

Abstract
The incorporation of genomic data into personal medical records poses many challenges to patient privacy. In response, various systems for preserving patient privacy in shared genomic data have been developed and deployed. Although these systems de-identify the data by removing explicit identifiers (e.g., name, address, or Social Security number) and incorporate sound security design principles, they suffer from a lack of formal modeling of inferences learnable from shared data. This report evaluates the extent to which current protection systems are capable of withstanding a range of re-identification methods, including genotype–phenotype inferences, location–visit patterns, family structures, and dictionary attacks. For a comparative re-identification analysis, the systems are mapped to a common formalism. Although there is variation in susceptibility, each system is deficient in its protection capacity. The author discovers patterns of protection failure and discusses several of the reasons why these systems are susceptible. The analyses and discussion within provide guideposts for the development of next-generation protection methods amenable to formal proofs.
The biomedical community currently finds itself in the midst of a genomics revolution. Genomic data, combined with increasing computational capabilities, provide opportunities for health care that until recently were severely limited. Beyond gross diagnostics, mounting evidence suggests genomic variation influences disease susceptibility and the ability to metabolize drugs. As a result, genomic data are increasingly collected, stored, and shared in research and clinical environments.1
The sharing and application of person-specific genomic data pose complex privacy issues and are considered the foremost challenges to the biomedical community.2,3 Many people fear knowledge gleaned from their genome will be misused, be abused, or instigate social stigma for themselves or familial relations.4,5 This fear is exacerbated by the HIPAA Privacy Rule, under which genomic data are not specified as an identifying patient attribute.6 As such, genomic data may be released for public research purposes under HIPAA's safe harbor provision.* Yet, when genomic data are not publicly available, recipients may be subject to data use agreements. Although legally binding, there is no guarantee genomic data will be used according to specification. Thus, it is best that privacy laws are complemented with technology to assist in the enforcement of protections.
Privacy protection technologies for genomic data must address the question, “How can person-specific DNA be shared, such that a recipient can not sufficiently associate the DNA to its explicit identity (i.e., name, Social Security number, etc.)?” Although genome variation uniquely characterizes an individual,7 there exists no public registrar that maps genomes to names of individuals. Over the last several years, many genomic data privacy protection systems have implicitly relied on this premise. These systems tend to separate DNA from explicit identifiers through methods ranging from simple removal of identifiers to strong cryptographic protocols.†
This report addresses the extent to which current privacy-enhancing technologies for genomic data are susceptible to compromise. Specifically, this work studies computational attacks that leverage information learned from shared genomic data and additional resources for linkage to named individuals. None of the systems analyzed is impregnable to re-identification. Rather, there exist patterns of flaws due to neglect of inferences that can be made from genomic data itself and the environments in which the data are shared.
The remainder of this report is organized as follows. In the following section, background on several published protection strategies for genomic data are provided. Each system is represented and discussed in a structured relational notation for comparative analysis. Next, computational re-identification methods for testing the protection systems are defined. With protection and re-identification methods presented, susceptibility analyses are performed and patterns of protection failures are discussed. This work concludes with a discussion on the need for research into formal anonymity protection schemas for genomic data and how such developments may proceed.
Current Privacy Protection Systems
In this section, four types of genomic data privacy protection systems are reviewed. Briefly, we introduce the following relational formalism to represent the systems. Person-specific data are organized as a table T(A1, A2, …, An) of rows and columns. Each column Ai is a semantic attribute, such as “date of birth,” “DNA sequence,” or “zip code.” Each row is an n-tuple t[a1, a2,…, an], where ai corresponds to a specific value of the ith attribute. An identified table, T+, includes explicitly identifiable information, such as name or Social Security number. Conversely, a de-identified table, T−, is devoid of identifiable information. ▶ provides examples of tables and tuples. For example, the record t[Bradley Malin, 000-00-0000, BIGBM, actg] is a relevant tuple for table T(Name, Social Security Number, Pseudonym, DNA). Adversaries are never provided with DNA in an identified table, so the DNA-identity mapping is unknown prior to receiving the de-identified table.
Figure 1.
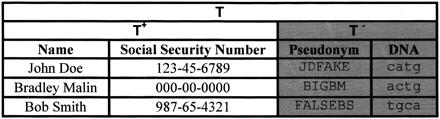
The table T(Name, Social Security Number, Pseudonym, DNA) is data collected by a specific location. T+(Name, Social Security Number) and T−(Pseudonym, DNA), are identified and de-identified tables, respectively.
De-identification
The first type of protection system, adopted in a wide range of communities and environments, is based on de-identification (DEID).8,9,10 The data holder classifies attributes into three types: explicit identifiers, quasi-identifiers, and nonidentifying. Explicit identifiers consist of information that can directly reveal, or allow for contact with, an individual, such as name or Social Security number. A quasi-identifying attribute does not reveal identity by itself but, in combination with other attributes, can be used to link to other sources with explicit identifying attributes. For example, Sweeney demonstrated the values for {date of birth, gender, and five-digit zip code} uniquely characterized over 87% of the United States population.11 Advocates of de-identification claim the corresponding identity of genomic data is sufficiently protected when explicit and quasi-identifying attributes are removed or generalized.
In DEID, the original table of a data holder takes the form T(Explicit-identifiers, Quasi-identifiers, Non-identifiers). When the data holder shares information, he removes Explicit-identifiers and generalizes values in Quasi-identifiers to prevent unique combinations. Thus, the data holder shares the dataset T′(Quasi-identifiers', Non-identifiers), where every value in the set of Quasi-identifiers' is derivative of its corresponding value in the original set of Quasi-identifiers. In many situations, a unique identifier is assigned to a patient for linkage purposes. For instance, in the Utah Resource for Genetic and Epidemiologic Research (RGE) system, the unique identifier is a random number.9 As a result, RGE data are released in a table T′(Quasi-identifying Attributes', Other Attributes, Random Number). ▶ depicts a data release for a DEID system.
Figure 2.
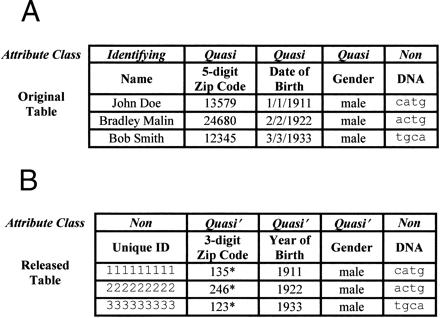
(A) Attributes of the original table are partitioned into explicit identifying (identifying), quasi-identifying (quasi), and nonidentifying (non). (B) Identifying attributes are removed, the quasi attributes are generalized (quasi'). A unique ID has been added.
Denominalization
Systems based on denominalization (DENOM) are similar to DEID, except they incorporate structured coding, often for familial relationships.12 In the original model, each patient is represented by six attributes {Individual, Family, Relation, Marriage, Sibling, Multiple}. Individual is a unique random number assigned to a patient, akin to the RGE system, which is used to manage the individual's clinical and biological samples. The remaining attributes correspond to genealogical information. Family is a random number assigned to every member of the same family. Relation corresponds to the relationship of an individual to another family member, such as child or parent. Sibling denotes the birth order of a child (i.e., oldest, next oldest, etc.). Marriage specifies which marriage a child was born into. Multiple specifies which family a tuple pertains to when the individual is classified under multiple families.
The individual and family codes are managed independently. In the system description, it is claimed different levels of anonymity are achieved through the suppression, or withholding, of various attributes. For example, biological samples are considered to be sufficiently anonymous when stripped of the latter five attributes.
Trusted Third Parties
The third system (TRUST), introduced by deCode Genetics, Inc., facilitates data transfers via a trusted third party (TTP) intermediary empowered with full data encryption/decryption capability.13 The full system consists of two protocols, both based on encryption and security. The first protocol facilitates discovery of research subjects, while the second specifies how biological samples are transferred to researchers. For brevity, we concentrate on the subject discovery protocol.‡
Researchers initiate the protocol by communicating a specific disease of interest to physicians attending the patient population. The physicians create and send a population-based list L{Name, Social Security Number, Additional Demographic Features§, Disease} to the TTP. The TTP applies a reversible encryption function f to the Social Security Number (SSN) to derive an alphabet-based pseudonym f(SSN). Next, the TTP sends researchers the encrypted data, minus explicit identifiers, as a list L′{f(SSN), Disease}. Upon reception, the researchers match L′ against f-encrypted genealogies linked to patient medical information. Based on these data, the researchers send a wish list of patients for further study, N{f(SSN)}, back to the TTP. Finally, the TTP decrypts, appends the proper identifying information, and forwards the list N′{name, SSN} to the appropriate attending physicians.
Semitrusted Third Parties
A fourth, and the most recent, system (SEMITRUST) was introduced by researchers at the University of Gent and affiliates.14 Akin to TRUST, this system also employs a third party, but one with restricted access to plaintext data, or a semitrusted third party (sTTP). The third party is permitted to hold and distribute encrypted data only.
For the first step of the SEMITRUST protocol, the data holder constructs a list of identified individuals and their corresponding genomic data L{Identity, DNA}. The data holder applies public-key encryption function h to the Identity attribute and sends L′{h(Identity), DNA} to the sTTP. Next, the sTTP applies its own public-key encryption function g to h(Identity) to create L″{g(h(Identity)), DNA}. In addition, the sTTP can act as a data broker for multiple data holders and can maintain a set of lists, A{g(hA(Identity)), DNA}, B{g(hB(Identity)), DNA}, …, Z{g(hZ(Identity)), DNA} for locations A, B, …, Z. When researchers query the sTTP for data, they are supplied with doubly encrypted lists. For additional data, researchers send requests onto the sTTP with a list of encrypted identities. In turn, the sTTP decrypts and sends the single-encrypted pairs onto the appropriate locations for additional data.
Re-identification Methods
In the following sections we briefly review four different types of re-identification techniques.
Family Structure
The first re-identification method (FAMILY) employs genealogical data accompanying genomic data. Genealogies, rich in depth and structure, permit the construction of complex familial relationships. Consider a simple family structure of two parents and one child. Since the parental genders are guaranteed, there exist 2 variants of this structure, since the child's gender is either male or female. When disease status is taken into account, it is represented as a Boolean variable; either an individual afflicted or not afflicted. In this aspect, all three family members can be represented as three attributes {Father, Mother, Child}, and there exist (father's disease status)*(mother's disease status)*(child's disease status)*(child's gender) = 2*2*2*2 = 16 possible family-disease combinations. In reality, pedigrees are much more robust than a simple nuclear family. For example, a three-generation family of two children per family permits on the order of 105 distinct variants of the family-disease structure and 106 individuals that could be uniquely characterized. The number of combinations∥ is larger when supplementary information, such as living status or medical/genetic features, is considered.16
The ability to determine unique family structures is only one part of the re-identification process. These structures must be linked to identifiable information, which, in many instances, is publicly available in the form of various genealogical databases. These databases are accessible both offline and via the World Wide Web. For example, genealogical records are available in many public databases, including <Ancestry.com>, <Infospace.com>, <RootsWeb.com>, <GeneaNet.com>, <FamilySearch.org>, and <Genealogy.com>.¶ From such data, it is not difficult to construct family structures and, with such information in hand, an adversary can link disease-labeled family structures to named individuals.
Genotype–Phenotype Inference
The second method relies on phenotype inferences extracted from the genomic data (GENPHEN). Given two tables X(A1, A2, …, An) and Y(B1, B2, …, Bm) a set of relations is constructed, and, when a unique match is found between the two, a re-identification is discovered. In the base case, this model is similar to the quasi-identifier–based linkage model used in Sweeney's earlier work with health data re-identification.14,17 For example, consider Health(Name, Address, Birthdate, Gender, Zip Code, Hospital Visit Date, Diagnosis, Treatment) and Genomic(Age, Gender, Hospital Visit Date, DNA). The set of extracted attribute relationships is {〈Birthdate, Age〉, 〈Gender, Gender〉, 〈Hospital Visit Date, Hospital Visit Date〉}, but the set of relationships is expanded when relationships between clinical and genomic data are known. It has been demonstrated there exist a minimum of 40 standardized diseases (via ICD-9 codes) to which DNA mutations in the genome are directly related.18 Furthermore, pharmacogenomics continues to uncover relationships between genomic variation and the ability to process drugs and treatments.2-3,19 Given such domain knowledge, it is possible to include {〈Diagnosis, DNA〉, 〈Treatment, DNA〉} relations.
Furthermore, extending Sweeney's original work, it is possible to build systems that utilize attributes not observed in clinical or genomic information for linkage. When more complete clinical information is available, nonstandard information, such as age of onset for progressive disorders, can be inferred. In previous research, we showed how this could be achieved with longitudinal clinical information and Huntington's disease. Our system was able to infer age of onset within a 3-year period and subsequently match DNA to clinical data.20 In its current implementation, this approach is applicable to any simple genetic disorder with defined clinical phenotypes.
An additional feature of the inference attack is it becomes more powerful with time. Since the goal of genomic medicine is to elicit the relationships between genomic data and clinical phenotype, the number of relations and specificity of such increase with advances in basic medical research. For example, the goal of the human genome diversity project and genomic anthropology is to pinpoint relationships between genomic variation and ethnicity. As a result, both the number and specificity of relations will expand, thus permitting an increasing capability for linkage.
Trails
The method of trail re-identification (TRAIL) utilizes location-specific information to match DNA to identity.21 Consider an environment with a set of locations, such as a set of hospitals, and a set of data subjects, such as a set of patients. Each location has the ability to collect multiple types of information, such as clinical and genomic data. To protect privacy when data are released, each hospital releases identified data and de-identified data separately. The first table released is T+(Demographic Information, Clinical Information), where Demographic Information contains identifiable data. The second table released, T−(DNA), consists of a list of genomic data samples.
An adversary retrieves data from a set of locations and creates two new tables, each one corresponding to location information for a particular data type. The first table consists of identified data, while the second consists of DNA data. The mapping of data to location is referred to as the data trail. In ▶, trails are depicted as Boolean vectors; either a data value is observed at a location (1) or not (0). Details on trail-matching algorithms and their application to real-world populations can be found in reference 21. In short, genomic data left behind by an individual are matched to explicitly identifiable data based on the patterns of trails between the tracks.
Figure 3.
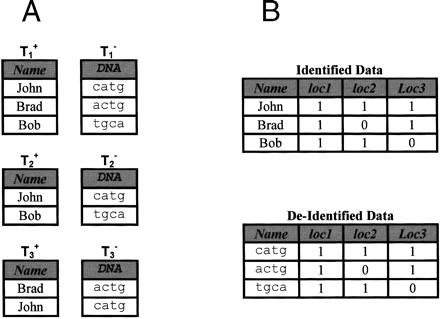
(A) Identified and de-identified data releases of locations loc1, loc2, and loc3. (B) Resulting identified and DNA tracks created. When re-identification is based on exact trail matching, John, Brad, and Bob are re-identified to catg, actg, and tgca, respectively.21
Dictionary Attack
The fourth re-identification method (DICTIONARY) is applicable when data are encrypted, or recoded, using nonrandom information. These methods, which obscure information, can provide the basis for further erosion of patient privacy, beyond that of a susceptibility to the re-identification methods presented above. Consider a set of hospitals H, where each hospital h∈H releases tables 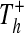 and
and 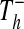 with attributes
with attributes 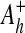 = {name, date of birth, gender, zip code, clinical data} and Ah− = {pseudonymh, DNA}. The attribute pseudonymh is generated through a reversible encryption function fh, such as public-key encryption fh(Identity, keyh) = pseudonymh, where Identity is a tuple of patient information [name, date of birth, gender, zip code]. An adversary can use a trail attack to re-identify some of the patients released from a set of data-releasing locations. Through re-identification, the adversary has constructed a table with the attributes {name, date of birth, gender, zip code, pseudonym1, pseudonym2, …, pseudonymH}, where pseudonymx is the pseudonym that hospital x uses for the identity of the patient. Thus, the adversary has achieved his goal of re-identifying the protected genomic data.
= {name, date of birth, gender, zip code, clinical data} and Ah− = {pseudonymh, DNA}. The attribute pseudonymh is generated through a reversible encryption function fh, such as public-key encryption fh(Identity, keyh) = pseudonymh, where Identity is a tuple of patient information [name, date of birth, gender, zip code]. An adversary can use a trail attack to re-identify some of the patients released from a set of data-releasing locations. Through re-identification, the adversary has constructed a table with the attributes {name, date of birth, gender, zip code, pseudonym1, pseudonym2, …, pseudonymH}, where pseudonymx is the pseudonym that hospital x uses for the identity of the patient. Thus, the adversary has achieved his goal of re-identifying the protected genomic data.
System Susceptibility Analyses
In this section, the general re-identification susceptibility for each of the protection methods is evaluated. The results are presented at a meta-level, such that either a system is considered susceptible or not susceptible. In ▶, a side-by-side comparison of protection model susceptibility is presented. Each of the protection models is susceptible to a minimum of three of the four re-identification attacks. Here, we discuss how each of the re-identification methods fares against the protection models in more detail.
Table 1.
General Susceptibility of Privacy Protection Models to Re-identification
| Re-identification Attack |
Privacy Protection System |
|||
|---|---|---|---|---|
| TRUST | SEMITRUST | DENOM | DEID | |
| FAMILY | Yes | No | Yes | Yes |
| TRAIL | No | Yes | No | Yes |
| GENPHEN | Yes | Yes | Yes | Yes |
| DICTIONARY | Yes | Yes | No | No |
Family Structure Susceptibility
The only model not susceptible to the family structure attack is the SEMITRUST system. Under this model, no familial relationships are considered in the genomic data. In specific cases, familial inferences may be possible, such as through haplotype analysis of DNA sequences. However, without more confidence regarding whether related family members are in the dataset, such analysis could create false family structures and familial relations.
It is interesting to note that the denominalization strategy behind DENOM strives to prevent the family attack almost explicitly. It provides protections by separating the individual from the family and using a local recoding of the identity. Yet, once this information is studied in a genealogical setting, the protections are minimal. Similarly, TRUST reveals genealogical information on a large scale, since this is how subject recruitment is performed.
In contrast, the RGE model of DEID is more difficult to analyze. As shown in ▶, the RGE model is susceptible to all re-identification attacks—although this may be somewhat deceiving. Since the RGE maintains a massive repository of diverse datasets, not all re-identification attacks can be performed on every dataset released. Thus, the analysis of re-identifiability for RGE released datasets is data dependent. Since RGE does have the ability to reveal genealogical information, and the only protection afforded to such data is de-identification and pseudonymization with random IDs, this model is susceptible to the family structure attack.
Trails Susceptibility
To construct a trail attack, two criteria must be satisfied. The first requirement is an individual's data are distributed over multiple locations. The second requirement is both genomic and identified data are available in partitions of the original collection. ▶ provides a characterization of which requirements the protection methods satisfy.
Table 2.
System Satisfiability of Trail Re-identification Criteria
| System | Multiple Locations | Partitioned Identified and DNA Data Available |
|---|---|---|
| TRUST | No | Yes |
| SEMITRUST | Yes | Yes |
| DENOM | No | Yes |
| DEID | Yes | Yes |
The TRUST model does not satisfy the multiple location criteria. No location-based information is revealed, nor is it necessary. In addition, the DENOM model is not susceptible, since under the current version, genomic data are collected at one location only. Yet, if this model is applied to a distributed environment, then the trail attack is a feasible re-identification route.
In comparison, it can be verified that the SEMITRUST model does satisfy both criteria and is susceptible. The RGE model of de-identification is susceptible as well, since genomic data could be requested from multiple sources. The health-specific information could be either supplied directly as a separate source or derived from various external resources, such as discharge information.
Genotype–Phenotype Susceptibility
This inference attack exploits relationships constructed between genomic data and known demographic or clinical information. As such, all four protection methods are susceptible to the attack, mainly due to the fact that the protection systems do not act directly on the genomic data. When considering simple versions of the inference attack, such as through direct ICD-9 linkage, with genomic data by itself, as is the case with the SEMITRUST model, this attack is dependent on the specificity of the known relationships between genomic data and clinical phenotype.
It is apparent that these methods can leak relationships that, although useful for research purposes and correlation studies, can allow for unique linkages to be constructed between identified and genomic data. This does not imply such relationships should not be inferable from shared data—rather the contrary. Yet, such inferences must be learnable or communicated in such a way that identities to which the data correspond can not be determined. The concept of revealing inferences without revealing identity will be addressed below.
Dictionary Susceptibility
The model most susceptible to DICTIONARY is a single pseudonymization function, in which pseudonyms are derived from patient-specific information. Since the RGE model uses random IDs for pseudonyms, a direct dictionary attack can not be achieved, regardless of the number of people re-identified through other means. In contrast, the other three systems are susceptible. The TRUST and SEMITRUST models are susceptible to a cryptographic dictionary attack. As an increasing number of people are re-identified, an adversary can collect a set of SSN, pseudonym pairs. Given enough pairs, the adversary may learn the key of the pseudonymizing function. In TRUST, the adversarial role can be played by any data requester. However, in the SEMITRUST model, this is not possible because the pseudonyms supplied to the researchers are doubly encrypted. Although nonrandom, it is virtually impossible to discern the effects of the originating location's pseudonymizing function from the semitrusted third party's (sTTP). Yet, in the event the sTTP is corrupt, it can leverage the fact that it receives single-encrypted pseudonyms from each of the submitting sources and attempt its own dictionary attack.
A modified version of the dictionary attack can be used to exploit familial relationship information released under the DENOM model. Given sufficient information to reconstruct and re-identify a certain amount of familial information, the recoding of familial relations can reveal additional information that may not have been learned in the family-structure attack, such as temporal information in the genealogy. For example, when a family has multiple children, the fifth cell of the family code denotes what order of birth a sibling is. Moreover, under the coding schema, this information is distinguishable for men, where the system uses even numbers, and women, where odd numbers are employed.
Compounding Re-identification
Many of the re-identification attacks presented in this report are complementary. As a result, they can be combined to assemble more robust re-identification methods. For example, FAMILY can be used in combination with GENPHEN to construct more informative family structures, or with DICTIONARY when additional information about familial relationships is known. Moreover, an iterative process of alternating re-identification methods can be employed. Since different re-identification methods exploit different types of information, an adversary could use one method to re-identify a certain number of individuals in the population, then a second method to re-identify individuals not re-identified by the first or until certain confounding entities were removed from consideration. This process can continue with as many methods as necessary, or repeat with the same methods, until no more re-identifications are possible.
Discussion
To an extent, the re-identification methods used in this study can be used to evaluate privacy protection technologies beyond those specifically designed for genomic data. The sole re-identification method directly dependent on genomic data is the GENPHEN attack, yet at its foundation, this method was based on the explicit representation of inferences between data types. As such, it is adaptable for other types of data relations. However, a note of caution: before re-identification susceptibility for additional types of data can be claimed, a careful analysis of the social setting and attendant protections must be made. Although linkage of data types may be possible, it must be validated that such data are equally accessible. With respect to genomic data, the status as a lesser-protected data type allows for re-identification using the above methods.
Given the current state of privacy protection systems, there exists a need for a new type of genomic data privacy protection model. In this sense, the results of this evaluation are a call to arms. Researchers must develop privacy protection methods that incorporate guarantees about the afforded protections. New methods must account for multiple environments of data sharing as well as the type of inferences that can be gleaned from the shared data themselves. These methods must be developed in a more scientific and logical manner, with formal proofs about the protection capabilities and limitations afforded by the specific method. Although proofs may be difficult to derive in the face of uncertainties about the sharing environment, especially when the data hold latent knowledge to be learned at a later point in time, researchers can validate their approaches against known re-identification attacks in a logical manner.
Pseudonyms and Linkage
Based on the system analyses above, it is apparent the application of pseudonymization and naïve de-identification alone are not sufficient as proofs of identity protection. Mainly, this is because current systems tend to be narrow in their consideration of what is inferable from genomic data as well as what additional information is available for relating genomic data to identified data. Yet, this does not imply pseudonyms and third-party solutions are worthless in the pursuit of genomic data privacy protection. Rather, to some extent, these systems do provide a level of privacy protection and additional functionality for data sharing. First, pseudonyms serve as a first-order protector and deterrent. It is conceivable that an adversary, who approaches re-identification in a noncomputational manner, will be deterred by the simple obscuring of explicitly identifiable information. Second, datasets devoid of linkage capabilities severely limit the types of research that can be performed. It is often the case in which researchers may need to request additional information about a subject. Third, a subject may wish to remove their data from a research study or audit how their data have been accessed. Yet, if a pseudonym, or linkage value, is to be used as a primary key, it must be chosen appropriately. It should not be based on personal demographics as is currently the case with the TRUST and SEMITRUST models. A pseudonym based on this type of information is susceptible to various attacks, such as DICTIONARY. Consequently, the RGE form of DEID and the DENOM models are more secure in their protection of linkage capabilities, with respect to pseudonym usage.
Accounting for Genomic Data
A common reason for re-identification susceptibility is the uniqueness of data that permit matching. One promising direction for research is the construction and analysis of systems based on formal computational models, such as k-anonymity.22 Under the model of k-anonymity, every released record is indistinguishable from k-1 other records in the release. Within the genomics community, the k-anonymity model, under the term binning, has recently been adapted for the protection of single nucleotide polymorphism (SNP) data.23 For example, consider the employment of the DNA generalization hierarchy in ▶ for purines (R) and pyrimadines (Y). If we wish to generalize the nucleotides C and G together, we only need to generalize up one level, and release R and R. To relate A and T, we must generalize to the indeterminate character N.
Figure 4.
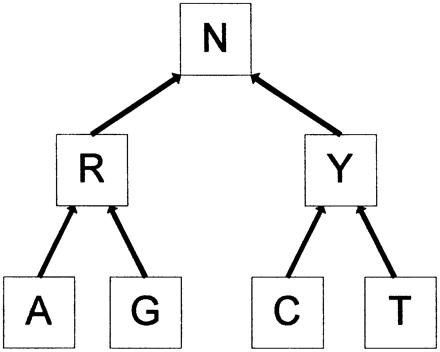
SNP generalization hierarchy for purines and pyrimadines.
Although it has not been presented as a full system or for general genomic data, the binning method is a feasible solution and worthwhile area of study for genomic privacy protection. This is especially so, since such models are amenable to proofs of withstanding various re-identification attacks. However, this research is in a nascent stage, and there are several deficiencies in the current binning model that researchers can build upon for more robust protection models. First, this model is restricted to SNP data and not more general genomic data. For a privacy protection system to function in the real world, it must be able to account for complex genomic features, such as nucleotide repeat structures and complex mutations.
Second, current binning models measure the amount of information lost via protection using an information theoretic perspective. While this is one way to characterize information loss, it does not take into account what the data are to be used for. Although formal protection methods, such as k-anonymity, advocate the direct manipulation of data values, there is no guarantee it will hinder applications or data usefulness. For example, in the statistics community, there has been much research into the design of formal protection methods that influence individual records but permit the recovery of aggregate statistics.24,25 More relevant to the genomics community, however, is recent research in privacy-preserving data mining, in which the privacy-preserving methods are being validated with objective functions, such that logical rules or classifiers can be constructed with formal privacy guarantees about the data values shared.26,27 The development of genomic data privacy methods, which incorporate models of utility, is an open and fruitful direction of research.
From an opposing perspective, researchers can not remain content with their proofs and experiments. New re-identification attacks will be developed by those in the academic community as well as adversaries outside the public realm. As such, researchers must continue to innovate and develop new methods of re-identification for testing their protection techniques. These methods may be new types of inferential or location-based techniques or completely new models yet to be discovered. Without the development of new protection and re-identification methods, researchers will continue to rely upon unfounded and possibly dangerous methods of privacy protection. The development of new identity protection strategies is paramount for continued data sharing and innovative research studies.
Conclusion
This research provided an analysis of the re-identification susceptibility of genomic data privacy protection methods for shared data. The results prove the current set of privacy protection methods do not guarantee the protection of the identities of the data subjects. This work stresses that a new direction in the research and advancement of anonymity protection methods for genomic data must be undertaken. The next generation of privacy protection methods must account for both social and computational interactions that occur in complex data sharing environments. In addition, privacy protection methods must provide proofs about what protections can and cannot be afforded to genomic data, as well as the limits of research with protected data. The development of new identity protection strategies is paramount for continued data sharing and innovative research.
Supported by a National Science Foundation IGERT Program grant and the Data Privacy Laboratory in the Institute for Software Research International, a department in the School of Computer Science at Carnegie Mellon University. The opinions expressed in this research are solely those of the author and do not necessarily reflect those of the National Science Foundation.
The author thanks Alessandro Acquisti, Michael Shamos, Latanya Sweeney, Jean Wylie, and the members of the Data Privacy Library at Carnegie Mellon University for their insightful comments and discussion.
Footnotes
For example, the PopSet database at the National Center for Biotechnology Information contains publicly available DNA sequence data, which are not subject to oversight by an Institutional Review Board.
The reader is directed toward references 9–12 for examples of computational data privacy in the biomedical community.
Details on the second protocol and its mapping to this paper's formalism are available in reference 19.
The set of attributes Additional Demographic Features corresponds to demographic attributes deemed useful by deCode.
Details of the combinatorics for more complex combinations of family-disease structures are provided elsewhere.15
At the time of writing, the website http://www.rat.de/kuijsten/navigator/ provided links to a number of genealogical resources.
References
- 1.Altman RB. Bioinformatics in support of molecular medicine. Proc AMIA Annu Fall Symp. 1998:53–61. [PMC free article] [PubMed]
- 2.Altman RB, Klein TE. Challenges for biomedical informatics and pharmacogenomics. Annu Rev Pharmacol Toxicol. 2002;42:113–33. [DOI] [PubMed] [Google Scholar]
- 3.Vaszar LT, Cho MK, Raffin TA. Privacy issues in personalized medicine. Pharmacogenomics. 2003;4:107–12. [DOI] [PubMed] [Google Scholar]
- 4.Rothstein MA. Genetic secrets: protecting privacy and confidentiality in the genetic era. Yale University Press, New Haven, 1997.
- 5.Hall MA, Rich SS. Patients' fear of genetic discrimination by health insurers: the impact of legal protections. Genet Med. 2000;2:214–21. [DOI] [PubMed] [Google Scholar]
- 6.U.S. Department of Health and Human Services. 45 CFR, Parts 160–164. Standards for privacy of individually identifiable health information, Final Rule. Federal Register: 2002;67(157):53182–273. [PubMed] [Google Scholar]
- 7.Lin Z, Owen AB, Altman RB. Genomic research and human subject privacy. Science. 2004;305:183. [DOI] [PubMed] [Google Scholar]
- 8.Burnett L, Barlow-Stewart K, Pros AL, Aizenberg H. The “GeneTrustee”: a universal identification system that ensures privacy and confidentiality for human genetic databases. J Law M. 2003;10(4):506–13. [PubMed] [Google Scholar]
- 9.Wylie JE, Mineau GP. Biomedical databases: protecting privacy and promoting research. Trends Biotechnol. 2003;21:113–6. [DOI] [PubMed] [Google Scholar]
- 10.Churches T. A proposed architecture and method of operation for improving the protection of privacy and confidentiality in disease registers. BMC Med Res Methodol. 2003;3:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sweeney L. Weaving technology and policy together to maintain confidentiality. J Law Med Eth. 1997;25:98–111. [DOI] [PubMed] [Google Scholar]
- 12.Gaudet D, Arsnault S, Belanger C, et al. Procedure to protect confidentiality of familial data in community genetics and genomics research. Clin Genet. 1999;55:259–64. [DOI] [PubMed] [Google Scholar]
- 13.Gulcher JR, Kristjansson K, Gudbjartsson H, Stefanson K. Protection of privacy by third-party encryption in genetic research. Eur J Hum Genetics. 2000;8:739–42. [DOI] [PubMed] [Google Scholar]
- 14.de Moor GJ, Claerhout B, de Meyer F. Privacy enhancing technologies: the key to secure communication and management of clinical and genomic data. Meth Info Med. 2003;42:148–53. [PubMed] [Google Scholar]
- 15.Malin B. Why pseudonyms don't anonymize: a computational re-identification analysis of genomic data privacy protection systems. Technical Report LIDAP-WP19, Data Privacy Laboratory, Carnegie Mellon University, Pittsburgh, PA, 2003.
- 16.Gulcher JR, Kong A, Stefanson K. The genealogic approach to human genetics. Cancer J. 2001;7(1):61–8. [PubMed] [Google Scholar]
- 17.Sweeney L. Uniqueness of Simple Demographics in the U.S. Population. Technical Report LIDAP-WP4. Data Privacy Laboratory, Carnegie Mellon University, Pittsburgh, PA, 2000.
- 18.Malin B, Sweeney L. Determining the identifiability of DNA database entries. Proc AMIA Annu Fall Symp. 2000:547–51. [PMC free article] [PubMed]
- 19.Hess P, Cooper D. Impact of pharmacogenomics on the clinical laboratory. Mol Diagn. 1999;4:289–98. [DOI] [PubMed] [Google Scholar]
- 20.Malin B, Sweeney L. Inferring genotype from clinical phenotype through a knowledge based algorithm. Pac Symp Biocomp. 2002:41–52. [DOI] [PubMed]
- 21.Malin B, Sweeney L. How (not) to protect genomic data privacy in a distributed network: using trail re-identification to evaluate and design anonymity protection systems. J Biomed Inf. 2004;37:179–92. [DOI] [PubMed] [Google Scholar]
- 22.Sweeney L. Achieving k-anonymity privacy protection using generalization and suppression. International Journal of Uncertainty, Fuzziness, and Knowledge-based Systems. 2002;10:571–88. [Google Scholar]
- 23.Lin Z, Hewitt M, Altman RB. Using binning to maintain confidentiality of medical data. In Proc AMIA Annu Fall Symp. 2002:454–8. [PMC free article] [PubMed]
- 24.Duncan GT, Fienberg S. A Markov perturbation method for tabular data, turning administrative systems into information systems, IRS Methodology Report Series 5. 1997;223–31.
- 25.Domingo-Ferrer J, Torra V. Disclosure control methods and information loss for microdata. In Confidentiality, Disclosure and Data Access: Theory and Practical Applications for Statistical Agencies, Amsterdam: North-Holland, 2002;93–112.
- 26.Agrawal R, Srikant R. Privacy preserving data mining. Proc ACM Symp Principles of Database Systems. 2000:439–50.
- 27.Agrawal D, Aggarwal C. On the design and quantification of privacy preserving data mining algorithms. Proc ACM Symp Principles of Database Systems. 2001:247–55.