

Abstract
Mcl-1, a Bcl-2 family member, is highly expressed in a variety of human cancers and is believed to enhance tumorigenic potential and chemotherapy resistance through the inhibition of apoptosis and senescence. We previously reported that Mcl-1′s regulation of chemotherapy-induced senescence (CIS) is dependent on its ability to prevent reactive oxygen species (ROS) generation. In this report, we demonstrate that Mcl-1-regulated CIS requires not only ROS, but specifically mitochondrial ROS, and that these events are upstream of activation of the DNA damage response, another necessary step toward senescence. Mcl-1′s anti-senescence activity also involves the unique ability to inhibit ROS formation by preventing the upregulation of pro-oxidants. Specifically, we found that NADPH oxidases (NOXs) are regulated by Mcl-1 and that NOX4 expression in particular is a required step for CIS induction that is blocked by Mcl-1. Lastly, we illustrate that by preventing expression of NOX4, Mcl-1 limits its availability in the mitochondria, thereby lowering the production of mitochondrial ROS during CIS. Our studies not only define the essential role of Mcl-1 in chemoresistance, but also for the first time link a key pro-survival Bcl-2 family member with the NOX protein family, both of which have significant ramifications in cancer progression.
Keywords: senescence, cancer therapy, Mcl-1, reactive oxygen species, NOX4
INTRODUCTION
Bcl-2 family members are among the most important pro-oncogenic proteins in all forms of cancer [1, 2]. While the anti-apoptotic family members were thought to be somewhat interchangeable, relying heavily on a common binding cleft that sequesters and inhibits pro-apoptotic Bcl-2 family members and other pro-apoptotic proteins, studies investigating drugs targeting this binding cleft found important variations amongst these proteins [3, 4]. Mcl-1 in particular has proven difficult to target, and identifying an effective small molecule inhibitor of this cleft (or alternative methods of reducing Mcl-1 expression) is the subject of ongoing research [5–7]. Most studies suggest unique physical characteristics of the Mcl-1 anti-apoptotic binding cleft account for this difficulty in targeting [8].
We recently showed that Mcl-1 contains an additional unique domain that is distinct among Bcl-2 family members and critical for its known ability to inhibit chemotherapy-induced senescence (but not apoptosis) [9, 10]. This observation may explain why current targeted therapies do not completely inhibit Mcl-1 activity, as they do not account for this domain. We have further demonstrated that in cells lacking p53 (which are normally senescence-resistant), downregulation of Mcl-1 recapitulates senescence mechanisms downstream of p53, including upregulation of p21 and induction of reactive oxygen species (ROS). However, much of Mcl-1′s non-apoptotic, anti-senescence activities remain poorly understood.
In the current study, we demonstrate that targeting Mcl-1 during senescence-inducing doxorubicin treatment in otherwise senescence-resistant cells causes activation of the ROS-dependent DNA damage response (DDR), which is critical for the induction of senescence in cancer [11]. Consistent with our previous discoveries, we find that Mcl-1′s unique senescence-inhibiting domain is responsible for abrogating ROS production [10]. We further show that mitochondrial ROS is necessary for the induction of senescence. Although other Bcl-2 family members can regulate ROS, we found that Mcl-1 has a unique ability to prevent ROS not by upregulating anti-oxidants, as is the case for Bcl-2, but by preventing the upregulation of pro-oxidants [12]. We observe that Mcl-1 prevents the expression of NADPH oxidases (NOXs). Mcl-1′s inhibition of NOX4 specifically, limits its availability to translocate to the mitochondria and induce ROS. To our knowledge, this is the first documentation of any Bcl-2 family member regulating this class of proteins. As molecules like NOX4 and ROS themselves have critical functions in neoplasia, the ramifications of juxtaposing these pro-tumor factors will have great impact in better understanding how cancer evades current therapeutic regimens [13].
RESULTS
Mcl-1 inhibits DNA damage response (DDR) components to prevent CIS
To better understand how Mcl-1 regulates chemotherapy-induced senescence, we started by examining a well-known component of this process, the activation of the DNA damage response (DDR), [14] Previous studies have demonstrated that Mcl-1 regulates and prevents DNA damage directly at the site of DNA breaks during apoptosis, as evidenced by co-immuno-precipitation of Mcl-1 with molecules such as γ-H2AX [15]. However, in our model of CIS, Mcl-1 prevents accumulation of these DNA damage factors, which would preclude direct binding as the only role for Mcl-1 [9, 10]. Figure 1A reveals that under CIS conditions induced by low-dose doxorubicin (DOX+) in CIS resistant, HCT116 p53−/− (shcontrol) cells, there is little expression of activated DDR components (phospho-ATM, ATR CHK1/2). However, in Mcl-1 knock-down cells (shMcl-1, CIS-sensitive), doxorubicin treatment causes significant up regulation of all four DDR factors studied (Figure 1B). To test whether these DDR factors are important in the initiation of CIS due to the lack of Mcl-1, we treated the same paired cell lines (CIS sensitive and resistant) with 2 inhibitors of the DDR: caffeine, which inhibits both ATR and ATM; and the ATM-specific inhibitor KU-55933. Figure 1A and 1B reveals that both inhibitors were effective at preventing activation of all DDR components studied. Although ATM and ATR can regulate separate DDR pathways, our findings are consistent with recent reports that activated ATM can initiate ATR. [16] Using multiple assays for senescence we then found that in Mcl-1 knock-down cells (CIS-sensitive), both caffeine and KU-55933 were effective at preventing the induction of CIS (Figure 1C–1G).
Figure 1. DDR components are important in Mcl-1-regulated senescence.

HCT116 p53−/− cells stably transfected with either a shControl (CIS resistant, Mcl-1 proficient) or shMcl-1 (CIS sensitive, Mcl-1 deficient), were treated with 100 ng/ml doxorubicin or left untreated in the presence or absence of caffeine (ATM/ATR inhibitor) or KU-55933 (ATM specific inhibitor). (A and B) Western blot detection of ATM-1981P, ATR-S428P Chk1-S345P, and Chk2-T68P under chemotherapy conditions. β-Actin (Actin) was used as a loading control. (C–G) Quantitative analysis of CIS in HCT116p53−/− shControl or shMcl-1 cells as assessed by β-gal activity (C), PML (D and E), and γ-H2AX nuclear body (F and G) formation in the presence or absence of the indicated ATM/ATR inhibitors. *P < 0.05, comparing those given DDR inhibitors + doxorubicin to doxorubicin alone. (H) The effects of DDR factor inhibitors on ROS generation. Cells were treated as in Figure A and B, and change in intracellular ROS production was determined using the Amplex Red reagent as described in the Material and Methods. Error bars represent ± S.D. Graphical data are inclusive of at least three independent experiments.
We have previously demonstrated that ROS production is a key step in the induction of senescence [9]. As such, we next sought to determine whether DDR activation occurs before or after ROS production during CIS by observing whether DDR-inhibitor treatment could affect ROS production in Mcl-1 knock-down cells. Figure 1H demonstrates that although DDR inhibitor treatment prevents CIS, ROS production is not affected. These data indicate that Mcl-1′s inhibition of ROS production, critical for CIS abrogation, is upstream of DDR activation.
A novel internal domain of Mcl-1 is required for its anti-ROS activity
Anti- apoptotic Bcl-2 family members function in part by binding to pro-apoptotic BH3-only molecules through a canonical binding cleft [17]. Using extensive mutagenesis, we recently identified four specific residues within an undefined loop domain of Mcl-1 that are important for anti-CIS function both in vitro and in vivo. For example, an alanine substitution at residue 198 greatly reduced Mcl-1′s anti-CIS activities, while a similar substitution at residue 201 or a deletion of Mcl-1′s three BH domains (Δ208–350) did not [10]. Here we examined the effects of these mutants on Mcl-1 mediated anti-ROS activity after chemotherapy. These constructs were transiently expressed in HCT116 p53−/− cells with stable knock-down of Mcl-1 (CIS-sensitive). The expression of the constructs was confirmed by western blotting (Figure 2A). In untreated cells, expression of the mutants (or empty vector) did not cause a significant change in baseline ROS production (Figure 2B). After treatment with doxorubicin, however, increased ROS production was detected only in the mutant containing the alanine substitution at residue 198 (P198A) (Figure 2C). This is in contrast to constructs containing the R201A substitution or C-terminal BH3 domain deletion (Δ208–350), which had ROS levels similar to those in cells expressing wild type Mcl-1. These results show Mcl-1′s ability to regulate ROS production under CIS conditions depends on residues in the loop domain (and not its canonical C-terminal anti-apoptotic domain) also known to regulate its anti-CIS function [10].
Figure 2. The Mcl-1 P198A residue required for inhibition of chemotherapy-induced ROS generation.

HCT116 p53−/− shMcl-1 cells were transiently transfected with vector control, wild type Mcl-1, or the following Mcl-1 mutants: P198A, R201A and Δ208–350 and were either left untreated or treated with doxorubicin. The ROS levels then were assessed with Amplex Red at the indicated time points. (A) Western blot of Mcl-1 protein levels after transfection of the indicated constructs. Cells expressing the indicated mutants were left untreated (B) or treated with doxorubicin (C). The data in (C) show that the expression of P198A residue but not the deletion of the BH3 region of Mcl-1 in HCT116 p53−/− shMcl-1 cells augmented chemotherapy-induced ROS production. The last time points (80 min) of ROS production measurements has been compered for the statistical differences between Mcl-1 mutants. Data are representative of three separate experimental cultures and transfections done in different days. Error bars represent ± S.D.
Mitochondrial ROS is significantly involved in induction of senescence
Having determined that ROS production is upstream of DDR activation during CIS, we next determined the source of ROS generation within the cells. In cancer, ROS can result from activation of oncogenes, aberrant metabolism, and mitochondrial dysfunction [18–22]. We evaluated the effects of various pharmacological inhibitors on the rate of ROS production under CIS conditions. The inhibitors we chose target enzymes that produce both non-mitochondrial ROS: allopurinol (xanthine oxidase inhibitor), NG-monomethyl-L-arginine (NMMA, a nitric oxide synthase inhibitor), metyrapone (a cytochrome P-450 inhibitor); and mitochondrial ROS: rotenone (a complex I inhibitor) and malonate (succinate dehydrogenase inhibitor). Figure 3A and 3C show that inhibition of both mitochondrial and non-mitochondrial ROS had no effect on the overall levels of ROS in CIS resistant cells after chemotherapy treatment. In CIS-sensitive cells, doxorubicin treatment results in dramatically higher levels of ROS, which are not affected by non-mitochondrial inhibitors (Figure 3B). Mitochondrial ROS inhibitors, however, are able to significantly lower ROS production in CIS-sensitive cells (Figure 3D). Because rotenone and malonate affect mitochondrial electron chain transport complexes (ETC) I and II respectively, and because ETC complex III is also known as a major source of mitochondrial ROS, we conducted a similar study using antimycin A and found that it's use had little effect on mitochondrial ROS production.(Supplementary Figure 1). This observation is consistent with previously published studies demonstrating that the Qi subunit of complex III affected by antimycin A inhibition is downstream of the Qo subunit, which is the a site of superoxide generation that results in hydrogen peroxide generation within the mitochondria, and suggests that rotenone and malonate act by preventing electron flow to complex III where it can be used to generate ROS [23]. These results clearly demonstrate that mitochondria are involved in chemotherapy-induced ROS production in CIS sensitive cells. We further examined whether mitochondrial ROS inhibitors affect downstream markers of senescence. As shown in Figure 3E, there is a significant decrease in γ-H2AX and PML nuclear body formation in response to mitochondria complex inhibitors, demonstrating that ROS generated by mitochondria is a necessary component of CIS development.
Figure 3. Mcl-1 acts to suppress mitochondrial ROS production thereby inhibiting CIS.
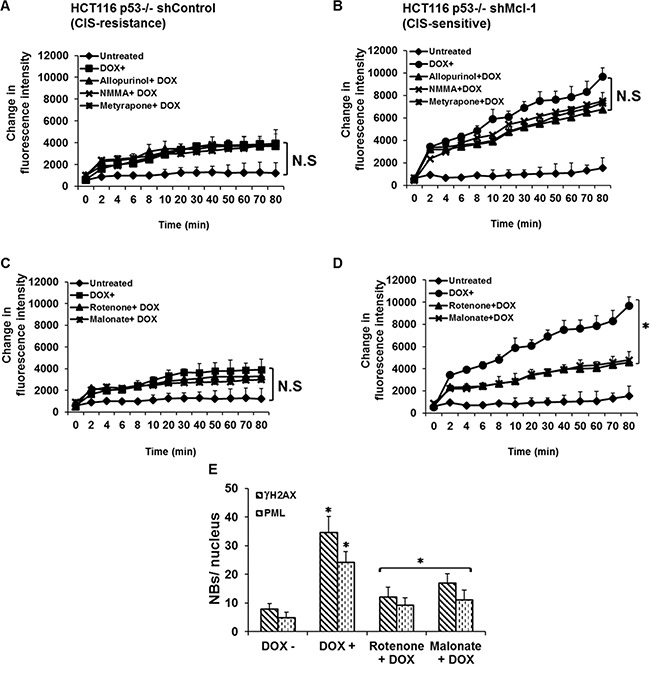
(A, B) Effects of cytoplasmic ROS-generating enzymes inhibitors on CIS. Cells were pretreated with or without the inhibitors: allopurinol (xanthine oxidase inhibitor); NG-monomethyl-L-arginine (NMMA) (a nitric oxide synthase) or metyrapone (a cytochrome P-450 inhibitor) before or after doxorubicin treatment. The data in A and (C) show that inhibition of mitochondrial and cytoplasmic ROS had no significant effect on the overall levels of ROS in CIS resistant cells after chemotherapy treatment. In CIS-sensitive cells, doxorubicin treatment results in dramatically higher levels of ROS in a time-dependent manner, which are not affected by cytoplasmic inhibitors (B). However, the Mitochondrial ROS inhibitors significantly reduced ROS production in CIS-sensitive cells (D). (E) Quantitative analysis of CIS in Mcl-1-deficient cells with or without the mitochondrial ROS-generating enzyme inhibitors as assessed by γH2AX and PML nuclear body formation. *P < 0.05, comparing those given the indicated inhibitors + doxorubicin to doxorubicin alone. Error bars represent ± S.D. Data are inclusive of at least three independent experiments.
Mcl-1 has no effect on anti-oxidants for its anti-CIS activities
Previous reports have demonstrated that other Bcl-2 family members are capable of inhibiting ROS generation through up-regulation of anti-oxidants or stabilization of the mitochondria [12, 24]. To assess if Mcl-1 regulates ROS generation through a similar up-regulation of anti-oxidants, we treated CIS-resistant/Mcl-1 proficient cells (HCT116 p53−/−) with chemical inhibitors of most major anti-oxidants, including: 3-AT (catalase), 2-MT (superoxide dismutase), and MSA (glutathione peroxidase) with or without doxorubicin. None of these inhibitors had a significant effect on Mcl-1′s anti-ROS function or senescence induction under CIS conditions as measured by PML and γ-H2AX foci formation or Ki67 staining (Figure 4A–4C). These results indicate that Mcl-1′s ability to prevent ROS-mediated CIS does not occur by up regulating anti-oxidants as is the case for Bcl-2. Knowing that the major processes leading to ROS generation are tightly regulated by a balance of anti- and pro-oxidants, we next tested the effect of diphenyleneiodonium (DPI), an inhibitor of the pro-oxidant family of NADPH oxidases (NOXs), as well as N-acetylcysteine (NAC), an anti-oxidant which we previously showed can prevent CIS [9]. Impressively, DPI (similar to NAC) caused a robust abrogation of ROS generation in Mcl-1 deficient cells as compared with the Mcl-1 proficient cells during CIS conditions after 24 hours of culture with doxorubicin, a time point that significant differences in ROS production can be observed (Figure 4D). DPI and NAC not only had a significant negative effect on ROS production, but also inhibited the induction of senescence (Figure 4E and 4F) in Mcl-1 deficient cells (CIS-sensitive) to the levels that were similar to CIS-resistant, Mcl-1 proficient cells. These data indicate that under CIS conditions, Mcl-1 inhibits the pro-oxidant side of ROS production, unlike other Bcl-2 family members.
Figure 4. Mcl-1 does not inhibit CIS through up-regulation of anti-oxidant molecules.

(A) HCT116 p53−/− shControl or shMcl-1 cells were pretreated with inhibitor of catalase (3-AT); SOD (2-MT) or GPx (MSA) with or without doxorubicin. ROS levels were assessed with Amplex Red at the indicated time points. (B and C) Quantitative analysis of CIS in HCT116 p53−/− shMcl-1 cells as assessed by PML and γ-H2AX nuclear body formation (B) and Ki67 staining (C). (D–F) Reactive oxygen species inhibitors block CIS. HCT116 p53−/− shcontrol or shMcl-1 cells were pretreated with or without N-acetylcysteine (NAC) or diphenyleneiodonium (DPI) in the presence or absence of doxorubicin for 24 hours. (D) Effects of NAC and DPI on the ROS generation. (E and F) Quantitative analysis of CIS with or without ROS-generating inhibitors as assessed by PML and γ-H2AX nuclear body formation (E) and Ki67 staining (F). Significant differences are compared with untreated control versus doxorubicin treated as well as untreated versus doxorubicin plus inhibitors (*p ≤ 0.05). Error bars represent ± S.D.
Mcl-1 prevents ROS generation and CIS through inhibition of NOX4 expression
Although DPI is widely used to inhibit various NOXs, it can have broad inhibition of other flavoenzymes [25, 26]. Thus we next set out to both confirm and identify which NOX proteins induce ROS during CIS. We first assessed the mRNA expression of the five major NADPH oxidase family members by real-time RT-qPCR. Figure 5A shows major increases in NOX1 and NOX4 after doxorubicin treatment in Mcl-1 deficient cells only. Further, in the same cells, NOX4 protein expression was preferentially up-regulated during CIS as shown by western blot analysis (Figure 5B). These results prompted us to examine whether senescence resistance could be restored in Mcl-1 deficient cells through the knock-down of NOX4 expression. Two siRNAs (one shown) were designed to specifically silence NOX4 expression, and were used in shcontrol and shMcl-1 cells (as well as a scramble control). Successful knockdown of NOX4 with these specific siRNAs was confirmed at the protein (Figure 5C) and mRNA levels (Figure 5D). Under CIS conditions in sensitive cells (Mcl-1 deficient), knock-down of NOX4 was sufficient to significantly abrogate both ROS production (Figure 5E) and senescence induction (Figure 5F one of three senescence assays shown). Notably, knock-down of NOX1 by siRNA (Supplementary Figure 2A) did not affect senescence induction in sensitive cells (Supplementary Figure 2B, one of three senescence assays shown).
Figure 5. Mcl-1 prevents accumulation of NOX4, protecting cells from ROS-mediated CIS.

(A) Differential expression of the major NADPH oxidase (NOX) family members during CIS conditions. Cells were treated with doxorubicin or left untreated in the presence or absence of DPI. Levels of Nox1-5 mRNA were determined by real-time quantitative PCR (RT-qPCR). (B) NOX1 and NOX4 protein expression assed by western blot. (C–F) Mcl-1 prevents accumulation of NOX4 and protects cells from ROS-mediated CIS. Cells were transfected with scramble siRNA (siScr) as control or with NOX4 siRNA (siNOX4). After doxorubicin treatment, the level of NOX4 was quantified by western blotting (C). Cells transfected as in Figure C were treated with or without doxorubicin or doxorubicin + DPI after which the level of NOX4 mRNA was quantified by real-time qPCR analysis (D), ROS levels (E), and PML nuclear body formation (F). (G) Expression of NOX4 protein in mitochondrial and cytosolic fractions of Mcl-1 proficient (shControl) or deficient (shMcl-1) cells under CIS conditions. Fraction purity was verified by staining for Tom20 as a mitochondrial marker and loading control. β-actin was used as a loading control for the cytosolic fraction. Significant differences are compared with untreated control versus doxorubicin treated as well as untreated versus doxorubicin plus inhibitors (*p ≤ 0.05). Error bars represent ± S.D. Quantitative data are inclusive of at least three independent experiments. (H) Mitochondria ROS measurement by MitoSOX. Representative images of MitoSOX fluorescence. Microscopic imaging demonstrated markedly increased in mitochondrial fluorescence intensity of MitoSOX in NOX4 siScr cells treated with DOX for 24 h. The mitochondrial ROS generation where largely prevented by NOX4 siRNA (Figure 5H).
In Figure 3 we showed the critical nature of mitochondrial ROS in our model of CIS. As NOX4 was previously reported to localize to the mitochondria, we also examined its subcellular localization in our system of CIS [27–29]. Mitochondrial and cytosol fractions were prepared from CIS- sensitive and resistant cells under doxorubicin treatment and probed for NOX4. The purity of the fractionation procedure was confirmed by the presence or absence of Tom20 (a mitochondrial marker), which was also used as a protein loading control in the mitochondrial fraction, versus β-actin used in the cytosolic fraction (actin). Immunoblot analysis using NOX4 specific antibody shows that NOX4 is predominantly up regulated in the mitochondrial fraction in CIS-sensitive cells under doxorubicin treatment in the absence of Mcl-1 (Figure 5G, right panel). NOX4 levels were observed to a lesser extent in the cytosol. (Figure 5G, left panel). Thus in the absence of Mcl-1 in cells undergoing CIS, not only is NOX4 upregulated, but it is largely present in the mitochondria, explaining the critical ROS production observed. Interestingly, these studies also demonstrated that under doxorubicin treatment in the presence of Mcl-1 (ShControl cells), no detectable NOX4 is found in the mitochondria despite moderate expression in the cytosol, indicating a role for Mcl-1 in regulating the trafficking of NOX4 to the mitochondria, in addition to its effect on NOX4 expression. Finally, in order to demonstrate that the presence of NOX4 results specifically in increased mitochondrial ROS, we used MitoSOX, a fluorescent indicator of mitochondrial superoxide generation, to observe that knock-down of NOX4 expression in shMcl-1 cells results in decreased mitochondrial ROS generation after doxorubicin treatment (Figure 5H).
Thus we now show for the first time a direct link between a Bcl-2 family member and a specific NADPH oxidase and illustrate that Mcl-1 has a unique ability to inhibit ROS production by preventing the up regulation of the pro-oxidant NOX4, ultimately leading to CIS resistance. There may be other NOXs that Mcl-1 regulates, but these data are of great import given the key role both Mcl-1 and NOX4 play in carcinogenesis [30, 31].
DISCUSSION
The targeting of survival proteins like Mcl-1 and the other Bcl-2 family members for cancer therapy is the subject of ongoing scientific and commercial interest [32]. While this interest has resulted in advances in treatment of hematogenous cancers, particularly in the inhibition of Bcl-2 and Bcl-xL in lymphoma, effective inhibitor treatments of solid tumors remains a challenge, particularly the effective targeting of Mcl-1 [33]. Indeed, the latest generation Bcl-2/Bcl-xL inhibitors are active at concentrations 10 fold lower than similarly designed Mcl-1 inhibitor, though a new generation of Mcl-1 inhibitor is under development that has higher activity [7, 33–38]. While initially defined as simply pro- or anti-apoptotic proteins, Bcl-2-family proteins are now known to interact with many other proteins in the cell, and contribute to the regulation of mitochondrial and endoplasmic reticulum function, autophagy, calcium homeostasis, and senescence [5, 39–44]. We have recently demonstrated that Mcl-1 is unique among the Bcl-2 family in that it can inhibit chemotherapy-induced senescence, even in the absence of p53, and that this inhibition occurs through a distinct molecular domain from its anti-apoptosis structure [9, 10].
In the current study, we expand upon our understanding of Mcl-1′s regulation of senescence by identifying its effects on well-known components of this process. We started by examining CIS induction of the DNA damage response (DDR). Although Mcl-1′s anti-apoptotic functions were previously linked to inhibition of the DDR by direct binding to DDR components, our data contrast with those results under CIS conditions in that we find Mcl-1 expression leads to lower expression of these components [15, 45]. We also demonstrate that chemicals designed to inhibit ATM and ATR can restore senescence resistance in cells made sensitive to CIS through knock-down of Mcl-1. By demonstrating that Mcl-1 inhibits the activation of major upstream DDR components (ATM, ATR, Chk1/2) and expression of downstream components like γ-H2AX, the very same proteins Mcl-1 was described to bind to, it is now clear that Mcl-1 may have a dual role: to directly bind and block activated components of the DDR (during apoptosis conditions), and to prevent them from ever being expressed (during senescence conditions) [15].
Our previous and current studies also highlighted that CIS pathways require generation of ROS and Mcl-1′s ability to inhibit CIS was dependent on its anti-ROS functions. ROS production in cancer is known to induce both cellular proliferation and injury [46–49]. Our study, however, showed that under senescence-inducing conditions, ROS production is upstream of other senescence-related events, including the DDR. Our observation that ROS production precedes the DDR during CIS is consistent with other studies showing that ATM can function as a redox sensor after DNA damage [50]. Thus Mcl-1′s inhibition of ROS prevents cellular injury and the DDR cascade.
We recently demonstrated that there is also a specific and unique domain within Mcl-1 that regulates CIS, distinct from those regulating apoptosis. Specifically there are 4 key residues in a loop domain (for instance P198) that, if mutated, will abrogate Mcl-1′s anti-senescent functions [10]. By re-expressing mutant Mcl-1 constructs in Mcl-1-deficient, CIS sensitive cells and measuring ROS production under CIS conditions, we were able to show that the P198 residue, (but not any of its C-terminal anti-apoptotic Bcl-2 homology domains) is necessary for anti-ROS function. Thus our data demonstrating that this internal loop domain is required to prevent ROS production are consistent with Mcl-1′s CIS-regulating activities.
Having identified that ROS is upstream of the DDR we delved further into the characteristics of its production. The generation of ROS is closely regulated by the balance of anti-oxidant and pro-oxidant actions, and members of the Bcl-2 family are known to affect generation of ROS under conditions of cellular stress [51, 52]. For instance, several Bcl-2 family members have been characterized as activators of anti-oxidants [50, 53, 54]. In contrast, we find that Mcl-1 affects ROS generation not by up regulation of anti-oxidants, but instead by regulating the expression of pro-oxidants, and specifically NOX4. To our knowledge, this is a unique finding among the Bcl-2 family. The NOX protein family are highly studied in cancer due to their role in inducing specific types and quantities of ROS, and the balance of their activities in relation to the pro-survival activities of the Bcl-2 family are not completely understood. Based on our data, Mcl-1 can be considered a regulator of this important pro-oncogenic class of molecules. Interestingly, we noted that doxorubicin treatment of shMcl-1 cells also results in the expression of NOX1, although subsequent knock-down of NOX1 resulted no effect on senescence. Both NOX1 and NOX4 expression are controlled by similar regulatory elements, so it is not surprising that expression of both genes are increased in our CIS model [55]. However, NOX1 appears to be a general cancer promoting factor, especially in CRC while NOX4 by itself was not shown to promote CRC progression [56, 57]. Additionally, of the two only NOX4 is known to induce liberation of H2O2 [28]. Thus, it is likely that NOX1 is upregulated to promote cancer progression during stress situations, while NOX4 is a factor that produces ROS which contributes to cellular injury and senescence [57]. How Mcl-1 specifically regulates the expression of NOX4 is under active investigation by our lab.
Our studies focused on production of H2O2 as the relevant species of ROS generated during the course of CIS. The reason for this is two-fold: 1) previous studies have indicated that while NOX4 generates significant levels of hydrogen peroxide, associated increases in superoxide radical generation are difficult to detect; and 2) CIS involves processes in various cellular compartments in addition to the mitochondria, including the DDR and gene expression changes in the nucleus, and H2O2 is a more stable molecule that is able to diffuse freely throughout the cell [58, 59].
While previous studies have found that inhibition of complexes I, II, and III of the mitochondrial electron transport chain all result in increased superoxide generation (especially complexes I and III), it is only inhibition of the Qi site of complex III that results in increased hydrogen peroxide generation in intact mitochondria [23, 60–62]. These findings are similar to our own studies in doxorubicin-treated, shMcl-1 cells where use of roteneone and malonate decreased hydrogen peroxide ROS generation while antimycin A increased it; indicating that ROS generated from complex III likely represents the principle site of ROS generation during CIS. Whether the mitochondrial ROS represents an up-stream event that sets up a feed-forward process that leads to the traffic of NOX4 to the mitochondria further enhancing ROS production has yet to be determined.
Interestingly, we found that doxorubicin treatment alone was able to measurably increase cellular ROS, even in the presence of various ROS inhibitors or knock-down of NOX4. Doxorubicin is known to localize to the mitochondria, where it is directly reduced into superoxide radicals by interaction with complex I [63]. It may be that doxorubicin-treated cancer cells have an initial induction of ROS that triggers downstream processes of senescence and further ROS production, but in CIS-resistant cells these events (including further ROS production) are blocked by both the expression of Mcl-1 and absence of p53 - both common features of cancer. In normal cells, senescence-inducing therapies would not be hindered as there is no upregulation of Mcl-1 or loss of tumor suppressor genes. For example, one of the significant side-effects related to doxorubicin treatment is cardiac toxicity, which often limits its clinical effectiveness [64, 65]. In particular, NOX2 and NOX4 are both highly expressed in heart tissue, and contribute to ROS-related pathologies including tissue damage during congestive heart failure and cardiovascular disease, especially in aged tissue [66, 67]. The association of NOX4 with elevated ROS and age-related senescence in the heart is particularly interesting in that NOX4 overexpression induces cellular senescence in a similar manner to our model of doxorubicin-induced CIS, and doxorubicin can induce senescence in cardiac cells [68–70]. More relevant to Mcl-1 biology, the absence of Mcl-1 in cardiac tissue specific mouse genetic knock-out models is associated with abnormal mitochondrial function, inability to regulate autophagy, and cardiac failure [71, 72]. Although these studies did not evaluate the role of Mcl-1 and ROS in cardiac injury, other models of heart failure have demonstrated this association [73, 74].
It is well established that Bcl-2 family members which do not inhibit CIS do inhibit ROS generation through stabilization of the mitochondria [24]. In our study we found that the most important ROS during CIS, were those generated in the mitochondria. Importantly, the mitochondrion is the organelle most commonly associated with Mcl-1 apoptosis-related activities [5]. We now add to the list of Mcl-1′s unique stabilization of the mitochondria abilities by illustrating its unique capability to not only prevent NOX4 expression, but NOX4′s subcellular translocation to the organelle and subsequent ROS generation. This observation is consistent with other studies that have demonstrated NOX4 must be present in the mitochondria to induce oxidative stress and processes similar to senescence, especially in cancer cells [28, 29].
In summary, our work has expanded an understanding of Mcl-1′s ability to inhibit ROS and CIS. While ROS generation often contributes to transformation and oncogenesis, high levels are detrimental to cellular function, inducing apoptosis and senescence [75]. Specific targeting and enhancing of ROS production and/or inhibiting anti-ROS mechanisms are of increasing interest in designing the next generation of cancer therapeutics [22]. Cancer cells may have high baseline NOX family expression and ROS allowing for their genetic instability and cancer progression. Other cancer cells like we employ only have increased NOX expression and ROS production upon chemotherapy treatment. Considering that targeting NOX family members and/or ROS production is a bone fide and known strategy in cancer therapy development, understanding the gatekeepers of these events is critical to best tailor therapy for patient outcomes. Our study provides strong evidence that Mcl-1 is such a gatekeeper that could allow cells to keep NOX4 in check to prevent critical overloading of ROS and events like senescence. Moreover, as overcoming senescence is not just critical for cancer progression and treatment resistance, but impacts carcinogenesis in general, these results have major ramifications for cancer research and treatment strategies.
MATERIALS AND METHODS
Cell lines and culture conditions
HCT116 human colon cancer lines (p53−/−) were generously provided by Bert Vogelstein (Johns Hopkins University). The cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with penicillin/streptomycin, non-essential amino acids, and 10% fetal bovine serum (FBS). HCT116 p53−/− shControl and HCT116 p53−/− shMcl-1 cells are derivatives of HCT116 p53−/− that stably express a transcript-specific short hairpin RNA (shRNA) that either knocks-down endogenous Mcl-1 expression or contains an irrelevant control RNA (Open Biosystems) and have been described previously [9]. All cell cultures were incubated at 37°C in a humidified incubator containing 5% CO2.
Drug treatments
The drugs used were purchased from the following providers and used at the concentrations indicated: doxorubicin (100 ng/ml, Sigma); ATM specific inhibitor, KU-55933 (20 uM, Calbiochem); ATM and ATR inhibitor, caffeine (5 mM, MP Biomedical); anti-oxidant inhibitors: 3-amino-1,2,4-triazole (3-AT, a catalase inhibitor, 20 uM, Sigma-Aldrich); 2-methoxyestradio (2-MT, a SOD inhibitor, 10 uM, Sigma-Aldrich); mercaptosuccinic acide (MSA, a GPx inhibitor, 15 uM, Sigma-Aldrich); pro-oxidant inhibitor, a NADPH oxidase inhibitor diphenyleneiodonium (DPI, 30 uM, Sigma-Aldrich); antioxidant N-acetyl-L-cysteine (NAC, 5 mM, Sigma-Aldrich); cytoplasmic ROS-generating enzyme inhibitors: allopurinol (xanthine oxidase inhibitor, 100 uM, Sigma-Aldrich); NG-monomethyl-L-arginine (NMMA, a nitric oxide synthase, 100 uM, Sigma-Aldrich); metyrapone (a cytochrome P-450 inhibitor, 500 uM, Sigma-Aldrich); the mitochondrial ROS-generating enzyme inhibitors: rotenone (a complex I inhibitor, 10 uM, Sigma-Aldrich), malonate (succinate dehydrogenase inhibitor, 5 uM, Sigma-Aldrich), or antimycin A ( a complex III inhibitor, 20 uM, Sigma Aldrich).
Plasmid transfections
Transient plasmid transfection in HCT116 p53−/− shMcl-1 cells was performed using Lipofectamine 2000 (Life Technologies) according to the manufacturer's instructions. Briefly, 2 × 105 cells/well in 6 well plates or 1 × 105 cells/well in 6 well plates on poly-L- lysine coated glass coverslips were transiently transfected with 0.5 ug of wild-type Mcl-1, P198A, R201A, Δ208–350 expressing constructs, or empty pcDNA3.1 vector (Invitrogen). Medium was changed after 24 hours and then cells were incubated for 48 hours prior to verifying transgene expression by western blotting. 48 hours post-transfection, cells were left untreated or treated in fresh media containing doxorubicin to induce senescence. For knock-down of NADPH oxidase 4 (NOX4) and (NOX1) by siRNA, HCT116 p53−/−shControl or shMcl-1 cells were transfected with NOX4 and NOX1 specific siRNA or with a negative control scramble siRNA (siScr) (Santa Cruz Biotechnology) and were cultured for 48 hours. After 24 hours doxorubicin (100 ng/ml) treatment, the gene and protein expression of NOX4 and NOX1 were quantified by real time quantitative PCR (RT-qPCR) and western blotting.
Immunoblotting
Western blotting analyses were performed as described previously (Demelash et al. 2015). The membranes were visualized using enhanced chemiluminescence (ECL) reagents (GE Healthcare) or WesternBright Quantum kit (Advansta, Menlo Park, California, USA). The following antibodies (at the indicated dilutions) were used in this study: anti-NOX1 (rabbit, 1:500, Santa Cruz Biotechnology); anti-NOX4 (rabbit, 1:1000, Santa Cruz Biotechnology); anti-Mcl-1 (rabbit, 1:1000); anti-phospho-ATM Ser198 (ATM-S1981p, rabbit, 1:500); anti-phospho-ATR Ser428p (ATR-S rabbit, 1:500); anti-phospho-Chk1Ser345p (rabbit, 1:500); anti-phospho-Chk2T68p (rabbit 1:500). Antibodies were purchased from Cell Signaling Technology. Mouse anti-β-actin (Santa Cruz Biotechnology) at a dilution of 1:10000 was used as loading control.
Immunofluorescence
Immunofluorescence was performed as described previously [10]. Anti-PML (mouse, 1:100 dilution) was purchased from Santa Cruz Biotechnology. Anti-γH2AX (Ser-139, mouse, 1:100) was purchased from BioLegend, and anti-Ki67 (mouse, 1:100) was from BD Biosciences. Cells were incubated with a goat anti-mouse (clone Poly 4043) or a donkey anti-rabbit (clone Poly 4064) secondary antibody conjugated with Cy3 (BioLegend, San Diego, CA) for 1 h in the dark, washed with PBS, and mounted on microscope slides using Vectashield mounting medium containing DAPI for fluorescence (Vector Laboratories, Burlingame, CA). Images were captured on a Leica SP2 confocal microscope using the appropriate filter sets.
ROS measurement
Changes in intracellular ROS production were determined using the Amplex Red reagent (10-acetyl-3, 7 dihydroxyphenoxazine) using the protocols provided by the manufacturer (Molecular Probes, Eugene, OR). Approximately 10.000 HCT116 p53−/−shControl or shMcl-1 cells/well were allowed to grow in 96 well plates for 24 hours. The following day, the media were replaced and the cells were pre-incubated for 1 hour with ATM specific inhibitor, KU-55933; ATM and ATR inhibitor, caffeine; anti-oxidant inhibitors: mercaptosuccinic acid (MSA, a GPx inhibitor); 3-amino-1,2,4-triazole (3-AT, a catalase inhibitor) or 2-methoxyestradio (2-MT, a SOD inhibitor); a pro-oxidant inhibitor, a NADPH oxidase inhibitor (DPI); the antioxidant N-acetyl-L-cysteine (NAC); cytoplasmic ROS-generating enzyme inhibitors: allopurinol (xanthine oxidase inhibitor); NG-monomethyl-L-arginine (NMMA, a nitric oxide synthase), metyrapone (a cytochrome P-450 inhibitor); the mitochondrial ROS-generating enzyme inhibitors: rotenone (a complex I inhibitor) or malonate (Succinate dehydrogenase inhibitor). The cells were then incubated in the presence or absence of doxorubicin. Fifty microliters of the Amplex Red reaction mixture (100 μM Amplex Red, and 0.2 U/ml horseradish peroxidase) was added to each well followed by 30 min incubation at 37°C. Amplex Red conversion to resorufin was measured at 590 nm emission and 560 nm excitation using a microplate reader. To measure specific mitochondrial generated ROS, MitoSOX Red was used as described in the manufacturer's protocol (Invitrogen). The fluorescent probe MitoSOX selectively reacts with superoxide in the mitochondria. Phenol red-free medium was used to avoid dye interference.
Real-time qPCR
Total RNA was extracted from HCT116 p53−/− shControl or shMcl-1 cells using the RNeasy Mini Kit (Qiagen). cDNA was synthesized from 1 μg of RNA using the Super Script III First-strand Synthesis System Kit (Invitrogen) according to the manufacturer's instructions. mRNA levels were determined by Real-time quantitative PCR (RT-qPCR) using SYBR Green and Taq Master Mix kit (Qiagen) with a set of primers specific for the human NOX family (NOX1 to NOX5). The threshold cycle (Ct) value of the target gene was normalized to the expression of reference gene GAPDH to obtain a 2 −ΔΔCt value. All reactions were performed in triplicate.
β-galactosidase senescence assays
Following 6 days of treatment or no treatment with drugs, cells were assayed for senescence-associated beta-galactosidase (SA-β-gal) expression as previously described [76]. Briefly, cells were washed and fixed with 2% PFA (Fisher Scientific) for 5 minutes at room temperature. Cells were then incubated in the dark for up to 16 hours in a staining solution containing 1 mg/ml X-Gal (Gold Biotechnology) in dimethylformamide (Acros Organics), 40 mM of a 0.2 M citric acid/Na phosphate buffer pH 6.0, 5 mM potassium ferrocyanide (Sigma), 5 mM potassium ferricyanide (Sigma), 150 mM sodium chloride, and 2 mM magnesium chloride. Stained cells were then visualized under an inverted bright-field microscope. Ten representative fields were randomly selected for the quantification of β-gal positive cells as a percentage of the total cell number.
PML and γH2AX foci quantification
Ten representative fields were randomly selected for the quantification of PML and γH2AX nuclear body formation (NBs). The numbers of foci present in each cell nucleus were manually counted in 30 transfected and drug treated cells as well as in transfected but not drug treated cells using a Leica DM5500 B florescent microscope at 40× oil immersion.
Cell proliferation assays
The proliferative capacity of cells was determined by Ki67 immunohistochemical staining. Ten representative fields were randomly selected for the quantification of Ki67-positive cells. The numbers of Ki67-positive cells were manually counted in drug treated or untreated cells using a Leica DM5500 B florescent microscope at 40× oil immersion.
Mitochondria and cytosol fractionation
Mitochondria and cytosol fractions were obtained using the Mitochondria Isolation Kit (Product No. 89874) for cultured cells from Thermo Scientific. Briefly, after 48 hours with or without 100 ng/ml doxorubicin treatment, 2 × 107 HCT116 p53−/− shControl and HCT116 p53−/− shMcl-1 cells were harvested by centrifuging at ~850 × g for 2 minutes, and mitochondria were isolated following the protocol provided by the kit. Mitochondria pellets and whole cell lysates were lysed in RIPA buffer. After removing the insoluble material by 14,000 × g centrifugation, protein from mitochondria, and cytosol was quantified by Thermo Scientific BCA Assay kit (Product No. 23225). For gel electrophoresis, 30 ug of total protein was loaded per lane and separated by SDS-PAGE, and then transferred to PVDF membrane (Bio-Rad). For immunoblot experiments, the membranes were sequentially blotted with anti-NOX4 (Santa Cruz), anti-Tom20 (Santa Cruz), mouse β-actin (Santa Cruz) primary antibodies, and horseradish peroxidase-conjugated secondary antibody (Bio-Rad), followed by chemiluminescence visualization (PerkinElmer Life Sciences).
Statistical analysis
The significance of difference between two groups were determined using Student's t test. Statistical evaluation of the data with multiple treatments was performed by one-way ANOVA for multiple comparisons. In all cases, P values of < 0.05 were considered significant. Data are presented as mean ± standard deviation (SD) from at least three independently and separately conducted experiments.
SUPPLEMENTARY MATERIALS FIGURES AND TABLES
Abbreviations
- CIS
Chemotherapy-induced senescence
- DDR
DNA damage response
- DPI
diphenyleneiodonium
- NAC
N-acetylcysteine
- NMMA
NG-monomethyl-L-arginine
- ROS
Reactive oxygen species
Footnotes
Authors’ contributions
AD and BG conceived the studies and wrote the paper. AD performed the experiments with contribution from LL. LP contributed to preparation of manuscript. All authors reviewed the results and approved the final version of the manuscript.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest with the contents of this article.
FUNDING
This work was supported by NIH grant R01CA132796 (to B.R.G).
REFERENCES
- 1.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, Ligon AH, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Juin P, Geneste O, Gautier F, Depil S, Campone M. Decoding and unlocking the BCL-2 dependency of cancer cells. Nat Rev Cancer. 2013;13:455–465. doi: 10.1038/nrc3538. [DOI] [PubMed] [Google Scholar]
- 3.Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DC. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 4.Billard C. BH3 mimetics: status of the field and new developments. Mol Cancer Ther. 2013;12:1691–1700. doi: 10.1158/1535-7163.MCT-13-0058. [DOI] [PubMed] [Google Scholar]
- 5.Perciavalle RM, Stewart DP, Koss B, Lynch J, Milasta S, Bathina M, Temirov J, Cleland MM, Pelletier S, Schuetz JD, Youle RJ, Green DR, Opferman JT. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol. 2012;14:575–83. doi: 10.1038/ncb2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bruncko M, Wang L, Sheppard GS, Phillips DC, Tahir SK, Xue J, Erickson S, Fidanze S, Fry E, Hasvold L, Jenkins GJ, Jin S, Judge RA, et al. Structure-guided design of a series of MCL-1 inhibitors with high affinity and selectivity. J Med Chem. 2015;58:2180–2194. doi: 10.1021/jm501258m. [DOI] [PubMed] [Google Scholar]
- 7.Wei G, Margolin AA, Haery L, Brown E, Cucolo L, Julian B, Shehata S, Kung AL, Beroukhim R, Golub TR. Chemical Genomics Identifies Small-Molecule MCL1 Repressors and BCL-xL as a Predictor of MCL1 Dependency. Cancer Cell. 2012;21:547–562. doi: 10.1016/j.ccr.2012.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Placzek WJ, Sturlese M, Wu B, Cellitti JF, Wei J, Pellecchia M. Identification of a novel Mcl-1 protein binding motif. J Biol Chem. 2011;286:39829–39835. doi: 10.1074/jbc.M111.305326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bolesta E, Pfannenstiel LW, Demelash A, Lesniewski ML, Tobin M, Schlanger S, Nallar SC, Papadimitriou JC, Kalvakolanu DV, Gastman BR. Inhibition of Mcl-1 Promotes Senescence in Cancer Cells: Implications for Preventing Tumor Growth and Chemotherapy Resistance. Mol Cell Biol. 2012;32:1879–92. doi: 10.1128/MCB.06214-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Demelash A, Pfannenstiel LW, Tannenbaum CS, Li X, Kalady MF, DeVecchio J, Gastman BR. Structure-Function Analysis of the Mcl-1 Protein Identifies a Novel Senescence-regulating Domain. J Biol Chem. 2015;290:21962–21975. doi: 10.1074/jbc.M115.663898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, Takatsu Y, Melamed J, d’Adda di Fagagna F, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–1018. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 12.Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 13.Guida M, Maraldi T, Beretti F, Follo MY, Manzoli L, De Pol A. Nuclear Nox4-derived reactive oxygen species in myelodysplastic syndromes. Biomed Res Int. 2014;2014:456937. doi: 10.1155/2014/456937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sulli G, Di Micco R, d’Adda di Fagagna F. Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat Rev Cancer. 2012;12:709–720. doi: 10.1038/nrc3344. [DOI] [PubMed] [Google Scholar]
- 15.Jamil S, Stoica C, Hackett TL, Duronio V. MCL-1 localizes to sites of DNA damage and regulates DNA damage response. Cell Cycle. 2010;9:2843–2855. doi: 10.4161/cc.9.14.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cuadrado M, Martinez-Pastor B, Murga M, Toledo LI, Gutierrez-Martinez P, Lopez E, Fernandez-Capetillo O. ATM regulates ATR chromatin loading in response to DNA double-strand breaks. J Exp Med. 2006;203:297–303. doi: 10.1084/jem.20051923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Letai AG. Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nat Rev Cancer. 2008;8:121–132. doi: 10.1038/nrc2297. [DOI] [PubMed] [Google Scholar]
- 18.Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ Fearon ER, Sundaresan M, Finkel T, Goldschmidt-Clermont PJ. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science. 1997;275:1649–1652. doi: 10.1126/science.275.5306.1649. [DOI] [PubMed] [Google Scholar]
- 19.Rodrigues MS, Reddy MM, Sattler M. Cell cycle regulation by oncogenic tyrosine kinases in myeloid neoplasias: from molecular redox mechanisms to health implications. Antioxid Redox Signal. 2008;10:1813–1848. doi: 10.1089/ars.2008.2071. [DOI] [PubMed] [Google Scholar]
- 20.Kamata H, Hirata H. Redox regulation of cellular signalling. Cell Signal. 1999;11:1–14. doi: 10.1016/s0898-6568(98)00037-0. [DOI] [PubMed] [Google Scholar]
- 21.Petros AM, Olejniczak ET, Fesik SW. Structural biology of the Bcl-2 family of proteins. Biochim Biophys Acta. 2004;1644:83–94. doi: 10.1016/j.bbamcr.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 22.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 23.Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. 2003;278:36027–36031. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- 24.Gottlieb E, Vander Heiden MG, Thompson CB. Bcl-x(L) prevents the initial decrease in mitochondrial membrane potential and subsequent reactive oxygen species production during tumor necrosis factor alpha-induced apoptosis. Mol Cell Biol. 2000;20:5680–5689. doi: 10.1128/mcb.20.15.5680-5689.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wind S, Beuerlein K, Eucker T, Muller H, Scheurer P, Armitage ME, Ho H, Schmidt HH, Wingler K. Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. Br J Pharmacol. 2010;161:885–898. doi: 10.1111/j.1476-5381.2010.00920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Latchoumycandane C, Marathe GK, Zhang R, McIntyre TM. Oxidatively truncated phospholipids are required agents of tumor necrosis factor alpha (TNFalpha)-induced apoptosis. J Biol Chem. 2012;287:17693–17705. doi: 10.1074/jbc.M111.300012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Block K, Gorin Y, Abboud HE. Subcellular localization of Nox4 and regulation in diabetes. Proc Natl Acad Sci USA. 2009;106:14385–14390. doi: 10.1073/pnas.0906805106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Graham KA, Kulawiec M, Owens KM, Li X, Desouki MM, Chandra D, Singh KK. NADPH oxidase 4 is an oncoprotein localized to mitochondria. Cancer Biol Ther. 2010;10:223–231. doi: 10.4161/cbt.10.3.12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci USA. 2010;107:15565–15570. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crosas-Molist E, Bertran E, Sancho P, Lopez-Luque J, Fernando J, Sanchez A, Fernandez M, Navarro E, Fabregat I. The NADPH oxidase NOX4 inhibits hepatocyte proliferation and liver cancer progression. Free Radic Biol Med. 2014;69:338–347. doi: 10.1016/j.freeradbiomed.2014.01.040. [DOI] [PubMed] [Google Scholar]
- 31.Caja L, Sancho P, Bertran E, Iglesias-Serret D, Gil J, Fabregat I. Overactivation of the MEK/ERK pathway in liver tumor cells confers resistance to TGF-{beta}-induced cell death through impairing up-regulation of the NADPH oxidase NOX4. Cancer Res. 2009;69:7595–7602. doi: 10.1158/0008-5472.CAN-09-1482. [DOI] [PubMed] [Google Scholar]
- 32.Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, Kipps TJ, Anderson MA, Brown JR, Gressick L, Wong S, Dunbar M, Zhu M, et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med. 2015. [DOI] [PMC free article] [PubMed]
- 33.Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, Kipps TJ, Anderson MA, Brown JR, Gressick L, Wong S, Dunbar M, Zhu M, et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med. 2016;374:311–322. doi: 10.1056/NEJMoa1513257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gandhi L, Camidge DR, Ribeiro de Oliveira M, Bonomi P, Gandara D, Khaira D, Hann CL, McKeegan EM, Litvinovich E, Hemken PM, Dive C, Enschede SH, Nolan C, et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol. 2011;29:909–916. doi: 10.1200/JCO.2010.31.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rudin CM, Hann CL, Garon EB, Ribeiro de Oliveira M, Bonomi PD, Camidge DR, Chu Q, Giaccone G, Khaira D, Ramalingam SS, Ranson MR, Dive C, McKeegan EM, et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res. 2012;18:3163–3169. doi: 10.1158/1078-0432.CCR-11-3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang H, Xue J, Hessler P, Tahir SK, Chen J, Jin S, Souers AJ, Leverson JD, Lam LT. Genomic analysis and selective small molecule inhibition identifies BCL-X(L) as a critical survival factor in a subset of colorectal cancer. Mol Cancer. 2015;14:126. doi: 10.1186/s12943-015-0397-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Phillips DC, Xiao Y, Lam LT, Litvinovich E, Roberts-Rapp L, Souers AJ, Leverson JD. Loss in MCL-1 function sensitizes non-Hodgkin’s lymphoma cell lines to the BCL-2-selective inhibitor venetoclax (ABT-199) Blood Cancer J. 2015;5:e368. doi: 10.1038/bcj.2015.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, Chanrion M, Kelly GL, Gong JN, Moujalled DM, Bruno A, Csekei M, Paczal A, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538:477–482. doi: 10.1038/nature19830. [DOI] [PubMed] [Google Scholar]
- 39.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 40.Zhang LN, Li JY, Xu W. A review of the role of Puma, Noxa and Bim in the tumorigenesis, therapy and drug resistance of chronic lymphocytic leukemia. Cancer Gene Ther. 2013;20:1–7. doi: 10.1038/cgt.2012.84. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Y, Pfannenstiel LW, Bolesta E, Montes CL, Zhang X, Chapoval AI, Gartenhaus RB, Strome SE, Gastman BR. Interleukin-7 inhibits tumor-induced CD27-CD28- suppressor T cells: implications for cancer immunotherapy. Clin Cancer Res. 2011;17:4975–4986. doi: 10.1158/1078-0432.CCR-10-3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X, Olberding KE, White C, Li C. Bcl-2 proteins regulate ER membrane permeability to luminal proteins during ER stress-induced apoptosis. Cell Death Differ. 2011;18:38–47. doi: 10.1038/cdd.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bonneau B, Prudent J, Popgeorgiev N, Gillet G. Non-apoptotic roles of Bcl-2 family: the calcium connection. Biochim Biophys Acta. 2013;1833:1755–1765. doi: 10.1016/j.bbamcr.2013.01.021. [DOI] [PubMed] [Google Scholar]
- 44.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 45.Jamil S, Mojtabavi S, Hojabrpour P, Cheah S, Duronio V. An essential role for MCL-1 in ATR-mediated CHK1 phosphorylation. Mol Biol Cell. 2008;19:3212–3220. doi: 10.1091/mbc.E07-11-1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 47.Poljsak B, Suput D, Milisav I. Achieving the balance between ROS and antioxidants: when to use the synthetic antioxidants. Oxid Med Cell Longev. 2013;2013:956792. doi: 10.1155/2013/956792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zingoni A, Cecere F, Vulpis E, Fionda C, Molfetta R, Soriani A, Petrucci MT, Ricciardi MR, Fuerst D, Amendola MG, Mytilineos J, Cerboni C, Paolini R, et al. Genotoxic Stress Induces Senescence-Associated ADAM10-Dependent Release of NKG2D MIC Ligands in Multiple Myeloma Cells. J Immunol. 2015;195:736–748. doi: 10.4049/jimmunol.1402643. [DOI] [PubMed] [Google Scholar]
- 49.Ghosh AK, Rai R, Park KE, Eren M, Miyata T, Wilsbacher LD, Vaughan DE. A small molecule inhibitor of PAI-1 protects against Doxorubicin-induced cellular senescence: molecular basis. Oncotarget. 2016;7:72443–72457. doi: 10.18632/oncotarget.12494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deng X, Kornblau SM, Ruvolo PP, May WS., Jr Regulation of Bcl2 phosphorylation and potential significance for leukemic cell chemoresistance. J Natl Cancer Inst Monogr. 2001. pp. 30–37. [DOI] [PubMed]
- 51.Kang J, Chong SJ, Ooi VZ, Vali S, Kumar A, Kapoor S, Abbasi T, Hirpara JL, Loh T, Goh BC, Pervaiz S. Overexpression of Bcl-2 induces STAT-3 activation via an increase in mitochondrial superoxide. Oncotarget. 2015;6:34191–34205. doi: 10.18632/oncotarget.5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Premkumar DR, Jane EP, Agostino NR, DiDomenico JD, Pollack IF. Bortezomib-induced sensitization of malignant human glioma cells to vorinostat-induced apoptosis depends on reactive oxygen species production, mitochondrial dysfunction, Noxa upregulation, Mcl-1 cleavage, and DNA damage. Mol Carcinog. 2013;52:118–133. doi: 10.1002/mc.21835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ellerby LM, Ellerby HM, Park SM, Holleran AL, Murphy AN, Fiskum G, Kane DJ, Testa MP, Kayalar C, Bredesen DE. Shift of the cellular oxidation-reduction potential in neural cells expressing Bcl-2. J Neurochem. 1996;67:1259–1267. doi: 10.1046/j.1471-4159.1996.67031259.x. [DOI] [PubMed] [Google Scholar]
- 54.Koziel K, Smigelskaite J, Drasche A, Enthammer M, Ashraf MI, Khalid S, Troppmair J. RAF and antioxidants prevent cell death induction after growth factor abrogation through regulation of Bcl-2 proteins. Exp Cell Res. 2013;319:2728–2738. doi: 10.1016/j.yexcr.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Manea A, Tanase LI, Raicu M, Simionescu M. Transcriptional regulation of NADPH oxidase isoforms, Nox1 and Nox4, by nuclear factor-kappaB in human aortic smooth muscle cells. Biochem Biophys Res Commun. 2010;396:901–907. doi: 10.1016/j.bbrc.2010.05.019. [DOI] [PubMed] [Google Scholar]
- 56.Chiera F, Meccia E, Degan P, Aquilina G, Pietraforte D, Minetti M, Lambeth D, Bignami M. Overexpression of human NOX1 complex induces genome instability in mammalian cells. Free Radic Biol Med. 2008;44:332–342. doi: 10.1016/j.freeradbiomed.2007.09.018. [DOI] [PubMed] [Google Scholar]
- 57.Meitzler JL, Antony S, Wu Y, Juhasz A, Liu H, Jiang G, Lu J, Roy K, Doroshow JH. NADPH oxidases: a perspective on reactive oxygen species production in tumor biology. Antioxid Redox Signal. 2014;20:2873–2889. doi: 10.1089/ars.2013.5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 59.Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC, Knaus UG. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal. 2006;18:69–82. doi: 10.1016/j.cellsig.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 60.Wojtovich AP, Smith CO, Haynes CM, Nehrke KW, Brookes PS. Physiological consequences of complex II inhibition for aging, disease, and the mKATP channel. Biochim Biophys Acta. 2013;1827:598–611. doi: 10.1016/j.bbabio.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Turrens JF, Boveris A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J. 1980;191:421–427. doi: 10.1042/bj1910421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Okun JG, Lummen P, Brandt U. Three classes of inhibitors share a common binding domain in mitochondrial complex I (NADH: ubiquinone oxidoreductase) J Biol Chem. 1999;274:2625–2630. doi: 10.1074/jbc.274.5.2625. [DOI] [PubMed] [Google Scholar]
- 63.Davies KJ, Doroshow JH. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J Biol Chem. 1986;261:3060–3067. [PubMed] [Google Scholar]
- 64.Weiss RB. The anthracyclines: will we ever find a better doxorubicin? Semin Oncol. 1992;19:670–686. [PubMed] [Google Scholar]
- 65.Shan K, Lincoff AM, Young JB. Anthracycline-induced cardiotoxicity. Ann Intern Med. 1996;125:47–58. doi: 10.7326/0003-4819-125-1-199607010-00008. [DOI] [PubMed] [Google Scholar]
- 66.Brandes RP, Weissmann N, Schroder K. NADPH oxidases in cardiovascular disease. Free Radic Biol Med. 2010;49:687–706. doi: 10.1016/j.freeradbiomed.2010.04.030. [DOI] [PubMed] [Google Scholar]
- 67.Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res. 2010;106:1253–1264. doi: 10.1161/CIRCRESAHA.109.213116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Geiszt M, Kopp JB, Varnai P, Leto TL. Identification of renox, an NAD(P)H oxidase in kidney. Proc Natl Acad Sci USA. 2000;97:8010–8014. doi: 10.1073/pnas.130135897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shiose A, Kuroda J, Tsuruya K, Hirai M, Hirakata H, Naito S, Hattori M, Sakaki Y, Sumimoto H. A novel superoxide-producing NAD(P)H oxidase in kidney. J Biol Chem. 2001;276:1417–1423. doi: 10.1074/jbc.M007597200. [DOI] [PubMed] [Google Scholar]
- 70.Piegari E, De Angelis A, Cappetta D, Russo R, Esposito G, Costantino S, Graiani G, Frati C, Prezioso L, Berrino L, Urbanek K, Quaini F, Rossi F. Doxorubicin induces senescence and impairs function of human cardiac progenitor cells. Basic Res Cardiol. 2013;108:334. doi: 10.1007/s00395-013-0334-4. [DOI] [PubMed] [Google Scholar]
- 71.Wang X, Bathina M, Lynch J, Koss B, Calabrese C, Frase S, Schuetz JD, Rehg JE, Opferman JT. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev. 2013;27:1351–1364. doi: 10.1101/gad.215855.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thomas RL, Roberts DJ, Kubli DA, Lee Y, Quinsay MN, Owens JB, Fischer KM, Sussman MA, Miyamoto S, Gustafsson AB. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev. 2013;27:1365–1377. doi: 10.1101/gad.215871.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lin CL, Tseng HC, Chen RF, Chen WP, Su MJ, Fang KM, Wu ML. Intracellular zinc release-activated ERK-dependent GSK-3beta-p53 and Noxa-Mcl-1 signaling are both involved in cardiac ischemic-reperfusion injury. Cell Death Differ. 2011;18:1651–1663. doi: 10.1038/cdd.2011.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Subramaniam V, Golik P, Murdock DG, Levy S, Kerstann KW, Coskun PE, Melkonian GA, Wallace DC. MITOCHIP assessment of differential gene expression in the skeletal muscle of Ant1 knockout mice: coordinate regulation of OXPHOS, antioxidant, and apoptotic genes. Biochim Biophys Acta. 2008;1777:666–675. doi: 10.1016/j.bbabio.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer. 2014;14:709–721. doi: 10.1038/nrc3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, Peacocket M, Campisi J. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.