

Mating system and genetic variation impede the spread of gene drives, which target natural populations of disease-vectoring insects.
Keywords: gene drive, CRISPR/Cas9, Population Genetics, population suppression, population engineering, selfish gene, immune to drive, genetic variation, inbreeding, nonrandom mating
Abstract
Synthetic gene drives based on CRISPR/Cas9 have the potential to control, alter, or suppress populations of crop pests and disease vectors, but it is unclear how they will function in wild populations. Using genetic data from four populations of the flour beetle Tribolium castaneum, we show that most populations harbor genetic variants in Cas9 target sites, some of which would render them immune to drive (ITD). We show that even a rare ITD allele can reduce or eliminate the efficacy of a CRISPR/Cas9-based synthetic gene drive. This effect is equivalent to and accentuated by mild inbreeding, which is a characteristic of many disease-vectoring arthropods. We conclude that designing such drives will require characterization of genetic variability and the mating system within and among targeted populations.
INTRODUCTION
Gene drives based on site-specific nuclease technologies—hybrid meganucleases, zinc finger nucleases, transcription activator–like effector nucleases, and clustered regularly interspaced short palindromic repeats/CRISPR-associated 9 (CRISPR/Cas9)—potentially offer a significant improvement on sterile-male release methods for suppressing populations of agricultural pests and disease-vectoring insects (1–3), allowing for more rapid transformation of populations with fewer releases of modified insects (4). The RNA-guided CRISPR/Cas9 system permits the construction of custom synthetic gene drives potentially capable of spreading agriculturally valuable and medically beneficial genomic alterations made in laboratory organisms through wild populations (5). Although initial studies of CRISPR-based gene drives in Drosophila melanogaster (6), Anopheles stephensi (7), and Anopheles gambiae (8) have demonstrated efficient drive propagation in laboratory-reared inbred populations, it is not clear how these gene drives will work in genetically diverse wild populations, where features like population structure, epistasis, and competition affect their evolutionary dynamics.
To generate a CRISPR/Cas9 gene drive (Fig. 1), a founder population is first created by targeted insertion of a drive cassette into a specified locus. This is achieved by transformation of the organism of interest with a plasmid encoding the Cas9 nuclease and the Cas9-targeting guide RNA (gRNA), both flanked by regions of DNA corresponding to the genomic sequences flanking the Cas9 target site. Following targeted cleavage of the locus of interest by Cas9, the drive construct is integrated at high frequency into the disrupted locus via homology-directed repair (HDR), generating heterozygotes. The remaining wild-type allele is then targeted and cleaved via gRNA and Cas9 expressed from the drive allele, leading to its conversion to a drive allele and homozygosity for the drive.
Fig. 1. Schematic of the propagation of a CRISPR/Cas9-based gene drive.
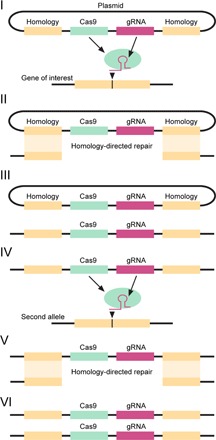
(i) A plasmid expressing Cas9 and gRNA flanked by DNA homologous to the sequences flanking the gRNA target site is expressed in the organism. Cas9 cleaves DNA at the gRNA-specified target site. (ii) The drive cassette, containing Cas9 and gRNA, is integrated into the target locus via HDR, resulting in (iii) heterozygosity for the drive allele. (iv) The remaining wild-type allele is targeted by Cas9 and gRNA expressed from the drive allele, leading to (v) HDR-mediated copying of the drive allele into the wild-type locus and (vi) homozygosity for the drive allele.
Gene drives work by segregation distortion (9) or “super Mendelian” inheritance, wherein heterozygous individuals either transmit a desired gene in >90% of their gametes instead of the 50% Mendelian expectation or are transformed into homozygotes. Natural examples include segregation distorter in D. melanogaster (10), Medea in Tribolium castaneum (11), the t-haplotype in Mus musculus (12), and spore killer in Neurospora crassa (13). In each natural case, the biased production of gametes of one allele is associated with some loss of fitness, so some heterozygotes persist, which has allowed the drives to be discovered and studied. A viability fitness loss of only 25% [selection coefficient (s) of −0.25] in both sexes or a fertility loss of 50% in one sex is sufficient to counter even the most extreme transmission bias, preventing the spread of a driven allele (14). The upper limits to fitness loss are twice as high in germline gene drive models (15), where individuals heterozygous at fertilization are converted to homozygotes, likely due to the activity of maternally loaded Cas9 and gRNA (7). Although all models to date assume random mating, a small level of inbreeding significantly slows the rate of spread by diminishing the frequency of heterozygotes (see below). Because any gene in the genome that suppresses drive is favored by natural selection, Charlesworth and Charlesworth (14) concluded that “a newly evolved distorter system will be inefficient.” Does this concern apply to laboratory-engineered CRISPR/Cas9 gene drives proposed for release into wild populations for purposes of population suppression? Here, we use evolutionary genetic analysis incorporating standing genetic variation and inbreeding in the model insect and agricultural pest T. castaneum to address this question.
RESULTS
For the purpose of assessing the effects of naturally occurring genetic variation on CRISPR/Cas9-based drive efficiency, we consider three separate regions of Cas9 target sites: the protospacer-adjacent motif (PAM), the seed region, and the outer protospacer. The PAM (often NGG, recognized by the commonly used Streptococcus pyogenes Cas9) is required for Cas9 to recognize and cleave its target site (16). Thus, conversion of either G to another base in the NGG PAM would be severely deleterious to drive copying, and we therefore call these variants in the PAM immune-to-drive (ITD) variants. The five bases immediately adjacent to the PAM, which are collectively known as the seed region (17), are responsible for nucleation of the gRNA:DNA hybrid, which is necessary for stable Cas9 binding and subsequent cleavage. Genome-wide characterization of Cas9 binding by chromatin immunoprecipitation and high-throughput sequencing has shown binding but little cleavage at targets with seed region mutations (17, 18) as well as substantially decreased Cas9 binding in some cases (18, 19). Seed mutations are thus expected to severely compromise drive copy, and for our modeling purposes, we treat them as ITDs. In the protospacer outside of the seed region, Cas9 exhibits a relatively relaxed specificity (20). Thus, single-nucleotide polymorphisms (SNPs) in this region would be expected to contribute minimally to drive resistance.
To assess the potential for standing genetic variation to contribute to drive resistance, we obtained the genomic sequence of high-quality gRNA-targeting regions from each of the four T. castaneum populations—Bhopal (India), Jerez (Spain), Purdue (Indiana), and Lima (Peru)—for exons in three potentially drive-relevant genes. The first, w, which encodes an ABC transmembrane transporter involved in eye pigmentation in D. melanogaster (21), was chosen because it has been used as a landing site for an antimalarial gene drive in A. stephensi (7) and its disruption imparts a moderate fitness cost in D. melanogaster (22) and A. stephensi (7). We found two SNPs in the selected w gRNA, one in the outer protospacer, present at low frequency in the Jerez population but at moderate frequency in the Purdue population, and one in the seed region, present at low frequency in the Purdue population (Fig. 2). Because of the sequence flexibility of the outer protospacer, the first SNP would not be expected to substantially contribute to impaired Cas9 recognition and cleavage. However, the second SNP, in the seed region, could severely reduce Cas9 association and subsequent cleavage and so is considered an ITD. We next considered a gRNA in Ace2, which is involved in development, female fertility, and insecticide resistance in T. castaneum (23) and thus expected to carry a heavy fitness penalty when disrupted. A drive targeting Ace2 might therefore conceivably be used for direct population suppression. We found two SNPs in the Ace2 gRNA. The first SNP, in the outer protospacer and thus expected to have little effect on drive spread, is present at moderate frequencies in the India, Purdue, and Purdue populations (Fig. 2). The second Ace2 gRNA SNP is present at low frequency in the Purdue population and at moderate frequency in the Peru population. This SNP lies at a critical base, the second G of the PAM (Fig. 2), and so would be expected to completely abrogate Cas9 target site recognition and cleavage. However, it has recently been reported that NGA is the most effective noncanonical PAM for cleavage by S. pyogenes Cas9 (24), so this variant may not totally impair recognition and cleavage. However, because Cas9 cleavage even with an NGG PAM is not necessarily 100% effective and the NGA PAM is less robust in cell-based CRISPR assays, we treat this NGG-to-NGA substitution as a PAM-disabling ITD. The last gRNA targeted the gene encoding the T. castaneum homolog of the highly conserved sperm flagellar protein 1, TC010993 (25). TC010993 mutation would be expected to severely reduce male fertility and might therefore be an attractive target for reproductive suppression of a population of interest. The TC010993 gRNA harbored a rare SNP present only in the Peru population in the outer protospacer region that would likely have little or no impact on drive spread (Fig. 2). Although there are no ITDs in any of the three gRNA-targeting regions in the India population, other work has shown that this population is reproductively incompatible with populations from North and South America (26).
Fig. 2. Genetic variation in Cas9 target sequences.

Each component of the Cas9 target sequence is highlighted according to its potential to affect Cas9 cleavage [red, PAM (complete abrogation of cleavage); yellow, seed (reduced cleavage efficiency); green, outer protospacer (little to no effect on cleavage efficiency)]. The frequency of each SNP in the analyzed population is given below each gRNA sequence. Note that all regions harbor SNP variants in some population and an obstructing variant in the critical PAM region of Ace2 occurs at a frequency of 0.375 in the Peru population.
To see how these SNP variants affect gene drive and population suppression, we modified the recursion formula for q, the CRISPR-like drive gene frequency [given in Eq. 1 of the study of Unckless et al. (15)], to allow for inbreeding and ITDs. The wild-type allele frequency is p, and that of the ITD variant is r, where (q + p + r) = 1. In Eq. 1, wild-type and ITD homozygous fitness is 1, individuals homozygous for the drive construct have a fitness of (1 − s), and ITD/drive and wild-type/drive heterozygotes have a fitness of (1 − hs), where h is the degree of dominance. The rate at which wild-type alleles are converted to drive alleles in heterozygotes equals c, where 0 ≤ c, s ≤ 1. All individuals converted from heterozygotes to homozygotes have a fitness of (1 − s). We let F be the proportion of individuals mating, nonrandomly, within their own type; for example, 2q(1 − q) (1 − F) is the frequency of heterozygotes weighted by the proportion of individuals mating nonrandomly. Half of the heterozygotes lost by inbreeding are added to the frequency of each respective homozygote; for example, q2 becomes q2 + q(1 − q)F. The frequency of the drive allele in the next generation is now
| (1) |
where the mean fitness W equals 1 – s{q2 + rqF + 2rqh(1 – F) + 2pqc(1 – F) + 2pqh(1 – F) (1 – c)}. When F = r = 0, q′ and W reduce to the formulas of Unckless et al. (15). Mean fitness of the population after the introduction of gene drive also measures the degree of population suppression. We thus modeled propagation of a CRISPR/Cas9-based drive based on each of the gRNAs described above. We considered each drive to be highly efficient (c = 0.95) and near-dominant (h = 0.98). Our assumption of high drive efficiency is based on the reported high efficiencies of these drives in D. melanogaster (6), A. stephensi (7), and A. gambiae (8). We considered each population to be randomly mating (F = 0). The TC010993 drive is an example of an ideal situation, in which a highly deleterious gene drive (s = 0.8) is introduced at a frequency of 0.7 into a randomly mating wild population harboring no ITDs. In this case, the drive is fixed after eight generations, effectively suppressing the population (Fig. 3A). However, when an ITD is present at low frequency (Iinit = 0.01) in a randomly mating population, similar to a drive scenario using the Ace2 gRNA described above, the frequency of the drive rapidly declines while ITD frequency increases, until the drive frequency is <0.01 and the ending ITD frequency Ifinal is ~0.45 after 10 generations (Fig. 3B), as calculated by
| (2) |
Fig. 3. ITDs limit drive propagation.
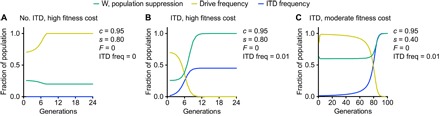
(A) In the absence of an ITD and inbreeding, a strongly deleterious drive rapidly spreads to fixation. (B) Addition of an ITD at a frequency of 0.01 severely impairs propagation of a strongly deleterious drive and leads to its removal from the population, as well as a substantial increase in ITD frequency. (C) In the presence of an ITD at a frequency of 0.01, a moderately deleterious drive remains at high frequency for several tens of generations but is eventually eliminated from the population. The populations considered in this figure are considered to be randomly mating (F = 0).
It is important to note that resistance to the drive increases, as measured by ITD frequency, until the drive is effectively eliminated from the population. This suggests that the population would be substantially more refractory to additional releases of individuals carrying this drive. We note that the ITD frequency of 0.01 is lower than the frequency of the Ace2 gRNA ITD in either of the populations in which it is present, suggesting that our estimate is conservative in this case. Last, we considered a scenario in which a gene expected to have lower fitness consequences is disrupted, perhaps with an antipathogen drive, as has been described in A. stephensi (7), which would be analogous to a drive based on the w gRNA we selected. We modeled this drive using all the above-described parameters, except that s = 0.4 to represent a modest fitness effect. In this example, the drive is present at a frequency of >0.9 for 63 generations but declines to <0.01 after 91 generations (Fig. 3C), indicating that a low-frequency ITD can eventually halt the spread of a drive with a moderate organismal fitness penalty. We also observed a marked increase in ITD frequency during drive elimination, consistent with our results using a highly deleterious drive.
We next tested the effect of inbreeding on drive propagation. Although rates of inbreeding (F) in T. castaneum have not been determined, these measurements have been made for mosquitoes and range from not significant to 0.8 (27–29). For our model, we chose a conservative estimate of F = 0.15. In the absence of ITDs, as in the TC010993-like scenario described above, this degree of inbreeding resulted in a rapid crash of drive frequency, leading to a frequency of <0.01 in six generations (Fig. 4A). Combination of an ITD at a frequency of 0.01, similar to our Ace2 example, with inbreeding at a rate of 0.15 had a negligible effect on the speed at which the drive crash occurred (Fig. 4B). Notably, inbreeding also limited the spread of the ITD relative to the corresponding no-inbreeding scenario (Fig. 3B), likely due to the rapidity of the cessation of drive spread (Fig. 4B). Last, introduction of inbreeding into the w-like situation described in Fig. 3C led to a drastic acceleration of drive loss, with drive frequency reaching <0.01 in 39 rather than 91 generations (Fig. 4C).
Fig. 4. Inbreeding impairs drive spread.
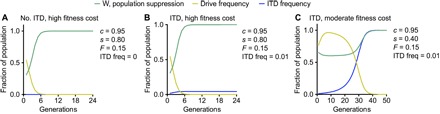
(A) Inbreeding at a rate of 0.15 in the absence of an ITD leads to rapid cessation of the spread of a strongly deleterious drive and its removal from the population. (B) Addition of an ITD at a frequency of 0.01 to the population inbreeding at a rate of 0.15 again leads to rapid loss of the strongly deleterious drive from the population, as well as a moderate increase in ITD frequency. (C) In the presence of an ITD at a frequency of 0.01, a moderately deleterious drive remains high for several generations but is effectively eliminated (frequency, <0.01) after 39 generations.
In the examples given above, we assumed specific values for the starting parameters c (drive efficiency), s (fitness penalty of the drive), and F (degree of inbreeding). We therefore sought to examine the general effects of varying each parameter on drive and ITD frequency. We first considered s, the fitness penalty of a drive, in the context of a randomly mating population with a low-frequency (0.01) ITD. As indicated by our results using s = 0.4 and 0.8 (Fig. 2), an increase in the fitness penalty of the drive leads to accelerated elimination from the population. By plotting the generation of drive elimination (considered to be the generation at which the drive frequency is first <0.01) versus s, we observed an exponential decrease in the number of generations to drive elimination as s is increased (Fig. 5A). We also found that, for values of s from 0.1 to 0.7, ITD frequency is ~1 at the generation of drive elimination (Fig. 5A). This is consistent with our analysis of a drive with a moderate fitness penalty (Fig. 3C) and suggests that although a drive with a lesser fitness cost will persist in the population for longer, it will have the secondary effect of rendering the population resistant to repeated releases of the drive. We performed a similar analysis for inbreeding, varying the value of F from 0 to 1. As suggested by our analysis in Fig. 4, increased inbreeding led to a more rapid drive elimination but reduced the frequency of ITDs in the population (Fig. 5B). Last, we considered modulation of c, drive efficiency, in the context of a randomly mating population. Less efficient drives persisted longer in the population but led to an increased ITD frequency, whereas more efficient drives decreased ITD frequency at the expense of persistence in the population (Fig. 5C).
Fig. 5. Analysis of systematic variation of model parameters.
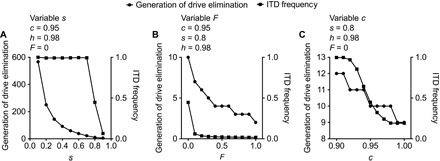
(A) Plot showing the effect of varying the drive fitness penalty s from 0.1 to 0.9 on the generation at which the drive is eliminated (considered to be the generation at which drive frequency is first <0.01) and ITD frequency. (B) Plot showing the effect of varying the inbreeding coefficient F from 0 to 1 on the generation at which the drive is eliminated and ITD frequency. (C) Plot showing the effect of varying the drive efficiency c from 0.9 to 1 on the generation at which the drive is eliminated and ITD frequency. For the plots shown in (B) and (C), we assumed s = 0.8.
DISCUSSION
We have modeled the effects of standing genetic variation and inbreeding on CRISPR/Cas9-based gene drives using T. castaneum as a model. Our results suggest that even a low-frequency ITD can severely limit the spread of a highly deleterious drive and cause its elimination from a population. A key result of our analysis is that ITDs were selected for and present at a higher frequency in the population following drive elimination, substantially increasing the resistance of the population to future attempts to propagate this drive. We found that ITDs have a less pronounced effect on drives with a lower fitness cost to the organism but are still capable of eliminating the drive from a population over time. This is particularly relevant to antipathogen drives inserted into loci thought to have little fitness effect on an organism, such as the insertion of an antimalarial drive into the white locus of A. stephensi (6), emphasizing the importance of minimizing fitness effects on the host. We note that our assumptions of high drive efficiency and near dominance favor drive transmission in our model and that a reduction in either of these parameters accelerates drive loss. We found that in the case of a strongly deleterious gene drive, moderate inbreeding caused a very rapid loss of the drive regardless of the presence of an ITD. Furthermore, Bull (30) has shown that gene drive can favor a gene that increases the tendency to inbreed, so the use of a constant level of inbreeding in our modeling is conservative. Inbreeding also accelerated the loss of a moderately deleterious drive, but the drive was stable for several generations before declining.
Our analyses suggest that standing genetic variation and inbreeding could have wide-ranging and sometimes severe consequences on the efficiency of drive propagation in a wild population. Inbreeding, which in our analyses had the most profound impact on drive propagation, may be most effectively overcome by saturation of the target population with drive individuals through repeated, high-volume releases. To minimize the potential impacts of ITDs on drive spread, we propose that a survey of genetic diversity at the locus of interest in the target population is a necessary first step in designing a CRISPR-based gene drive. If a target region of a locus of interest is highly variable, it may be prudent to select a different region of the gene for disruption to avoid the potential complications of ITDs described here. It is also important to note that although CRISPR/Cas9-based gene drives have been reported to be highly efficient, they do generate short insertions and deletions (indels) at a low frequency (6–8). These mutations would render the gRNA target site resistant to further editing and thus represent induced ITDs that would then be selected for, similar to the naturally occurring ITDs, when their fitness cost is not too high.
Although standing genetic variation in a wild target population has the potential to severely compromise CRISPR/Cas9-based gene drives, recent advances in characterization of additional engineered and natural Cas9 variants may offer potential solutions to the problem of ITDs in a region of interest. For example, molecular evolution of S. pyogenes Cas9 yielded variants with non-NGG PAM specificities (31). Furthermore, Cas9 variants from different bacterial species with different PAM specificities have been described and used for genome editing (31–33), as has Cpf1, an alternative CRISPR nuclease with a non-NGG PAM specificity (34). If the PAM specificity can be suitably matched to the variation of the target population, these alternative CRISPR nucleases would not be susceptible to the problems highlighted in our model.
Our results raise significant practical concerns for the design of CRISPR/Cas9-based gene drives intended for use in the wild. Analysis of genetic variation in the target population is essential, as is, if possible, characterization of mating structure to assess the potential impact of inbreeding on drive spread. To this end, it will be of great interest to test the predictions of our modeling in diverse laboratory populations before the implementation of any CRISPR/Cas9-based gene drive in a wild population.
MATERIALS AND METHODS
gRNA design
Exon sequences for w, Ace2, and TC010993 (Tcas3 assembly) were obtained from BeetleBase (http://beetlebase.org/blast/blast.html) (35) and input into CRISPRdirect (https://crispr.dbcls.jp) (36) using the Tcas3 assembly for the specificity check. Only high-quality gRNA sequences (defined as having a single 20-nucleotide oligomer + PAM and a single 12-nucleotide oligomer + PAM match) were selected for SNP analysis.
Populations and sample preparation
A total of 96 individuals were randomly selected from four wild-caught laboratory populations of T. castaneum [Bhopal, India; Lima, Peru; Lafayette, IN (Purdue); and Jerez, Spain]. Each population was established from individuals collected from local populations of T. castaneum found in granaries and markets. All populations were collected and maintained at population sizes of greater than 250 individuals to reduce the potential for inbreeding and loss of genetic variation in the laboratory. To compare the genetic variation within and between populations, DNA was extracted and purified using the cetyl trimethylammonium bromide (CTAB) method (37) from 24 randomly selected individuals from each population. Each individual DNA sample was uniquely barcoded using the Nextera Index Kit (Illumina). Libraries with an insert size of 300 base pairs were constructed using the Nextera DNA Library Preparation Kit (Illumina) with a modified protocol (38). Three individual DNA samples were discarded because of low yield after library preparation and fragment size selection steps.
Sequencing and mapping
Libraries were sequenced for 150 cycles in paired-end mode on an Illumina HiSeq 2500 at the Hubbard Center for Genome Studies, University of New Hampshire (Durham, NH). We sequenced 93 individuals (24 individuals each from the India, Peru, and Purdue populations and 21 individuals from the Jerez population) to a coverage depth of ~2 to 3×, with an average of ~50× total coverage for each population (Jerez, 37.675 ± 0.005; Purdue, 51.935 ± 0.009; Peru, 50.286 ± 0.008; India, 63.003 ± 0.009). The FASTQ files were filtered using Cutadapt (https://cutadapt.readthedocs.io/en/stable/) to remove low-quality sequence (Q < 20) and trimmed to remove library adaptors. The resulting files were processed for mapping using Fix Pairings (https://github.com/svm-zhang/make_life_easier). The paired-end reads for each individual were aligned to the Tcas3 reference genome using BWA 0.7.6a (39). The resulting SAM files were converted to BAM and mpileup files using SAMtools (40). The individual whole-genome sequence data were pooled by population, and mpileup files were synced using PoPoolation2 (https://sourceforge.net/projects/popoolation2/) (41). From this, we obtained a direct major and minor allele count for every SNP in our target loci. To further reduce the contribution of sequencing error, we used a technical replicate to filter out discordant sites. We defined discordant sites as positions within the genome where the alleles called in each of the two technical replicates did not agree (for example, in one technical replicate, we observed both T and A allele calls at a given position but observed only T alleles at the same position in the other technical replicate).
Acknowledgments
We thank S. Zhang, H. Long, and J.-F. Gout for assistance with sequencing data analysis. Funding: This work was supported by Indiana University startup funds (G.E.Z.) and NIH R01 GM084238 (M.J.W.). The sequencing was supported by an NSF Doctoral Dissertation Improvement Grant (1311167 to M.J.W and A.L.D). Author contributions: D.W.D., D.J.S., G.E.Z., and M.J.W. conceived the study; A.L.D. constructed sequencing libraries; D.W.D. and A.L.D. analyzed sequencing data and called SNPs; G.E.Z. performed gRNA analysis; and G.E.Z. and M.J.W. wrote the paper. All authors read and approved the final manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Full sequencing data will be described elsewhere (D.W.D., A.L.D., and M.L.W.’s manuscript is in preparation) and deposited into the Sequence Read Archive upon publication. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
REFERENCES AND NOTES
- 1.Vanderplank F. L., Hybridization between Glossina species and suggested new method for control of certain species of tsetse. Nature 154, 607–608 (1944). [Google Scholar]
- 2.Bushland R. C., Lindquist A. W., Knipling E. F., Eradication of screw-worms through release of sterilized males. Science 122, 287–288 (1955). [DOI] [PubMed] [Google Scholar]
- 3.Knipling E. F., Sterile-male method of population control. Science 130, 902–904 (1959). [DOI] [PubMed] [Google Scholar]
- 4.Robert M. A., Okamoto K. W., Gould F., Lloyd A. L., Antipathogen genes and the replacement of disease-vectoring mosquito populations: A model-based evaluation. Evol. Appl. 7, 1238–1251 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Esvelt K. M., Smidler A. L., Catteruccia F., Church G. M., Concerning RNA-guided gene drives for the alteration of wild populations. eLife 3, e03401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gantz V. M., Bier E., The mutagenic chain reaction: A method for converting heterozygous to homozygous mutations. Science 348, 442–444 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gantz V. M., Jasinskiene N., Tatarenkova O., Fazekas A., Macias V. M., Bier E., James A. A., Highly efficient Cas9-mediated gene drive for population modification of the malaria vector mosquito Anopheles stephensi. Proc. Natl. Acad. Sci. U.S.A. 112, E6736–E6743 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hammond A., Galizi R., Kyrou K., Simoni A., Siniscalchi C., Katsanos D., Gribble M., Baker D., Marois E., Russell S., Burt A., Windbichler N., Crisanti A., Nolan T., A CRISPR-Cas9 gene drive system targeting female reproduction in the malaria mosquito vector Anopheles gambiae. Nat. Biotechnol. 34, 78–83 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lewontin R. C., Population genetics. Annu. Rev. Genet. 1, 37–70 (1967). [DOI] [PubMed] [Google Scholar]
- 10.Hiraizumi Y., Crow J. F., Heterozygous effects on viability, fertility, rate of development, and longevity of Drosophila chromosomes that are lethal when homozygous. Genetics 45, 1071–1083 (1960). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wade M. J., Beeman R. W., The population dynamics of maternal-effect selfish genes. Genetics 138, 1309–1314 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dobrovolskaia-Zavadskaia N., Sur la mortification spontanée de la queue chez la souris nouveau-née et sur l’existence d’un caractére héréditaire “non viable”. C. R. Soc. Biol. 97, 114–116 (1927). [Google Scholar]
- 13.Turner B. C., Perkins D. D., Spore killer, a chromosomal factor in neurospora that kills meiotic products not containing it. Genetics 93, 587–606 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.B. Charlesworth, D. Charlesworth, Elements of Evolutionary Genetics (Roberts and Company Publishers, 2010). [Google Scholar]
- 15.Unckless R. L., Messer P. W., Connallon T., Clark A. G., Modeling the manipulation of natural populations by the mutagenic chain reaction. Genetics 201, 425–431 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sternberg S. H., Redding S., Jinek M., Greene E. C., Doudna J. A., DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507, 62–67 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu X., Scott D. A., Kriz A. J., Chiu A. C., Hsu P. D., Dadon D. B., Cheng A. W., Trevino A. E., Konermann S., Chen S., Jaenisch R., Zhang F., Sharp P. A., Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 32, 670–676 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuscu C., Arslan S., Singh R., Thorpe J., Adli M., Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 32, 677–683 (2014). [DOI] [PubMed] [Google Scholar]
- 19.O’Geen H., Henry I. M., Bhakta M. S., Meckler J. F., Segal D. J., A genome-wide analysis of Cas9 binding specificity using ChIP-seq and targeted sequence capture. Nucleic Acids Res. 43, 3389–3404 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu X., Kriz A. J., Sharp P. A., Target specificity of the CRISPR-Cas9 system. Quant. Biol. 2, 59–70 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mackenzie S. M., Brooker M. R., Gill T. R., Cox G. B., Howells A. J., Ewart G. D., Mutations in the white gene of Drosophila melanogaster affecting ABC transporters that determine eye colouration. Biochim. Biophys. Acta 1419, 173–185 (1999). [DOI] [PubMed] [Google Scholar]
- 22.Krstic D., Boll W., Noll M., Influence of the white locus on the courtship behavior of Drosophila males. PLOS ONE 8, e77904 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu Y., Park Y., Gao X., Zhang X., Yao J., Pang Y.-P., Jiang H., Zhu K. Y., Cholinergic and non-cholinergic functions of two acetylcholinesterase genes revealed by gene-silencing in Tribolium castaneum. Sci. Rep. 2, 288 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y., Ge X., Yang F., Zhang L., Zheng J., Tan X., Jin Z.-B., Qu J., Gu F., Comparison of non-canonical PAMs for CRISPR/Cas9-mediated DNA cleavage in human cells. Sci. Rep. 4, 5405 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chan S. W., Fowler K. J., Choo K. H. A., Kalitsis P., Spef1, a conserved novel testis protein found in mouse sperm flagella. Gene 353, 189–199 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Drury D. W., Jideonwo V. N., Ehmke R. C., Wade M. J., An unusual barrier to gene flow: Perpetually immature larvae from inter-population crosses in the flour beetle, Tribolium castaneum. J. Evol. Biol. 24, 2678–2686 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Urbanelli S., Bellini R., Carrieri M., Sallicandro P., Celli G., Population structure of Aedes albopictus (Skuse): The mosquito which is colonizing Mediterranean countries. Heredity 84, 331–337 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Donnelly M. J., Cuamba N., Charlwood J. D., Collins F. H., Townson H., Population structure in the malaria vector, Anopheles arabiensis Patton, in East Africa. Heredity 83, 408–417 (1999). [DOI] [PubMed] [Google Scholar]
- 29.Vazeille M., Mousson L., Rakatoarivony I., Villeret R. G., Rodhain F., Duchemin J.-B., Failloux A.-B., Population genetic structure and competence as a vector for dengue type 2 virus of Aedes aegypti and Aedes albopictus from Madagascar. Am. J. Trop. Med. Hyg. 65, 491–497 (2001). [DOI] [PubMed] [Google Scholar]
- 30.Bull J. J., Lethal gene drive selects escape through inbreeding. Evol. Med. Public Health 2017, 1–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kleinstiver B. P., Prew M. S., Tsai S. Q., Topkar V. V., Nguyen N. T., Zheng Z., Gonzales A. P. W., Li Z., Peterson R. T., Yeh J.-R. J., Aryee M. J., Joung J. K., Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523, 481–485 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ran F. A., Cong L., Yan W. X., Scott D. A., Gootenberg J. S., Kriz A. J., Zetsche B., Shalem O., Wu X., Makarova K. S., Koonin E. V., Sharp P. A., Zhang F., In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Komor A. C., Badran A. H., Liu D. R., CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell 168, 20–36 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zetsche B., Gootenberg J. S., Abudayyeh O. O., Slaymaker I. M., Makarova K. S., Essletzbichler P., Volz S. E., Joung J., van der Oost J., Regev A., Koonin E. V., Zhang F., Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-cas system. Cell 163, 759–771 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim H. S., Murphy T., Xia J., Caragea D., Park Y., Beeman R. W., Lorenzen M. D., Butcher S., Manak J. R., Brown S. J., BeetleBase in 2010: Revisions to provide comprehensive genomic information for Tribolium castaneum. Nucleic Acids Res. 38, D437–D442 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naito Y., Hino K., Bono H., Ui-Tei K., CRISPRdirect: Software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics 31, 1120–1123 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doyle J. J., Doyle J. L., A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15 (1987). [Google Scholar]
- 38.Baym M., Kryazhimskiy S., Lieberman T. D., Chung H., Desai M. M., Kishony R., Inexpensive multiplexed library preparation for megabase-sized genomes. PLOS ONE 10, e0128036 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H., Durbin R., Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. 1000 Genome Project Data Processing Subgroup , The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kofler R., Pandey R. V., Schlötterer C., PoPoolation2: Identifying differentiation between populations using sequencing of pooled DNA samples (Pool-Seq). Bioinformatics 27, 3435–3436 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]