

Abstract
Three fundamental questions in biology are how do individual cells differentiate to form tissues, how do tissues function in a coordinated and flexible fashion, and which gene regulatory mechanisms support these processes. Single cell genomics open new ways to tackle these questions by combining the comprehensive nature of genomics with the microscopic resolution required to describe complex multi-cellular systems. Early single cell genomic studies provides us with remarkably rich phenomenology of heterogeneous cellular states, but transforming observational studies to models of dynamics and causal mechanism in tissues poses new challenges and require stronger integration between theoretical, computational and experimental frameworks.
Multicellular organisms evolved sophisticated strategies for cooperation between cells, such that a single genome codes for multiple specialized and complementary functional programs that maximizes fitness when working together1–4. Compartmentalization at several levels – from cells, to tissues, to organs – leads to functional diversification of cells and systems, with the same underlying genome. Physical copies of the genome are embedded in cells to allow them to maintain a semi-autonomous decision-making process, through selective management of small molecules, RNA and protein concentrations within cytoplasmic and nuclear compartments. Theoretically, this allows genomes to break the inherent symmetry imposed by the precise duplication of DNA content in multi-cellular species. In particular, it allows cellular differentiation to take place by progressive acquisition of specific intra-cellular molecular compositions, enable epigenetic mechanisms to emerge and implement cellular memory. At a higher layer, inter-cellular signaling, extracellular structures, and environmental cues are used to form complex spatial structures into which cells (and their genomes) are physically embedded. This creates additional levels of compartmentalization to facilitate the encoding of complex and structured tissues.
A Linnaean framework is a major part of the present theoretical foundation to cellular heterogeneity, defining taxonomies over cell types and aiming to map this hierarchical classification to the organism’s ontogeny. In this context, the composition of single cells within tissue and compartments has historically been characterized using microscopy and florescence activated cell sorting (FACS). While these techniques have high spatial or cellular resolution, they have relied by necessity on a limited number of molecular or visual markers, giving rise to classification schemes that are difficult to study globally and quantitatively, even when combined with downstream genome wide analysis. Nevertheless, molecular taxonomies are being built extensively through accumulation of large image and gene expression repositories5–10, and have proven successful both in some somatic tissues with little cell differentiation in the adult (e.g., the retina11), and in those that dynamically and continuously re-differentiate throughout the life span (e.g., hematopoiesis10,12).
However, despite continued progress in enhancing the palette of available markers and the throughput of microscopy and molecular tissue profiling, the Linnaean framework may be inherently inadequate when extending to whole tissues and organisms. In particular, the approach becomes ambiguous whenever complex mixtures of molecular behaviors are tightly coupled spatially and functionally in ways that make it impossible to separate them physically. Hierarchical classification is also not naturally applicable when the molecular mechanisms driving continuous effects, such as during a developmental continuum, differentiation bifurcation, plastic transitions between multiple cell “types”, morphogen gradients, or pathological, irregular deterioration of cellular programs. Indeed, in developmental biology, especially when dealing with embryonic differentiation, models for continuous change are well developed, and understanding how such processes diverge to form terminally differentiated stable states is a major challenge. Even the precise definition of a cell type is far from being agreed upon, and different fields define this key notion differently.
The recent advents in single cell genomics – allowing the profiling of the genetic and molecular state of ever growing numbers of individual cells – could also open the way for new, data-driven definition of cell identity and function, less encumbered conceptually by strict a priori hierarchies and less dependent practically on pre-defined markers. Thus, genomics - which emerged as a science taking advantage of the consistency of genomic DNA within individuals - is rapidly repurposed to handle complex genomic functions that ultimately occurs within single cells. This could help formulate afresh key axes – temporal dynamics, spatial organization, and molecular mechanisms – that inform and control cell identity. The major challenge for this emerging field is now to bring together the power of quantitative and comprehensive genomics with microscopic resolution, in order to replace coarse-grained, step-wise and deterministic models for cell and tissue function with more quantitative and predictive models.
Single cell genomics: state of the art
Genome-wide transcriptional profiling and epigenomics opened the way to comprehensive measurements of cells’ molecular state, in lieu of strategies based on selected markers. Until recently, comprehensive genomic analysis has relied either on pooling heterogeneous mixtures or on first sorting sub-populations and then profiling them. Bulk profiling only provides mixture averages, enabling genome-wide screening for regulated genes and detection of compositional changes between experiments, but not direct identification of the cellular programs composing the samples under study. Analysis of sorted populations, on the other hand, was limited to known sub-populations and sorting panels, while masking intra-sample variation.
Over the past few years, several genomics, transcriptomics and epigenomics assays were reconfigured to allow analysis of single cells. It is now possible to collect genome-wide profiles of RNA13–17, DNA18–23, histone modifications24,25, chromatin accessibility26,27, DNA methylation28–31, nuclear lamina interactions32, and chromosomal contacts33, as well as single cell protein signatures34. Initial studies focused on improving the assays’ throughput (number of cells), robustness (performance with samples of varying quality), complexity (the number of distinct molecules captured from each cell), and accuracy (level of noise). Single cell genomics has already produced a phenomenology of cellular heterogeneity at unprecedented detail in several systems16,17,26,35–54.
Single cell RNA-seq (scRNA-seq) has been at the forefront of these methods, in particular in terms of throughput. Whereas initial studies analyzed a handful to a hundred cells13,14,17,55, a series of technological advances, from robotics16,56 to microfluidics39,57 to reverse emulsion and hydrogel droplets increased assay throughput to tens or even hundreds of thousands of cells in an experiment58,59. There were also advances in techniques for acquiring cells and processing minuscule amounts of RNA14,60, improving the robustness of scRNA-seq on small samples, such as biopsies54,61, fixed cells62, and even avoiding tissue dissociation altogether by isolating nuclei48,63. It is still not routine to apply scRNA-seq to clinical samples, but it is certainly within reach38,54.
In contrast to throughput and robustness, the complexity and noise of scRNA-Seq remain difficult to characterize and optimize. Naively, scRNA-Seq aims to measure a complete census of mRNA molecules in each cell with minimal error. But as individual cells have variable, and a priori unknown, mRNA content, and since other cellular features may affect mRNA recovery, it is not currently possible to estimate the assay’s performance using e.g., replicate experiments. Instead, complexity estimates are based on some independent assumptions of mRNA content in cells (which we estimate between 105–106 molecules per cell, when excluding rRNA from measured estimates of total RNA64,65) and noise estimations are performed following analysis of variance for transcripts with presumed lower variability, or using spiked-in controls (reviewed in 66). Unique Molecular Identifiers (UMIs)16,17,56,58,59,67 and techniques for error correction (reviewed in 66) greatly reduced the level of technical noise, by addressing PCR duplicates, detecting cross-cell contamination, and defining molecule counts for downstream statistical models. Nevertheless, scRNA-Seq still not only samples cells from tissues, but also of molecules within cells.
Sampling – which initially appears as an unfortunate limitation, is a remarkably powerful approach to design efficient experiments, when applied correctly. The optimal scRNA sampling strategy of a cell population or tissue depends on the question at hand. Recent analyses16,17,39,52,58,59,68 have suggested that since the marginal utility of sequencing scRNA-seq libraries decreases rapidly with sequencing depth, aiming for a larger number of cells with fewer reads per cell may be better suited for cell type identification and classification. When a very large number of cells are profiled, clusters of cells with similar RNA distributions can be identified and pooled to form idealized models of single cell transcription with resolution that is limited only by the number of cells. Conversely, grouping sampled cells into fine granularity clusters cannot be achieved unless some minimal amount of RNA is captured from each cell. Higher sampling depth may also be required for analyzing the regulatory relationship between genes within single cells (below).
Throughput, robustness and complexity are also being optimized for single cell epigenomics assays. Currently, such assays analyze dozens to hundreds of cells, with partial automation. Complexity is particularly challenging in epigenomics assays, which must target single copy molecules per cell and, unlike scRNA-seq, cannot buffer partial sampling by analysis of high copy number molecules. Two pooling strategies have been applied to circumvent low-complexity data. First, pooling single cells25–27,47 allows effective analysis even when molecule recovery rate is between 1% to 10%. Second, pooling signals in the same cell across multiple related loci25,26 (e.g., those known to be bound by the same transcription factor) can help recover a cell’s epigenetic state even when coverage per locus is sparse.
Importantly, DNA methylation, histone modifications, chromatin accessibility, and chromosome 3D organization – each carry unique information that is not available from scRNA-seq, even at its maximal complexity and throughput. For example, changes in chromatin organization may precede and foreshadow later differentiation events, before these are recognizable at the level of RNA expression47,69, and may be a more stable fingerprint of a cell’s type and stable status. DNA methylation landscapes may reflect developmental potential and regulatory element activity in ways that cannot be inferred for the instantaneous RNA levels of the cell. Emerging strategies can measure multiple types of profiles simultaneously within the same single cell, thus helping to match the chain of events from DNA through regulatory mechanism, to RNA, protein and phenotype. Current studies have combined DNA and RNA70–72, RNA and a signature of proteins73–76, and RNA and DNA methylation72, and additional pair- and multi-way combinations will likely arise soon.
Thus, single cell genomics data can now include tens of thousands of high quality scRNA-seq profiles along with hundreds of single cell epigenomic profiles, possibly for the same cells. These data provide the ultimate Linnaean toolkit, supporting unbiased and comprehensive classification of cells into sub-populations, and simultaneously defining genome-wide transcription and epigenomic states for each of the detected “cell-types”. But going beyond such data and its classification and into mechanistic models of genome and tissue regulation, we must first interpret and measure single cells in their temporal and spatial contexts.
The temporal axis: inferring dynamics
Biological processes are dynamic at multiple time scales, from fast responses to environmental stimuli (minutes to hours), through cell differentiation (hours to days) to pathogenesis (up to years). Characterizing the series of regulatory events that underlie such dynamic processes can be surprisingly challenging, because cells are rarely perfectly synchronous and their dynamics is non-deterministic (Fig. 1). Previously, genome scale studies of regulatory dynamics required either successful synchronization, or the ability to isolate specific sub-populations of cells at distinct functional points along the process (Fig. 1a). However, robust genomic profiling of even a process like the cell cycle remained challenging77,78.
Figure 1. The temporal axis.
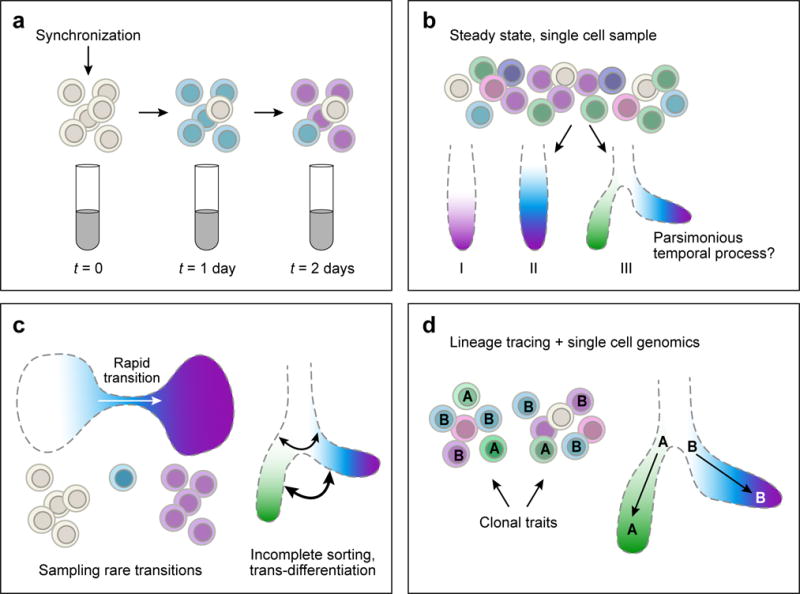
(a) Bulk assays sample cell populations that progressively lose synchrony, limiting the precise inference of the temporal dynamics. (b) Sampling of a single heterogenous mixture of single cells in different states can be used to infer temporal dynamics based on a maximum parsimony principle, where the sampled cells are organized along a linear or tree-like process, such that differences in the molecular profiles of the sampled cells are captured fatefully by the inferred topology. (c) The maximum parsimony approach for inferring temporal dynamics from single cell samples is challenged when transition between frequent states are rare (left), or when cells undergo complex, non-hierarchical, or non-deterministic dynamics (right). In either case, the maximum parsimony model for the data may become under-determined. (d) Adding anchor points, such as known stem cell states or differentiated states, may help distinguishing between different alternative dynamical models in single cell data. Experimental information on the clonal relationship within a single cell sample can lead to correct identification of a bifurcation process as illustrated schematically here.
Single cell genomics partially alleviates these limitations. While current genomic techniques mostly remain incompatible with live-cell tracking, sampling cells in an asynchronously progressing population allows in principle to computationally devise models for cellular dynamics at any time scale that is represented in the cell population under study79. Single cell genomics thus suggests a universal, computationally-driven, approach for inferring dynamics in genome function and regulation.
A basic principle in the inference of cellular dynamics from single cell data is maximum parsimony. Based on this principle, out of all possible models of dynamics that connect the observed cellular states, the one involving minimal transcriptional changes is preferred. This principle is effective for inferring cellular dynamics that proceed directionally and irreversibly along a succession of focal points through coordinated changes in gene expression. In such cases, it should be possible to infer the ‘ordering’ of cells, and the positions of key branching or differentiation points. Moreover, since each sampled single cell is positioned along the functional process, the ‘residence time’ in a phase of the process relates to the proportion of cells sampled from that phase79. If the number of cells is substantial, even very transient (and hence rare) states can be positioned confidently within an inferred trajectory. Thus, biological asynchrony becomes an asset, and for short-term processes that recur continuously (e.g., hematopoiesis), the entire dynamical process can be effectively sampled in a single experiment with very deep sampling of single cells35,37,40,80–85 (reviewed in Ref. 66).
A series of studies35,37,40,80–85 have now used these principles to infer dynamics from single cell profiles, while assuming linear35,40,48,86, cyclic45,87, or bifurcating37,80–84 trajectories. For example, one early method, Wanderlast, used single cell multiplex protein measurements from mass cytometry (CyTOF) data to build a trajectory of B cell differentiation35. Related approaches successfully ordered cells from scRNA-seq data sampled during multiple time points along adipocyte differentiation in vitro40 or neurogenesis in vivo48,86. For the cell cycle, while bulk profiles of synchronized cell populations have proven challenging to compare across systems77,78, a single cell ‘cycle’ can be readily reconstructed, and appears robustly conserved in cell lines and tissues, in both human and mouse45,54,58,88,89. The most recent methods have also reconstructed bifurcations with some encouraging progress37,80–84. Cellular dynamics was also inferred from epigenomic data; this is particularly important when major cell-cell variation is present, as in studies of chromosomal conformation in cycling cells90 (Peter Fraser and A. Tanay, personal communication).
However, de-novo inference of chains of events or branching structure of a dynamical process is computationally challenging even for deeply sampled processes, and it may quickly become under-determined by the data, especially if little or no prior knowledge exists to determine anchor points. In particular, the principle of maximum parsimony may not always be appropriate. Analogous to difficulties in phylogenetic reconstruction under a model considering lateral-transfer of genes91, the possibility of trans-differentiation, plasticity, or incomplete cell lineage sorting calls for the integration of new computational and experimental approaches with the current parsimony-based reconstruction algorithms. Moreover, any given cell undergoes multiple dynamic processes simultaneously (responds to a nutrient, undergoes cell division, is at a point in its differentiation), and methods are needed to delineate these processes and distinguish convergent programs from commitment and differentiation. It will be important to peg the inferred dynamical models to other, independent, measures of the process, such as morphological92 or genetic features. For example, in B cells, trajectory analysis identified a very early putative population of comprising only 0.007% of all analyzed cells. The temporal position of this new population could be confirmed by assaying the status of the IgH locus. Indeed, in T and B lymphocytes, TcR and BcR/Ig sequences provide a unique tool, because their genetic status can be determined from scRNA-Seq data directly53,54,93.
Inference of cellular dynamics will be greatly enhanced by advances in assays that measure both DNA and RNA in the same cell. Cell lineage maps – which relate sampled extant cells through the cell division events and progenitor cells that gave rise to them – can be inferred from genetic information, either through engineered tools in model organisms94, or through the natural accrual of mutations at each DNA replication event95,96. Coupled with the functional identity of a cell, for example through scRNA-Seq, it should be possible to further derive cell fate maps, determining which earlier cell types give rise to later ones, a cornerstone of developmental biology97.
Understanding temporal dynamics of single cells is important for shedding light on human disease, especially cancer, where it will often require to infer cellular histories rather than measure them directly. Tumorigenesis is highly dynamic and involves both genetic and epigenetic changes in the context of a heterogeneous environment98. However, only one or a few snapshots are obtained of a patient’s tumor. Simultaneous measurements of both the genetic and functional state of each malignant cell in a tumor biopsy, as well as of the cells in the microenvironment, would provide an extraordinary opportunity to track a tumor’s evolution, understand metastasis and monitor and predict response to therapy. Emerging studies have shown the promise of this and related approaches in melanoma54, glioblastoma38,99, breast cancer23, leukemia101, and oligodendroglioma100.
The spatial axis: measurement and inference
Although physiological processes take place in tissues, and spatial organization is critical to tissue function, most single cell genomics approaches currently dissociate specimens and cannot maintain a registry of the cells’ 3D organization. While a general solution is not yet fully realized, this is an area of intense study, with several emerging solutions56,102–110. One class of approaches48,56,102–104 relegates the spatial resolution problem to the computational model (Fig. 2). The cell’s transcriptome carries the imprint of its location, and by combining single cell profiles with a reference map of a small number of marker genes, several studies have mapped back single cell profiles to their spatial position48,56,102–104. For example, a study in early fish embryos56, used a reference map of legacy in situ expression data for a few dozen markers (each measured independently, and not at single cell resolution) together with single cell RNA-Seq from the same developmental stage, to map cells into 100-cell spatial “bins”. Because the expression patterns of cells in the embryo is a super-position of spatial gradients and early cell type specification, this also allowed distinguishing cell type signatures that are independent of the spatial position per se. A similar computational strategy103 mapped punctate spatial patterns in a worm brain. Other studies have shown similar imprint of both discrete regions and morphogen gradients in single cell profiles from early mouse embryos104 to the hippocampus48.
Figure 2. The spatial axis.
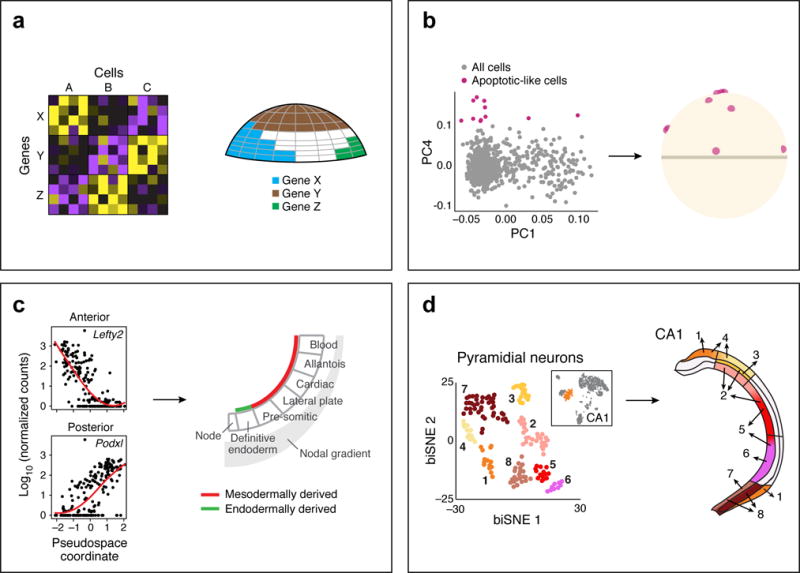
(a) Spatial mapping takes as input single cell profiles (left) and a reference map of the spatial expression patterns of a small number of landmark genes. The expression of the landmark genes in the cells is used to determine the spatial position of the entire cell. (b–d) Examples of successful spatial mapping in the early fish embryo (b) (adapted from 56), where a few cells, with a distinct apoptotic-like profile, were mapped to a salt-and-pepper pattern; in the early mesoderm, where single cell expression can define an anterior-posterior pseudospace (adapted from 104); and in the hippocampus (d) where pyramidal neuron cell clusters from the CA1 region map along lateral-medial and anterior-posterior axis (adapted from 48).
These examples suggest that even experimental designs where cruder information (e.g., from microdissection) is combined with single cell profiles can help provide rich spatial genomics. However, the compatibility of single cell genomics methods with such dissection must be improved to minimize batch bias and improve consistency across sections. More fundamentally, computational spatial reconstruction as currently implemented relies on the idea of canonical tissue organization in embryogenesis or organogenesis, such that multiple experiments can faithfully reproduce the same structure. We hypothesize that even in less constrained developmental, differentiation or pathological scenarios reproducible spatial structure can be used for computational analysis. For example, histopathology suggests that there are higher order features, at different length scales, which are preserved across samples, and indeed are the basis of clinical pathology. In order to devise universal strategies for spatial analysis in single cell genomics, new and flexible computational approaches that can work at multiple scales (e.g., identifying spatial motifs) would have to be integrated with improved experimental techniques that directly interrogate spatial structures.
To meet these challenges, techniques for genome-scale analysis of single cells in tissue sections in situ are evolving rapidly. Multiplexing RNA fluorescence in situ hybridization (RNA-FISH), for example using MERFISH105, reliably measures the expression and spatial position of thousands of different transcripts in multiplex in situ. Other techniques have produced encouraging proof-of-concept data of in situ RNA sequencing in preserved tissue sections and in cells106,109. It is also possible to determine the spatial expression of dozens of proteins in multiplex, by coupling either laser or ion beam ablation of the tissue with mass cytometry measurements of each ablated ‘pixel’107,108. Recent studies (Bernd Bodenmiller, personal communications) have used such imaging mass cytometry107 to analyze breast cancer tissues, and group cells into types not by their intrinsic profiles, but by the neighborhood that they inhabit. When using these types of methods, the notion of cell type may be generalized to include spatial context, or even be redefined completely as a spatial feature on top of which gene regulatory mechanisms and programs must be superimposed.
One major implication of a tissue’s spatial structure is the relative localization of cells of specific types and the molecular composition of physical contacts between them. Direct cellular contacts are particularly important when modeling intra-cellular regulatory mechanisms. While a complete 3D map of a tissue can theoretically also characterize all cell-cell junctions, approaches are being developed to directly measure these, for example by assessing small, tightly coupled bone marrow microniches of two cells (Alexander van Oudenaarden, personal communication). When restricting the spatial organization problem to a question on the distribution of contacts between cell types, it may be possible to devise computational strategies analyzing correlations between existence and fractions of specific cell types across many samples. This can be used to generate hypotheses on the mutual dependency and cellular interactions within complex niches, as has been recently shown in tumors54.
The mechanism axis: modeling gene regulation
Moving beyond phenomenology, new approaches are needed for modeling the regulatory mechanisms underlying the observed repertoire of cellular behaviors. Despite intensive research, systematic dissection of the regulatory mechanisms has remained a substantial challenge. Single cell genomics provides new opportunities to combine observational, mechanistic and perturbational approaches for inference and modeling of regulatory mechanisms within and between cells (Fig. 3). Given the natural cellular resolution of the data, studies in this emerging domain are initially focusing at the intra-cellular level, often restricted to transcriptional regulation. We first discuss some these approaches, and then turn to the challenges of embedding intra-cellular regulation within inter-cellular contexts in the outlook.
Figure 3. The mechanism axis.
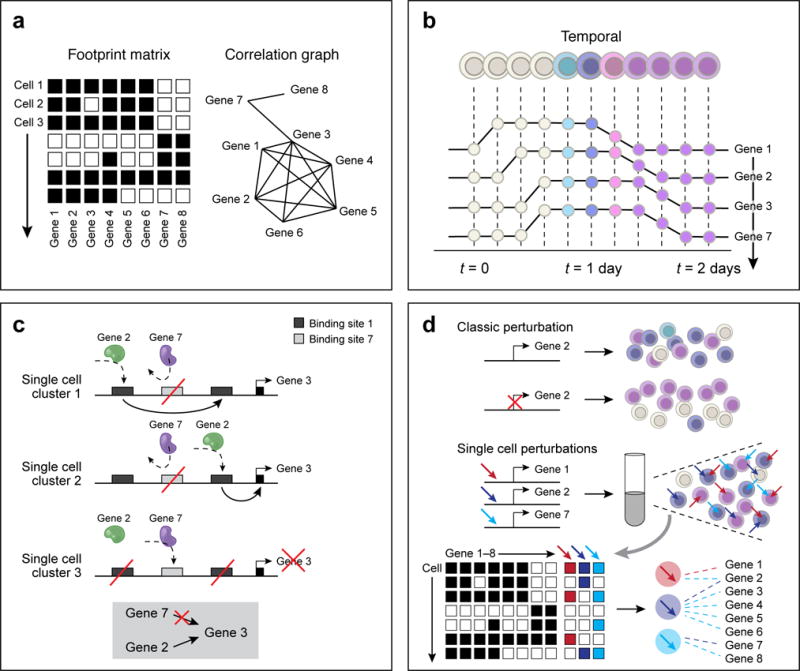
(a) Inference through co-variation across single cells. Using expression profiles for regulators and targets (left, columns) across cells (rows), a correlation graph (right) is constructed between genes and identify candidate regulators. With increasing number of cells, the correlation approach can help exclude putative regulatory relationships, if they are inconsistent with observing states. In the schematic example, gene 7 is unlikely to regulate genes 1,2,4,5, or 6, but may regulate genes 3 and 8. (b) Inference through temporally resolved single cell data. Putative regulatory interactions are identified as time lags between the activity profiles of regulators and their potential targets. In the schematic example, the data suggest that gene 7 is unlikely to regulate gene 3. (c) Refinement of regulatory models with epigenetic information. A schematic depiction of a regulatory region around gene 3, including three putative enhancer elements that are targeted by two putative regulators, encoded by genes 2 and 7. Pooled single cell epigenomics data identifies two states: (1) gene 3 is active and targeted by gene 2, and (2) gene 3 is inactive and targeted by gene 7, suggesting together, that gene 2, but not gene 7, is activating gene 3. (d) Causal inference of regulatory models by perturbations. Top: Perturbation experiments of specific genes, followed by single cell profiling help determine causal relationships. Bottom: Perturbations performed in a pool, combinatorially for multiple loci, followed by single cell monitoring of both the perturbation and its effect on transcription, with enhanced power for causal inference.
The observational approach for inferring gene regulatory mechanism was classically applied through inference of correlations between molecular profiles of mRNA levels and/or epigenetic marks across a diverse set of bulk samples (e.g., different tissue types, cell types, or stimuli). Such analyses were confounded by the inherently mixed state within profiled cellular ensembles, which could generate indirect correlations or hide other important regulatory interactions. In contrast, single cell analysis is highly powered for co-variation analysis at the intra-cellular level (Fig. 3a). The most basic approach takes a large number of single cell profiles that capture multiple transcriptional states and generates candidate regulatory interactions by computing gene-gene correlations. Such analyses thus identify candidate regulators controlling cell types by the correlation between their expression pattern and the profiles of genes defining the types’ transcriptional signatures. At higher resolution, single cell analysis refines the definition of types and sub-types, and allows sensitive identification of correlation or lack of correlation within small cellular niches, thus allowing us to progressively exclude more spurious gene-gene putative interaction. Such analyses helped predict regulators controlling cell types in immune cells16, epithelial cells44, and neurons48,52,111,112.
But even at the single cell level, correlation does not imply causation, and predictive modeling of gene regulation must rely on perturbations of the system and on integration of mechanistic constraints into the inferred models. First, there is inherent variation between cells even within a seemingly homogeneous population, due to extrinsic noise66,113 or asynchrony in response17, such that every individual cell can be viewed as its own perturbation system. In this case, observational strategies (e.g., correlation) can theoretically, and even practically, support causal inference. For example, an early study used co-variation analysis across 15 dendritic cells after stimulation with LPS17, to recover a module of co-expressed anti-viral genes and correctly associate Stat2 and Irf7 as its regulators. A similar approach detected Irf7 as a regulator of a common in vivo response to LPS in several dendritic cell subtypes, each of which implementing a distinct basal transcriptional program16. In other systems, even in steady state, cells in a population may naturally span a spectrum of transcriptional states rather than a few discrete modes, corresponding to some phenotypic variation. For example, cell cycle regulated gene modules can be detected45,89, or modeled separately from programs controlling cellular differentiation46. In another application, single cell profiling recovered a spectrum of transcriptional states corresponding to Th17 pathogenicity and auto-immunogenic potential and identified gene modules and putative regulators associated with this spectrum, which were experimentally validated in animal models43. The computational sophistication of the techniques used for inferring regulatory models from single cell data is bounded by the size of the data set and the reliability of the readout. Very large numbers of single cells were shown to support more quantitative modeling in Mass Cytometry data36, suggesting that a similar approach may soon become applicable for single cell genomics.
Temporal resolution can play a key role in inferring regulatory mechanisms from observational data. For dynamic processes occurring on a time scale longer than transcription itself, such as stable induction of cell-type specific transcription factors, it is possible to use datasets ordered along temporal trajectories and significantly enhance the power of causal inference by identifying time-lags between the activity profiles of early and late regulators associated with specific transitions. This approach has been applied to predict and validate a role for the IL7/Stat5 pathway in early B cell development in vivo35, for different transcriptional regulators in myoblast differentiation in vitro40, and for neurogenesis during embryonic development86 and in the adult48. However, many regulatory processes occur at much shorter time scale, or involve post transcriptional and translational mechanisms that are not observed at the transcriptional level, thereby restricting our ability to infer regulatory relationship from observations even in time-resolved single cell data sets.
The incorporation of epigenomics data, especially at the single cell level, into models of gene regulation can power up a causal inference framework, by adding considerations that are not represented in observations of transcriptional states. Population and reference epigenomics may help bound the census of potential regulatory interactions by linking TFs with binding sites, or enhancer elements with target genes, helping to exclude putative regulatory relationships that are incompatible with the epigenomic mechanistic constraints. Single cell epigenomics, independently or simultaneously with RNA profiles, can define the regulatory landscape in a genome with an even better resolution, linking epigenetic activity at particular regulatory elements with the activities at other elements, or with the RNA output of target genes. Such epigenetic activity can be identified by methods for assessing chromosome accessibility26,27 or DNA hypomethylation28,31. Inference of gene regulation from single cell epigenomics is still limited by the depth and breadth of the data, and computational approaches allowing pooling of a large number of epigenomic profiles to compute robust correlations25–27,31 must be further developed. Such approaches may eventually help us develop quantitative mechanistic models of transcription, and uncover the molecular basis of intrinstic and extrinsic factors that drive transcriptional variability and noise.
Ultimately, combining single cell genomics with experimental perturbation of the systems under study provides the most direct avenue for causal inference. Analysis of classical knockout models ex vivo17,39 or in vivo46, allowed for example to validate and refine the regulatory interactions between factors, such as Cebpa and Cebpe during myeloid cell differentiation46. Moreover, modern high throughput perturbation methods, especially those based on CRISPR technology, can be combined with single cell genomics to perform causal analysis at unprecedented scale and resolution. This can be achieved by coupling existing CRISPR screens with a single cell RNA-seq readout, providing intimate dissection of the molecular response to perturbations and going beyond pre-defined, lower-content phenotypes. Such combination require massive throughput from scRNA-seq technology, and reading the (multiplexed) perturbation with the profiling method. Finally, perturbation screens can be designed specifically to perturb regulatory systems and test hypotheses derived by mechanistic models of single cell gene regulation.
Outlook
Efforts for mapping and classification of cellular programs in human and in model organisms are becoming increasingly ambitious, aiming at a comprehensive atlas of cell types and subtypes in organs and organisms. This opens remarkable opportunities to go beyond descriptive studies of cell type and state and to develop mechanistic-predictive models of regulatory programs. Reference maps are an essential starting point for inference and testing of predictive models. Mechanistic models, in turn, make it possible to dissect, annotate and contextualize large reference maps. Thus, a comprehensive atlas of normal cellular states in human or mouse will open the way for, but will not substitute, the need for predictive and mechanistic models. This is because each disease, and each individual’s genetic variants, span a new system and possible new variation on the reference state, which must be characterized. In the context of an extensive reference atlas, however, and given that some features appear quite robust to inter-individual variation, the focus of gene regulatory modeling need not be complete de novo inference of states from e.g., sequence or epigenomes, but on the ability to infer the effect of small perturbations, given the availability of measured states with very similar characteristics. Thus, modeling how known cellular states are perturbed may shed critical light on diseases mechanisms, while providing a tractable path to predictive model of cellular function, despite the incredible computational complexity of the cellular process.
By way of metaphor, the existence of a “periodic-table” of cell states may be sufficient for understanding the “physics” of individual cells, but not the “chemistry” by which they combine in tissues. Thus, as single cell genomics is revolutionizing our ability to map cell states and infer mechanisms for intra-cellular gene regulation (the “periodic elements”), another grand challenge remains to integrate such individual cellular states into models of functioning tissues (the “chemistry”). Cells are the building blocks of tissues, and the emerging techniques we discuss above may allow their mapping within 3D space, or the tracking of their immediate cellular neighborhoods and lineage trees. Methods to measure and model key intercellular molecular markers, such as metabolites, signaling molecules and components of the extracellular matrix will be critical in order to assemble single cells into cohesive models of tissues. As with cell state models, descriptive tissue models are only a starting point towards understanding and predicting the higher order organization and function of tissue. While the complexity of the processes and the molecular decisions made by communities of interacting cells may appear daunting, it is likely that there exists an intermediate molecular-anatomical level of “tissue modules” with distinct functionalities1. We thus envision a framework that combines information on spatial proximity, molecular communication between cells, and the functional impact of those interactions on cell states to identify and study recurrent multi-cell modules in tissues. Such modules may consist, as previously proposed1, of cells with complementary functions, including the core specialized cell of the tissue (e.g., adipocyte cells in the fat; myocytes in muscle; epithelial cells in the gut mucosa, etc.) along with accessory cells providing key support functions1. If and when such modules can be characterized and studied experimentally, single cell genomics may lead toward a real revolution in our fundamental understanding of biology.
Acknowledgments
We thank L. Gaffney for help with artwork. AR is an HHMI Investigator. AT is a Kimmel investigator and is supported by FAMRI and the European Research Council. AR is a member of the SAB of ThermoFisher and Syros Pharmaceuticals and a consultant to Driver Group.
References
- 1.Okabe Y, Medzhitov R. Tissue biology perspective on macrophages. Nature immunology. 2016;17:9–17. doi: 10.1038/ni.3320. [DOI] [PubMed] [Google Scholar]
- 2.Mayr E. The Growth of Biological Thought: Diversity, Evolution, and Inheritance. Belknap Press; 1982. [Google Scholar]
- 3.Szathmary E, Maynard-Smith J. The Major Transitions in Evolution. Oxford; Univeristy Press: 1995. [Google Scholar]
- 4.Gould SJ. Ontogeny and Phylogeny. Belknap Press; 1977. [Google Scholar]
- 5.Richardson L, et al. EMAGE mouse embryo spatial gene expression database: 2014 update. Nucleic acids research. 2014;42:D835–844. doi: 10.1093/nar/gkt1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oh SW, et al. A mesoscale connectome of the mouse brain. Nature. 2014;508:207–214. doi: 10.1038/nature13186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller JA, et al. Transcriptional landscape of the prenatal human brain. Nature. 2014;508:199–206. doi: 10.1038/nature13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hawrylycz MJ, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489:391–399. doi: 10.1038/nature11405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bakken TE, et al. A comprehensive transcriptional map of primate brain development. Nature. 2016;535:367–375. doi: 10.1038/nature18637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jojic V, et al. Identification of transcriptional regulators in the mouse immune system. Nature immunology. 2013;14:633–643. doi: 10.1038/ni.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanes JR, Masland RH. The types of retinal ganglion cells: current status and implications for neuronal classification. Annual review of neuroscience. 2015;38:221–246. doi: 10.1146/annurev-neuro-071714-034120. [DOI] [PubMed] [Google Scholar]
- 12.Chao MP, Seita J, Weissman IL. Establishment of a normal hematopoietic and leukemia stem cell hierarchy. Cold Spring Harbor symposia on quantitative biology. 2008;73:439–449. doi: 10.1101/sqb.2008.73.031. [DOI] [PubMed] [Google Scholar]
- 13.Ramskold D, et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nature biotechnology. 2012;30:777–782. doi: 10.1038/nbt.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Picelli S, et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nature methods. 2013;10:1096–1098. doi: 10.1038/nmeth.2639. [DOI] [PubMed] [Google Scholar]
- 15.Hashimshony T, Wagner F, Sher N, Yanai I. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell reports. 2012;2:666–673. doi: 10.1016/j.celrep.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 16.Jaitin DA, et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science. 2014;343:776–779. doi: 10.1126/science.1247651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shalek AK, et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013;498:236–240. doi: 10.1038/nature12172. [DOI] [PMC free article] [PubMed] [Google Scholar]; Refs 13–17 are early reports on development and scaling of single cell RNA-seq.
- 18.Zong C, Lu S, Chapman AR, Xie XS. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science. 2012;338:1622–1626. doi: 10.1126/science.1229164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu X, et al. Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor. Cell. 2012;148:886–895. doi: 10.1016/j.cell.2012.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Navin N, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leung ML, Wang Y, Waters J, Navin NE. SNES: single nucleus exome sequencing. Genome biology. 2015;16:55. doi: 10.1186/s13059-015-0616-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hou Y, et al. Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell. 2012;148:873–885. doi: 10.1016/j.cell.2012.02.028. [DOI] [PubMed] [Google Scholar]
- 23.Gao R, et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nature genetics. 2016 doi: 10.1038/ng.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]; Refs 18–13 introduce and develop single cell genome sequencing, with applications for cancer evolution
- 24.Rotem A, et al. High-Throughput Single-Cell Labeling (Hi-SCL) for RNA-Seq Using Drop-Based Microfluidics. PloS one. 2015;10:e0116328. doi: 10.1371/journal.pone.0116328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rotem A, et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nature biotechnology. 2015;33:1165–1172. doi: 10.1038/nbt.3383. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper develops single cell ChIP-Seq and the idea of single cell pooling for analysis of sparse epigenomic data
- 26.Buenrostro JD, et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523:486–490. doi: 10.1038/nature14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cusanovich DA, et al. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science. 2015;348:910–914. doi: 10.1126/science.aab1601. [DOI] [PMC free article] [PubMed] [Google Scholar]; Refs 26–27 develop two variants of single cell ATAC-seq and discuss applications for identifying subtypes and pool loci.
- 28.Smallwood SA, et al. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nature methods. 2014;11:817–820. doi: 10.1038/nmeth.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mooijman D, Dey SS, Boisset JC, Crosetto N, van Oudenaarden A. Single-cell 5hmC sequencing reveals chromosome-wide cell-to-cell variability and enables lineage reconstruction. Nature biotechnology. 2016;34:852–856. doi: 10.1038/nbt.3598. [DOI] [PubMed] [Google Scholar]; This paper is using restriction enzymes to profile hydroxymethylation and perform very-short term lineage reconstruction.
- 30.Guo H, et al. Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome research. 2013;23:2126–2135. doi: 10.1101/gr.161679.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Farlik M, et al. Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics. Cell reports. 2015;10:1386–1397. doi: 10.1016/j.celrep.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; Refs 28,30–31 develop variants for single cell DNA methylation profiling, and are dealing with sparse single cell epigenome analysis
- 32.Kind J, et al. Genome-wide maps of nuclear lamina interactions in single human cells. Cell. 2015;163:134–147. doi: 10.1016/j.cell.2015.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagano T, et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 2013;502:59–64. doi: 10.1038/nature12593. [DOI] [PMC free article] [PubMed] [Google Scholar]; A proof of concept report on single cell Hi-C, demonstrating variability of T cell chromosomal architectures.
- 34.Bendall SC, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–696. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bendall SC, et al. Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell. 2014;157:714–725. doi: 10.1016/j.cell.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krishnaswamy S, et al. Conditional density-based analysis of T cell signaling in single-cell data. Science. 2014;346:1250689. doi: 10.1126/science.1250689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marco E, et al. Bifurcation analysis of single-cell gene expression data reveals epigenetic landscape. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E5643–5650. doi: 10.1073/pnas.1408993111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patel AP, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shalek AK, et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature. 2014;510:363–369. doi: 10.1038/nature13437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trapnell C, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nature biotechnology. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arvey A, et al. Genetic and epigenetic variation in the lineage specification of regulatory T cells. eLife. 2015;4:e07571. doi: 10.7554/eLife.07571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Avraham R, et al. Pathogen Cell-to-Cell Variability Drives Heterogeneity in Host Immune Responses. Cell. 2015;162:1309–1321. doi: 10.1016/j.cell.2015.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gaublomme JT, et al. Single-Cell Genomics Unveils Critical Regulators of Th17 Cell Pathogenicity. Cell. 2015;163:1400–1412. doi: 10.1016/j.cell.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grun D, et al. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature. 2015;525:251–255. doi: 10.1038/nature14966. [DOI] [PubMed] [Google Scholar]
- 45.Kowalczyk MS, et al. Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome research. 2015;25:1860–1872. doi: 10.1101/gr.192237.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paul F, et al. Transcriptional Heterogeneity and Lineage Commitment in Myeloid Progenitors. Cell. 2015;163:1663–1677. doi: 10.1016/j.cell.2015.11.013. [DOI] [PubMed] [Google Scholar]
- 47.Corces MR, et al. Lineage-specific and single cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nature genetics. 2016 doi: 10.1038/ng.3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Habib N, et al. Div-Seq: Single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science. 2016 doi: 10.1126/science.aad7038. [DOI] [PMC free article] [PubMed] [Google Scholar]; Adapating RNA-seq to single cell nuclei, with applications to rare cells, or other challenging samples.
- 49.Matcovitch-Natan O, et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science. 2016 doi: 10.1126/science.aad8670. [DOI] [PubMed] [Google Scholar]
- 50.Olsson A, et al. Single-cell analysis of mixed-lineage states leading to a binary cell fate choice. Nature. 2016 doi: 10.1038/nature19348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Proserpio V, et al. Single-cell analysis of CD4+ T-cell differentiation reveals three major cell states and progressive acceleration of proliferation. Genome biology. 2016;17:103. doi: 10.1186/s13059-016-0957-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shekhar K, et al. Comprehensive classification of retinal bipolar neurons by single-cell transcriptomics. Cell. 2016 doi: 10.1016/j.cell.2016.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stubbington MJ, et al. T cell fate and clonality inference from single-cell transcriptomes. Nature methods. 2016;13:329–332. doi: 10.1038/nmeth.3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tirosh I, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kumar RM, et al. Deconstructing transcriptional heterogeneity in pluripotent stem cells. Nature. 2014;516:56–61. doi: 10.1038/nature13920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. Nature biotechnology. 2015;33:495–502. doi: 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Treutlein B, et al. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature. 2014;509:371–375. doi: 10.1038/nature13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Macosko EZ, et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Klein AM, et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 2015;161:1187–1201. doi: 10.1016/j.cell.2015.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]; Refs 26–27 enhance throughput in single cell RNA-seq using droplet technology.
- 60.Hashimshony T, et al. CEL-Seq2: sensitive highly-multiplexed single-cell RNA-Seq. Genome biology. 2016;17:77. doi: 10.1186/s13059-016-0938-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nichterwitz S, et al. Laser capture microscopy coupled with Smart-seq2 for precise spatial transcriptomic profiling. Nat Commun. 2016;7:12139. doi: 10.1038/ncomms12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thomsen ER, et al. Fixed single-cell transcriptomic characterization of human radial glial diversity. Nature methods. 2016;13:87–93. doi: 10.1038/nmeth.3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lake BB, et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science. 2016;352:1586–1590. doi: 10.1126/science.aaf1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Milo R, Jorgensen P, Moran U, Weber G, Springer M. BioNumbers–the database of key numbers in molecular and cell biology. Nucleic acids research. 2010;38:D750–753. doi: 10.1093/nar/gkp889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Han F, Lillard SJ. In-situ sampling and separation of RNA from individual mammalian cells. Analytical chemistry. 2000;72:4073–4079. doi: 10.1021/ac000428g. [DOI] [PubMed] [Google Scholar]
- 66.Wagner A, Regev A, Yosef N. Uncovering the vectors of cellular identity with single-cell genomics. Nature biotechnology. 2016 doi: 10.1038/nbt.3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Islam S, et al. Quantitative single-cell RNA-seq with unique molecular identifiers. Nature methods. 2014;11:163–166. doi: 10.1038/nmeth.2772. [DOI] [PubMed] [Google Scholar]
- 68.Heimberg G, Bhatnagar R, El-Samad H, Thomson M. Low Dimensionality in Gene Expression Data Enables the Accurate Extraction of Transcriptional Programs from Shallow Sequencing. Cell Syst. 2016;2:239–250. doi: 10.1016/j.cels.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lavin Y, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159:1312–1326. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Macaulay IC, et al. G&T-seq: parallel sequencing of single-cell genomes and transcriptomes. Nature methods. 2015;12:519–522. doi: 10.1038/nmeth.3370. [DOI] [PubMed] [Google Scholar]
- 71.Dey SS, Kester L, Spanjaard B, Bienko M, van Oudenaarden A. Integrated genome and transcriptome sequencing of the same cell. Nature biotechnology. 2015;33:285–289. doi: 10.1038/nbt.3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Angermueller C, et al. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nature methods. 2016;13:229–232. doi: 10.1038/nmeth.3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Frei AP, et al. Highly multiplexed simultaneous detection of RNAs and proteins in single cells. Nature methods. 2016;13:269–275. doi: 10.1038/nmeth.3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Darmanis S, et al. Simultaneous Multiplexed Measurement of RNA and Proteins in Single Cells. Cell reports. 2016;14:380–389. doi: 10.1016/j.celrep.2015.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Albayrak C, et al. Digital Quantification of Proteins and mRNA in Single Mammalian Cells. Molecular cell. 2016;61:914–924. doi: 10.1016/j.molcel.2016.02.030. [DOI] [PubMed] [Google Scholar]
- 76.Genshaft AS, et al. Multiplexed, targeted profiling of single-cell proteomes and transcriptomes in a single reaction. Genome biology. 2016 doi: 10.1186/s13059-016-1045-6. [DOI] [PMC free article] [PubMed] [Google Scholar]; Refs 26–27 introduce techniques for similtaneous profiling of RNA and DNA, RNA and DNA methylation, or RNA and proteins.
- 77.Lu Y, et al. Combined analysis reveals a core set of cycling genes. Genome biology. 2007;8:R146. doi: 10.1186/gb-2007-8-7-r146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bar-Joseph Z, et al. Genome-wide transcriptional analysis of the human cell cycle identifies genes differentially regulated in normal and cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:955–960. doi: 10.1073/pnas.0704723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kafri R, et al. Dynamics extracted from fixed cells reveal feedback linking cell growth to cell cycle. Nature. 2013;494:480–483. doi: 10.1038/nature11897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Setty M, et al. Wishbone identifies bifurcating developmental trajectories from single-cell data. Nature biotechnology. 2016;34:637–645. doi: 10.1038/nbt.3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Moignard V, et al. Decoding the regulatory network of early blood development from single-cell gene expression measurements. Nature biotechnology. 2015;33:269–276. doi: 10.1038/nbt.3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Haghverdi L, Buttner M, Wolf FA, Buettner F, Theis FJ. Diffusion pseudotime robustly reconstructs lineage branching. Nature methods. 2016 doi: 10.1038/nmeth.3971. [DOI] [PubMed] [Google Scholar]
- 83.Haghverdi L, Buettner F, Theis FJ. Diffusion maps for high-dimensional single-cell analysis of differentiation data. Bioinformatics. 2015;31:2989–2998. doi: 10.1093/bioinformatics/btv325. [DOI] [PubMed] [Google Scholar]
- 84.Chen J, Schlitzer A, Chakarov S, Ginhoux F, Poidinger M. Mpath maps multi-branching single-cell trajectories revealing progenitor cell progression during development. Nat Commun. 2016;7:11988. doi: 10.1038/ncomms11988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Angerer P, et al. destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics. 2016;32:1241–1243. doi: 10.1093/bioinformatics/btv715. [DOI] [PubMed] [Google Scholar]
- 86.Shin J, et al. Single-Cell RNA-Seq with Waterfall Reveals Molecular Cascades underlying Adult Neurogenesis. Cell Stem Cell. 2015;17:360–372. doi: 10.1016/j.stem.2015.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gut G, Tadmor MD, Pe’er D, Pelkmans L, Liberali P. Trajectories of cell-cycle progression from fixed cell populations. Nature methods. 2015;12:951–954. doi: 10.1038/nmeth.3545. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ref 80–87 introduce techniques for inferring dynamics from steady state sampling of single cell profiles.
- 88.Proserpio V, et al. Single-cell analysis of CD4+ T-cell differentiation reveals three major cell states and progressive acceleration of proliferation. Genome biology. 2016;17:103. doi: 10.1186/s13059-016-0957-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Buettner F, et al. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nature biotechnology. 2015;33:155–160. doi: 10.1038/nbt.3102. [DOI] [PubMed] [Google Scholar]
- 90.Naumova N, et al. Organization of the mitotic chromosome. Science. 2013;342:948–953. doi: 10.1126/science.1236083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Koonin EV. Horizontal gene transfer: essentiality and evolvability in prokaryotes, and roles in evolutionary transitions. F1000Research. 2016;5 doi: 10.12688/f1000research.8737.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kellogg RA, Tay S. Noise facilitates transcriptional control under dynamic inputs. Cell. 2015;160:381–392. doi: 10.1016/j.cell.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 93.Afik S, et al. Targeted reconstruction of T cell receptor sequence from single cell RNA-sequencing links CDR3 length to T cell differentiation state. bioRxiv. 2016 doi: 10.1093/nar/gkx615. doi: http://dx.doi.org/10.1101/072744. [DOI] [PMC free article] [PubMed]
- 94.McKenna A, et al. Whole organism lineage tracing by combinatorial and cumulative genome editing. Science. 2016 doi: 10.1126/science.aaf7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Reizel Y, et al. Cell lineage analysis of the mammalian female germline. PLoS genetics. 2012;8:e1002477. doi: 10.1371/journal.pgen.1002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shlush LI, et al. Cell lineage analysis of acute leukemia relapse uncovers the role of replication-rate heterogeneity and microsatellite instability. Blood. 2012;120:603–612. doi: 10.1182/blood-2011-10-388629. [DOI] [PubMed] [Google Scholar]
- 97.Sulston JE, Horvitz HR. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Developmental biology. 1977;56:110–156. doi: 10.1016/0012-1606(77)90158-0. [DOI] [PubMed] [Google Scholar]
- 98.Alizadeh AA, et al. Toward understanding and exploiting tumor heterogeneity. Nat Med. 2015;21:846–853. doi: 10.1038/nm.3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang J, et al. Clonal evolution of glioblastoma under therapy. Nature genetics. 2016;48:768–776. doi: 10.1038/ng.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tirosh I, et al. Single-cell RNA-seq supports a developmental hierarchy in IDH-mutant oligodendroglioma. Nature. 2016 doi: 10.1038/nature20123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li S, et al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat Med. 2016;22:792–799. doi: 10.1038/nm.4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Durruthy-Durruthy R, et al. Reconstruction of the mouse otocyst and early neuroblast lineage at single-cell resolution. Cell. 2014;157:964–978. doi: 10.1016/j.cell.2014.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Achim K, et al. High-throughput spatial mapping of single-cell RNA-seq data to tissue of origin. Nature biotechnology. 2015;33:503–509. doi: 10.1038/nbt.3209. [DOI] [PubMed] [Google Scholar]
- 104.Scialdone A, et al. Resolving early mesoderm diversification through single-cell expression profiling. Nature. 2016;535:289–293. doi: 10.1038/nature18633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen KH, Boettiger AN, Moffitt JR, Wang S, Zhuang X. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science. 2015;348:aaa6090. doi: 10.1126/science.aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lee JH, et al. Highly multiplexed subcellular RNA sequencing in situ. Science. 2014;343:1360–1363. doi: 10.1126/science.1250212. [DOI] [PMC free article] [PubMed] [Google Scholar]; Refs 105, 106 develop multiplexed RNA-FISH or in situ sequencing to allow spatial mapping of a large number of diffreent transcripts at single cell resolution.
- 107.Giesen C, et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nature methods. 2014;11:417–422. doi: 10.1038/nmeth.2869. [DOI] [PubMed] [Google Scholar]
- 108.Angelo M, et al. Multiplexed ion beam imaging of human breast tumors. Nat Med. 2014;20:436–442. doi: 10.1038/nm.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ke R, et al. In situ sequencing for RNA analysis in preserved tissue and cells. Nature methods. 2013;10:857–860. doi: 10.1038/nmeth.2563. [DOI] [PubMed] [Google Scholar]
- 110.Stahl PL, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016;353:78–82. doi: 10.1126/science.aaf2403. [DOI] [PubMed] [Google Scholar]
- 111.Zeisel A, et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science. 2015;347:1138–1142. doi: 10.1126/science.aaa1934. [DOI] [PubMed] [Google Scholar]
- 112.Tasic B, et al. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nature neuroscience. 2016;19:335–346. doi: 10.1038/nn.4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Stewart-Ornstein J, Weissman JS, El-Samad H. Cellular noise regulons underlie fluctuations in Saccharomyces cerevisiae. Molecular cell 45. 2012:483–493. doi: 10.1016/j.molcel.2011.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]