

Abstract
Multiple myeloma (MM) is a hematological malignancy that remains incurable, with relapse rates greater than 90%. The main limiting factor for the effective use of chemotherapies in MM is the serious side effects caused by these drugs. The emphasis in cancer treatment has shifted from cytotoxic, non-specific chemotherapies to molecularly targeted and rationally designed therapies showing greater efficacy and fewer side effects. Traditional chemotherapy has shown several disadvantages such as lack of targeting capabilities, systemic toxicity and side effects; low therapeutic index, as well as, most anticancer drugs have poor water solubility. Nanoparticle delivery systems (NPs) are capable of targeting large doses of chemotherapies into the target area while sparing healthy tissues, overcoming the limitations of traditional chemotherapy.
Here, we review the current state-of-the-art in nanoparticle-based strategies designed to treat multiple myeloma. Many nanoparticle delivery systems have been studied for myeloma using non-targeted NPs (liposomes, polymeric NPs, and inorganic NPs), triggered NPs, as well as targeted NPs (VLA-4, ABC drug transporters, bone microenvironment targeting). The results in preclinical and clinical studies are promising; however, there remains much to be learned in the emerging field of nanomedicine in myeloma.
Keywords: Multiple myeloma, Nanoparticles, Passive Targeting, Triggered Targeting, Targeted Targeting
1. INTRODUCTION
Multiple myeloma (MM) is a hematological malignancy of plasma cells localized in the bone marrow. Despite recent advances in therapy, MM remains incurable with relapse rates greater than 90% (1, 2). The main limiting factor for the effective use of chemotherapies in MM is the serious side effects caused by these drugs. The development of specific and targeted therapies based on nanoparticle delivery systems in MM is under investigation.
Over the last decades, a large number of nanoparticle delivery systems have been designed for cancer therapy. The emphasis in cancer treatment has shifted from cytotoxic, non-specific chemotherapies to molecularly targeted and rationally designed therapies showing greater efficacy and fewer side effects (3). Many drugs are hydrophobic and poorly water-soluble; properties which help them to penetrate the cell membrane and reach intracellular targets, however, its therapeutic application is associated with poor absorption and low bioavailability (4-7). Therefore, NPs mainly aim to minimize drug degradation upon administration, prevent undesirable side effects, and increase drug bioavailability in the pathological area (8-10).
This review will provide an insight into the advances in nanoparticle delivery systems focused on oncology therapeutics and in particular in MM.
2. NANOPARTICLE DELIVERY SYSTEMS IN CANCER
Current treatment for cancer relies on chemotherapy as a major strategy. Traditional chemotherapy has shown several disadvantages such as lack of targeting capabilities, affecting normal healthy tissues; non-specific distribution, producing systemic toxicity and side effects; low therapeutic index, and most anticancer drugs have poor water solubility (11-13). Nanoparticle delivery systems are aimed to target higher doses of active agents into the tumor areas while sparing healthy tissues, overcoming the limitations of traditional chemotherapy (9, 10). The delivery of anticancer drugs through a nanoparticle delivery system offers multiple attractive opportunities (Table 1), including: 1) improved stability and delivery of poorly soluble in water drugs, permitting re-evaluation of drugs with poor pharmacokinetics, high cytotoxicity or poor cellular uptake (14); 2) controlled release in a specific location triggered by an specific stimuli, protection of the drug from harsh environment (acidic environment, proteases, or lysosomes of cells) extending half-life of the drug in the circulation, and controlled release over time to achieve a drug dose within a therapeutic window (10, 15-18); 3) targeting properties can be enhanced to deliver drugs specifically to a cell or tissue, reducing systemic side effects, increasing the concentration of the drug in the target area, and increasing therapeutic index for the chemotherapeutic agent (19); and 4) nanoparticle delivery systems offer a multifunctional approach to deliver combination therapy of drugs, as well as, imaging agents to improve detection, imaging, and treatment of cancer (20, 21).
Table 1.
Comparison of traditional chemotherapy and nanoparticle delivery systems properties.
| Property | Traditional Chemotherapy | Nanoparticle delivery systems |
|---|---|---|
|
| ||
| Drug solubility | Poor; affecting pharmacokinetics | Improved; increased stability |
|
| ||
| Controlled release | None | Controlled release time and location, and protection from environment |
|
| ||
| Targeting abilities | Non-specific targeting; non-specific distribution; systemic side effects; low drug concentration in target area; low therapeutic index | Specific targeting; lower systemic side effects; improved drug concentration in target area; improved therapeutic index |
|
| ||
| Multi-functionality | None | Combination therapy for detection, imaging and treatment |
Nanoparticle delivery systems are very diverse in function of shapes, sizes, and surface-properties (Figure 1): Liposomes: are vesicles made of a bilayer of amphiphilic lipids enclosing a hydrophilic core, which can carry hydrophilic drugs within the aqueous core area while hydrophobic drugs within the hydrophobic region of the bilayer(22) (Figure 1a). Polymeric NPs: are particles prepared from polymers, they can be biodegradable or non-biodegradable, synthetic or natural and the drug is dissolved, entrapped, encapsulated or attached to the matrix (23) (Figure 1b). Micelles: are artificial vesicles similar to liposomes but made of a monolayer of amphiphilic lipids enclosing a hydrophobic core, which can carry hydrophobic anticancer agents (Figure 1c). Dendrimers: are repetitively branched molecules consisting of radially symmetric molecules of tree-like arms or branches (Figure 1d). Polymersomes: are artificial vesicles made of a bilayer of synthetic amphiphilic block copolymers enclosing a hydrophilic core, which can carry hydrophilic drugs within the aqueous core, therefore presenting a similar structure to liposomes (Figure 1e). Inorganic NPs are particles formed by the crystallization of inorganic salts, forming a three-dimensional arrangement with linked atoms (24) (Figure 1f).
Figure 1. A summary of nanoparticles for drug delivery in cancer.
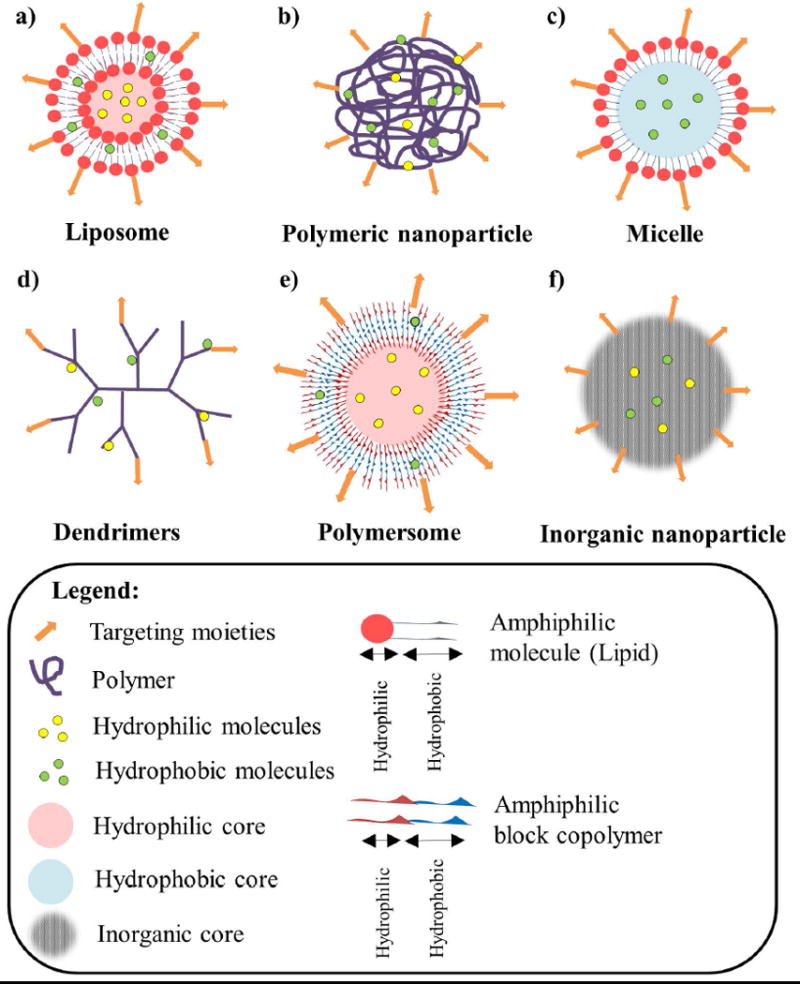
a) Liposomes are vesicles made of a hydrophobic bilayer of amphiphilic lipids (composed of a hydrophilic head group and a hydrophobic tail) enclosing a hydrophilic core, which can carry hydrophilic drugs within the aqueous core area while hydrophobic drugs within the hydrophobic region of the bilayer; b) Polymeric NPs are made of polymers and can encapsulate hydrophilic and hydrophobic molecules. c) Micelles are made of a hydrophobic monolayer of amphiphilic lipids enclosing a hydrophobic core, which can carry hydrophobic anticancer agents. d) Dendrimers are repetitively branched molecules consisting of radially symmetric molecules of tree-like arms or branches, which can encapsulate hydrophilic and hydrophobic molecules. e) Polymersomes are artificial vesicles made of a bilayer of synthetic amphiphilic block copolymers enclosing a hydrophilic core, which can carry hydrophilic drugs within the aqueous core and hydrophobic drugs within the hydrophobic region of the bilayer. f) Inorganic NPs are particles formed by the crystallization of inorganic salts, forming a three-dimensional arrangement with linked atoms, which can encapsulate hydrophilic and hydrophobic molecules. All the previous nanoparticles can be modified to contain specific targeting moieties for active targeting strategies.
The delivery of anticancer drugs through nanoparticle delivery systems can be achieved in three ways: take advantage of pathophysiological conditions of the tumors (passive targeting) (Figure 2), induction of drug release once in the tumor through triggered or inducible approaches (triggered targeting) (Figure 3), or addition of high-affinity ligands to target the tumor to the surface of nanoparticle delivery systems (active targeting) (Figure 4).
Figure 2. Schematic of passive (non-targeted) nanoparticles in cancer.
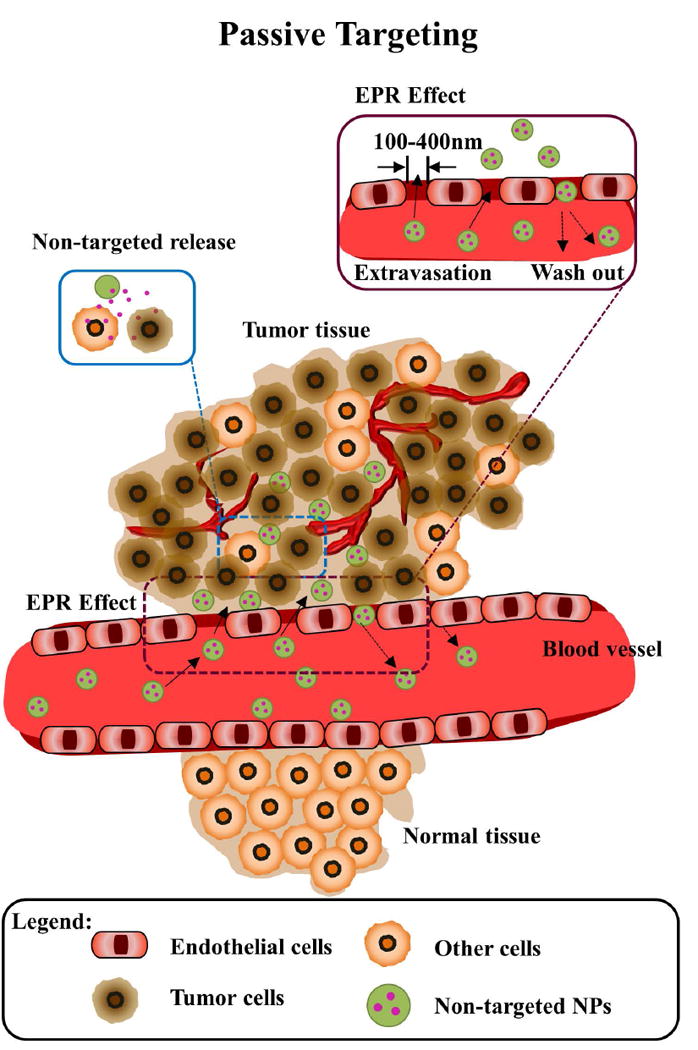
Normal tissues lack of gaps between adjacent endothelial cells discarding nanoparticles to extravasate from the vasculature. Tumor tissues present large gaps (100-400 nm) between the endothelial cells on the tumor vasculature. The enhanced permeability and retention (EPR) effect allows untargeted nanoparticles of appropriate size to bind, internalize and release drugs into cancer (brown) and non-cancer cells (orange) cells due to unspecific surface adsorption inside tumor tissues. A significant part of nanoparticles can be washed out back to the blood circulation, resulting in low tumor accumulation.
Figure 3. Schematic of triggered (inducible) nanoparticles in cancer.
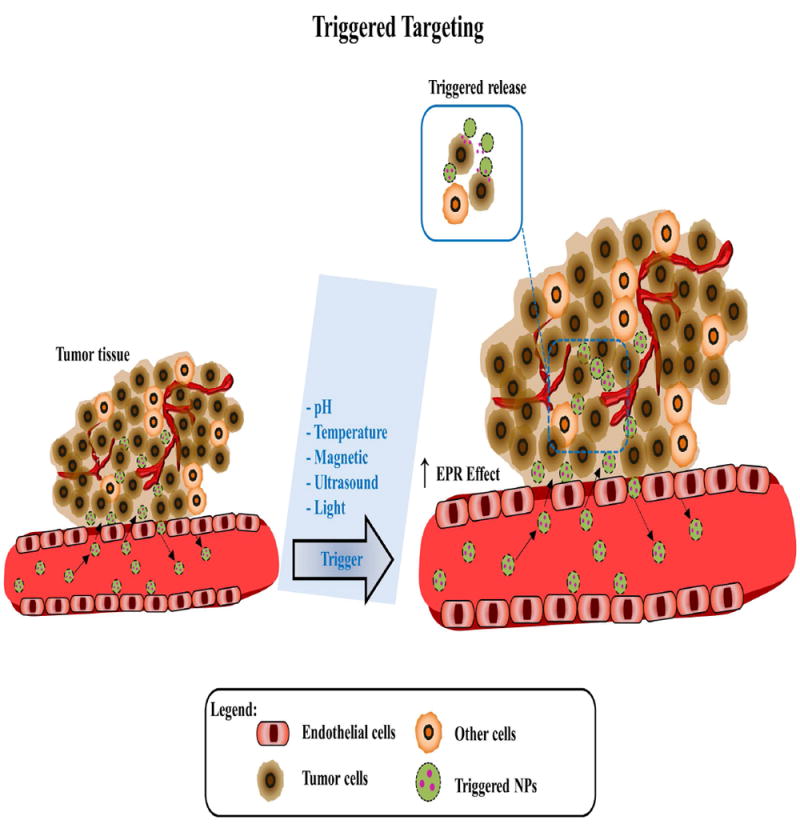
Triggered nanoparticles extravasate from the vasculature accumulate in the tumors due to increase EPR effect by biological factors of the tumor microenvironment or external factors (pH, temperature, magnetic field, ultrasound, and light exposure). The release is triggered by those factors into cancer cells (brown).
Figure 4. Schematic of active (targeted) nanoparticles in cancer.
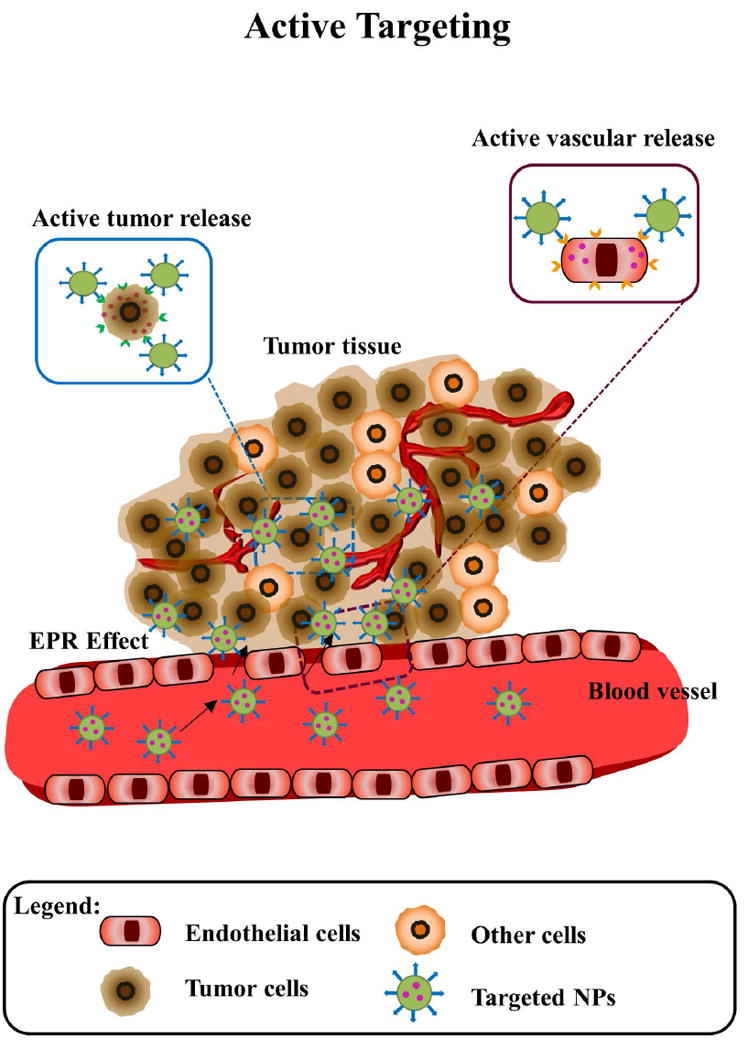
Nanoparticles extravasate from the vasculature due to EPR effect, then targeted nanoparticles bind and internalize into tumor tissues, the retention and uptake of these nanoparticles in cancer cells (brown) is augmented due to specific antigen-antibody/ ligand-receptor interactions, and wash out of nanoparticles is reduced.
Passive targeting
Non-targeted NPs exploits passive targeting approaches taking advantage of pathophysiological conditions of the tumor microenvironment for specific delivery, such as enhanced permeation and retention (EPR) effect: most solid tumors have blood vessels with aberrant architecture and extensive production of vascular permeability factors stimulating extravasation within tumor tissues, in addition to lack of lymphatic drainage. Therefore, tumors exhibit enhanced vascular permeability, which will ensure a sufficient supply of nutrients and oxygen to tumor tissues for rapid growth (25-27). The tumor vasculature is formed by poorly aligned defective endothelial cells with wide fenestrations (up to 4 μm), leading to EPR effect, which facilitates transport of macromolecules (>40 kDa) and small-sized NPs (10 - 400 nm) into tumors (28-30). EPR is the gold-standard technique for drug delivery systems for cancer treatment (Figure 2). However, large tumors do not exhibit the EPR effect and show less accumulation of macromolecules in central areas (31).
Triggered targeting
A selective and controlled delivery of drugs in the tumors is still a challenge. The introduction of stimuli-responsive mechanisms to drug delivery systems help to provide better solutions for tumor drug targeting strategies. NPs can be induced to release the drugs once in the tumor through triggered or inducible approaches (32, 33). Several factors (such as pH (34-37), temperature (38-41), magnetic field (42, 43), ultrasound (44, 45) or light exposure (46, 47)) have been shown to increase the accumulation of NPs and/or enhance the release of the drugs from the NPs in the tumor (Figure 3).
Active targeting
The EPR effect and PEG-stealth have improved biodistribution and circulation of nanoparticle delivery systems. Stimuli-triggered NPs also have improved controlled release in the tumor tissue. However, better methods are required to increase the number of NPs that accumulate in the target tissue and reduce the amount of NPs that are concentrated in organs such as liver, spleen, and bone marrow due to clearance by mononuclear phagocytic system (MPS). The main approach in the active targeting consists of ligand-based targeting, these ligands facilitate binding to a marker or receptor overexpressed in the targeted tumor cells, triggering receptor-mediated endocytosis, or targeting tumor microenvironment extracellular matrix or surface receptors on tumor blood vessel endothelial cells (48-54) (Figure 4).
Even though nanoparticles have shown very promising results in cancer, there are two main disadvantages of NPs, which need to be overcome for successful usage: 1) Clearance: while NPs smaller than 6 nm are rapidly cleared by the kidneys, larger NPs either accumulate in a lesion or are cleared by the mononuclear phagocytic system (MPS) accumulating in lymph nodes, spleen, and liver (55, 56). A strategy to improve circulation half-life consist on the conjugation of hydrophilic polymers such as polyethylene glycol (PEG) to the NPs surface, which confers them long-circulation due to stealth properties by reducing opsonization (57, 58). 2) Toxicity: Nanoparticles toxicity depends not only on their chemical composition, but also on the composition of any chemicals adsorbed onto their surfaces (59). As a solution, the surfaces of nanoparticles can be modified to make them less harmful.
3. NANOPARTICLE DELIVERY SYSTEMS FOR MYELOMA
Multiple myeloma (MM) is an incurable disease characterized by the proliferation of plasma cells in the bone marrow (60), and represents approximately 20% of deaths from hematological malignancies (61). Several chemotherapeutic regimens have been used in the management of MM, including anthracycline antibiotics (doxorubicin), glucocorticoids (dexamethasone and prednisone), and nitrogen mustard alkylating agents (melphalan) (62-66). The introduction of novel agents, such as immunomodulatory drugs (IMiDs) thalidomide, lenalidomide, and pomalidomide; and the proteasome inhibitors (PIs) bortezomib and carfilzomib has significantly improved survival in MM (66-69). Despite the introduction of the previous novel therapies, more than 90% of MM patients relapse or become refractory to those treatments (1, 2).
The main limiting factor for the effective use of chemotherapies in MM is the serious side effects caused by these drugs. The use of the PIs bortezomib and carfilzomib has led to significant improvement of the survival of MM patients (66). However, treatment with bortezomib is limited by its neurotoxicity, especially in the peripheral nerves, which leads to sensory axonal neuropathy (70). Carfilzomib is a second generation proteasome inhibitor, but the safety data from a meta-analysis reported thrombocytopenia, anemia, fatigue, nausea, and diarrhea as the most common adverse events, with dose-limiting neutropenia or peripheral neuropathy (71). Immunomodulatory drugs are emerging promising therapies in MM which showed synergistic effects when they are added to current treatments (69). Nevertheless, one-fourth of patients discontinued thalidomide because of toxicity, including peripheral neuropathy, constipation, somnolence, and fatigue as common side-effects (72). Moreover, cutaneous adverse neutropenia, deep vein thrombosis, infection, and hematologic cancer were observed in patients treated with lenalidomide (73, 74). Dose limiting neutropenia, thrombocytopenia, neuropathy, and deep vein thrombosis were common adverse effects observed in patients treated with pomalidomide (75).
Nanoparticle delivery systems offer a new strategy to increase the efficacy of the treatment and reduce side effects in normal tissues by delivering drugs to the target tissue, in this case the bone marrow niche in which myeloma cells developed. Table 2 and 3 provides an insight into the preclinical and clinical advances in nanoparticle delivery systems used in multiple myeloma.
Table 2.
Preclinical nanoparticle delivery systems in MM.
| Type | Formulation | Drug | In vitro efficacy | In vivo efficacy | Biodistribution/ Toxicity | Ref. |
|---|---|---|---|---|---|---|
| Non- targeted | Liposome | Bortezomib | Proteasome inhibition, induction of apoptosis, and cytotoxicity | Dose: 1 mg/kg twice i.v. Tumor growth inhibition | Reduced body weight loss No biodistribution | Ashley, J.D., et al., J Med Chem, 2014. 57(12): p. 5282-92. |
| Anti-estrogens | N/A | Dose: 12 mg/kg/week i.v. Tumor growth inhibition | N/A | Maillard, S., et al., J Steroid Biochem Mol Biol, 2005. 94(1-3): p. 111-21. | ||
| Polymeric NPs (PLGA-PEG) | Thymoquinone | Anti-proliferative effect | N/A | N/A | Ravindran, J., et al., Biochem Pharmacol, 2010. 79(11): p. 1640-7 | |
| Polymeric NPs (Chitosan) | Camptothecin | Cytotoxicity | Dose: 2.5 mg/kg thrice i.v. Tumor growth inhibition and PST | N/A | Li, Z., et al., Oncol Rep, 2012. 27(4): p. 1035-40 | |
| Micelles | Carfilzomib | Improved stability and cytotoxicity | N/A | N/A | Ao, L., et al., J Pharmacol Exp Ther, 2015. 355(2): p. 168- 73. | |
| Inorganic NPs (Silica) | Snake venom from Walterinnesia aegyptia | Induction of apoptosis, and cytotoxicity | Dose: 1 μg/kg/day s.c. in the tumor site Enhanced anticancer efficacy | N/A | Sayed, D., et al., Oxid Med Cell Longev, 2012. p. 386286. Al-Sadoon, M.K., et al., Cell Immunol, 2013. 284(1-2): p. 129-38. | |
| Inorganic NPs (Iron oxide) | Placitaxel | Identification of CD138- CD34- tumor stem cells | Dose: 0.6 - 2 mg/kg once a week for 2 weeks Tumor growth inhibition of CD138- CD34- tumor stem-like cells | N/A | Yang, C., et al., Int J Nanomedicine, 2013. 8: p. 1439-49. | |
| Triggered | Magnetic NPs | Doxorubicin | N/A | Dose: 0.13 mg/kg + magnetic field Tumor growth inhibition | No significance toxicity No biodistribution | Hayashi, K., et al., Theranostics, 2014. 4(8): p. 834-44. |
| Targeted | VLA-4 | Doxorubicin | Induction of cytotoxicity and apoptosis, overcame CAM-DR | Dose: 6 mg/kg thrice Tumor growth inhibition and higher accumulation in the tumor | Reduced body weight loss | Kiziltepe, T., et al., Blood Cancer J, 2012. 2(4): p. e64 |
| Carfilzomib | Induction of apoptosis and cytotoxicity | Dose: 5 mg/kg twice a week i.v. Tumor growth inhibition and higher accumulation at the tumor | Reduced body weight loss | Ashley, J.D., et al., J Control Release, 2014. 196: p. 113-21 | ||
| MYC inhibitor | Induction of apoptosis and cytotoxicity | Dose: 0.145 mg/ml thrice a week i.v. Survival benefits | N/A | Soodgupta, D., et al., Mol Cancer Ther, 2015. 14(6): p. 1286- 94 | ||
| ABC drug transporters | Placitaxel | Inhibition of CD138- CD34- tumor stem-like cells growth | Dose: 10 μg mAb and 8 μg placitaxel for 4 weeks Tumor growth inhibition and PST in CSC models | Lower toxic side effects | Yang, C., et al.,Nanomedicine, 2014. 9(1): p. 45-60. & J Biomed Nanotechnol, 2014. 10(2): p. 336-44. | |
| Bone micro- environment | Bortezomib | Targeted NPs presented good binding, and induction of apoptosis | Dose: 0.5 mg/kg twice a week i.p. Tumor growth inhibition and PST | Biodistribution by imaging No side effects studies | Swami, A., et al., Proc Natl Acad Sci U S A, 2014. 111 (28): p. 10287-92 |
Legend: NPs, Nanoparticles; N/A, Not available; i.v., intravenous; s.c., subcutaneous; i.t., intratumoral; i.p., intraperitoneal; PST, prolonged survival time; CAM-DR, cell adhesion mediated drug-resistance; ABC, ATP-binding cassette; CSC, cancer stem cells.
Table 3.
Clinical nanoparticle delivery systems in MM
| Type | Phase | Study design | Results | Ref. |
|---|---|---|---|---|
| Non-targeted. PEGylated liposomal doxorubicin | Phase I | Advanced hematologic malignancies patients received PegLD (day 4 at 30 mg/m2) in combination with bortezomib (days 1, 4, 8, and 11 from 0.90 to 1.50 mg/m2). | Common toxicities were Grade 3 or 4. The MTD was 1.50 and 30 mg/m2 of bortezomib and PegLD, respectively. Antitumor activity was seen against multiple myeloma, 36% CR or near-CR, and another 36% PR. | Orlowski, R.Z., et al., Blood, 2005. 105(8): p. 3058-65. |
| Phase III | Relapsed or refractory multiple myeloma patients received either intravenous bortezomib 1.3 mg/m2 on days 1, 4, 8, and 11 of an every-21-day cycle with PegLD 30 mg/m2 on day 4 or bortezomib alone. | TTP was significantly prolonged in the combination arm (median TTP = 9.3 months) compared with bortezomib monotherapy (median TTP = 6.5 months) | Orlowski, R.Z., et al., J Clin Oncol, 2007. 25(25): p. 3892-901. | |
| Phase II | Newly diagnosed multiple myeloma patients received intravenous PegLD (40 mg/m2), vincristine (2.0 mg, Day 1), and oral or intravenous dexamethasone (40 mg per day for 4 days) every 4 weeks for six or more cycles and/or for two cycles after the best response. | The most common toxicities were Grade 3. The overall response rate was 88%: 12% CR. TPP was 23.1 months, with 2-year and 3-year progression-free survival rates of 42% and 23%, respectively. The patient survival rate at 3 years was 67%. | Hussein, M.A., et al., Cancer, 2002. 95(10): p. 2160-8. |
Legend: PegLD, PEGylated liposomal doxorubicin; MTD, maximum tolerated dose; CR, complete response; PR, partial response; TTP, time to progression
3. 1. Non-targeted therapies
Non-targeted approaches exploit passive targeting based on the pathophysiological conditions of the myeloma microenvironment for specific delivery of the drugs. Most of the nanoparticle delivery systems that have been studied for myeloma used non-targeted NPs.
-
Liposomes have been used to encapsulate bortezomib, a proteasome inhibitor with high neurotoxicity. Due to the poor water solubility of bortezomib, instead of loading the drug in the aqueous core (76), direct conjugation of bortezomib to the surface of liposomes via a reversible boronic ester linkage was used (77). Liposomal bortezomib NPs showed size ranges of 100 nm with high reproducibility and encapsulation efficiency of 80%. In vitro studies were performed to study proteasome inhibition, apoptosis, and cell viability. Liposomal bortezomib NPs inhibited proteasome activity, induced apoptosis and cytotoxicity on MM cells. In the in vivo studies SCID mice were injected subcutaneously with MM cells, treated with free drug or liposomal bortezomib NPs intravenously (i.v.) on days 1 and 4 at a dose of 1 mg/kg bortezomib equivalent, and analyzed for tumor progression and systemic toxicity. The results revealed that liposomal bortezomib NPs were efficacious in tumor growth inhibition, and reducing systemic side effects measured by body weight loss. Free drug group showed >20% weight loss and moribundity on day 7, which led to required sacrificed of the mice. In case of liposomal bortezomib NPs there was <10% loss in body mass during the 2 week study period (77) (Table 2). However, even though reduced side effects, distribution studies should have been done to verify the location of the NPs.
Liposomes have been also used for incorporation of anti-estrogens to prevent oral administration of an anti-estrogen being widely distributed to the whole body and reaching unwanted tissues. Liposomes size was around 100 nm and encapsulation efficiency of >90%. Loaded liposomes were administered i.v. at a dose of 12 mg anti-estrogen/kg/week in a MM xenograft model. Loaded liposomes induced the arrest of tumor growth in contrary to free anti-estrogen or to empty liposomes (78) (Table 2). Further studies of the drug delivery of anti-estrogens in tumors which express estrogen receptors will be required, as well as, distribution of these NPs and systemic toxicities should be investigated.
The first nanoparticle delivery system approved by the FDA for clinical use in multiple myeloma was a PEGylated liposomal doxorubicin. It has been used use in combination with other anti-myeloma therapeutics (bortezomib, or vincristine and dexamethasone). Relapsed or refractory multiple myeloma patients received PEGylated liposomal doxorubicin (Tibotec Therapeutics) administered on day 4 at 30 mg/m2 and bortezomib given on days 1, 4, 8, and 11 from 0.90 to 1.50 mg/m2. Time to progression (TTP) was significantly prolonged in the combination arm (median TTP = 9.3 months) compared with bortezomib monotherapy (median TTP = 6.5 months) (79-81) (Table 3). Newly diagnosed multiple myeloma patients received intravenous PEGylated liposomal doxorubicin (40 mg/m2), vincristine (2.0 mg, Day 1), and oral or intravenous dexamethasone (40 mg per day for 4 days) every 4 weeks for six or more cycles and/or for two cycles after the best response. The overall response rate was 88% with 12% complete remission. TPP was 23.1 months, with 2-year and 3-year progression-free survival rates of 42% and 23%, respectively (82) (Table 3). These results are very encouraging for clinical efficacy of NPs on MM, although further follow-up results need to be supplemented.
-
Polymeric NPs such as PLGA-PEG NPs have been developed to encapsulate thymoquinone, an anti-inflammatory and anti-cancer natural product derived from the medicinal spice black cumin, which has problems of bioavailability (83). PLGA-PEG NPs encapsulating thymoquinone showed size around 200 nm with homogeneous distribution and encapsulation efficiency of 94%. Thymoquinone PLGA-PEG NPs had anti-proliferative effect on MM cells and these NPs were more potent than free drug sensitizing leukemic cells to TNF-α and paclitaxel-induced apoptosis (84) (Table 2). However, this study is very preliminary, only in vitro studies were realized. In vivo studies to prove efficacy and distribution will demonstrate the potential of thymoquinone NPs in myeloma.
Other polymeric NPs are nanocolloids based on N,N,N-trimethyl chitosan have been developed to encapsulate camptothecin, a potent anticancer agent of plant origin, which is extremely water insoluble (85). In vitro cytotoxicity showed no statistical difference between loaded nanocolloids and free drug. However, loaded nanocolloids more effectively inhibited tumor growth and prolonged survival time than free drug in vivo. In this case the murine Balb/c myeloma model was treated with i.v. injections of loaded nanocolloids (2.5 mg/kg), free drug (2.5 mg/kg), or nanocolloids (25 mg/kg) every 3 days for 15 days (86) (Table 2). Further studies of distribution and systemic toxicities should be examined.
Micelles composed of biodegradable block copolymers of PEG and poly-(caprolactone) (PCL) were developed to improve the metabolic stability of the proteasome inhibitor carfilzomib against enzyme-mediated degradation and delivering of this poorly water soluble drug in a controlled manner (87). Drug loading efficiency was around 2 – 4% and encapsulation efficiency around 20 – 40%. Micelles showed improved stability profiles with at least 50% of the active carfilzomib remaining after 20 minutes of incubation in mouse liver homogenates. Micelles were tested in RPMI-8226 cell line and shown comparable anticancer effect compared with free carfilzomib (88) (Table 2). However, this study is very preliminary, only in vitro studies were realized. In vivo studies to confirm improved stability of carfilzomib, and efficacy and distribution of the micelles will demonstrate the potential of this approach in myeloma.
-
Inorganic NPs have been developed for the treatment of myeloma. Silica NPs have been combined with snake venom from Walterinnesia aegyptia, a natural toxin with antitumor potential (89). The particle size was 300 nm and the combination of the silica NPs with the snake venom was tested on cells from 5 myeloma patients and XG2 cell line. This combination decreased viability and induced apoptosis (90). In a follow-up publication enhanced anticancer efficacy was detected in a murine MM model with loaded NPs subcutaneously (s.c.) injected (1 μg/kg/day) in the tumor site compared to treatment with NPs or vehicle (91) (Table 2). Nevertheless, distribution of silica NPs combined with snake venom, systemic toxicities, and comparison to standard care of treatment in MM should be addressed in the future.
Moreover, iron oxide NPs have been investigated in MM. While paclitaxel is an effective anticancer drug with poor solubility in water, Abraxane® (Celgene, Summit, NJ, USA) is a water-soluble commercially available nanoparticle albumin-bound paclitaxel-loaded Fe3O4 nanoparticle (92), approved by the FDA for the treatment of metastatic breast cancer. Paclitaxel Fe3O4 NPs were used to treat CD138- CD34- tumor stem-like cells in multiple myeloma-bearing mice. The NPs size was 7 nm, with good stability and sustained-drug release. Tumor growth was more inhibited when treated with placitaxel Fe3O4 NPs (0.6 - 2 mg/kg once a week for 2 weeks) compared to NPs alone or drug alone, as well as detection of induced apoptosis of tumor cells in treated mice (93) (Table 2). Nonetheless, distribution of these NPs, effect on CD138+ non-stem cell-like tumors, and systemic toxicities should be investigated.
The main disadvantages of non-targeted NPs are associated to problems in pharmacokinetic and pharmacodynamics, including their dependency on abnormal leakiness of blood vessels and lack of specificity. Therefore, new techniques are needed to improve the accumulation of NPs at the disease site.
3. 2. Triggered therapies
The combination of magnetic hyperthermia treatment through magnetic NPs and remote-control drug release from the same NPs using heat as the trigger to release the drugs have been used in MM. Doxorubicin magnetically responsive NPs were injected intratumorally (i.t.) 5 mg/kg (0.13 mg/kg doxorubicin equivalent) into mice bearing subcutaneous xenograft tumors and then exposed to the magnetic field. Combination of chemotherapy and magnetic hyperthermia NPs decreased tumor volume gradually, until reaching zero tumor volume at day 45 without further recurrence (94) (Table 2). In addition, no significant toxicity was found. These results with i.t injection showed promising results, i.v injection as more general approach and related-distribution should be tested in the future. One limitation of this approach is the ability to treat only the tumor site with well-defined localization in case of further clinical evaluation.
3. 3. Targeted therapies
In general, targeted NPs used the same nanoparticle delivery systems of non-targeted therapies, but in this case NPs contain a targeted-ligand to help NPs to be directed towards tumor cells, tumor blood vessel endothelial cells, or the tumor microenvironment. The strategies used in MM to date are the following:
-
Very Late Antigen-4 (VLA-4) is an integrin receptor expressed on cancer of hematopoietic origin, such as MM, and is a key adhesion molecule in MM associated with cell adhesion mediated drug-resistance (CAM-DR). Therefore, VLA-4 is a relevant surface receptor to target MM cells (95, 96). The use of VLA-4 targeted NPs has been explored to enhance the targeting of VLA-4 expressing MM cells. VLA-4 antagonist peptide and pH-sensitive doxorubicin were incorporated in lipid NPs. These NPs had a size of 20 nm, induced cytotoxicity and apoptosis on MM cells in vitro, and overcame CAM-DR. In vivo, 6 mg/kg doxorubicin equivalent nanoparticles were injected on days 1, 3 and 5 on mice with palpable tumors. Free drug group showed >15% weight loss and moribundity on day 7, which led to required sacrifice of the mice. In case of targeted NPs there was around 10% loss in body mass. Targeted NPs demonstrated enhancement in tumor growth inhibition compared to non-targeted NPs, as well as, significant higher accumulation in the tumor (97) (Table 2).
Another study has developed liposomal carfilzomib NPs, such as the ones previously described for bortezomib (77), enriched with a VLA-4 antagonist peptide. NPs were stable and reproducible with size 70 nm, and loading efficiency of 98%. In vitro studies showed that VLA-4 targeted liposomal carfilzomib NPs were cytotoxic for MM cells and induced apoptosis. In vivo, SCID mice with subcutaneously injected tumors were treated with i.v injection at a dose of 5 mg/kg of carfilzomib free or in NPs on days 1, 2, 8, and 9, and analyzed for tumor progression and systemic toxicity. The results revealed that targeted liposomal carfilzomib NPs were efficacious in tumor growth inhibition, and reducing systemic side effects measured by body weight loss. Free drug group showed >15% weight loss and moribundity on day 4, which led to required sacrificed of the mice. In case of liposomal carfilzomib NPs (targeted and non-targeted) there was <10% loss in body mass. Non-targeted NPs demonstrated less enhancement in tumor growth inhibition compared to targeted liposomal carfilzomib NPs, which had preferential accumulation at the MM tumor (98) (Table 2).
A recent publication in 2015 combined VLA-4 targeted NPs with an inhibitor of MYC-MAX dimerization (MYC transcription factor is an oncoprotein activated in MM), which had poor bioavailability and rapid metabolism (99). VLA-4 targeted perfluorocarbon NPs and polysorbate micelles were developed with size of 200 and 20 nm, respectively. In a KaLwRij metastatic mouse model NPs were injected i.v on days 3, 5, 7, 10, 12, and 14 at an equivalent dose of 0.145 mg/ml of inhibitor. The smaller targeted NPs loaded with the inhibitor conferred survival benefits compared to free drug, larger targeted NPs, or non-targeted NPs (100) (Table 2).
The use of VLA-4 integrin targeting strategies has led to very promising results with several different drug combinations. However, VLA4-4 expression on human myeloma cells is very heterogeneous, with even negligible expression in some MM cells, as well VLA-4 is expressed in many other non-myeloma cells (97, 100). Prescreening of myeloma cell surface biomarkers expression on a personalized medicine approach will be required in case of clinical approach.
ATP-binding cassette (ABC) drug transporters, such as ABCG2 (breast cancer resistance protein 1), enable cancer cells to not be affected by the cytotoxic effects of chemotherapy that kill most cells in a tumor (101). It has been shown that MM expresses ABC transporters (102-104). Previously described paclitaxel Fe3O4 NPs (93) were targeted to the ABCG2 transporter overexpressing MM cancer stem cells with monoclonal antibodies (mAbs). Multiple myeloma cancer stem cells (CSC) mouse model was locally treated once a week with equivalent of 10 μg mAb and 8 μg paclitaxel NPs for 4 weeks. Targeted NPs inhibited tumor growth, increased survival by inducing apoptotic pathways, and showed less toxic side effects in comparison with the single-agent treatments (105, 106) (Table 2). Targeted CSC therapy using mAb in combination with nanoparticle delivery systems is a promising strategy to target MM CSCs for refractory MM patients. This is because the therapeutic strategy has the specific action on the MM CSCs and the proven therapeutic efficacy at lower drug dosages as well as preventing the paclitaxel efflux from cells for generating the cytotoxic effects on the MM CSCs. Further investigation is required in this promising targeted approach.
Bone microenvironment targeting, aimed to target the bone mineral component of the bone marrow microenvironment instead myeloma cells directly. Seventy percent of the bone is made up of the inorganic mineral hydroxyapatite, which includes several types of calcium forms. Bisphosphonates-based drugs preferentially stick to calcium and bind to it upon systemic administration (107). Alendronate (a bisphosphonate) targeted PLGA-PEG NPs encapsulating bortezomib have been engineered to target selectively the bone material in the bone marrow. NPs were elaborated through nanoprecipitation and single emulsion with size 75 nm and 195 nm, and 5.4% and 24% encapsulation efficiency, respectively. In vitro, targeted NPs indicated good binding to hydroxyapatite and induction of apoptosis in same way that non-targeted NPs loaded with bortezomib. In vivo biodistribution after 24h of intraperitoneally (i.p) injection showed increase retention of targeted NPs in spleen, femurs and skull, with around 9-fold increase of targeted compared to non-targeted NPs in bone sections. SCID mice were injected with MM cells and after 21 days, treatment was injected i.p twice a week with 0.5 mg/kg bortezomib equivalent. Targeted and non-targeted NPs inhibited MM growth in vivo and increased survival in the same way that free drug. In addition, pre-treatment thrice a week for 21 days with 0.3 mg/kg bortezomib equivalent before injection of MM cells inhibited myeloma growth and increased survival compared to free drug (108) (Table 2). Nonetheless, non-significant differences were found between targeted and non-targeted NPs. The study showed that bortezomib, as a pretreatment regimen, modified the bone microenvironment, and loaded NPs enhanced survival and decreased tumor burden. However, biodistribution studies in organs such as lung, liver and kidney should be tested, and the reduction of off-side effects provided.
4. FUTURE DIRECTIONS
The application of NPs to improve delivery of drugs is a revolutionary approach to advance the treatment of cancer and many other diseases. The emphasis of NPs focus on the delivery of drugs more efficiently to reduce side effects. Several liposomes and polymeric NPs have been approved by the FDA for clinical use in cancer, and many others are under investigation in several clinical trials, including indications such as solid tumors, and even hematological malignancies (acute myeloid leukemia) (12, 109). NPs offer the option to deliver therapeutics with improved pharmacokinetics, safety profiles, and biodistribution, allowing us to reconsider chemotherapies previously discarded due to lack of targeting capabilities, non-specific distribution, and side effects.
Contributions of NPs to Multiple Myeloma treatment
Nanoparticle delivery systems can improve MM treatment in two main ways: 1) increase the specificity of treatments based on improve potency and efficacy by hitting the right target, the bone marrow niche in which myeloma cells developed. 2) At the same time, NPs offer the advantage to reduce side effects in normal tissues by delivering drugs to the target tissue. This will be allowed through delivery of higher doses to the MM cells and lower the doses to other tissues. Therefore, the ability to improve potency or efficacy of drug treatments combined with the reduction of side effects may improve disease outcomes besides suppressing drug adverse effects.
Challenges of NPs for Multiple Myeloma treatment
The EPR effect is an important factor for delivery of nanoparticle systems in general, which has been confirmed mainly for solid tumors. Although encouraging results have been shown with liposomes, polymeric NPs, micelles, and inorganic NPs using non-targeted approaches (82, 86, 88, 93), MM is a hematological malignancy and it is still not clear that the EPR effect will have a role in how non-targeted NPs exploit passive targeting based on the pathophysiological conditions of the myeloma microenvironment.
Targeted approaches containing targeted-ligands against a marker or receptor overexpressed in the targeted tumor cells is challenging in MM. The characterization of MM by a single surface marker is being questioned over the last decade. CD138, the gold-standard surface marker to detect MM cells, is shown to be affected by drug exposure and hypoxia (110). Other secondary antibodies used in combinations such as CD38, CS1 and CD20 are highly expressed in many other cell types (111). Other markers such as VLA4-4 is expressed in several cell types and the expression in MM cells is very heterogeneous (97, 100). The lack of a constitutively expressed unique surface marker in MM limits the specific active targeting of NP systems.
In addition, myeloma is a complex disease involving multiple pathways and mutations. Inhibition of a pathway by a single drug may be insufficient to achieve therapeutic efficacy. As a result, combination therapy will let to use drugs at lower doses, reducing cytotoxic effects but increasing efficacy due to the synergistic effect of several agents targeting different pathways (112). The effectivity of combination therapy by NPs will depend on the ability of NPs to carry multiple therapeutic agents with different physicochemical properties and pharmacological behaviors, which adds more challenges to the technical development of NPs in MM.
Strategies to improve NPs for MM treatment
NPs offer multifunctionality, combining both diagnostics and therapeutics, combination known as theranosis. For successful theranosis, the efficient delivery of imaging agents and drugs is critical to provide sufficient imaging signal or drug concentration in the targeted disease site (113, 114). This will allow for monitoring drug delivery and image-guided therapy of the target site. In addition, the investigation of new molecular targets will help the ability to improve delivery at the tumor level. These benefits could include advances in detection, imaging, and treatment of myeloma. On the other hand, NPs can be designed to target multiple antitumor moieties at the same time to maximize the accuracy of tumor targeting. In this case, further investigation of myeloma cell surface markers is required to identify alternative targets with adequate expression levels.
Another consideration will be to contemplate the disadvantages of current in vitro testing techniques to effectively evaluate the behavior of nanoparticle delivery systems. The discrepancy between preclinical and clinical outcomes can be attributed to the failure of classic two dimensional culture models to accurately recapitulate the complex biology of MM and drug responses observed in patients. Three-dimensional (3D) culture systems are gaining strength as in vitro systems to assess and predict drug sensitivity in myeloma (115-117). 3D in vitro systems could help to evaluate NPs as drug delivery systems and predict better the in vivo performance of NPs.
In summary, the promising results in myeloma preclinical studies will lead to further investigation, and in a short period of time nanoparticle-based therapies will undergo more clinical investigation. Safety of these devices will be an important consideration before moving forward. Therefore, there remains much to be learned in the emerging field of nanomedicine in myeloma.
5. CONCLUSIONS
The main limiting factor for the effective use of chemotherapies in MM is the serious side effects caused by these drugs. The development of specific and targeted therapies based on nanoparticle delivery systems in MM is under investigation offering a new strategy to increase the efficacy of the treatment and reduce side effects in normal tissues by delivering drugs to the target tissue. Many nanoparticle delivery systems have been studied for myeloma using non-targeted NPs (liposomes, polymeric NPs, and inorganic NPs), triggered NPs, as well as targeted NPs (VLA-4, ABC drug transporters, bone microenvironment targeting). The promising results in myeloma preclinical and clinical studies will lead to further investigation. Therefore, there remains much to be learned in the emerging field of nanomedicine in myeloma.
Acknowledgments
This research was partially supported by the 2016 Multiple Myeloma Research Foundation (MMRF) Research Fellow Award and the National Center for Advancing Translational Sciences (NCATS) of the National Institutes of Health (NIH) and the National Cancer Institute (NCI) of the NIH under Award Number U54CA199092.
Footnotes
CONFLICTS OF INTEREST:
Dr. Azab receives research support from Verastem, Selexys, Karyopharm, Cell Works, Cleave Bioscience, Glycomimetics, and Vasculox; and is the founder and owner of Targeted Therapeutics LLC and Cellatrix LLC. Dr. de la Puente is co-founder of Cellatrix LLC. Moreover, Drs. Azab and de la Puente have a provisional patent application describing a novel nanoparticulate drug-delivery system for multiple myeloma.
References
- 1.Ludwig H, Milosavljevic D, Zojer N, Faint JM, Bradwell AR, Hubl W, Harding SJ. Immunoglobulin heavy/light chain ratios improve paraprotein detection and monitoring, identify residual disease and correlate with survival in multiple myeloma patients. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2013;27(1):213–9. doi: 10.1038/leu.2012.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nair RR, Gebhard AW, Emmons MF, Hazlehurst LA. Emerging strategies for targeting cell adhesion in multiple myeloma. Adv Pharmacol. 2012;65:143–89. doi: 10.1016/B978-0-12-397927-8.00006-3. [DOI] [PubMed] [Google Scholar]
- de Bono JS, Ashworth A. Translating cancer research into targeted therapeutics. Nature. 2010;467(7315):543–9. doi: 10.1038/nature09339. [DOI] [PubMed] [Google Scholar]
- 4.Yokogawa K, Nakashima E, Ishizaki J, Maeda H, Nagano T, Ichimura F. Relationships in the structure-tissue distribution of basic drugs in the rabbit. Pharmaceutical research. 1990;7(7):691–6. doi: 10.1023/a:1015803202857. [DOI] [PubMed] [Google Scholar]
- 5.Rickerby DG. Nanotechnological medical devices and nanopharmaceuticals: the European regulatory framework and research needs. Journal of nanoscience and nanotechnology. 2007;7(12):4618–25. [PubMed] [Google Scholar]
- 6.Berkner S, Schwirn K, Voelker D. Nanopharmaceuticals - tiny challenges for the environmental risk assessment of pharmaceuticals. Environmental toxicology and chemistry / SETAC. 2015 doi: 10.1002/etc.3039. [DOI] [PubMed] [Google Scholar]
- 7.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced drug delivery reviews. 2001;46(1-3):3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 8.Wilczewska AZ, Niemirowicz K, Markiewicz KH, Car H. Nanoparticles as drug delivery systems. Pharmacol Rep. 2012;64(5):1020–37. doi: 10.1016/s1734-1140(12)70901-5. [DOI] [PubMed] [Google Scholar]
- 9.Etheridge ML, Campbell SA, Erdman AG, Haynes CL, Wolf SM, McCullough J. The big picture on nanomedicine: the state of investigational and approved nanomedicine products. Nanomedicine : nanotechnology, biology, and medicine. 2013;9(1):1–14. doi: 10.1016/j.nano.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moghimi SM, Hunter AC, Murray JC. Nanomedicine: current status and future prospects. The FASEB Journal. 2005;19(3):311–30. doi: 10.1096/fj.04-2747rev. [DOI] [PubMed] [Google Scholar]
- 11.Ferrari M. Cancer nanotechnology: opportunities and challenges. Nature reviews Cancer. 2005;5(3):161–71. doi: 10.1038/nrc1566. [DOI] [PubMed] [Google Scholar]
- 12.Prabhu RH, Patravale VB, Joshi MD. Polymeric nanoparticles for targeted treatment in oncology: current insights. International journal of nanomedicine. 2015;10:1001–18. doi: 10.2147/IJN.S56932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanna V, Pala N, Sechi M. Targeted therapy using nanotechnology: focus on cancer. International journal of nanomedicine. 2014;9:467–83. doi: 10.2147/IJN.S36654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langer R. Drug delivery and targeting. Nature. 1998;392(6679 Suppl):5–10. [PubMed] [Google Scholar]
- 15.Wang Y, Shim MS, Levinson NS, Sung H-W, Xia Y. Stimuli-Responsive Materials for Controlled Release of Theranostic Agents. Advanced Functional Materials. 2014;24(27):4206–20. doi: 10.1002/adfm.201400279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng R, Meng F, Deng C, Klok HA, Zhong Z. Dual and multi-stimuli responsive polymeric nanoparticles for programmed site-specific drug delivery. Biomaterials. 2013;34(14):3647–57. doi: 10.1016/j.biomaterials.2013.01.084. [DOI] [PubMed] [Google Scholar]
- 17.Luo Y, Wang TT, Teng Z, Chen P, Sun J, Wang Q. Encapsulation of indole-3-carbinol and 3,3’-diindolylmethane in zein/carboxymethyl chitosan nanoparticles with controlled release property and improved stability. Food Chem. 2013;139(1-4):224–30. doi: 10.1016/j.foodchem.2013.01.113. [DOI] [PubMed] [Google Scholar]
- 18.Cu Y, Booth CJ, Saltzman WM. In vivo distribution of surface-modified PLGA nanoparticles following intravaginal delivery. J Control Release. 2011;156(2):258–64. doi: 10.1016/j.jconrel.2011.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu FX, Karnik R, Wang AZ, Alexis F, Levy-Nissenbaum E, Hong S, Langer RS, Farokhzad OC. Targeted nanoparticles for cancer therapy. Nano Today. 2007;2(3):14–21. [Google Scholar]
- 20.Montet X, Weissleder R, Josephson L. Imaging pancreatic cancer with a peptide-nanoparticle conjugate targeted to normal pancreas. Bioconjugate chemistry. 2006;17(4):905–11. doi: 10.1021/bc060035+. [DOI] [PubMed] [Google Scholar]
- 21.Nasongkla N, Bey E, Ren JM, Ai H, Khemtong C, Guthi JS, Chin SF, Sherry AD, Boothman DA, Gao JM. Multifunctional polymeric micelles as cancer-targeted, MRI-ultrasensitive drug delivery systems. Nano Lett. 2006;6(11):2427–30. doi: 10.1021/nl061412u. [DOI] [PubMed] [Google Scholar]
- 22.Bangham AD, Standish MM, Watkins JC. Diffusion of univalent ions across the lamellae of swollen phospholipids. Journal of molecular biology. 1965;13(1):238–52. doi: 10.1016/s0022-2836(65)80093-6. [DOI] [PubMed] [Google Scholar]
- 23.Chan JM, Valencia PM, Zhang L, Langer R, Farokhzad OC. Polymeric nanoparticles for drug delivery. Methods Mol Biol. 2010;624:163–75. doi: 10.1007/978-1-60761-609-2_11. [DOI] [PubMed] [Google Scholar]
- 24.Kim T, Hyeon T. Applications of inorganic nanoparticles as therapeutic agents. Nanotechnology. 2014;25(1):0957–4484. doi: 10.1088/0957-4484/25/1/012001. [DOI] [PubMed] [Google Scholar]
- 25.Folkman J. Tumor angiogenesis: therapeutic implications. The New England journal of medicine. 1971;285(21):1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 26.de la Puente P, Muz B, Azab F, Azab AK. Cell trafficking of endothelial progenitor cells in tumor progression. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(13):3360–8. doi: 10.1158/1078-0432.CCR-13-0462. [DOI] [PubMed] [Google Scholar]
- 27.Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, Jain RK, McDonald DM. Openings between defective endothelial cells explain tumor vessel leakiness. The American journal of pathology. 2000;156(4):1363–80. doi: 10.1016/S0002-9440(10)65006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Advances in enzyme regulation. 2001;41:189–207. doi: 10.1016/s0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 29.Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Advanced drug delivery reviews. 2011;63(3):131–5. doi: 10.1016/j.addr.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 30.Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nature reviews Clinical oncology. 2010;7(11):653–64. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagamitsu A, Greish K, Maeda H. Elevating blood pressure as a strategy to increase tumor-targeted delivery of macromolecular drug SMANCS: Cases of advanced solid tumors. Japanese Journal of Clinical Oncology. 2009;39(11):756–66. doi: 10.1093/jjco/hyp074. [DOI] [PubMed] [Google Scholar]
- 32.Nakayama M, Akimoto J, Okano T. Polymeric micelles with stimuli-triggering systems for advanced cancer drug targeting. J Drug Target. 2014;22(7):584–99. doi: 10.3109/1061186X.2014.936872. [DOI] [PubMed] [Google Scholar]
- 33.Jhaveri A, Deshpande P, Torchilin V. Stimuli-sensitive nanopreparations for combination cancer therapy. J Control Release. 2014;190:352–70. doi: 10.1016/j.jconrel.2014.05.002. [DOI] [PubMed] [Google Scholar]
- 34.Vaupel P, Kallinowski F, Okunieff P. Blood Flow, Oxygen and Nutrient Supply, and Metabolic Microenvironment of Human Tumors: A Review. Cancer research. 1989;49(23):6449–65. [PubMed] [Google Scholar]
- 35.Harguindey S, Orive G, Luis Pedraz J, Paradiso A, Reshkin SJ. The role of pH dynamics and the Na+/H+ antiporter in the etiopathogenesis and treatment of cancer. Two faces of the same coin--one single nature. Biochim Biophys Acta. 2005;25(1):1–24. doi: 10.1016/j.bbcan.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 36.Rofstad EK. Microenvironment-induced cancer metastasis. Int J Radiat Biol. 2000;76(5):589–605. doi: 10.1080/095530000138259. [DOI] [PubMed] [Google Scholar]
- 37.Yatvin MB, Kreutz W, Horwitz BA, Shinitzky M. pH-sensitive liposomes: possible clinical implications. Science. 1980;210(4475):1253–5. doi: 10.1126/science.7434025. [DOI] [PubMed] [Google Scholar]
- 38.Kong G, Braun RD, Dewhirst MW. Characterization of the effect of hyperthermia on nanoparticle extravasation from tumor vasculature. Cancer research. 2001;61(7):3027–32. [PubMed] [Google Scholar]
- 39.Hirsch LR, Stafford RJ, Bankson JA, Sershen SR, Rivera B, Price RE, Hazle JD, Halas NJ, West JL. Nanoshell-mediated near-infrared thermal therapy of tumors under magnetic resonance guidance. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(23):13549–54. doi: 10.1073/pnas.2232479100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng FY, Su CH, Wu PC, Yeh CS. Multifunctional polymeric nanoparticles for combined chemotherapeutic and near-infrared photothermal cancer therapy in vitro and in vivo. Chem Commun (Camb) 2010;46(18):3167–9. doi: 10.1039/b919172k. [DOI] [PubMed] [Google Scholar]
- 41.Dudar TE, Jain RK. Differential response of normal and tumor microcirculation to hyperthermia. Cancer research. 1984;44(2):605–12. [PubMed] [Google Scholar]
- 42.Torchilin V. Multifunctional and stimuli-sensitive pharmaceutical nanocarriers. European Journal of Pharmaceutics and Biopharmaceutics. 2009;71(3):431–44. doi: 10.1016/j.ejpb.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tartaj P, Del Puerto Morales M, Veintemillas-Verdaguer S, González-Carreño T, Serna CJ. The preparation of magnetic nanoparticles for applications in biomedicine. Journal of Physics D: Applied Physics. 2003;36(13):R182–R97. [Google Scholar]
- 44.Bohmer MR, Klibanov AL, Tiemann K, Hall CS, Gruell H, Steinbach OC. Ultrasound triggered image-guided drug delivery. Eur J Radiol. 2009;70(2):242–53. doi: 10.1016/j.ejrad.2009.01.051. [DOI] [PubMed] [Google Scholar]
- 45.Husseini GA, Pitt WG. Micelles and nanoparticles for ultrasonic drug and gene delivery. Advanced drug delivery reviews. 2008;60(10):1137–52. doi: 10.1016/j.addr.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao Y. Rational design of light-controllable polymer micelles. Chem Rec. 2007;7(5):286–94. doi: 10.1002/tcr.20127. [DOI] [PubMed] [Google Scholar]
- 47.Lee HI, Wu W, Oh JK, Mueller L, Sherwood G, Peteanu L, Kowalewski T, Matyjaszewski K. Light-induced reversible formation of polymeric micelles. Angew Chem Int Ed Engl. 2007;46(14):2453–7. doi: 10.1002/anie.200604278. [DOI] [PubMed] [Google Scholar]
- 48.Zhang XQ, Xu X, Bertrand N, Pridgen E, Swami A, Farokhzad OC. Interactions of nanomaterials and biological systems: Implications to personalized nanomedicine. Advanced drug delivery reviews. 2012;64(13):1363–84. doi: 10.1016/j.addr.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Neri D, Bicknell R. Tumour vascular targeting. Nature reviews Cancer. 2005;5(6):436–46. doi: 10.1038/nrc1627. [DOI] [PubMed] [Google Scholar]
- 50.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nature nanotechnology. 2007;2(12):751–60. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 51.Acharya S, Dilnawaz F, Sahoo SK. Targeted epidermal growth factor receptor nanoparticle bioconjugates for breast cancer therapy. Biomaterials. 2009;30(29):5737–50. doi: 10.1016/j.biomaterials.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 52.Minko T. Drug targeting to the colon with lectins and neoglycoconjugates. Advanced drug delivery reviews. 2004;56(4):491–509. doi: 10.1016/j.addr.2003.10.017. [DOI] [PubMed] [Google Scholar]
- 53.Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Molecular pharmaceutics. 2008;5(4):505–15. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martinez A, Muniz E, Teijon C, Iglesias I, Teijon JM, Blanco MD. Targeting tamoxifen to breast cancer xenograft tumours: preclinical efficacy of folate-attached nanoparticles based on alginate-cysteine/disulphide-bond-reduced albumin. Pharmaceutical research. 2014;31(5):1264–74. doi: 10.1007/s11095-013-1247-5. [DOI] [PubMed] [Google Scholar]
- 55.Albanese A, Tang PS, Chan WC. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annual review of biomedical engineering. 2012;14:1–16. doi: 10.1146/annurev-bioeng-071811-150124. [DOI] [PubMed] [Google Scholar]
- 56.Longmire M, Choyke PL, Kobayashi H. Clearance properties of nano-sized particles and molecules as imaging agents: considerations and caveats. Nanomedicine (Lond) 2008;3(5):703–17. doi: 10.2217/17435889.3.5.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gref R, Minamitake Y, Peracchia MT, Trubetskoy V, Torchilin V, Langer R. Biodegradable long-circulating polymeric nanospheres. Science. 1994;263(5153):1600–3. doi: 10.1126/science.8128245. [DOI] [PubMed] [Google Scholar]
- 58.Sutton D, Nasongkla N, Blanco E, Gao J. Functionalized micellar systems for cancer targeted drug delivery. Pharmaceutical research. 2007;24(6):1029–46. doi: 10.1007/s11095-006-9223-y. [DOI] [PubMed] [Google Scholar]
- 59.De Jong WH, Borm PJ. Drug delivery and nanoparticles:applications and hazards. International journal of nanomedicine. 2008;3(2):133–49. doi: 10.2147/ijn.s596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ludwig H. Advances in biology and treatment of multiple myeloma. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2005;2(16 Suppl):ii106–12. doi: 10.1093/annonc/mdi717. [DOI] [PubMed] [Google Scholar]
- 61.Kyle RA, Gertz MA, Witzig TE, Lust JA, Lacy MQ, Dispenzieri A, Fonseca R, Rajkumar SV, Offord JR, Larson DR, Plevak ME, Therneau TM, Greipp PR. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clinic proceedings Mayo Clinic. 2003;78(1):21–33. doi: 10.4065/78.1.21. [DOI] [PubMed] [Google Scholar]
- 62.Barlogie B, Hall R, Zander A, Dicke K, Alexanian R. High-dose melphalan with autologous bone marrow transplantation for multiple myeloma. Blood. 1986;67(5):1298–301. [PubMed] [Google Scholar]
- 63.Barlogie B, Smith L, Alexanian R. Effective Treatment of Advanced Multiple Myeloma Refractory to Alkylating Agents. New England Journal of Medicine. 1984;310(21):1353–6. doi: 10.1056/NEJM198405243102104. [DOI] [PubMed] [Google Scholar]
- 64.Gertz MA, Kalish LA, Kyle RA, Hahn RG, Tormey DC, Oken MM. Phase III study comparing vincristine, doxorubicin (Adriamycin), and dexamethasone (VAD) chemotherapy with VAD plus recombinant interferon alfa-2 in refractory or relapsed multiple myeloma. An Eastern Cooperative Oncology Group study. American journal of clinical oncology. 1995;18(6):475–80. doi: 10.1097/00000421-199512000-00003. [DOI] [PubMed] [Google Scholar]
- 65.Alexanian R, Barlogie B, Dixon D. High-Dose Glucocorticoid Treatment of Resistant Myeloma. Annals of Internal Medicine. 1986;105(1):8–11. doi: 10.7326/0003-4819-105-1-8. [DOI] [PubMed] [Google Scholar]
- 66.de la Puente P, Azab AK. Contemporary drug therapies for multiple myeloma. Drugs Today (Barc) 2013;49(9):563–73. doi: 10.1358/dot.2013.49.9.2020941. [DOI] [PubMed] [Google Scholar]
- 67.Kumar SK, Rajkumar SV, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Zeldenrust SR, Dingli D, Russell SJ, Lust JA, Greipp PR, Kyle RA, Gertz MA. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111(5):2516–20. doi: 10.1182/blood-2007-10-116129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mitsiades CS, Hayden PJ, Anderson KC, Richardson PG. From the bench to the bedside: emerging new treatments in multiple myeloma. Best practice & research Clinical haematology. 2007;20(4):797–816. doi: 10.1016/j.beha.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.de la Puente P, Muz B, Azab F, Luderer M, Azab AK. Molecularly targeted therapies in multiple myeloma. Leukemia research and treatment. 2014;2014 doi: 10.1155/2014/976567. 976567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Richardson PG, Xie W, Mitsiades C, Chanan-Khan AA, Lonial S, Hassoun H, Avigan DE, Oaklander AL, Kuter DJ, Wen PY, Kesari S, Briemberg HR, Schlossman RL, Munshi NC, Heffner LT, Doss D, Esseltine DL, Weller E, Anderson KC, Amato AA. Single-agent bortezomib in previously untreated multiple myeloma: efficacy, characterization of peripheral neuropathy, and molecular correlations with response and neuropathy. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27(21):3518–25. doi: 10.1200/JCO.2008.18.3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thompson JL. Carfilzomib: a second-generation proteasome inhibitor for the treatment of relapsed and refractory multiple myeloma. The Annals of pharmacotherapy. 2013;47(1):56–62. doi: 10.1345/aph.1R561. [DOI] [PubMed] [Google Scholar]
- 72.Offidani M, Corvatta L, Marconi M, Malerba L, Mele A, Olivieri A, Brunori M, Catarini M, Candela M, Capelli D, Montanari M, Rupoli S, Leoni P. Common and rare side-effects of low-dose thalidomide in multiple myeloma: focus on the dose-minimizing peripheral neuropathy. European journal of haematology. 2004;72(6):403–9. doi: 10.1111/j.1600-0609.2004.00238.x. [DOI] [PubMed] [Google Scholar]
- 73.Yang B, Yu RL, Chi XH, Lu XC. Lenalidomide treatment for multiple myeloma: systematic review and meta-analysis of randomized controlled trials. PloS one. 2013;8(5):e64354. doi: 10.1371/journal.pone.0064354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Patrizi A, Venturi M, Dika E, Maibach H, Tacchetti P, Brandi G. Cutaneous adverse reactions linked to targeted anticancer therapies bortezomib and lenalidomide for multiple myeloma: new drugs, old side effects. Cutaneous and ocular toxicology. 2014;33(1):1–6. doi: 10.3109/15569527.2013.787086. [DOI] [PubMed] [Google Scholar]
- 75.Kumar A, Porwal M, Verma A, Mishra AK. Impact of pomalidomide therapy in multiple myeloma: a recent survey. J Chemother. 2014 doi: 10.1179/1973947814Y.0000000201. 1973947814Y0000000201. [DOI] [PubMed] [Google Scholar]
- 76.Chang HI, Yeh MK. Clinical development of liposome-based drugs: formulation, characterization, and therapeutic efficacy. International journal of nanomedicine. 2012;7:49–60. doi: 10.2147/IJN.S26766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ashley JD, Stefanick JF, Schroeder VA, Suckow MA, Kiziltepe T, Bilgicer B. Liposomal bortezomib nanoparticles via boronic ester prodrug formulation for improved therapeutic efficacy in vivo. Journal of medicinal chemistry. 2014;57(12):5282–92. doi: 10.1021/jm500352v. [DOI] [PubMed] [Google Scholar]
- 78.Maillard S, Ameller T, Gauduchon J, Gougelet A, Gouilleux F, Legrand P, Marsaud V, Fattal E, Sola B, Renoir JM. Innovative drug delivery nanosystems improve the anti-tumor activity in vitro and in vivo of anti-estrogens in human breast cancer and multiple myeloma. The Journal of steroid biochemistry and molecular biology. 2005;94(1-3):111–21. doi: 10.1016/j.jsbmb.2004.12.023. [DOI] [PubMed] [Google Scholar]
- 79.Orlowski RZ, Nagler A, Sonneveld P, Blade J, Hajek R, Spencer A, San Miguel J, Robak T, Dmoszynska A, Horvath N, Spicka I, Sutherland HJ, Suvorov AN, Zhuang SH, Parekh T, Xiu L, Yuan Z, Rackoff W, Harousseau JL. Randomized phase III study of pegylated liposomal doxorubicin plus bortezomib compared with bortezomib alone in relapsed or refractory multiple myeloma: combination therapy improves time to progression. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2007;25(25):3892–901. doi: 10.1200/JCO.2006.10.5460. [DOI] [PubMed] [Google Scholar]
- 80.Orlowski RZ, Voorhees PM, Garcia RA, Hall MD, Kudrik FJ, Allred T, Johri AR, Jones PE, Ivanova A, Van Deventer HW, Gabriel DA, Shea TC, Mitchell BS, Adams J, Esseltine DL, Trehu EG, Green M, Lehman MJ, Natoli S, Collins JM, Lindley CM, Dees EC. Phase 1 trial of the proteasome inhibitor bortezomib and pegylated liposomal doxorubicin in patients with advanced hematologic malignancies. Blood. 2005;105(8):3058–65. doi: 10.1182/blood-2004-07-2911. [DOI] [PubMed] [Google Scholar]
- 81.Voorhees PM, Orlowski RZ, Mulkey F, Watson P, Geyer S, Sanford BL, Bennett E, Chanan-Khan AA, Bloomfield CD, Larson RA. Long-term outcomes for newly-diagnosed multiple myeloma patients treated with pegylated liposomal doxorubicin and bortezomib: final results of CALGB (Alliance) 10301, a multicentre phase II study. British journal of haematology. 2015 doi: 10.1111/bjh.13592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hussein MA, Wood L, Hsi E, Srkalovic G, Karam M, Elson P, Bukowski RM. A Phase II trial of pegylated liposomal doxorubicin, vincristine, and reduced-dose dexamethasone combination therapy in newly diagnosed multiple myeloma patients. Cancer. 2002;95(10):2160–8. doi: 10.1002/cncr.10946. [DOI] [PubMed] [Google Scholar]
- 83.Al-Ali A, Alkhawajah AA, Randhawa MA, Shaikh NA. Oral and intraperitoneal LD50 of thymoquinone, an active principle of Nigella sativa, in mice and rats. Journal of Ayub Medical College, Abbottabad : JAMC. 2008;20(2):25–7. [PubMed] [Google Scholar]
- 84.Ravindran J, Nair HB, Sung B, Prasad S, Tekmal RR, Aggarwal BB. Thymoquinone poly (lactide-co-glycolide) nanoparticles exhibit enhanced anti-proliferative, anti-inflammatory, and chemosensitization potential. Biochemical pharmacology. 2010;79(11):1640–7. doi: 10.1016/j.bcp.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 85.Hatefi A, Amsden B. Camptothecin delivery methods. Pharmaceutical research. 2002;19(10):1389–99. doi: 10.1023/a:1020427227285. [DOI] [PubMed] [Google Scholar]
- 86.Li Z, Li X, Cao Z, Xu Y, Lin H, Zhao Y, Wei Y, Qian Z. Camptothecin nanocolloids based on N,N,N-trimethyl chitosan: efficient suppression of growth of multiple myeloma in a murine model. Oncology reports. 2012;27(4):1035–40. doi: 10.3892/or.2012.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang Z, Yang J, Kirk C, Fang Y, Alsina M, Badros A, Papadopoulos K, Wong A, Woo T, Bomba D, Li J, Infante JR. Clinical Pharmacokinetics, Metabolism, and Drug-Drug Interaction of Carfilzomib. Drug Metabolism and Disposition. 2013;41(1):230–7. doi: 10.1124/dmd.112.047662. [DOI] [PubMed] [Google Scholar]
- 88.Ao L, Reichel D, Hu D, Jeong H, Kim KB, Bae Y, Lee W. Polymer Micelle Formulations of Proteasome Inhibitor Carfilzomib for Improved Metabolic Stability and Anticancer Efficacy in Human Multiple Myeloma and Lung Cancer Cell Lines. The Journal of pharmacology and experimental therapeutics. 2015;355(2):168–73. doi: 10.1124/jpet.115.226993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Badr G, Al-Sadoon MK, El-Toni AM, Daghestani M. Walterinnesia aegyptia venom combined with silica nanoparticles enhances the functioning of normal lymphocytes through PI3K/AKT, NFkappaB and ERK signaling. Lipids in health and disease. 2012;11:27. doi: 10.1186/1476-511X-11-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sayed D, Al-Sadoon MK, Badr G. Silica nanoparticles sensitize human multiple myeloma cells to snake (Walterinnesia aegyptia) venom-induced apoptosis and growth arrest. Oxidative medicine and cellular longevity. 2012;2012:386286. doi: 10.1155/2012/386286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Al-Sadoon MK, Rabah DM, Badr G. Enhanced anticancer efficacy of snake venom combined with silica nanoparticles in a murine model of human multiple myeloma: molecular targets for cell cycle arrest and apoptosis induction. Cellular immunology. 2013;284(1-2):129–38. doi: 10.1016/j.cellimm.2013.07.016. [DOI] [PubMed] [Google Scholar]
- 92.Elsadek B, Kratz F. Impact of albumin on drug delivery--new applications on the horizon. J Control Release. 2012;157(1):4–28. doi: 10.1016/j.jconrel.2011.09.069. [DOI] [PubMed] [Google Scholar]
- 93.Yang C, Wang J, Chen D, Chen J, Xiong F, Zhang H, Zhang Y, Gu N, Dou J. Paclitaxel-Fe3O4 nanoparticles inhibit growth of CD138(-) CD34(-) tumor stem-like cells in multiple myeloma-bearing mice. International journal of nanomedicine. 2013;8:1439–49. doi: 10.2147/IJN.S38447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hayashi K, Nakamura M, Miki H, Ozaki S, Abe M, Matsumoto T, Sakamoto W, Yogo T, Ishimura K. Magnetically responsive smart nanoparticles for cancer treatment with a combination of magnetic hyperthermia and remote-control drug release. Theranostics. 2014;4(8):834–44. doi: 10.7150/thno.9199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hideshima T, Richardson P, Anderson KC. Novel therapeutic approaches for multiple myeloma. Immunological reviews. 2003;194:164–76. doi: 10.1034/j.1600-065x.2003.00053.x. [DOI] [PubMed] [Google Scholar]
- 96.Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS. Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood. 1999;93(5):1658–67. [PMC free article] [PubMed] [Google Scholar]
- 97.Kiziltepe T, Ashley JD, Stefanick JF, Qi YM, Alves NJ, Handlogten MW, Suckow MA, Navari RM, Bilgicer B. Rationally engineered nanoparticles target multiple myeloma cells, overcome cell-adhesion-mediated drug resistance, and show enhanced efficacy in vivo. Blood cancer journal. 2012;2(4):e64. doi: 10.1038/bcj.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ashley JD, Stefanick JF, Schroeder VA, Suckow MA, Alves NJ, Suzuki R, Kikuchi S, Hideshima T, Anderson KC, Kiziltepe T, Bilgicer B. Liposomal carfilzomib nanoparticles effectively target multiple myeloma cells and demonstrate enhanced efficacy in vivo. J Control Release. 2014;196:113–21. doi: 10.1016/j.jconrel.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 99.Clausen DM, Guo J, Parise RA, Beumer JH, Egorin MJ, Lazo JS, Prochownik EV, Eiseman JL. In vitro cytotoxicity and in vivo efficacy, pharmacokinetics, and metabolism of 10074-G5, a novel small-molecule inhibitor of c-Myc/Max dimerization. The Journal of pharmacology and experimental therapeutics. 2010;335(3):715–27. doi: 10.1124/jpet.110.170555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Soodgupta D, Pan D, Cui G, Senpan A, Yang X, Lu L, Weilbaecher KN, Prochownik EV, Lanza GM, Tomasson MH. Small Molecule MYC Inhibitor Conjugated to Integrin-Targeted Nanoparticles Extends Survival in a Mouse Model of Disseminated Multiple Myeloma. Mol Cancer Ther. 2015;14(6):1286–94. doi: 10.1158/1535-7163.MCT-14-0774-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dou J, Gu N. Emerging strategies for the identification and targeting of cancer stem cells. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2010;31(4):243–53. doi: 10.1007/s13277-010-0023-y. [DOI] [PubMed] [Google Scholar]
- 102.Nooter K, Stoter G. Molecular mechanisms of multidrug resistance in cancer chemotherapy. Pathology, research and practice. 1996;192(7):768–80. doi: 10.1016/S0344-0338(96)80099-9. [DOI] [PubMed] [Google Scholar]
- 103.Turner JG, Gump JL, Zhang C, Cook JM, Marchion D, Hazlehurst L, Munster P, Schell MJ, Dalton WS, Sullivan DM. ABCG2 expression, function, and promoter methylation in human multiple myeloma. Blood. 2006;108(12):3881–9. doi: 10.1182/blood-2005-10-009084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Abraham J, Salama NN, Azab AK. The role of P-glycoprotein in drug resistance in multiple myeloma. Leukemia & lymphoma. 2015;56(1):26–33. doi: 10.3109/10428194.2014.907890. [DOI] [PubMed] [Google Scholar]
- 105.Yang C, Xiong F, Wang J, Dou J, Chen J, Chen D, Zhang Y, Luo S, Gu N. Anti-ABCG2 monoclonal antibody in combination with paclitaxel nanoparticles against cancer stem-like cell activity in multiple myeloma. Nanomedicine (Lond) 2014;9(1):45–60. doi: 10.2217/nnm.12.216. [DOI] [PubMed] [Google Scholar]
- 106.Yang C, He X, Song L, Zhan X, Zhang Y, Dou J, Gu N. Gamma-Fe2O3 nanoparticles increase therapeutic efficacy of combination with paclitaxel and anti-ABCG2 monoclonal antibody on multiple myeloma cancer stem cells in mouse model. Journal of biomedical nanotechnology. 2014;10(2):336–44. doi: 10.1166/jbn.2014.1730. [DOI] [PubMed] [Google Scholar]
- 107.Zhang S, Gangal G, Uludag H. ‘Magic bullets’ for bone diseases: progress in rational design of bone-seeking medicinal agents. Chemical Society reviews. 2007;36(3):507–31. doi: 10.1039/b512310k. [DOI] [PubMed] [Google Scholar]
- 108.Swami A, Reagan MR, Basto P, Mishima Y, Kamaly N, Glavey S, Zhang S, Moschetta M, Seevaratnam D, Zhang Y, Liu J, Memarzadeh M, Wu J, Manier S, Shi J, Bertrand N, Lu ZN, Nagano K, Baron R, Sacco A, Roccaro AM, Farokhzad OC, Ghobrial IM. Engineered nanomedicine for myeloma and bone microenvironment targeting. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(28):10287–92. doi: 10.1073/pnas.1401337111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xu X, Ho W, Zhang X, Bertrand N, Farokhzad O. Cancer nanomedicine: from targeted delivery to combination therapy. Trends in molecular medicine. 2015;21(4):223–32. doi: 10.1016/j.molmed.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Muz B, de la Puente P, Azab F, Luderer MJ, King J, Vij R, Azab AK. A CD138-independent strategy to detect minimal residual disease and circulating tumour cells in multiple myeloma. British journal of haematology. 2016;173(1):70–81. doi: 10.1111/bjh.13927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.San Miguel JF, Gutierrez NC, Mateo G, Orfao A. Conventional diagnostics in multiple myeloma. European journal of cancer (Oxford, England 1990) 2006;42(11):1510–9. doi: 10.1016/j.ejca.2005.11.039. [DOI] [PubMed] [Google Scholar]
- 112.Kamaly N, Xiao Z, Valencia PM, Radovic-Moreno AF, Farokhzad OC. Targeted polymeric therapeutic nanoparticles: design, development and clinical translation. Chemical Society reviews. 2012;41(7):2971–3010. doi: 10.1039/c2cs15344k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Koo H, Huh MS, Sun IC, Yuk SH, Choi K, Kim K, Kwon IC. In vivo targeted delivery of nanoparticles for theranosis. Accounts of chemical research. 2011;44(10):1018–28. doi: 10.1021/ar2000138. [DOI] [PubMed] [Google Scholar]
- 114.Brigger I, Dubernet C, Couvreur P. Nanoparticles in cancer therapy and diagnosis. Advanced drug delivery reviews. 2002;54(5):631–51. doi: 10.1016/s0169-409x(02)00044-3. [DOI] [PubMed] [Google Scholar]
- 115.Kirshner J, Thulien KJ, Martin LD, Debes Marun C, Reiman T, Belch AR, Pilarski LM. A unique three-dimensional model for evaluating the impact of therapy on multiple myeloma. Blood. 2008;112(7):2935–45. doi: 10.1182/blood-2008-02-142430. [DOI] [PubMed] [Google Scholar]
- 116.Calimeri T, Battista E, Conforti F, Neri P, Di Martino MT, Rossi M, Foresta U, Piro E, Ferrara F, Amorosi A, Bahlis N, Anderson KC, Munshi N, Tagliaferri P, Causa F, Tassone P. A unique three-dimensional SCID-polymeric scaffold (SCID-synth-hu) model for in vivo expansion of human primary multiple myeloma cells. Leukemia. 2011;25(4):707–11. doi: 10.1038/leu.2010.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.de la Puente P, Muz B, Gilson RC, Azab F, Luderer M, King J, Achilefu S, Vij R, Azab AK. 3D tissue-engineered bone marrow as a novel model to study pathophysiology and drug resistance in multiple myeloma. Biomaterials. 2015;73:70–84. doi: 10.1016/j.biomaterials.2015.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]