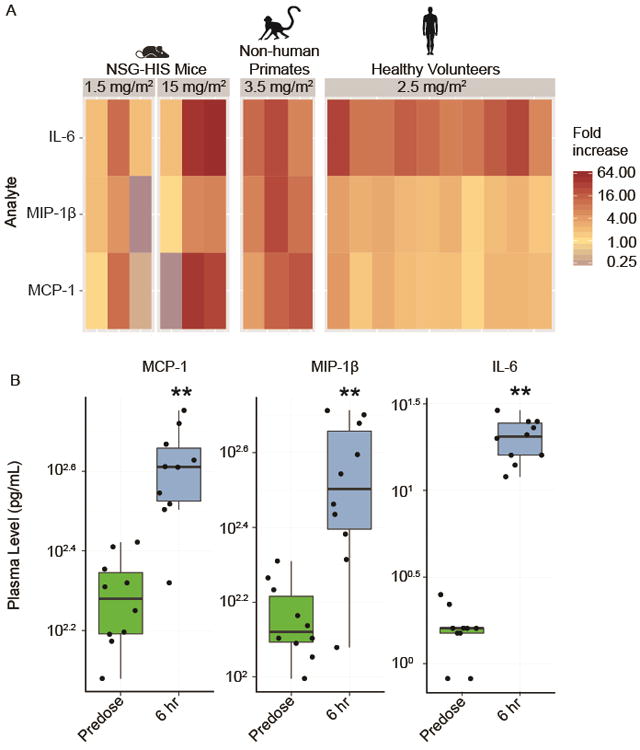
The NSG-HIS mouse model is suitable for the preclinical assessments of motolimod. (A) The in vivo plasma biomarker response to motolimod, administered at the respective doses shown, measured at 6 hours, is compared across NSG-HIS mice (n=3/group), non-human primates (n=3/group), and healthy human volunteers (n=10/group; 5 males and 5 females). Multiple cytokines and chemokines indicative of TLR8 stimulation and immune activation were evaluated using HumanMAP®. Representative analytes showing fold increase changes are shown. (B) In healthy volunteers (n=10), significant upregulation in several analytes is shown 6 hours after dosing with motolimod (p≤0.01, one-sided Wilcoxon signed-rank test) as denoted by asterisks (**).
PLD and motolimod achieve potent immunomodulation in NSG-HIS mice
Given the immunomodulatory effects of doxorubicin (22, 23) and prior evidence of positive interactions with innate immunity activation (24), we next tested whether concomitant administration of PLD would attenuate motolimod-induced TLR8 activation in vivo. NSG-HIS mice were administered IP PLD once followed by SC motolimod on the same day or 2–5 days later. Whether a concurrent or sequential dose schedule was utilized, the acute response in plasma cytokines and chemokines and in human monocytes that was previously observed with motolimod monotherapy was not significantly attenuated by the addition of PLD (Figure 3A and Supplementary Figure S3). Given the slow release of doxorubicin in the TME and to maximize the timing of drug interactions, we adopted a staggered schedule for all subsequent murine experiments, with administration of PLD (50 mg/m2) on day 1, followed by motolimod at either 1.5 or 15 mg/m2 on days 5, 7, and 9 of each 14-day cycle. This combination was administered for 3 cycles. In pilot experiments, both dose levels of motolimod were biologically active, eliciting cytokine and chemokine responses comparable to those shown in Figures 1 and 2, with no acute toxicities. The 1.5 mg/m2 dose level was selected for use in our subsequent studies as it more closely approximated the relevant human dose levels identified in a previous phase 1 study of motolimod (10).
Figure 3.
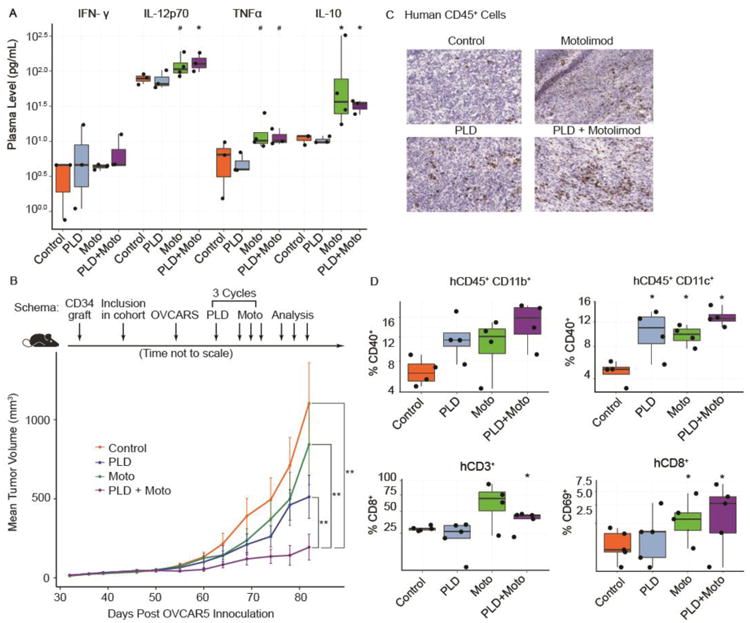
Preclinical studies of motolimod (Moto) plus pegylated liposomal doxorubicin (PLD) in NSG-HIS mice. (A) NSG-HIS mice were given either PLD alone (intraperitoneally, 50 mg/m2), motolimod alone (subcutaneously 2 days after PLD dosing, 1.5 mg/m2), the combination, or vehicle control. Plasma was collected 6 hours after dosing and levels of cytokines and chemokines were assessed using the HumanMAP® panel. Select biomarkers are shown. Symbols denote p-values compared to control (*=p≤0.05, #=p≤0.1; 1-sided Wilcoxon rank-sum test). (B) Tumor-bearing NSG-HIS mice inoculated with the human ovarian cancer cell line OVCAR5 were randomized to receive vehicle control, PLD, Moto, or the combination. Tumor growth was assessed at multiple time points post-inoculation. The combination of PLD + Moto showed highly significant improvement compared to control, PLD, and Moto. Two of the 10 mice in the PLD + Moto arm had no measurable tumors at the last 2 time points. Two asterisks (**) denote p≤0.01; 2-sided log-linear mixed-effect model. (C) IHC was used to assess the presence of human (h)CD45+ tumor infiltrating leukocytes. (D) The combination of PLD plus motolimod resulted in significant increases in hCD11b+ and hCD11c+ cells expressing hCD40 (top panel). The combination also yielded increases in the frequency of hCD8+ T cells expressing hCD69 (bottom panel). Treatment group(s) (n=5/group) with significant magnitude increase(s) with respect to the control (p≤0.05; 1-sided Wilcoxon rank-sum test) are identified with an asterisk (*).
PLD plus motolimod effectively control tumors in NSG-HIS mice
The potential anti-tumor effects of PLD plus motolimod in ovarian cancer was next evaluated using NSG-HIS mice bearing HLA-matched human ovarian tumor / human bone marrow chimeric xenografts. NSG-HIS mice reconstituted with CD34+ cord blood cells from HLA-A2+ donor neonates (NSG-HIS-A2) were inoculated with subcutaneous HLA-A2+ OVCAR5 tumors. Tumor grafts were accepted at a rate > 95% and treatment began when tumors were approximately 50 mm3 (approximately day 28). As expected, mice treated with PLD alone showed reduced tumor growth compared to untreated animals. Motolimod alone did not significantly affect tumor growth relative to untreated mice. However, the combination of PLD and motolimod markedly reduced tumor growth compared to either agent alone, including 2 of 10 mice who experienced complete disappearance of their tumors (Figure 3B).
To test whether the drugs mobilized immunity against tumors, we examined tumors for overall human CD45+ leukocyte infiltration. Rare human CD45+ cells were detected in OVCAR5 tumors of untreated mice by immunohistochemical (IHC) analysis, indicating a baseline low-level tumor inflammation. Mice treated with either PLD or motolimod monotherapy or with the combination exhibited a marked increase in CD45+ cells (Figure 3C). Consistent with activation of human tumor macrophages and DCs, there was an increase in the frequency of CD40-expressing human CD11b+ and CD11c+ cells in tumors following treatment with either drug alone or the combination relative to controls (Figure 3D, top). An increase in human CD8+ TILs was also seen in mice receiving motolimod alone or motolimod in combination with PLD, while activation of CD8+ T lymphocytes, judged by the upregulation of CD69, was noted following treatment with motolimod in combination with PLD (Figure 3D, bottom). Thus, the addition of PLD did not attenuate the effect of motolimod, and together the two drugs activated tumor-infiltrating monocytes and T cells, which was associated with significant tumor control.
PLD plus motolimod cooperate in antitumor innate and adaptive immunity mechanisms
Activated macrophages secrete high amounts of TNFα, which can induce apoptosis in susceptible cells expressing the TNF receptor 1 (TNFR1) (25). We confirmed by western blot that OVCAR5 cells express TNFR1 (Supplementary Figure S4). Given the potent activation of tumor macrophages by PLD plus motolimod, we hypothesized that the therapeutic interaction could be partly explained by PLD rendering tumor cells susceptible to death induced by TNFα. In agreement with prior observations (26), OVCAR5 cells were initially resistant to TNFα even at high levels (Figure 4A). However, cells pre-exposed to PLD became sensitive to TNFα killing in vitro. Consistent with their increased susceptibility to TNFα-induced cell death, and in agreement with prior evidence (27), treatment of OVCAR5 cells with PLD significantly reduced cellular FADD-like IL-1β-converting enzyme-inhibitory protein (c-FLIP), an inhibitor of procaspase-8 recruitment, which prevents apoptosis induced by TNF family death ligands (Figure 4B) (28).
Figure 4.
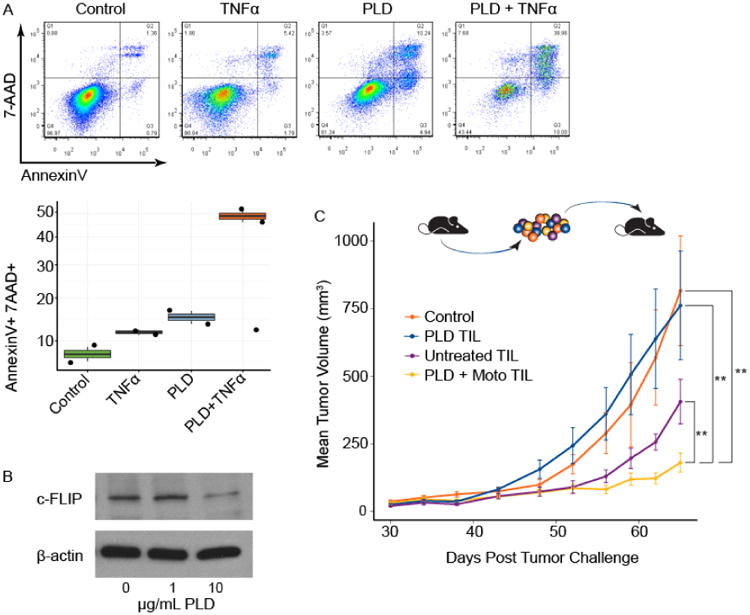
Innate and adaptive immune-mediated interactions underlie the effect of the motolimod plus PLD combination. (A) OVCAR5 cells were evaluated in vitro for sensitivity to TNFα. Cells were left untreated (control) or were exposed to TNFα or PLD alone, or were exposed to TNFα after pre-exposure to PLD. After treatment, cell death was measured using a flow cytometry-based annexin V/7-AAD staining protocol. For each treatment, representative flow cytometry plots are shown with corresponding quantitative measurements of the percentage of apoptotic and necrotic cells. (B) Western blot analysis of c-FLIP expression in isolated OVCAR5 tumor cells at baseline and after treatment with PLD at 1 or 10 μg/ml. (C) To test the ability of tumor-infiltrating lymphocytes (TILs) to restrict tumor growth in NSG-HIS-A2 mice, ex vivo expanded TILs from mice previously treated with PLD alone (PLD TIL), PLD + motolimod (PLD + Moto TIL) or from untreated mice (Untreated TIL) were transferred adoptively to treatment-naïve NSG-HIS-A2 mice bearing established OVCAR5 tumors (n=10/group). NSG-HIS-A2 mice that received no TILs served as the control. Tumor volume was assessed periodically post transfer; animals receiving PLD + motolimod TIL had highly significant reduction in tumor volume compared with control, PLD alone and Untreated TIL. Two asterisks (**) denote p≤0.01; 2-sided log-linear mixed-effect model.
In view of the observed increase in activated human CD8+ TILs following PLD plus motolimod, we also asked whether TILs were in part responsible for the tumor growth restriction in this model. Human TILs were expanded ex vivo from each treatment group using IL-2 and tested against OVCAR5 cells, using TILs from the same human donor in each experiment. Consistent with the significant CD69 upregulation seen in TILs following combination treatment, TILs from mice treated with PLD plus motolimod exhibited high levels of cytotoxicity against OVCAR cells ex vivo, while TILs from mice treated with monotherapy exhibited modest cytotoxicity by chromium release assay ex vivo (Supplementary Figure S5). There was no cytotoxicity from TILs expanded from control mice. These results were consistent among different human CD34+ cell donors. To further test the contribution of TILs to restrict tumor growth, TILs were harvested from tumors at the end of treatment from each experimental group and from control mice, expanded ex vivo using IL-2, and adoptively transferred to treatment-naïve NSG-HIS-A2 mice bearing established OVCAR5 tumors. TILs from animals treated with PLD plus motolimod conferred a highly significant anti-tumor effect (Figure 4C), suggesting that the increased efficacy of the PLD plus motolimod combination was in part also due to the cytolytic activity of TILs.
Translation of the PLD plus motolimod combination into the clinic
Based on the above preclinical data, a phase 1b study of PLD plus motolimod was initiated in women with recurrent or persistent ovarian cancer following platinum-based chemotherapy (Study GOG-9925). A standard dose-escalation design was used to evaluate the combination of 40 mg/m2 PLD with 3 dose levels of motolimod (2.5, 3.0, and 3.5 mg/m2), which were selected based on the results of a previous phase 1 dose-seeking study of single-agent motolimod (doses of 0.1 to 3.9 mg/m2) in adults with advanced cancer (10). PLD was administered IV on day 1 of a 28-day cycle, followed by SC motolimod on days 3, 10, and 17. Cycles of therapy were repeated until disease progression or unacceptable toxicity.
Following enrollment of the first and second cohorts (PLD + 2.5 mg/m2 motolimod [n=3], and PLD + 3.0 mg/m2 motolimod [n=3], respectively), study accrual was suspended due to a global shortage in the commercial supply of PLD. After a 6-month disruption, PLD availability returned and enrollment proceeded (cohort 3; PLD + 3.5 mg/m2 motolimod [n=7]). During the interim absence of PLD, there was a need to identify alternate combinations wherein motolimod could augment the activity of existing chemotherapeutics. Because paclitaxel is an alternate standard-of-care chemotherapy for this patient population, the protocol was amended to include a second treatment arm evaluating motolimod plus paclitaxel. In this regimen, a single-dose level of motolimod (3.0 mg/m2) was combined with paclitaxel (80 mg/m2). Both agents were administered on days 1, 8, and 15 of a 28-day cycle. Cycles were repeated until disease progression or unacceptable toxicity.
Twenty subjects were evaluated on Study GOG-9925: 13 treated with PLD plus motolimod, and 7 treated with paclitaxel plus motolimod. The majority of participants (n=16, 80%) were Caucasian and aged 50–69 years (80%) (Table S1).
Motolimod in combination with PLD or paclitaxel was generally safe and well tolerated. There were no DLTs and no serious, unexpected drug-related adverse events. There was no evidence of toxicity interactions between motolimod and PLD or motolimod and paclitaxel. No significant drug-related gastrointestinal, neurologic, or cardiac toxicities were observed. Clinical tolerability of motolimod was better at the 2.5 and 3.0 mg/m2 dose levels compared to 3.5 mg/m2. The most common AEs on each treatment regimen are provided in Table 1. Eleven of the 20 subjects experienced at least 1 AE ≥ grade 3 in severity; AEs in 7 of these subjects were considered related to combination therapy. Treatment-related events ≥ grade 3 observed in subjects receiving PLD plus motolimod included: neutropenia (n=3); anemia (n=2); mucositis oral (n=2); lymphopenia, palmar-plantar erythrodysesthesia syndrome, urinary tract infection, and leukopenia (n=1 each); and chills, fever, and vomiting (all in one subject). All of these events were attributed to PLD except for the chills, fever and vomiting. Treatment-related events ≥ grade 3 observed in subjects receiving paclitaxel plus motolimod included: bladder infection, fatigue, febrile neutropenia, neutropenia, and leukopenia (all reported in 1 subject and attributed to paclitaxel).
Table 1. Adverse Events and Laboratory Abnormalities Reported in ≥ 20% of Subjects.
| PLD + motolimod n = 13 | paclitaxel + motolimod n = 7 | |||
|---|---|---|---|---|
|
| ||||
| Adverse Event | All Grades n (%) | Grade 3/4 n (%) | All Grades n (%) | Grade 3/4 n (%) |
| Abdominal pain | 7 (54) | 2 (15) | 3 (49) | 0 (0) |
| Alopecia | 3 (23) | 0 (0) | 2 (29) | 0 (0) |
| Anemia | 10 (77) | 2 (15) | 7 (100) | 0 (0) |
| Anorexia | 6 (46) | 0 (0) | 1 (14) | 0 (0) |
| Anxiety | 3 (23) | 0 (0) | 1 (14) | 0 (0) |
| Arthralgia | 6 (46) | 0 (0) | 3 (43) | 0 (0) |
| Back pain | 5 (39) | 0 (0) | 0 (0) | 0 (0) |
| Chills | 9 (69) | 1 (8) | 3 (49) | 0 (0) |
| Constipation | 7 (54) | 0 (0) | 2 (29) | 0 (0) |
| Cough | 3 (23) | 1 (8) | 2 (29) | 0 (0) |
| Diarrhea | 5 (39) | 0 (0) | 4 (57) | 1 (14) |
| Dyspnea | 4 (31) | 1 (8) | 3 (43) | 0 (0) |
| Fatigue | 13 (100) | 0 (0) | 7 (100) | 1 (14) |
| Fever | 11 (85) | 1 (8) | 3 (43) | 0 (0) |
| Headache | 9 (69) | 0 (0) | 1 (14) | 0 (0) |
| Hypertension | 4 (31) | 1 (8) | 1 (14) | 1 (14) |
| Injection site reaction | 12 (92) | 0 (0) | 5 (71) | 0 (0) |
| Mucositis oral | 7 (54) | 2 (15) | 0 (0) | 0 (0) |
| Myalgia | 2 (15) | 0 (0) | 2 (29) | 0 (0) |
| Nausea | 10 (77) | 1 (8) | 4 (57) | 0 (0) |
| Palmar-plantar erythrodysesthesia syndrome | 4 (31) | 1 (8) | 0 (0) | 0 (0) |
| Paresthesia | 4 (31) | 0 (0) | 0 (0.0) | 0 (0) |
| Peripheral sensory neuropathy | 6 (46) | 0 (0) | 4 (57) | 0 (0) |
| Vomiting | 10 (77) | 2 (15) | 5 (71) | 0 (0) |
| Weight loss | 2 (15) | 0 (0) | 2 (29) | 0 (0) |
| Laboratory Abnormality | ||||
| Hypoalbuminemia | 3 (23) | 2 (15) | 2 (29) | 0 (0) |
| Hypokalemia | 2 (15) | 1 (8) | 2 (29) | 0 (0) |
| Hyponatremia | 2 (15) | 1 (8) | 2 (29) | 1 (14) |
| Platelet count decreased | 4 (31) | 1 (8) | 0 (0) | 0 (0) |
| Neutrophil count decreased | 8 (62) | 3 (23) | 1 (14) | 1 (14) |
| White blood cell decreased | 8 (62) | 1 (8) | 1 (14) | 1 (14) |
Adverse events were graded according to the Common Terminology Criteria for Adverse Events (CTCAE).
The biological effects of PLD plus motolimod are similar in patients with ovarian cancer and NSG-HIS mice
Standard pharmacokinetic (PK) evaluations of each drug were performed to rule out any alternations in the metabolism and elimination due to drug-drug interactions. In both regimens, PK parameters associated with motolimod were within the range of previously reported values (Supplementary Figure S6A). Exposure to PLD was consistent with historical data (Supplementary Figure S6B). Plasma levels of paclitaxel were also within the expected range.
The pharmacodynamic effect of motolimod in combination with PLD or paclitaxel was evaluated in ovarian cancer patients by quantitative assessment of more than 90 cytokines, chemokines, and other immune inflammatory mediators. These data confirmed that motolimod was biologically active when combined with either PLD or paclitaxel, with the induction of multiple inflammatory biomarkers indicative of TLR8 stimulation and immune activation. Motolimod was biologically active at all 3 dose levels tested (Figure 5A). Specifically, motolimod plus PLD increased plasma levels of G-CSF, IL-6, MCP-1, and MIP-1β at one or more of the motolimod dose levels. Importantly, the magnitude of biomarker response was maintained over multiple treatments over 2 cycles, with no evidence of augmentation or attenuation (Figure 5B). Results with the paclitaxel regimen were similar (Supplementary Figure S7). Taken together, these results indicated that, as seen in the NSG-HIS mouse, the combination of cytotoxic chemotherapy plus motolimod did not attenuate the immunostimulatory effects of motolimod in humans.
Figure 5.
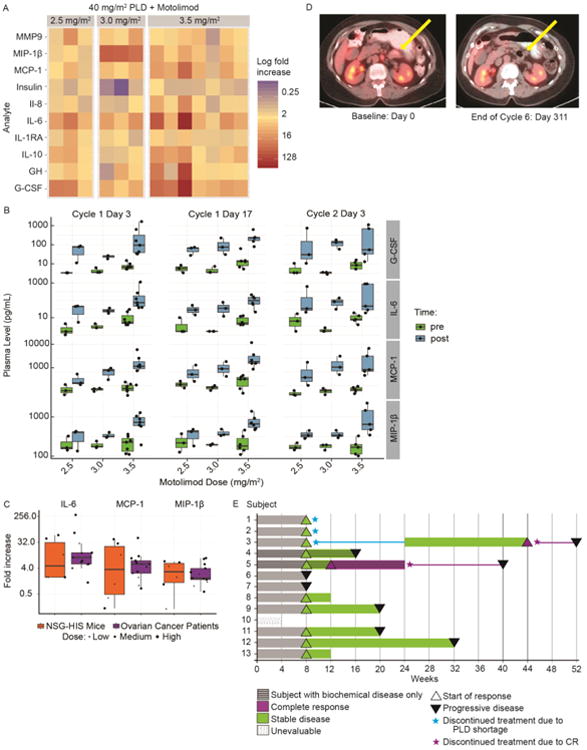
Biological effects of motolimod in humans. The in vivo cytokine response to motolimod was assessed in women with advanced ovarian cancer who were treated with 40 mg/m2 of PLD in combination with motolimod at 2.5 (n=3), 3.0 (n=3), or 3.5 mg/m2 (n=7). Plasma was collected prior to the initial dose of motolimod and 8 hours post-dose, and levels of selected cytokines and chemokines were assessed using the HumanMAP® panel. (A) Immune mediator concentrations were log (base 2) transformed, and fold changes were calculated for each analyte level by dividing the post-dose value by the corresponding value from the pre-dose sample. The corresponding heat map for mediators exhibiting a mean log fold increase of ≥ 1.3 over baseline is shown. (B) Mediators that broadly demonstrated a dose-related increase of ≥ 1.3-fold at 1 or more of the motolimod dose levels included G-CSF, IL-6, MIP-1β, and MCP-1, and this response was maintained over two treatment cycles. (C) To demonstrate the translational relevance of the anti-tumor response seen in NSG-HIS mice treated with the combination of PLD + motolimod, the murine plasma mediator response was compared to that observed in clinical participants. Many of the mediators induced in the plasma of women with ovarian cancer (n=13) were also induced in NSG-HIS mice (n=6) following motolimod administration. Overall, the magnitude of the mediator response (fold increase with respect to baseline/control) in mice dosed with 15 mg/m2 motolimod was comparable to what was seen in ovarian subjects at the 3.5 mg/m2 dose.
Response following PLD + motolimod in recurrent stage 3c ovarian cancer. (D) This subject with recurrent non-target (non-measurable) disease was enrolled on study GOG-9925 and received PLD 40 mg/m2 (day 1), followed by 2.5 mg/m2 of motolimod (days 3, 10, and 17) for 2 cycles without major incident. Her disease remained stable during a 5-month treatment-free interval necessitated by a shortage in the global supply of PLD. Thereafter she received 4 additional cycles of PLD plus motolimod and achieved a complete response (no evidence of disease). (E) Swim plot illustrating the course of the 13 subjects who received the PLD plus motolimod combination.
Next, we conducted comparative pharmacodynamic assessments in women enrolled in the GOG-9925 study and in NSG-HIS mice bearing ovarian tumor xenografts treated with PLD plus motolimod. In both clinical study participants and NSG-HIS mice, plasma levels of IL-6, MCP-1, and MIP-1β increased in a dose-related manner in response to motolimod (Figure 5C). The magnitude of the pharmacodynamic effects seen in NSG-HIS mice dosed with 15 mg/m2 motolimod plus PLD was comparable to the levels seen in study participants who received 3.5 mg/m2 motolimod plus PLD.
Clinical results with the combination of PLD plus motolimod
The antitumor activity of the PLD/motolimod combination was assessed as an exploratory objective of the GOG-9925 study. At the time of study entry, subjects had evidence of recurrent disease either by biochemical criteria (elevated CA-125) or by imaging (measurable or non-measurable disease on CT or MRI). Of the 13 subjects treated with PLD plus motolimod, one subject with non-measurable disease treated with 2.5 mg/m2 of motolimod achieved a complete response (CR) (Figure 5D). Interestingly, this subject received PLD plus 2.5 mg/m2 motolimod for two cycles on study, with SD and interval improvement on interval imaging. Due to the shortage of PLD, she was unable to receive additional study drugs and elected to forego additional anticancer therapy. A CA125 level within normal range by cycle 4 and a repeat PET/CT confirming ongoing stabilization of disease 5 months later in the absence of therapy suggest a durable benefit from the two cycles of PLD/motolimod. She subsequently received 4 additional cycles of PLD plus motolimod (6 cycles total) and achieved a CR, then elected to discontinue study treatment. Post-study follow-up showed the CR was maintained for 10 weeks without additional therapy. (Figure 5D). Another subject enrolled based on biochemical evidence of disease (see Methods) and treated at the 3.0 mg/m2 dose level achieved a biochemical CR after 3 cycles. She continued treatment for 3 additional cycles, for a total of 6 cycles, then elected to go off treatment. The CR endured for 16 additional weeks without anticancer therapy. Moreover, stable disease (SD) was seen in 8 subjects, including 1 subject with biochemical disease only; 3 additional subjects had progressive disease (PD) (Figure 5E). Overall, the response rate of subjects treated with PLD plus motolimod was 15% (n=2) and the disease control rate (DCR) was 69% (n=9; Table S2). Of the 7 subjects who received paclitaxel plus motolimod, one achieved a partial response (PR), 2 experienced SD, and 4 had PD, with a DCR of 43% (n=3; Table S2). Notably, the 2 subjects who achieved CR with PLD plus motolimod and the subject who achieved a PR with paclitaxel and motolimod achieved their best response after receiving ≥3 cycles of therapy.
Discussion
Cancer immunotherapy has resulted in major successes in a variety of solid tumors. Durable objective responses and long-term survival benefits have been achieved with T cell-based adoptive therapy as well as using antibodies that activate T cells in vivo by blocking key lymphocyte inhibitory receptors (29). Although the field of T cell activation has significantly advanced, activation of innate immunity still remains an elusive yet key goal of immunotherapy.
TLR8 is an innate sensor that may facilitate the transition from innate to adaptive immune responses owing to its ability to activate monocytes and mDC. Due to phylogenetic differences in TLR8 and the relative lack of activity in mice, TLR8 agonists have not been evaluated as cancer therapies to date. We therefore sought to assess for the first time, the application of a novel small molecule TLR8 agonist, motolimod, in combination with immunogenic chemotherapy using a humanized mouse model. Comparative pharmacodynamic studies confirmed that the human immune system in non-tumor bearing NSG-HIS mice responds to motolimod in a similar manner to non-human primates and normal human volunteers, with clear dose-dependent activation of plasma and immune cell biomarkers. Having identified an active dose range that closely recapitulated the optimal dose identified in a phase 1 study in cancer patients (10), we examined the combination of motolimod with PLD, a cytotoxic drug with favorable pharmacokinetic and immunomodulatory profiles. The NSG-HIS model proved a clinically relevant system for evaluating human immunotherapies such as motolimod and helped determine the pharmacodynamic effects and test the antitumor efficacy of the combination.
Clinical development of immunomodulatory agents that recognize human targets but not their murine counterparts is challenging. Although the NSG-HIS-A2 tumor model has limitations related to the partial HLA mismatch and multiple minor H mismatching between bone marrow and tumor allograft, it offers an acceptable immunocompetent surrogate mouse model with which to examine the effects of therapy on the TME. In spite of the potential allo-recognition, tumors engrafted successfully and grew in NSG-HIS-A2 mice, indicating local tolerance mechanisms enacted by the tumor. These tolerance mechanisms were effectively disrupted by the combination of PLD plus motolimod, which resulted in significant activation of tumor-infiltrating CD11b+ monocytes and CD11c+ DCs, which in turn were associated with activation of allo-reactive CD8+ T cells. A concomitant elevation of human Th1-polarizing and other inflammatory cytokines in the plasma documented effective systemic activation of immune cells with the PLD/motolimod combination, which preserved the potency of single agent motolimod therapy by eliciting IL-12 and TNFα responses. Additional distinct favorable features of the combination included enhanced IFNγ and lower levels of IL-10 relative to motolimod monotherapy.
Activated TILs were in part responsible for the observed therapeutic effect of the PLD/motolimod combination in this mouse model, which shows that PLD plus motolimod significantly reprogrammed the TME through activation of monocytes to overcome local T cell tolerance. It is also possible that tumor restriction was mediated in part by activated macrophages. Our in vitro experiments demonstrated a synergistic interaction between TNFα, a cytokine released by activated macrophages (and effector T cells) and PLD. Pretreatment of tumor cells with PLD was required to induce susceptibility to TNFα-mediated death, which was explained by down-regulation of c-FLIP induced in tumor cells stressed by PLD. An additional interaction could entail the upregulation of the pro-apoptotic molecule Bfk by both TNFα and cytotoxic chemotherapy (30). These results prove the principle of a positive interaction of the combination due to independent effects of PLD and motolimod on tumor cells and immune effector cells respectively. It is likely that in tumors TNFα is derived from both activated tumor monocytes as well as activated effector TILs.
Additional and complementary positive immunomodulatory mechanisms may be envisioned in human tumors in vivo, including the induction of immunogenic cell death induced by PLD, which could further contribute to proper antigen presentation by DCs, the production of T-cell recruiting chemokines by activated tumor macrophages, and a possible reprogramming of additional immunosuppressive monocyte populations such as myeloid-derived suppressive cells (MDSC), which should be further investigated if the combination proves effective in the clinic.
Based on the promising preclinical results, we developed the GOG-9925 phase 1 clinical study, adopting a phased administration schedule with motolimod initiated 2 days after PLD in each cycle. This schedule was selected to maximize the PLD-associated antigen-shedding effects on the tumor. Despite the absence of DLTs, the clinical tolerability of motolimod was considered better at 3.0 mg/m2 compared to 3.5 mg/m2. Since all dose levels were associated with robust pharmacodynamic responses, and evidence of antitumor activity was observed at the 2.5 and 3.0 mg/m2 dose levels, 3.0 mg/m2 was selected as the recommended phase 2 dose. Similar to observations in the mouse, PLD did not attenuate the immune signature of motolimod in patients. Similar biomarker responses were observed in ovarian cancer subjects as in the NSG-HIS mice, indicating the accuracy of pharmacodynamics predictions in the NSG-HIS mouse. Notable adverse events including thrombocytopenia, palmar-plantar erythrodysesthesia syndrome, and peripheral sensory neuropathy were typically attributed to PLD and/or prior therapy rather than to motolimod and were generally grade 1 or 2 in severity. Furthermore, the incidence of these events was similar to those reported in other trials of PLD in this setting (31-33). Finally, the clinical benefit observed with PLD plus motolimod suggests that the combination may improve clinical outcomes. The time to best response following treatment with motolimod plus chemotherapy was consistent with reports of delayed clinical response that have been observed with other immunotherapies such as ipilimumab (34, 35).
Although the Phase 1 safety data are encouraging, it is important to note that PLD shortage during the study likely limited the understanding of the clinical effect of the chemo-immunotherapy combination, and may have tainted the toxicity endpoints, as PLD is known to have delayed toxicity as well as efficacy. Based on these preclinical and initial clinical data, a randomized, placebo-controlled trial comparing PLD to PLD plus motolimod in platinum-resistant ovarian cancer was initiated (GOG-3003, NCT01666444). This trial enrolled nearly 300 women; data for the primary endpoint of overall survival are expected before the end of 2016. In addition, the immune effects elicited by the combination suggest that PLD/motolimod chemo-immunotherapy could be effectively combined with approaches further enhancing T cell function, such as checkpoint blockade immunotherapy. This hypothesis is being tested in a phase 1/2a study assessing the PLD/motolimod combination with an anti-PD-L1 neutralizing antibody in the same patient population (NCT02431559).
In summary, we followed an integrative pharmacologic approach that used traditional approaches, as well as a humanized mouse model to advance the clinical development of motolimod for cancer therapy. The NSG-HIS model mimicked human donors and non-human primates and proved useful in exploring combinations of motolimod with chemotherapy drugs, allowing identification of appropriate combination schedules and dissecting interactions at the level of the TME. In spite of its limitations, this model offers important advantages including experimental throughput and cost containment relative to non-human primates. In addition, this model solves a major limitation in cancer drug development by allowing assessment of the effects of drug combinations on the human peripheral immune system and in the TME. Further optimization of mice reconstituted with a human immune system and bearing human tumor grafts can be envisioned, including the use of autologous tumor and peripheral HSCs to obviate current limitations from alloreactive graft models.
Supplementary Material
Statement of Translational Relevance.
An integrative pharmacologic approach was used to identify a rational treatment approach for ovarian cancer by combining motolimod—a Tolllike receptor 8 (TLR8) agonist that stimulates human (but not mouse) innate immune responses—with pegylated liposomal doxorubicin (PLD), a chemotherapeutic drug that induces immunogenic cell death. Multi-species experiments were performed using human volunteers, non-human primates, NSG-HIS (“humanized immune system”) mice reconstituted with human CD34+ cells, and ovarian cancer patients. Study results demonstrated the pharmacodynamic effects of motolimod and PLD as well as providing insight into the mechanism of action of the combination and the impact on tumor growth. This integrative approach offers a new innovation to the rapidly expanding domain of immuno-oncology by providing additional methods to enhance clinical development of immunomodulatory agents with human specificity. The findings also have broad implications for the utility of NSG-HIS mice in clinical and translational human immunology.
Acknowledgments
We thank Gregory N. Dietsch, PhD, for providing the analysis of pharmacokinetic and pharmacodynamic samples, Raphael Gottardo, PhD, for providing statistical analyses of the preclinical data and Eilidh Williamson, PhD, for providing medical writing assistance.
Financial Support: This study was supported by National Cancer Institute grants: GOG Tissue Bank (U10 CA27469, U24 CA114793, and NRG Oncology Operations grant number U10 CA180868) as well as NRG SDMC grant U10 CA180822,) and the GOG Statistical and Data Center (CA37517). GOG-9925 was an NRG Oncology and Gynecologic Oncology Group study. Support was also provided by VentiRx Pharmaceuticals, Seattle, WA.
George Coukos has received research funding from VentiRx Pharmaceuticals, Boeringer-Ingelheim and Celgene and has a patent related to motolimod. Gwenn-äel Danet-Desnoyers has received research funding from Janssen Research and Development, Amgen, and Gilead and has a patent related to daratumamab. Gregory N. Dietsch, Kristi L. Manjarrez, and Robert M. Hershberg are employees of, and have equity ownership in, VentiRx Pharmaceuticals, and each have patents related to motolimod. Dr. Hershberg has received income from Celgene and Nanostring Technologies. Andrea Facciabene has a patent related to motolimod. Paula M. Fracasso is an employee of, and has equity ownership in, Bristol Myers Squibb. Bradley J. Monk has received income from Roche/Genentech, AstraZeneca, and Pfizer, and research support from Amgen, Genentech, Lilly, Array Biopharma, TESARO, and Morphotek. Joan L. Walker has received research support from Abbott Laboratories and Precision Therapeutics, Inc.
Footnotes
Potential Conflicts of Interest: Carol Aghajanian, William E. Brady, Heather A. Lankes, Katherine M. Bell-McGuinn, Alexander Malykhin, and Carolyn K. McCourt have no conflicts of interest to disclose.
References
- 1.Casares N, P M, Tesniere A, Ghiringhelli F, Roux S, Chaput N, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005;202:1691–701. doi: 10.1084/jem.20050915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zitvogel L, Kepp O, Senovilla L, Menger L, Chaput N, Kroemer G. Immunogenic tumor cell death for optimal anticancer therapy: the calreticulin exposure pathway. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16(12):3100–4. doi: 10.1158/1078-0432.CCR-09-2891. [DOI] [PubMed] [Google Scholar]
- 3.Aguilar-Cazares D, Meneses-Flores M, Prado-Garcia H, Islas-Vazquez L, Rojo-Leon V, Romero-Garcia S, et al. Relationship of dendritic cell density, HMGB1 expression, and tumor-infiltrating lymphocytes in non-small cell lung carcinomas. Applied immunohistochemistry & molecular morphology : AIMM / official publication of the Society for Applied Immunohistochemistry. 2014;22(2):105–13. doi: 10.1097/PAI.0b013e3182849808. [DOI] [PubMed] [Google Scholar]
- 4.Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13(9):1050–9. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 5.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–68. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O'Neill LA, Golenbock D, Bowie AG. The history of Toll-like receptors - redefining innate immunity. Nat Rev Immunol. 2013;13(6):453–60. doi: 10.1038/nri3446. [DOI] [PubMed] [Google Scholar]
- 7.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5(10):987–95. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 8.Jin Liu, C X, Li-Chung Hsu, Yunping Luo, Rong Xiang, Tsung-Hsien Chuang. A five-amino-acid motif in the undefined region of the TLR8 ectodomain is required for species-specific ligand recognition. Molecular Immunology. 2010;47:1083–90. doi: 10.1016/j.molimm.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu H, D G, et al. VTX-2337 is a novel TLR8 agonist that activates NK cells and augments ADCC. Clin Cancer Res. 2012;18:499–509. doi: 10.1158/1078-0432.CCR-11-1625. [DOI] [PubMed] [Google Scholar]
- 10.Northfelt DW, Ramanathan RK, Cohen PA, Von Hoff DD, Weiss GJ, Dietsch GN, et al. A phase I dose-finding study of the novel Toll-like receptor 8 agonist VTX-2337 in adult subjects with advanced solid tumors or lymphoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20(14):3683–91. doi: 10.1158/1078-0432.CCR-14-0392. [DOI] [PubMed] [Google Scholar]
- 11.Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of pegylated liposomal Doxorubicin: review of animal and human studies. Clinical pharmacokinetics. 2003;42(5):419–36. doi: 10.2165/00003088-200342050-00002. [DOI] [PubMed] [Google Scholar]
- 12.Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348(3):203–13. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 13.Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A. 2005;102(51):18538–43. doi: 10.1073/pnas.0509182102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–9. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 15.Coleman RL, Monk BJ, Sood AK, Herzog TJ. Latest research and treatment of advanced-stage epithelial ovarian cancer. Nature reviews Clinical oncology. 2013;10(4):211–24. doi: 10.1038/nrclinonc.2013.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Govindaraj RG, Manavalan B, Basith S, Choi S. Comparative analysis of species-specific ligand recognition in Toll-like receptor 8 signaling: a hypothesis. PLoS One. 2011;6(9):e25118. doi: 10.1371/journal.pone.0025118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dietsch GN, Randall TD, Gottardo R, Northfelt DW, Ramanathan RK, Cohen PA, et al. Late-Stage Cancer Patients Remain Highly Responsive to Immune Activation by the Selective TLR8 Agonist Motolimod (VTX-2337) Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21(24):5445–52. doi: 10.1158/1078-0432.CCR-15-0578. [DOI] [PubMed] [Google Scholar]
- 18.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, et al. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388(6638):190–5. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 19.Le Tourneau C, Lee JJ, Siu LL. Dose escalation methods in phase I cancer clinical trials. Journal of the National Cancer Institute. 2009;101(10):708–20. doi: 10.1093/jnci/djp079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gorden KB, G K, et al. Synthetic TLR agonists reveal functional differences between human TLR7 and TLR8. J Immunol. 2005;174:1259–68. doi: 10.4049/jimmunol.174.3.1259. [DOI] [PubMed] [Google Scholar]
- 21.Tanaka S, Saito Y, Kunisawa J, Kurashima Y, Wake T, Suzuki N, et al. Development of mature and functional human myeloid subsets in hematopoietic stem cell-engrafted NOD/SCID/IL2rgammaKO mice. J Immunol. 2012;188(12):6145–55. doi: 10.4049/jimmunol.1103660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alagkiozidis I, Facciabene A, Tsiatas M, Carpenito C, Benencia F, Adams S, et al. Time-dependent cytotoxic drugs selectively cooperate with IL-18 for cancer chemo-immunotherapy. Journal of translational medicine. 2011;9:77. doi: 10.1186/1479-5876-9-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simpkins F, Flores A, Chu C, Berek JS, Lucci J, 3rd, Murray S, et al. Chemoimmunotherapy using pegylated liposomal Doxorubicin and interleukin-18 in recurrent ovarian cancer: a phase I dose-escalation study. Cancer immunology research. 2013;1(3):168–78. doi: 10.1158/2326-6066.CIR-13-0098. [DOI] [PubMed] [Google Scholar]
- 24.Alagkiozidis I, Facciabene A, Carpenito C, Benencia F, Jonak Z, Adams S, et al. Increased immunogenicity of surviving tumor cells enables cooperation between liposomal doxorubicin and IL-18. Journal of translational medicine. 2009;7:104. doi: 10.1186/1479-5876-7-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaur U, Aggarwal BB. Regulation of proliferation, survival and apoptosis by members of the TNF superfamily. Biochemical pharmacology. 2003;66(8):1403–8. doi: 10.1016/s0006-2952(03)00490-8. [DOI] [PubMed] [Google Scholar]
- 26.Xiao CW, Yan X, Li Y, Reddy SA, Tsang BK. Resistance of human ovarian cancer cells to tumor necrosis factor alpha is a consequence of nuclear factor kappaB-mediated induction of Fas-associated death domain-like interleukin-1beta-converting enzyme-like inhibitory protein. Endocrinology. 2003;144(2):623–30. doi: 10.1210/en.2001-211024. [DOI] [PubMed] [Google Scholar]
- 27.Kelly MM, Hoel BD, Voelkel-Johnson C. Doxorubicin pretreatment sensitizes prostate cancer cell lines to TRAIL induced apoptosis which correlates with the loss of c-FLIP expression. Cancer Biol Ther. 2002;1(5):520–7. doi: 10.4161/cbt.1.5.169. [DOI] [PubMed] [Google Scholar]
- 28.Piao X, Komazawa-Sakon S, Nishina T, Koike M, Piao JH, Ehlken H, et al. c-FLIP maintains tissue homeostasis by preventing apoptosis and programmed necrosis. Science signaling. 2012;5(255):ra93. doi: 10.1126/scisignal.2003558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480–9. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van der Sluis TC, van Duikeren S, Huppelschoten S, Jordanova ES, Beyranvand Nejad E, Sloots A, et al. Vaccine-induced tumor necrosis factor-producing T cells synergize with cisplatin to promote tumor cell death. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21(4):781–94. doi: 10.1158/1078-0432.CCR-14-2142. [DOI] [PubMed] [Google Scholar]
- 31.Monk BJ, Herzog TJ, Kaye SB, Krasner CN, Vermorken JB, Muggia FM, et al. Trabectedin plus pegylated liposomal Doxorubicin in recurrent ovarian cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28(19):3107–14. doi: 10.1200/JCO.2009.25.4037. [DOI] [PubMed] [Google Scholar]
- 32.Colombo N, Kutarska E, Dimopoulos M, Bae DS, Rzepka-Gorska I, Bidzinski M, et al. Randomized, open-label, phase III study comparing patupilone (EPO906) with pegylated liposomal doxorubicin in platinum-refractory or -resistant patients with recurrent epithelial ovarian, primary fallopian tube, or primary peritoneal cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30(31):3841–7. doi: 10.1200/JCO.2011.38.8082. [DOI] [PubMed] [Google Scholar]
- 33.Vergote I, Finkler NJ, Hall JB, Melnyk O, Edwards RP, Jones M, et al. Randomized phase III study of canfosfamide in combination with pegylated liposomal doxorubicin compared with pegylated liposomal doxorubicin alone in platinum-resistant ovarian cancer. International journal of gynecological cancer : official journal of the International Gynecological Cancer Society. 2010;20(5):772–80. doi: 10.1111/igc.0b013e3181daaf59. [DOI] [PubMed] [Google Scholar]
- 34.McDermott D, Lebbe C, Hodi FS, Maio M, Weber JS, Wolchok JD, et al. Durable benefit and the potential for long-term survival with immunotherapy in advanced melanoma. Cancer treatment reviews. 2014;40(9):1056–64. doi: 10.1016/j.ctrv.2014.06.012. [DOI] [PubMed] [Google Scholar]
- 35.Hoos A, Eggermont AM, Janetzki S, Hodi FS, Ibrahim R, Anderson A, et al. Improved endpoints for cancer immunotherapy trials. Journal of the National Cancer Institute. 2010;102(18):1388–97. doi: 10.1093/jnci/djq310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dudley ME, W J, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother. 2003 Jul-Aug;26(4):332–42. doi: 10.1097/00002371-200307000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45(2):228–47. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.