

Abstract
Intensively managed agricultural pastures contribute to N2O and N2 fluxes resulting in detrimental environmental outcomes and poor N use efficiency, respectively. Besides nitrification, nitrifier-denitrification and heterotrophic denitrification, alternative pathways such as codenitrification also contribute to emissions under ruminant urine-affected soil. However, information on codenitrification is sparse. The objectives of this experiment were to assess the effects of soil moisture and soil inorganic-N dynamics on the relative contributions of codenitrification and denitrification (heterotrophic denitrification) to the N2O and N2 fluxes under a simulated ruminant urine event. Repacked soil cores were treated with 15N enriched urea and maintained at near saturation (−1 kPa) or field capacity (−10 kPa). Soil inorganic-N, pH, dissolved organic carbon, N2O and N2 fluxes were measured over 63 days. Fluxes of N2, attributable to codenitrification, were at a maximum when soil nitrite (NO2−) concentrations were elevated. Cumulative codenitrification was higher (P = 0.043) at −1 kPa. However, the ratio of codenitrification to denitrification did not differ significantly with soil moisture, 25.5 ± 15.8 and 12.9 ± 4.8% (stdev) at −1 and −10 kPa, respectively. Elevated soil NO2− concentrations are shown to contribute to codenitrification, particularly at −1 kPa.
Introduction
The concentration of nitrous oxide (N2O) in the atmosphere has increased since 1750 due to human activity with values surpassing the highest concentrations recorded in ice cores during the past 800,000 years, and exceeding the pre-industrial level by 20%1. Reductions in the anthropogenic forcing of Earth’s climate system and the recovery of the ozone layer would be enhanced if anthropogenic emissions of N2O were reduced1,2. However, the atmospheric N2O concentration continues to increase, predominately due to agricultural intensification, with 80% of the increase resulting from increased fertilizer use and manure applications for the purpose of food production3. Nitrous oxide emissions from grazed grasslands make a significant contribution to anthropogenic N2O emissions4,5 as a consequence of ruminant urine patches supplying nitrogen (N) substrate that is in excess of the pasture sward’s N requirement6,7. Emissions of N2O from pastures result from microbial transformations of N substrates applied via nitrification, nitrifier-denitrification, heterotrophic denitrification (hereafter referred to as denitrification unless otherwise stated), and codenitrification8–10. A further significant consequence of denitrifying mechanisms is the production and loss of dinitrogen (N2). Although environmentally benign, N2 losses lead to poor N use efficiency and reduced production, resulting in economic losses through the need to add further inorganic N. While reactive N (Nr) losses, such as nitrate (NO3−) leaching and ammonia (NH3) volatilization, are well researched, the loss of N2 from pasture systems is poorly studied and often only identified by default via the application of N balance methods11. For example, of the N applied to grasslands some 20–40% is typically unaccounted for and assumed to be lost as N211–13. Therefore, methods to reduce emissions of both N2O and N2 require a better understanding of the emission pathways.
Shoun et al.14 and Tanimoto et al.15 first described codenitrification after demonstrating, with 15N tracer, that N2O and N2 production occurred in a different manner to the routinely accepted pathways of nitrification and denitrification. It has been suggested that codenitrification results from microbially mediated N-nitrosation reactions14–16. Codenitrification is one of the least studied N loss pathways and its contribution to agricultural N2O and N2 emissions remains unclear17.
Codenitrification is a process that co-metabolises organic N compounds, such as amines, to produce N2O and/or N2, and is also referred to as biotic N-nitrosation16. Codenitrification involves the replacement of a hydrogen atom in an organic compound with a nitroso group (—N=O). Under near neutral to alkaline soil pH conditions, common to pasture soils, codenitrification may occur via enzymatic catalysis (Fig. 1), with enzymatic nitrosyl compounds (E-NO+ or E-NO) attracting nucleophilic compounds16,18. Nucleophiles involved in codenitrification include hydroxylamine, ammonium (NH4+), hydrazine, amino compounds, and ammonia (NH3). The resulting gas products formed, N2O or N2, contain one N atom originating from the inorganic-N (e.g. NO2−), and a second atom from the co-metabolised organic compound16,18. Significant rates of both partial and complete codenitrification are only likely to occur if nucleophile concentrations are at least one or two orders of magnitude greater than that of NO2− and NO16.
Figure 1.
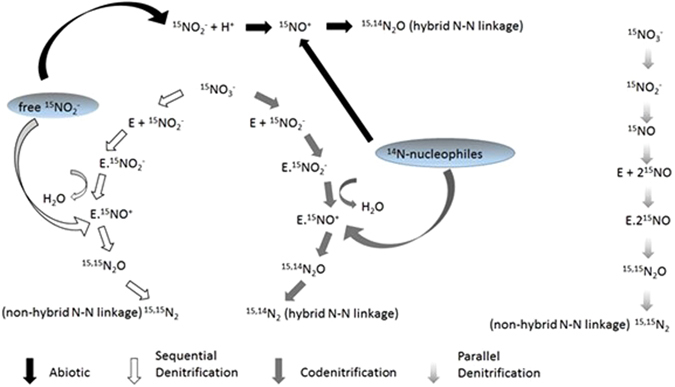
Simplified diagram (adapted from Spott et al.16, Weeg-Aerssens et al.18, Schmidt et al.55) showing abiotic denitrification, parallel denitrification, sequential denitrification and codenitrification pathways. During abiotic production an electrophile (e.g. the nitrosonium cation NO+ which is formed under acidic soil conditions) replaces the hydrogen atom of a nucleophile with a hybrid N-N bond formed following deprotonation. The parallel pathway results in a non-hybrid N-N bond as the result of two NO2− or two NO molecules being bound, simultaneously to one enzyme (E), which theoretically excludes the possibility of a nitrosation reaction occurring and the formation of a hybrid N-N bond55,56. However, a two-step process occurs in the sequential pathway when NO2− or NO molecules initially bind to an enzyme (E) followed by a free NO2−, or NO molecule, (originating from the original NO3− pool) reacting with the enzyme complexed N species to form a non-hybrid N-N bond. The two-step sequence also permits the enzyme complexed N species to function as an electrophile which is able to be to be attacked by nucleophiles producing a hybrid N-N bond. Nucleophiles able to partake in codenitrification reactions include amines, ammonium, hydrazine, and ammonia.
Heterotrophic denitrification results in the reduction of NO3− to N2 with nitrite (NO2−), nitric oxide (NO), and N2O obligate intermediaries19 (Fig. 1). Formation of the N2O molecule is recognized as occurring via parallel or sequential pathways16 and references therein. In the parallel pathway simultaneous bonding of two NO2− or two NO molecules to an enzyme, where both NO2− and NO are derived from the same NO3− source, creates a non-hybrid N-N bond, thus precluding the occurrence of codenitrification16. However, a two-step reaction, the sequential pathway, results in either NO2− or NO initially bonding with an enzyme, which in turn may react with either free NO2− or NO to form a non-hybrid N-N bond, or alternatively, this enzyme bound N can act as an electrophile and react with nucleophiles (e.g. amines) to form a hybrid N-N bond (Fig. 1). Consequently, hybrid N-N gas production, codenitrification, can occur simultaneously as a result of conventional denitrification (Fig. 1)16. Formation of hybrid N2 has also been reported to occur when NH3, hydrazine (N2H4) or amines are co-metabolised during codenitrification20.
Abiotic nitrosation is also a well-recognized phenomena21,22. In abiotic reactions, free NO2− derived from nitrification or denitrification processes is chemically transformed to produce the nitrosonium cation (NO+) under acidic conditions. The NO+ cation reacts with a nucleophile (e.g. amine) to produce a hybrid N-N linkage (Fig. 1)16 and references therein. This process differs from codenitrification since the formation of the NO+ electrophile is chemically dependent on the soil pH and involves free NO2− in the soil solution as the precursor. Nucleophiles involved in abiotic reactions include hydroxylamine, NH4+, hydrazine, amines, and NH3. However, relatively high soil pH values under grazed pasture conditions mean that the equilibrium concentrations of free nitrosating agents are generally inadequate for abiotic nitrosation to be significant16.
In grazed pastures ruminant urine deposition onto pasture soil temporarily elevates soil pH following urea hydrolysis, creating a urinary-N cascade that produces potential nucleophiles (e.g. NH4+ and NH3) at high concentrations. Simultaneously, enzyme bound nitrosating agents (E-NO+ or E-NO), may be formed during denitrification of nitrate (NO3−) or as supplied by NO2− or NO during processes such as nitrification of nitrifier-denitrification19. Thus urine patches are potentially conducive to codenitrification occurring. In the only in vivo study to date to focus on codenitrification, Selbie et al.23 confirmed the occurrence of codenitrification within ruminant urine-affected pasture soil with 95% of the N2 emitted over 123 days resulting from codenitrification, with N2 the dominant product, and where the codenitrified N2 was equivalent to 56% of the N applied. This experiment by Selbie et al.23 received regular rainfall and it may be that the dominance of codenitrified N2 over codenitrified N2O may have been the result of, as the authors suggest, hybrid N2O being converted to hybrid N2 via heterotrophic denitrification (Fig. 1). Conceptually, the recognized environmental constraints on denitrification should also apply to codenitrification16, since codenitrification depends on enzyme bound nitrosyl compounds, formed during denitrification, being present (Fig. 1). A key driver of denitrification is the soil’s oxygen status, and wetter soils result in higher levels of anaerobiosis since oxygen diffuses 1 × 104 times slower through water when compared to air24. Thus wetter soils should have higher rates of codenitrification. In order to test this hypothesis, and better understand the constraints and importance of codenitrification in pasture soils, we performed an experiment using either saturated soil or soil at field capacity to determine relative rates of codenitrification. The objective of the study was to investigate the effect of soil moisture on the rate of codenitrification from simulated urine applied to a free draining permanent grassland soil.
Results
Soil moisture, pH, DOC and inorganic-N
The −1 kPa and −10 kPa moisture treatments imposed resulted in average WFPS values (%±s.e.m) of 88.9 ± 1.1 and 48.5 ± 0.4, respectively. The relative gas diffusivity values at −1 and −10 kPa were 0.0028 and 0.2079, respectively. There was a significant interaction of soil moisture and sampling date (p < 0.001) on soil pH, DOC and inorganic N contents (Figs 2–4). Soil pH in the non-urea treatment was generally constant over time (Fig. 2) regardless of soil moisture treatment, averaging 5.49 ± 0.11 (Stdev). However, soil pH (p < 0.001) increased within 6 hours of urea application, and increased further, peaking at 8.57 ± 0.29 and 8.78 ± 0.09 in the −1 kPa and −10 kPa treatments, respectively, on day 3 before declining over time (Fig. 2). On days 21 and 35 the soil pH was lower in the −1 kPa treatment than in the −10 kPa treatment (p < 0.001) with the reverse occurring on day 63 (p < 0.05).
Figure 2.
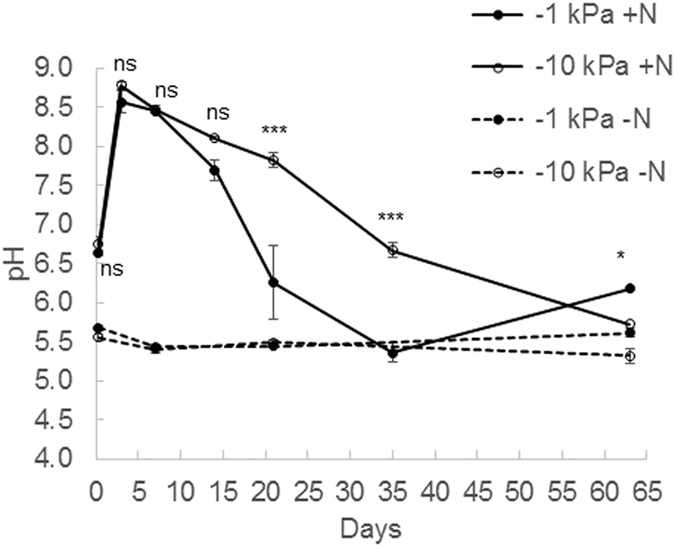
Changes in soil pH over time. Soil pH under near saturated (−1 kPa) or field capacity (−10 kPa) soil moisture conditions, following urea application (+N) or nil urea application (−N). Symbols are means (n = 4) with vertical error bars the standard error of the mean. Asterisks *,**,***indicate significant differences between moisture treatments under urea treatments at P < 0.05, P < 0.01, and P < 0.001, respectively.
Figure 4.
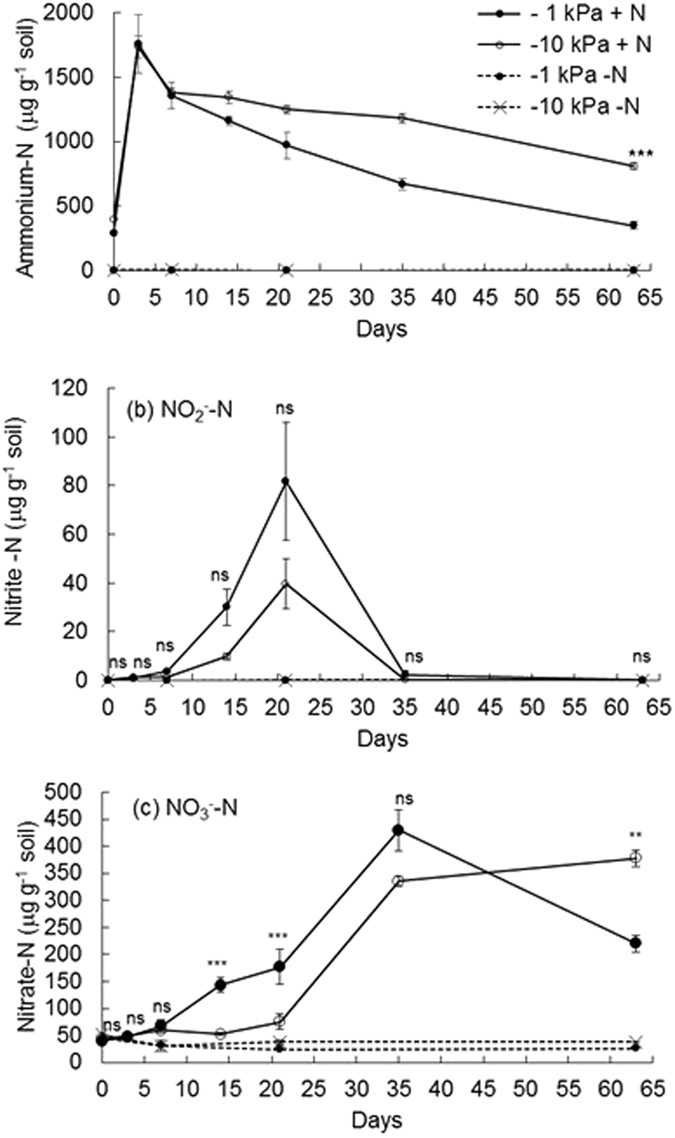
Changes in soil inorganic-N over time. Concentrations of extractable (a) ammonium-N (b) nitrite-N and (c) nitrate-N under near saturated (−1 kPa) or field capacity (−10 kPa) soil moisture conditions, following urea application (+N) or nil urea application (−N). Symbols are means (n = 4) with vertical error bars the standard error of the mean. Asterisks *,**,***indicate significant differences between moisture treatments under urea treatments at P < 0.05, P < 0.01, and P < 0.001, respectively.
Soil DOC was higher (P < 0.001) under the urea treatment throughout the experiment (Fig. 3) and within the urea treatment soil DOC concentrations were significantly lower at −1 kPa than at −10 kPa from day 3 to day 62 (Fig. 3). In the urea treatment soil DOC correlated strongly with soil pH at both −1 kPa (r = 0.79; p < 0.001) and −10 kPa (r = 0.89; p < 0.001).
Figure 3.
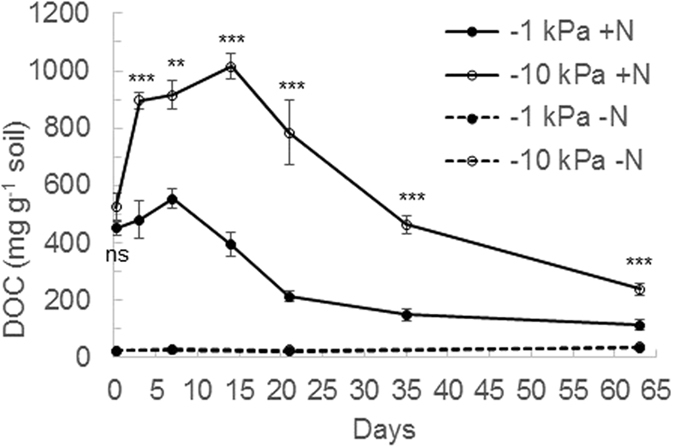
Changes in soil cold water extractable organic carbon (DOC) over time. Concentrations of soil DOC under near saturated (−1 kPa) or field capacity (−10 kPa) soil moisture conditions, following urea application (+N) or nil urea application (−N). Symbols are means (n = 4) with vertical error bars the standard error of the mean. Asterisks *,**,***indicate significant differences between moisture treatments under urea treatments at P < 0.05, P < 0.01, and P < 0.001, respectively.
Soil NH4+-N concentrations increased following urea application (Fig. 4), peaking at day 3 and then declining over time with a faster rate of decline in the −1 kPa treatment from day 14 (p < 0.05) such that soil NH4+-N concentrations were lower at −1 kPa on days 35 and 63 (Fig. 4). The 15N enrichment of the NH4+-N in the urea treatment declined from 44 to 37 atom% over the experiment with higher 15N enrichment on days 14, 21 and 35 in the −10 kPa treatment (Fig. 5). Concentrations of NO2−-N increased from day 7 under the urea treatment and peaked at day 21, with more NO2−-N present in the −1 kPa treatment, prior to returning to background levels at day 35 (Fig. 4). Concentrations of NO2−-N, extracted from the urea treatment, were only sufficient for 15N enrichment determinations on days 14 and 21, where the 15N enrichment was higher (p < 0.05) at −1 kPa than at −10 kPa on day 14, with no differences on day 21 (Fig. 5). Soil NO3−-N concentrations also began to increase at day 7 under the urea treatment and were consistently higher (p < 0.001) in the −1 kPa treatment on days 14 and 21. Soil NO3−-N concentrations peaked on day 35, before they declined to be less than those observed in the −10 kPa treatment (p < 0.01) at day 63 (Fig. 4). Changes in soil NO3−-15N enrichment reflected the concentration dynamics with 15N enrichment increasing faster at −1 kPa to 41 atom% 15N at day 21 while at −10 kPa the NO3−-15N enrichment was only 34 atom% 15N by day 63 (Fig. 5).
Figure 5.

Inorganic-N 15N enrichment over time. The 15N enrichment of the ammonium-N (NH4+-N), nitrite-N (NO2−-N) and nitrate-N (NO3−-N) are shown over time following 15N urea application at near saturated (−1 kPa) or field capacity (−10 kPa) soil moisture conditions. Symbols are means (n = 4) with vertical error bars the standard error of the mean.
N2O-N fluxes and 15N enrichment
Trends in daily N2O fluxes differed with treatment (Fig. 6). At −10 kPa in the absence of urea N2O-N fluxes were generally <5 μg m−2 h−1, with fluxes only greater than this (≤29 μg m−2 h−1) between day 0 and day 10 following treatment application (Fig. 6). Under the −1 kPa treatment, in the absence of urea, N2O-N fluxes also peaked after water application on day 2 at 498 μg m−2 h−1, before declining to ca 100 μg m−2 h−1 on day 12, where after N2O-N fluxes were constant until day 63, averaging 92 μg N2O-N m−2 h−1 between days 12 to 63 (Fig. 6). Adding urea at −10 kPa caused N2O-N fluxes to increase steadily from day 12 until they peaked at day 30 (449 μg m−2 h−1) where after they steadily declined to <10 μg m−2 h−1 by day 51 (Fig. 6). The highest N2O-N fluxes were observed at −1 kPa with urea addition, where a rapid increase in the flux occurred peaking at 11,603 μg m−2 h−1 on day 2, followed by a rapid decrease to 163 μg m−2 h−1 by day 7. Then the flux gradually increased until day 35 (9220 μg m−2 h−1) whereupon it too decreased to be 476 μg m−2 h−1by day 61 (Fig. 6).
Figure 6.

Nitrous oxide fluxes over time. Fluxes of N2O under near saturated (−1 kPa) or field capacity (−10 kPa) soil moisture conditions, following urea application (+N) or nil urea application (−N) where (a) −1 kPa +N (b) −10 kPa +N (c) −1 kPa –N and (d) −10 kPa –N show N2O fluxes over time. Note differing y-axis scales. Symbols are means (n = 4) with vertical error bars the standard error of the mean.
Soil moisture treatment influenced cumulative N2O-N fluxes (p < 0.001) with total emissions of 0.08 and 2.26 g N2O-N m−2 at −10 and −1 kPa, respectively, when averaged over plus and minus urea treatments. Similarly, application of urea increased cumulative N2O-N fluxes (p < 0.001) from 0.10 to 2.25 g N2O-N m−2 when averaged over soil moisture treatments. An interaction between soil moisture and N application (p < 0.002) resulted in higher cumulative N2O-N fluxes at −1 kPa when urea was applied equal to 3.99 g m−2 (Table 1). The N2O-N emission factors for the urea-N applied, allowing for non-N fluxes equated to 4.14% and 0.18% of N applied at −1 kPa and −10 kPa, respectively.
Table 1.
Mean cumulative N2O, N2DN and N2co emissions (g N m−2). P values are for the interaction between treatments.
| Urea-N | Moisture (kPa) | N2O | N2DN | N2co | |||
|---|---|---|---|---|---|---|---|
| +N | −1 | 3.99 | A | 8.61 | A | 1.92 | A |
| +N | −10 | 0.18 | B | 1.98 | A | 0.26 | B |
| −N | −1 | 0.16 | B | na | na | ||
| −N | −10 | −0.0003 | C | na | na | ||
| P value | 0.0321 | 0.0554 | 0.0437 |
Tukey-Kramer grouping: LS-means with the same letter are not significantly different, na not applicable. N2DN and N2co represent heterotrophic denitrification and codenitrification, respectively.
Upon urea application, the atom % 15N enrichment of the N2O emitted at −1 kPa increased steadily to reach a maximum value of 43.9 atom % 15N on day 25 before declining at a relatively slow rate to a value of 36.3 atom % 15N by day 59 (Fig. 7). With the exception of day 2, the atom % 15N enrichment of the N2O emitted at −1 kPa was higher than that emitted at −10 kPa (P < 0.05) on any given day. At −10 kPa the atom % 15N enrichment of the N2O flux was observed to increase abruptly at day 12, reaching a maximum of 32.8 on day 30 and thereafter declining relatively abruptly to remain at ca 10 atom % 15N (Fig. 7). Fluxes of N2O associated with codenitrification were low and only measurable on days 2, 5, 8 and 12 for the −1 kPa treatment and days 3, 5, 8, 12 and 16 for the −10 kPa treatment (Fig. 8). Highest fluxes were observed for the −1 kPa treatment (3637 μg N2O-N m−2 hr−1) comprising 20% of total N2O flux with emissions of codenitrified N2O subsequently reducing rapidly. Codenitrified N2O fluxes in the −10 kPa treatment were extremely low and never rose above 70 μg N2O-N m−2 hr−1).
Figure 7.

Nitrous oxide 15N enrichment over time. The 15N enrichment of the N2O molecule, over time, is shown for N2O evolved from soil under near saturated (−1 kPa) or field capacity (−10 kPa) conditions, following 15N urea application. Symbols are means (n = 4) with vertical error bars the standard error of the mean.
Figure 8.
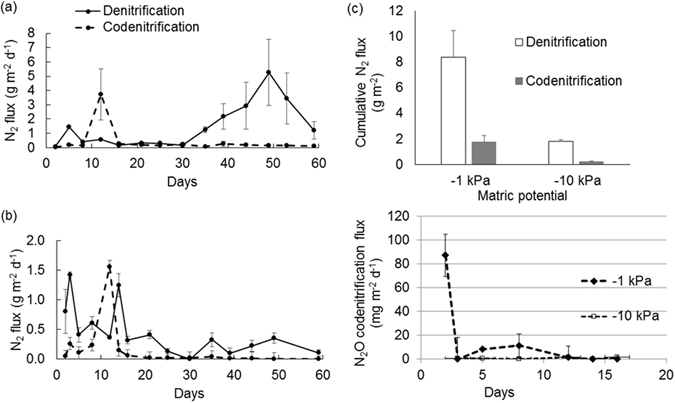
Denitrification and codenitrification fluxes over time. The codenitrification and denitrification fluxes, over time since 15N urea addition, are shown as daily N2 fluxes for (a) soil at −1 kPa (b) soil at −10 kPa and (c) as cumulative codenitrification and denitrification N2 fluxes, while (d) is the N2O codenitrification flux, over time since 15N urea addition, as daily N2O fluxes. Symbols are means (n = 4) with vertical error bars the standard error of the mean.
N2 fluxes and codenitrification after urea addition
The average daily denitrification fluxes were 1.48 (0.34) g N m−2 d−1 and 0.53 (0.07) g m−2 d−1 (s.e.m in brackets) at −1 and −10 kPa, respectively. At −1 kPa denitrification fluxes initially peaked at day 5 and then were higher after day 30, peaking on day 49 (Fig. 8). The decline in soil NO3− concentration after day 35 coincided with higher denitrification fluxes. At −10 kPa denitrification fluxes were highest after the initial wetting up following treatment application where after they generally declined (Fig. 8). Consequently, cumulative denitrification as N2 was higher (p = 0.055) at −1 kPa, totaling 8.61 g N m−2, than at −10 kPa where observed fluxes were 1.98 g N m−2 (Fig. 8).
The average daily codenitrification fluxes under urea treatments at −1 and −10 kPa were 0.38 (0.15) g N m−2 d−1 and −10 kPa 0.07 (0.01) g N m−2 d−1, respectively. Codenitrification fluxes peaked on day 12 regardless of soil kPa value, but were higher on day 10 at −1 kPa (Fig. 8). Average daily codenitrification fluxes were ca 5-fold higher at −1 kPa after day 30 than at −10 kPa. Consequently, cumulative codenitrification rates of N2 were also higher (p = 0.043) at −1 kPa (1.91 g N m−2) than at −10 kPa (0.26 g N m−2). Cumulative codenitrification, as a proportion of denitrification, did not vary as a result of soil matric potential equaling 25.5 ± 15.8% and 12.9 ± 4.8% (±stdev) at −1 and −10 kPa, respectively. The contribution of codenitrification as a proportion of total denitrification (codenitrification plus denitrification) also did not vary with soil matric potential, being 19.3 ± 10.4% and 11.3 ± 3.8% (±stdev) at −1 and −10 kPa, respectively.
Discussion
Inorganic-N pools and 15N enrichment
Following urea application to the soil the ensuing hydrolysis produces NH4+ and bicarbonate (HCO3−) ions. The HCO3− ions are further hydrolysed to produce hydroxide ions (OH−) and carbon dioxide25 and it is this second hydrolysis reaction that generated the observed increase in soil pH under the urea treatments (Fig. 2). Elevated soil pH also influences the equilibrium between NH4+ and ammonia (NH3): as soil pH becomes elevated (>7.0) concentrations of NH3 increase25. Urea-N not volatilized as NH3 may be transferred along the inorganic-N cascade via NH4+, NO2− and NO3−.
During nitrification microbes utilise NH4+ and oxidise it to NO2−. Elevated NH3 concentrations may inhibit NH4+ oxidation26,27. Thus the slower decline in the NH4+ concentration observed in the −10 kPa treatment, under urea, may have been due to NH3 inhibition of nitrification. In favour of this were both the relative gas diffusivity of the soil being 2 orders of magnitude higher at −10 kPa, which would have facilitated NH3 diffusion through the soil, and the soil pH remaining higher for longer (Fig. 2). The latter would have promoted the presence of NH3 for longer. A slower rate of decline in soil pH at −10 kPa also demonstrates nitrification was slower, since nitrification results in the net release of H+ ions19. Further evidence to support a slower rate of NH4+ oxidation can be found in the slower rate of increase in ammonium oxidizing bacteria (AOB) gene and transcript abundance28.
Elevated soil NO2− concentrations resulted from nitrification of NH4+ and their increase, from day 5 until day 20, occurred over a period when soil pH was sufficiently high to result in NH3 generation. Ammonia toxicity acts more strongly on nitrifier NO2− oxidation than nitrifier NH4+ oxidation29. It has been shown that solution-phase NH3 (slNH3) inhibits NO2− oxidation, as evidenced by strong relationships between cumulative slNH3 and cumulative NO2− and static copy numbers of the nxrA gene, which is associated with nitrite oxidoreductase, and as a consequence soil NO2− is strongly correlated with N2O production29. The high N2O fluxes that occurred, between ca. days 7 to 35, at both −1 and −10 kPa under urea, where the soil NO2− concentrations were elevated strongly demonstrates this, and it can be assumed slNH3 induced NO2− toxicity lead to the ensuing N2O emissions.
The higher NO3− concentrations observed under urea on days 14 and 21 at −1 kPa were a consequence of the more rapid nitrification rates in this treatment, while the lower NO3− concentration in this treatment observed at day 63 resulted from higher denitrification induced losses of NO3−, which is further supported by the increase in soil pH under this treatment, since denitrification results in a net release of OH− ions19.
The 15N enrichment of the NH4+ pool, under urea, shows that it was predominantly derived from the urea applied, regardless of soil moisture treatment. The fact the NH4+ pool 15N enrichment was initially ca. 5 atom% lower than the urea solution applied was likely due to the release of NH4+ as a consequence of the high soil pH solubilising soil organic matter, as demonstrated by the elevated DOC concentrations under the urea treatment. Solubilisation of soil organic matter is routinely observed following urine or urea application to soil30. The reason for the NO2− pool 15N enrichment being ca. half that observed in the NH4+ pool on days 14 and 21 at −1 kPa, shows antecedent soil N was also contributing to this pool which could have come from mineralization and subsequent oxidation of NH4+, despite the presence of NH3, since relatively low quantities of NH4+ would be needed to dilute the NO2− pool, or alternatively there may have been some denitrification of antecedent NO3− generating NO2−. The fact that the NO3− pool 15N enrichment aligned closely with that of the NO2− pool 15N enrichment at −10 kPa demonstrates NO2− was the dominant precursor to NO3− pool at −10 kPa. Furthermore, the slower rate of increase in the NO3− pool 15N enrichment at −10 kPa, when compared to −1 kPa, further supports the fact there was a slower rate of nitrification at −10 kPa. The increase in the NO3− pool 15N enrichment over time, in both the −1 and −10 kPa treatments, demonstrates the NO3− pool was initially dominated by antecedent soil NO3− as in fact occurred (Fig. 4c).
N2O fluxes and 15N enrichment
While simply wetting of the soil, as occurred under the non-urea treatment, induced N2O fluxes at −1 kPa, this wetting effect was not sufficient to generate the high N2O fluxes observed under urea from days 0 to 4. These high initial N2O fluxes under urea, as previously observed31, are due to the chemically induced anoxia that results from the hydrolysis reactions generating both NH3 and CO2, as demonstrated in situ32. Such high fluxes were not observed at −10 kPa during this period because the higher relative gas diffusivity of the soil at −10 kPa ensured the soil was not as anaerobic.
As noted above periods of high N2O flux between days 14 and 37 aligned with the presence of elevated NO2− concentrations. The atom % 15N enrichment of the N2O at −10 kPa was comparable with that of the NO2− pool at this time, further demonstrating that the N2O flux predominately originated from the NO2− pool, and because the 15N enrichment of the N2O declined as NO2− concentrations declined. Despite both the NO3− concentration and NO3− 15N enrichment both increasing after this time, this was not reflected in any increased N2O fluxes or its 15N enrichment because the higher relative gas diffusivity at −10 kPa made conditions unsuitable for the denitrification of NO3− 24.
However, at −1 kPa the N2O evolved predominately via denitrification of the NO3− pool up until ca. day 15 as demonstrated by the alignment of the N2O 15N enrichment with the NO3− pool 15N values. The higher N2O fluxes at −1 kPa between days 15 to 35 were ca. 15-fold higher due to the more anaerobic conditions and, as inferred above, are presumed to have occurred as a result of the relatively high NO2− concentrations over this period. However, the N2O 15N enrichment did not reflect that of the KCl extracted NO2− pool measured on days 14 and 21 at −1 kPa, but did reflect that of the NH4+ and NO3− pools on these days. Differences in the 15N enrichment of the KCl extracted NO2− and actual in situ 15N enrichment of the NO2− pool may possibly have arisen due to the method of treatment application where, in the −10 kPa treatment the urea solution infiltrated further and contacted a greater soil volume than at −1 kPa, as evidenced by the greater release of DOC at −10 kPa (Fig. 3), and which would have resulted in a more uniform NO2− pool. It is likely that, at −1 kPa, denitrification of antecedent NO3− occurred and that this generated sufficient NO2− to isotopically dilute the relatively small 15N enriched NO2− pool, derived from NH4+ and/or NO3−, when the soil was extracted. After day 35, the N2O 15N enrichment reflected that of the NO3− pool, and given the compatible conditions for denitrification, it can be assumed that denitrification of the NO3− pool dominated N2O production after day 30, and this assumption is supported by the elevated denitrification flux occurring after this time (Fig. 8).
N2 denitrification and codenitrification of N2 and N2O
As expected denitrification occurred at higher rates under the more anaerobic moisture treatment as a result of the lower Dp/Do conditions promoting denitrification in the presence of NO3− substrate.
The N transformations that ensued following urea hydrolysis, and hydrolysis itself, generated previously recognized codenitrification nucleophiles that include NH4+, NH3, and possibly organic-N compounds such as amines16. The latter might occur as a result of the dissolution of soil organic matter. While the enzymatically utilized NO2− and NO compounds, that form electrophiles, are generated during nitrification and denitrification19.
Codenitrification N2O fluxes were generally low for both treatments, with measurable values mainly associated with the initial soil wetting. Conversely, codenitrification to N2 was observed to peak on day 12, regardless of soil moisture, when NH3, NH4+ and NO2− were all present at an elevated soil pH (≥7.70), and at relatively high concentrations. Thus it is possible that either NH3 or NH4+ were undertaking the role of the nucleophile at this time, since the elevated pH (>5.5) would have prevented any significant abiotic nitrosation occurring via NO+ formation16. Recently, however, the formation of both N2O and N2, under both oxic and anoxic conditions, was reported in an in vitro experiment maintained at pH 6.2–6.9 where either live fungi or fungal necromass were incubated with glutamine and NO2− 33. A subsequent isotope experiment with glutamine and 15NO2− demonstrated the hybrid formation of N2 after an incubation period of >7 days, again under either oxic or anoxic conditions33. Hence, based on this recent study, even though the soil in the current study was at a pH (≥7.70) sufficient to prevent acidic pathways of abiotic hybrid N-N bonds forming, we cannot rule out the possibility that abiotic reactions, under alkaline conditions, contributed to the codenitrification flux measured in the current experiment.
Production of N2O or N2 via biotic codenitrification may result from the actions of archaea, bacteria or fungi. While archaea have been found to generate N2O through N-nitrosating hybrid formation34 they are unlikely to have been the dominant mechanism in the current study since archaea are thought to prefer low N conditions35,36 and urea addition resulted in lower ammonia oxidizing archaea gene copy numbers28. The codenitrification observed is most likely to be the result of fungi or bacterial activity. Delineation of the relative contributions made by fungi or bacteria to codenitrification is beyond the scope of the present study, however, future studies should aim to examine relative fungal and bacterial contributions.
Spott et al.16 conceptualized that the recognized constraints on denitrification might also apply to codenitrification, and thus higher codenitrification fluxes might be expected under more anaerobic conditions. The current results support this concept: after day 30 the higher daily codenitrification fluxes under the more anaerobic (−1 kPa) soil moisture conditions, when at the same time denitrification fluxes were higher, resulted in higher cumulative codenitrification fluxes. This reinforces the fact that NO2− and or NO play a key role in the codenitrification process. The NO molecule has been observed to readily diffuse within the soil profile37, at relatively high concentrations, during denitrification and this would result in reactions with nucleophiles.
Unlike the results of Selbie et al.23 codenitrification did not dominate the N2 fluxes observed in the current study. This could be the result of the experimental system used in the current study differing to that used by Selbie et al.23. Differences include the lack of a pasture turf and associated microbiology and root exudation, the use of sieved repacked soil that may also have altered the fungal-bacterial community structure or activity as a result of sieving, constant soil moisture contents as opposed to wetting and drying events, and the lack of other climatic variables such as wind and rainfall.
In particular, fungal populations may have been reduced on sieving, and given that fungal P450 NOR is implicated in supplying enzyme bound nitrosating agents this could have had a significant influence on the results38. Given that enzyme bound nitrosating agents produced during denitrification may also consist of metal-nitrosyl complexes16 any differences in soil Fe and Cu levels between studies may also explain the observed differences in codenitrification. Likewise, differences in the kinetic properties of different nucleophiles, combined with the ratio of NO or NO2− availability to nucleophile concentration, have also been shown to significantly impact on codenitrification/denitrification: lower Km and high nitrosyl donor/nucleophile ratios have been shown to reduce the level of codenitrification15,20.
This study confirms the role of anaerobic soil conditions in enhancing codenitrification fluxes under ruminant urine/urea deposition. It also demonstrates for the first time that high levels of NO2−, or other transitional N compounds ensuing from NO2−, that may occur during nitrification, are also able to contribute to codenitrification processes. To progress knowledge of codenitrification in grazed pastures more detailed studies are now required to both identify the microbial pathways operating and the relative importance of the possible nucleophiles and nitrosating agents that occur in grazed pastures.
Materials and Methods
Soil collection and experimental design
Soil was collected in early spring (March) from a permanently grazed dairy pasture at the Teagasc Moorepark Research Centre, County Cork, Ireland (8°15′W, 52°9′N). The top 5 cm of soil was removed and the A-horizon was sampled, 5–20 cm depth. Soil physical and textural characteristics are shown in Table 2. Cows had not grazed the pasture for over one month so recent urine deposition sites were avoided. The soil is classified as a Typical Brown earth from the Clashmore Series39, or as a Haplic Cambisol in the World Reference Database40. Field moist soil was then bagged and shipped to Lincoln University, New Zealand, following appropriate biosecurity protocols. It was then sieved (≤2 mm) to remove any stones, plant roots or earthworms. Sieved soil, with a gravimetric water content (θg) of 0.24 g water g−1 soil, was then packed into stainless steel rings (7.3 cm internal diameter, 7.4 cm deep) to a depth of 4.1 cm at a bulk density of 1.1 Mg m−3, the latter simulating the in situ soil bulk density. This resulted in a total porosity of 0.58 cm3 pores cm−3 soil. Packed soil cores were then arranged in a factorial experiment replicated four times.
Table 2.
Physical and textural characteristics of soil sampled.
| Depth | Bulk density (Mg m−3) | Porosity (%) | Texture | Sand (%) | Silt (%) | Clay (%) |
|---|---|---|---|---|---|---|
| 0–10 | 1.19 | 0.55 | Sandy loam | 53 | 31 | 16 |
| 10–20 | 1.28 | 0.52 | Sandy loam | 55 | 31 | 14 |
Treatments consisted of two levels of soil moisture, −1 kPa and −10 kPa simulating ‘near-saturation’ and ‘field-capacity’, respectively, and two levels of urea, (0 and 1000 kg N ha−1), replicated 4 times, with 7 destructive sampling times (112 cores in total). Preliminary tests showed that −1 and −10 kPa corresponded to 53% and 30% volumetric water content, or 91% and 52% water-filled pore space (WFPS). Soil cores were maintained at these water contents using tension tables41. Soil relative gas diffusivity values were calculated using the values for air-filled pore space and total porosity and the generalized-density corrected equation of Chamindu Deepagoda et al.42; Equation 9b. It is recognized that artificial urine simulation does not generate identical effects to ruminant urine43, that urea contributes >70% of the total urine-N pool6,44, and that this N source is predominately responsible for the subsequent dynamics and transformations of organic and inorganic N in the soil under ruminant urine patches. Thus, in order to apply the N treatments, soil cores were wetted up on the tension tables to a point where there remained the capacity to add a further 10 mL of liquid, without inducing drainage. Subsequently, in the plus N treatment, 10 mL of a urea solution (42 g urea-N L−1; 50 atom%, Cambridge Isotope Laboratories Inc., USA) was slowly applied to the soil surface, to avoid drainage, to mimic an extreme bovine urine deposition event with a potentially high N2 flux. Real urine could not be used since there was a need to have the urea-N highly enriched with 15N to detect N2 fluxes. In the nil N treatment 10 mL of deionized water was applied instead of a urea solution. Tension tables were maintained in a room with a mean temperature of 20 °C.
Soil chemical analyses
After treatment application and throughout the experiment, on days 0, 3, 7, 14, 21, 35, and 63, soil inorganic N concentrations were determined by destructively sampling 16 soil cores (2 levels of urea × two levels of soil moisture × 4 replicates). Soil cores were fully extracted, homogenized, and a subsample was taken to determine θg: by drying the soil at 105 °C for 24 hours. A flat surface pH electrode was used to determine soil pH (Broadley James Corp., Irvine, California). Then further soil subsamples were extracted (equivalent of 10 g dry soil: 100 mL 2 M KCl shaken for 1 hour) and filtered (Whatman 42) to determine soil inorganic-N. The NH4+-N, NO2−-N, and NO3−-N concentrations were analysed using flow injection analysis45. The 15N enrichment of NH4+-N was determined according to Stark and Hart46 while NO2−-15N and NO3−-15N enrichments were determined according to the methods of Stevens and Laughlin47. Concentrations of dissolved organic carbon (DOC) in the soil were measured according to Ghani et al.48 with analyses performed on a Shimadzu TOC analyzer (Shimadzu Oceania Ltd., Sydney, Australia).
Gas flux determinations
Nitrous oxide and N2 fluxes were regularly determined, from two days before until 63 days after treatment application using only the last batch of soil cores to be destructively analysed. This was performed by placing a soil core into a 1-L stainless steel tin fitted with a gas-tight lid and rubber septa. Samples for N2O flux determinations were taken upon lid closure and then after 15 and 30 minutes. A further sample was taken for N2O-15N enrichment and N2 flux determination after 3 hours, after which cores were returned to the tension tables. Gas samples were taken using a 20-mL glass syringe fitted with a 3-way tap and a 0.5 mm by 16 mm needle and placed in either 6 mL vials for the N2O flux determinations or 12 mL vials for the N2O-15N enrichment and N2 flux samples (Exetainer; Labco Ltd., Lampeter, UK). An automated gas chromatograph (8610; SRI Instruments, Torrance, CA), coupled to an autosampler (Gilson 222XL; Gilson, Middleton, WI), was used to determine N2O gas concentrations in the samples, as previously described49. A continuous-flow-isotope mass spectrometer (Sercon 20/20; Sercon, Chesire, UK) inter-faced with a TGII cryofocusing unit (Sercon, Chesire, UK), was used to determine the 15N enrichment of the N2O-N and N2-N gas samples50.
The ion currents (I) at mass to charge ratios (m/z) of 44, 45, and 46 facilitated the calculation of the N2O molecular mass ratios 45R (45I/44I) and 46R (46I/44I). The N2O sources were subsequently allocated to either the fraction derived from the denitrifying pool (d’D) of enrichment aD or the fraction derived from the pool or pools at natural abundance d’N = (1-d’D) using the method of Arah (1997). The ion currents at m/z 28, 29, and 30 permitted the N2 molecular ratios 29R (29I/28I) and 30R (30I/28I) to be quantified. Differences between the N2 molecular ratios of the enriched and ambient atmospheres were expressed as Δ29R and Δ30R The N2 flux was subsequently calculated using three methods:
-
(i)
The enrichment of the denitrifying pool (15XN) was calculated using Δ29R and Δ30R, and then the N2 flux51,
-
(ii)
Using only the Δ30R data with the assumption that the enrichment of the denitrifying pool was aD52 and the equation of Mulvaney53
-
(iii)
Using Δ29R and Δ30R to calculate the relative contributions of denitrification (N2DN), according to method (ii), and codenitrification (N2CO).
Increases in Δ29R and Δ30R may occur from denitrification but codenitrification contributes most to Δ29R where the ratio of Δ29R to Δ30R is always 27254. By assuming all Δ30R was the result of denitrification, method (ii), N2DN was calculated. Then using the ‘backsolver’ facility in Microsoft ExcelTM, the contribution of Δ29R to N2DN was determined. The difference between the total measured value of Δ29R and Δ29R determined for N2DN was assigned to codenitrification. Thus the fraction of the total number of moles of N2 in the headspace, resulting from codenitrification (dCD) were calculated as:
| 1 |
where p1 (0.9963) and q1 (0.0037) represent the atom fractions of 14N and 15N in the natural abundance pool, respectively, and p2 and q2 are the atom fractions of 14N and 15N in the enriched NO3− pool, respectively, from which codenitrification is assumed to occur. Using the headspace volume of the sample chamber, corrected for standard temperature and pressure, the mass of N2-N in the headspace was determined with the amount derived from denitrification or codenitrification ascertained by multiplying by dD or dCD, respectively.
Data analyses
Data were analysed using the Glimmix procedure within the SAS® software version 9.4 (SAS, 2014). Cumulative results were analysed for the +N treatment only. For all other variables, analyses was as N treatment × moisture × day or moisture × day factorials. Any repeated measurements over time were modelled using correlation structures and spatial covariance was used to model the unequally-spaced time measurements. Residual checks were made and, where required, log transformation was used to correct for skew and non-constant variance. Multiplicity adjustments were made for simple effects within interactions, as interest was primarily in comparisons within time points.
Acknowledgements
The authors gratefully acknowledge the assistance of Manjula Premaratne and Roger Cresswell in assisting with gas chromatography analyses and mass spectrometer analyses. This work was funded by the New Zealand Government through the New Zealand Fund for. Global Partnerships in Livestock Emissions Research to support the objectives of the Livestock Research Group of the Global Research Alliance on Agricultural Greenhouse Gases (Agreement number: 16084) awarded to SEM and the University of Otago. Charlotte Johns gratefully acknowledges funding received from the Teagasc Walsh Fellowship Scheme.
Author Contributions
C.d.K., K.R. and G.L. were the principal investigators for the project funding. T.C., K.R., G.L. and L.C. designed the experiment. C.J. conducted the measurements. T.C., K.R. and G.L. drafted the manuscript with C.d.K., S.E.M., DR, M.S.S., and L.B. providing assistance with data interpretation and manuscript preparation, while J.G. assisted with statistical interpretation.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
A correction to this article is available online at https://doi.org/10.1038/s41598-018-22645-7.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.IPCC, Summary for Policymakers. In Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, edited by Stocker, T. F. et al., pp. 1535 (Cambridge University Press, 2013).
- 2.Ravishankara AR, Daniel JS, Portmann RW. Nitrous Oxide (N2O): The Dominant Ozone-Depleting Substance Emitted in the 21st Century. Science. 2009;326:123–125. doi: 10.1126/science.1176985. [DOI] [PubMed] [Google Scholar]
- 3.Ciais, P. et al. Carbon and Other Biogeochemical Cycles. In: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change. (Cambridge University Press, Cambridge, United Kingdom, 2013).
- 4.Hargreaves PR, Rees RM, Horgan GW, Ball BC. Size and Persistence of Nitrous Oxide Hot-Spots in Grazed and Ungrazed Grassland. Environ. Nat. Resor. Res. 2015;5(4):1. [Google Scholar]
- 5.Oenema O, et al. Trends in global nitrous oxide emissions from animal production systems. Nutr. Cycl. Agroecosyst. 2005;72(1):51–65. doi: 10.1007/s10705-004-7354-2. [DOI] [Google Scholar]
- 6.Haynes RJ, Williams PH. Nutrient cycling and soil fertility in the grazed pasture ecosystem. Adv. Agron. 1993;49:119–199. doi: 10.1016/S0065-2113(08)60794-4. [DOI] [Google Scholar]
- 7.Selbie DR, Buckthought LE, Shepherd MA. The Challenge of the Urine Patch for Managing Nitrogen in Grazed Pasture Systems. Adv. Agron. 2015;129:229–292. doi: 10.1016/bs.agron.2014.09.004. [DOI] [Google Scholar]
- 8.Zhu X, Burger M, Doaneb TA, Howarth WR. Ammonia oxidation pathways and nitrifier denitrification are significant sources of N2O and NO under low oxygen availability. P Natl. Acad. Sci. USA. 2013;110(16):6328–6333. doi: 10.1073/pnas.1219993110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laughlin RJ, Stevens RJ. Evidence for fungal dominance of denitrification and codenitrification in a grassland soil. Soil Sci. Soc. Am. J. 2002;66(5):1540–1548. doi: 10.2136/sssaj2002.1540. [DOI] [Google Scholar]
- 10.Firestone, M. K. & Davidson, E. A. Microbiological Basis of NO and N2O Production and Consumption in Soil In Exchange of Trace Gases between Terrestrial Ecosystems and the Atmosphere, edited by Andreae, M. O. & Schimel, D. S., pp. 7–21(John Wiley & Sons Ltd, New York, 1989).
- 11.Allison FE. The enigma of soil nitrogen balance sheets. Adv. Agron. 1955;7:213–250. doi: 10.1016/S0065-2113(08)60339-9. [DOI] [Google Scholar]
- 12.Clough TJ, et al. Resolution of the N-15 balance enigma? Aust. J. Soil Res. 2001;39(6):1419–1431. doi: 10.1071/SR00092. [DOI] [Google Scholar]
- 13.Ball R, Keeney DR, Theobald PW, Nes P. Nitrogen balance in urine-affected areas of a New Zealand pasture. Agron. J. 1979;71:309–314. doi: 10.2134/agronj1979.00021962007100020022x. [DOI] [Google Scholar]
- 14.Shoun H, Kim DH, Uchiyama H, Sugiyama J. Denitrification by fungi. FEMS Microbiol. Lett. 1992;94:277–281. doi: 10.1111/j.1574-6968.1992.tb05331.x. [DOI] [PubMed] [Google Scholar]
- 15.Tanimoto T, Hatano K, Kim DH, Uchiyama H, Shoun H. Co-denitrification by the denitrifying system of the fungus Fusarium oxysporum. FEMS Microbiol. Lett. 1992;93:177–180. doi: 10.1111/j.1574-6968.1992.tb05086.x. [DOI] [Google Scholar]
- 16.Spott O, Russow R, Stange CF. Formation of hybrid N2O and hybrid N2 due to codenitrification: First review of a barely considered process of microbially mediated N-nitrosation. Soil Biol. Biochem. 2011;43:1995–2011. doi: 10.1016/j.soilbio.2011.06.014. [DOI] [Google Scholar]
- 17.Baggs, E. M. & Philippot, L. Microbial terrestrial pathways to nitrous oxide. In Nitrous Oxide and Climate Change (Earthscan, 2010).
- 18.Weeg-Aerssens E, Tiedje JM, Averill BA. Evidence from isotope labeling studies for a sequential mechanism for dissimilatory nitrite reduction. J Am. Chem. Soc. 1988;110:6851–6856. doi: 10.1021/ja00228a039. [DOI] [Google Scholar]
- 19.Wrage N, Velthof GL, van Beusichem ML, Oenema O. Role of nitrifier denitrification in the production of nitrous oxide. Soil Biol. Biochem. 2001;33:1723–1732. doi: 10.1016/S0038-0717(01)00096-7. [DOI] [Google Scholar]
- 20.Kim CH, Hollocher TC. Catalysis of nitrosyl transfer-reactions by a dissimilatory nitrite reductase (cytochrome-cd1) J. Biol. Chem. 1984;259:2092–2099. [PubMed] [Google Scholar]
- 21.Van Cleemput O, Samater AH. Nitrite in soils: accumulation and role in the formation of gaseous N compounds. Fert. Res. 1996;45:81–89. doi: 10.1007/BF00749884. [DOI] [Google Scholar]
- 22.Sen S, Chalk PM. Chemical interactions between soil N and alkaline-hydrolysing N fertilizers. Fert. Res. 1993;36:239–248. doi: 10.1007/BF00748702. [DOI] [Google Scholar]
- 23.Selbie, D. R. et al. Confirmation of co-denitrification in grazed grassland. Scientific Reports 5, Article number: 17361 (2015). [DOI] [PMC free article] [PubMed]
- 24.Balaine, N., Clough, T. J., Beare, M. H., Thomas, S. M. & Meenken, E. D. Soil Gas Diffusivity Controls N2O and N2 Emissions and their Ratio. Soil Sci. Soc. Am. J (2016).
- 25.Avnimelech Y, Laher M. Ammonia volatilization from soils: Equilibrium considerations. Soil Sci. Soc. Am. J. 1977;41:1080–1084. doi: 10.2136/sssaj1977.03615995004100060013x. [DOI] [Google Scholar]
- 26.Anthonisen AC, Loehr RC, Prakasam TBS, Srinath EG. Inhibition of nitrification by ammonia and nitrous acid. J. Water Pollut. Control Fed. 1976;48:835–852. [PubMed] [Google Scholar]
- 27.Park S, Bae W. Modeling kinetics of ammonium oxidation and nitrite oxidation under simultaneous inhibition by free ammonia and free nitrous acid. Proc. Biochem. 2009;44:631–640. doi: 10.1016/j.procbio.2009.02.002. [DOI] [Google Scholar]
- 28.Samad, M. S. et al. Response to nitrogen addition reveals metabolic and ecological strategies of soil bacteria. bioRxiv (107961) (2017). [DOI] [PubMed]
- 29.Venterea R, Clough TJ, Coulter JA, Breuillin-Sessoms F. Ammonium sorption and ammonia inhibition of nitrite-oxidizing bacteria explain contrasting soil N2O production. Science Reports. 2015;5:12153. doi: 10.1038/srep12153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clough TJ, et al. The mitigation potential of hippuric acid on N2O emissions from urine patches: An in situ determination of its effect. Soil Biol. Biochem. 2009;41:2222–2229. doi: 10.1016/j.soilbio.2009.07.032. [DOI] [Google Scholar]
- 31.Krol DJ, Forrestal PJ, Lanigan GJ, Richards KG. In situ N2O emissions are not mitigated by hippuric and benzoic acids under denitrifying conditions. Sci. Total Environ. 2015;511:362–368. doi: 10.1016/j.scitotenv.2014.12.074. [DOI] [PubMed] [Google Scholar]
- 32.Owens J, et al. Nitrous Oxide Fluxes, Soil Oxygen, and Denitrification Potential of Urine- and Non-Urine-Treated Soil under Different Irrigation Frequencies. J. Environ. Qual. 2016;45(4):1169–1177. doi: 10.2134/jeq2015.10.0516. [DOI] [PubMed] [Google Scholar]
- 33.Phillips RL, et al. Chemical formation of hybrid di-nitrogen calls fungal codenitrification into question. Scientific Reports. 2016;6:39077. doi: 10.1038/srep39077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stieglmeier M, et al. Aerobic nitrous oxide production through N-nitrosating hybrid formation in ammonia-oxidizing archea. ISME J. 2014;8:1135–1146. doi: 10.1038/ismej.2013.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Di HJ, et al. Nitrification driven by bacteria and not archaea in nitrogen-rich grassland soils. Nat. Geosci. 2009;2:621–624. doi: 10.1038/ngeo613. [DOI] [Google Scholar]
- 36.Sterngren, A. E., Hallin, S. & Bengtson, P., Archaeal Ammonia Oxidizers Dominate in Numbers, but Bacteria Drive Gross Nitrification in N-amended Grassland Soil Front. Microbiol. 6, doi:10.3389/fmicb.2015.01350, Article Number 1350 (2015). [DOI] [PMC free article] [PubMed]
- 37.Clough TJ, Rolston DE, Stevens RJ, Laughlin RJ. N2O and N2 gas fluxes, soil gas pressures, and ebullition events following irrigation of 15NO3−-labelled subsoils. Aust. J. Soil Res. 2003;41(3):401–420. doi: 10.1071/SR02104. [DOI] [Google Scholar]
- 38.Wasser IM, de Vries S, Moenne-Loccoz P, Schroder I, Karlin KD. Nitric oxide in biological denitrification: Fe/Cu metalloenzyme and metal complex NOx redox chemistry. Chem. Rev. 2002;102:1201–1234. doi: 10.1021/cr0006627. [DOI] [PubMed] [Google Scholar]
- 39.Gardiner, M. J. & Radford, T. Soil Associations of Ireland and Their Land Use Potential – Explanatory Bulletin to Soil Map of Ireland 1980 (The Agricultural Institute, Dublin, 1980).
- 40.WRB, IUSS Working Group WRB. 2006. World reference base for soil resources 2006. (Rome, 2006).
- 41.Romano, N., Hopmans, J. W. & Dane, G. H. Water retention and storage. In Methods of Soil Analysis, Part 4, Physical Methods, edited by G. C. Topp & G. H. Dane, pp. 692–698 (Soil Science Society of America, Madison, WI, 2002).
- 42.Chamindu Deepagoda TKK, et al. Generalized Density-Corrected Model for Gas Diffusivity in Variably Saturated Soils. Soil Sci. Soc. Am. J. 2011;75(4):1315–1329. doi: 10.2136/sssaj2010.0405. [DOI] [Google Scholar]
- 43.Kool DM, Hoffland E, Abrahamse SPA, van Groenigen JW. What artificial urine composition is adequate for simulating soil N2O fluxes and mineral N dynamics? Soil Biol. Biochem. 2006;38(7):1757–1763. doi: 10.1016/j.soilbio.2005.11.030. [DOI] [Google Scholar]
- 44.Bathhurst NO. The amino acids of sheep and cow urine. J. Agr. Sci. 1952;42:476–478. doi: 10.1017/S0021859600057385. [DOI] [Google Scholar]
- 45.Blakemore, L. C., Searle, P. L. & Daly, B. K. Methods for chemical analysis of soils. (Manaaki-Whenua Press, Lincoln, New Zealand, 1987).
- 46.Stark JM, Hart RH. Diffusion technique for preparing salt solutions, Kjeldahl digests, and persulfate digests for nitrogen-15 analysis. Soil Sci. Soc. Am. J. 1996;60:1846–1855. doi: 10.2136/sssaj1996.03615995006000060033x. [DOI] [Google Scholar]
- 47.Stevens RJ, Laughlin RJ. Determining nitrogen−15 in nitrite or nitrate by producing nitrous oxide. Soil Sci. Soc. Am. J. 1994;58:1108–1116. doi: 10.2136/sssaj1994.03615995005800040015x. [DOI] [Google Scholar]
- 48.Ghani A, Dexter M, Perrott KW. Hot-water extractable carbon in soils: a sensitive measurement for determining impacts of fertilisation, grazing and cultivation. Soil Biol. Biochem. 2003;35:1231–1243. doi: 10.1016/S0038-0717(03)00186-X. [DOI] [Google Scholar]
- 49.Clough TJ, Kelliher FM, Wang YP, Sherlock RR. Diffusion of N-15-labelled N2O into soil columns: a promising method to examine the fate of N2O in subsoils. Soil Biol. Biochem. 2006;38(6):1462–1468. doi: 10.1016/j.soilbio.2005.11.002. [DOI] [Google Scholar]
- 50.Stevens RJ, Laughlin RJ, Atkins GJ, Prosser SJ. Automated determination of nitrogen-15 labelled dinitrogen and nitrous oxide by mass spectrometry. Soil Sci. Soc. Am. J. 1993;57:981–988. doi: 10.2136/sssaj1993.03615995005700040017x. [DOI] [Google Scholar]
- 51.Mulvaney RL, Boast CW. Equations for determination of nitrogen-15 labelled dinitrogen and nitrous oxide by mass spectrometry. Soil Sci. Soc. Am. J. 1986;50:360–363. doi: 10.2136/sssaj1986.03615995005000020021x. [DOI] [Google Scholar]
- 52.Stevens RJ, Laughlin RJ. Lowering the detection limit for dinitrogen using the enrichment of nitrous oxide. Soil Biol. Biochem. 2001;33:1287–1289. doi: 10.1016/S0038-0717(01)00036-0. [DOI] [Google Scholar]
- 53.Mulvaney RL. Determination of 15N-labeled dinitrogen and nitrous oxide with triple-collector mass spectrometers. Soil Sci. Soc. Am. J. 1984;48:690–692. doi: 10.2136/sssaj1984.03615995004800030045x. [DOI] [Google Scholar]
- 54.Clough TJ, Stevens RJ, Laughlin RJ, Sherlock RR, Cameron KC. Transformations of inorganic-N in soil leachate under differing storage conditions. Soil Biol. Biochem. 2001;33(11):1473–1480. doi: 10.1016/S0038-0717(01)00056-6. [DOI] [Google Scholar]
- 55.Schmidt HL, Werner RA, Yoshida N, Well R. Is the isotopic composition of nitrous oxide an indicator for its origin from nitrification or denitrification?A theoretical approach from referred data and microbiological and enzyme kinetic aspects. Rapid Commun. Mass Spectrom. 2004;18:2036–2040. doi: 10.1002/rcm.1586. [DOI] [PubMed] [Google Scholar]
- 56.Ye RW, Torosuarez I, Tiedje JM, Averill BA. (H2O)-O-18 isotope exchange studies on the mechanism of reduction of nitric-oxide and nitrite to nitrous oxide by denitrifying bacteria - evidence for an electrophilic nitrosyl during reduction of nitric oxide. J. Biol. Chem. 1991;266:12848–12851. [PubMed] [Google Scholar]