

Abstract
The simplicity and effectiveness of calorie restriction (CR) in lifespan and healthspan extension have fascinated generations searching for the fountain of youth. CR reduces the level of oxidative stress and damage, which has been postulated in the free radical theory of aging as a major cause of aging and diseases of aging. This reduction has long been viewed as a result of passively slowing metabolism. Recent advances in nutrient sensing have provided molecular insights into the oxidative stress response and suggest that CR triggers an active defense program involving a cascade of molecular regulators to reduce oxidative stress. Physiological studies have provided strong support to oxidative stress in the development of aging-associated conditions and diseases, but also revealed the surprising requirement of oxidative stress to support normal physiological functions and in some context, even slow aging and prevent the progression of cancer. Deciphering the molecular mechanisms and physiological implications of the oxidative stress response during CR will increase our understanding of the basic biology of aging and pave the way for designing CR mimetics to improve healthspan.
Trends
In response to nutritional input and increased ROS levels, nutrient sensors are activated and reduce oxidative stress through diverse mechanisms.
In addition to acting as damaging agents, ROS also function as signaling molecules to support normal biological processes and physiological functions, such as stem and progenitor cell fate decision and innate immunity.
Oxidative stress is a reversible driver of the functional deterioration of stem cell aging.
Although oxidative stress has a pro-cancer role at the tumor initiation stage by promoting transformation, proliferation, metabolic reprogramming, and the establishment of the tumor microenvironment, cancer cells also hijack the oxidative stress responses to survive, progress, and metastasize.
In 1930’s, the fear for the adverse health consequences due to limited availability of food triggered the first calorie restriction (CR) experiment, which led to the surprising finding that CR increases lifespan in rodents [1]. Ever since this initial discovery, the link between nutrient uptake and longevity has been expanded across species. A growing number of studies in model organisms have extrapolated upon the effects of CR and fasting in decreasing the onset of age-associated pathologies [2–6]. The pro-longevity pro-health effect of CR was viewed as a result of the passive effect of food limitation and slow metabolism, resulting in the reduced production of reactive oxygen species (ROS) and prevention of oxidative stress [7]. However, it has become increasingly appreciated that the organismal effects of CR are actively regulated processes and CR triggers a robust defense program involving a cascade of molecular regulators to reduce oxidative stress [8–10]. Pathways regulating ROS signaling and antioxidant activity have thus become promising focal points for uncovering mechanisms through which nutrient sensing networks employ a pro-health phenotype.
Historically, ROS were thought to be produced as byproducts of cellular respiration and have deleterious effects in cells. Following the induction of Denham Harman’s free radical theory of aging, a large body of evidence has emerged that supports the notion that oxidative stress causes the deterioration of cellular integrity and tissue functions, and underlies the etiology of numerous diseases [11, 12]. However, recent studies also reveal ROS as signaling molecules essential to support normal physiological functions [13]. Some studies even caution that the effects of oxidative stress in aging and disease are context dependent.
In this review, we summarize the recent advances in the molecular understanding of the CR response to reduce oxidative stress and highlight the actions of nutrient sensors in coordinating the metabolic reprogramming and the oxidative stress response. Furthermore, we critically review the recent studies that illustrate the two faces of ROS as signaling molecules and damaging agents under physiological conditions, and discuss how cancer cells hijack the oxidative stress response mechanisms to promote tumorigenesis.
Nutrient Sensors and Oxidative Stress
Oxidative stress originates from an offsetting ratio of pro-oxidants to antioxidants, in favor of pro-oxidants. Upon nutrient deprivation, cells experience a metabolic switch from energy inefficient glycolysis to energy efficient oxidative phosphorylation [14–16]. At the organismal level, animals also switch their energy source from glycolysis to oxidative phosphorylation during fasting or CR [17]. This metabolic switch requires cells to be reliant on increased mitochondrial activity, which is associated with increased ROS production [18]. It is therefore plausible that nutrient sensors not only regulate intermediary metabolism to facilitate the metabolic switch but also the oxidative stress response to cope with the associated ROS production.
Regulation of Oxidative Stress by Sirtuins
The clearest evidence indicating nutrient sensors are critically required to reduce oxidative stress and damage during CR has come from studies of SIRT3 [9, 10]. SIRT3 belongs to the sirtuin family of nicotinamide adenine dinucleotide (NAD+) dependent deacylases. Their dependence on NAD+ links their activity to cellular metabolic status [19]. There are seven sirtuins in mammals, SIRT1-7, that localize to various cellular compartments. CR reduces the accumulation of oxidative damage and protects against the development of oxidative damage-related pathologies, such as hearing loss, in wild type (WT) mice [9, 10]. However, these protective effects are blunted in mice deficient in SIRT3, a mitochondrial sirtuin. It is interesting to note that the level of oxidative damage and hearing loss are indistinguishable in WT and SIRT3 knockout (KO) mice fed ad libitum, and SIRT3 expression level is induced by CR in WT mice [20, 21], indicating that the SIRT3 protective program is specifically turned on during CR to reduce oxidative stress. More broadly, these observations highlight a new paradigm for nutrient sensing and the oxidative stress response: instead of passively slowing the metabolic rate, CR initiates an active regulatory program to reduce oxidative stress, with nutrient sensors centrally positioned in such regulation.
It is striking that in addition to hearing loss, SIRT3 prevents a wide spectrum of aging-associated conditions and diseases. SIRT3 KO mice develop spontaneous cancer, insulin resistance, diet-induced obesity, inflammation, hyperlipidemia, steatohepatitis, reduced hematopoietic stem cell number and function, and sarcopenia [22–30]. Furthermore, SIRT3 prevents neuronal death in mouse models of Huntington’s disease [31]. It has been argued that induction of SIRT3 and reduction of oxidative stress contribute to the profound pro-health effects of CR, underscoring the free radical theory of aging.
Extensive biochemical studies have provided mechanistic insights into the oxidative stress response regulated by SIRT3. The critical antioxidant enzyme superoxide dismutase 2 (SOD2) is modified by acetylation in cells and SIRT3 targets key lysine residues on SOD2 for deacetylation and activation [9, 32]. SIRT3 also deacetylates isocitrate dehydrogenase 2 (IDH2), resulting in increased level of the reducing equivalent NADPH [10]. NADPH is used by glutathione reductase to convert oxidized glutathione (GSSG) to reduced glutathione (GSH), the cofactor used by glutathione peroxidase (GPX) to detoxify ROS. In addition to the antioxidants, SIRT3 also deacetylates a large number of mitochondrial enzymes and proteins controlling the major metabolic pathways, such as fatty acid oxidation, ketogensis, amino acid metabolism, acetyl CoA metabolism, oxidative phosphorylation, to coordinate the directionality and the rate of the metabolic flux upon changes in nutritional input [33]. The concurrent activation of the mitochondrial metabolic pathways and antioxidants by SIRT3 is a clear indication that cells have evolved mechanisms to reduce oxidative stress when mitochondrial activity is turned on upon nutrient deprivation.
Much data have accumulated to suggest that the nuclear SIRT1 mediates aspects of the CR response, including increased physical activity, disease protection, and lifespan extension [34–36]. Although there is no direct evidence indicating that SIRT1 mediates the CR response at least in part by reducing oxidative stress, biochemical studies have convincingly demonstrated that SIRT1 regulates cellular redox status by deacetylating the longevity factor forkhead box O 3a (FOXO3a), a transcription factor that governs the expression of several anti-oxidant genes [37, 38]. It is likely that SIRT1 reduces oxidative stress to influence cellular and physiological outcomes during CR. The convergence of SIRT1 and SIRT3 on the regulation of antioxidants suggests synergistic activation of antioxidants at the transcriptional and posttranslational levels to allow effective quenching of oxidative stress under the condition of CR.
Regulation of Oxidative Stress by AMPK, mTOR, and GCN2
Besides sirtuins, nutrient sensors that have been shown to be required for lifespan extension under CR regimens or fasting include AMP-activated protein kinase (AMPK) [39–42], the TOR signaling network [43, 44], and general amino acid control non-derepressible 2 (GCN2) [45, 46]. AMPK is an energy sensor [47], while TOR and GCN2 sense the availability of intracellular amino acids [48, 49]. These nutrient sensors have been intensively studies for metabolic regulation, but evidence for their regulation of oxidative stress has begun to emerge. It has recently been noted that cancer cells are dependent on AMPK to suppress oxidative stress and promote cell survival under energy stress conditions, such as glucose limitations, anchorage-independent growth and solid tumor formation [50, 51]. Aberrant regulation of the mTOR signaling network results in increased mitochondrial biogenesis and the ROS level in hematopoietic stem cells (HSCs) [52], while genetic deletion of GCN2 in antigen presenting cells or intestinal epithelial cells also results in increased ROS [53].
These observations further solidify the link between nutrient sensing and the oxidative stress response, and importantly, offer an opportunity to deepen the mechanistic understanding of this regulation. AMPK has been proposed to reduce oxidative stress by inhibiting acetyl-CoA carboxylases (ACC1 and ACC2), decreasing NADPH consumption in fatty-acid synthesis, and subsequently maintaining the NADPH level [50]. GCN2 and mTOR inhibition reduce oxidative stress by activating autophagy and recycling damaged mitochondria [53–55]. Thus, nutrient sensors could engage multiple ways to influence the activity of antioxidants, the integrity of mitochondria, and the metabolic flux to modulate ROS production and scavenging (Figure 1).
Figure 1. Calorie restriction triggers an active defense program to reduce oxidative stress and prevent aging and aging-associated diseases.
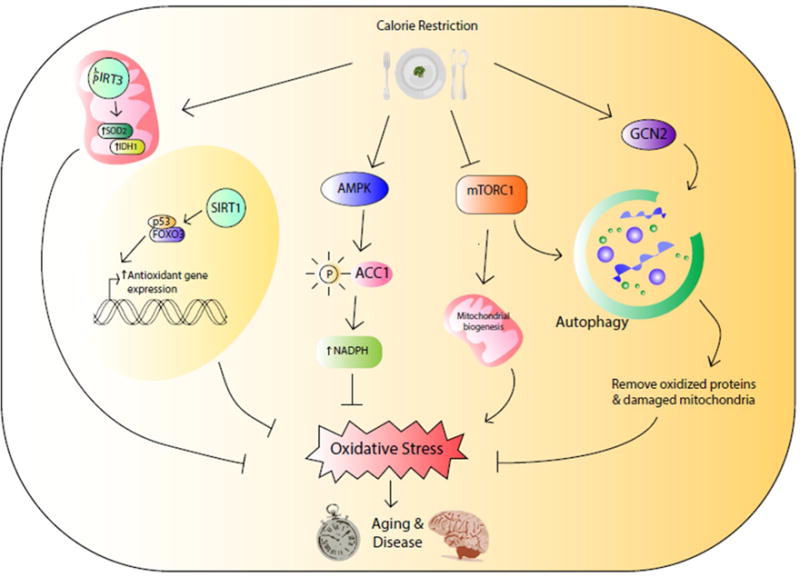
SIRT1 and SIRT3 reduce oxidative stress by regulating the transcriptional expression or posttranslational modification of antioxidants. AMPK reduces fatty acid synthesis and increases NADPH level to combat against oxidative stress. Inhibition of mTOR kinase suppresses mitochondrial biogenesis. Both inhibition of mTOR and activation of GCN2 facilitate the clearance of oxidized proteins and damaged mitochondria through autophagy.
Interestingly, evidence is emerging to suggest that in addition to nutritional input, nutrient sensors can also be activated in response to ROS. The expression of SIRT3 is induced by nutrient deprivation and ROS [56]. AMPK can also be activated by ROS [57–59]. Thus, in addition to the well-established function of AMPK in energy sensing and reestablishing energy homeostasis, AMPK also senses oxidative stress and induces the oxidative stress response. The intimate connection between nutrient sensing and oxidative stress sensing underscores a previously underappreciated crosstalk: the oxidative stress response is engaged during nutrient deprived conditions to dispose increased production of ROS associated with the metabolic switch to mitochondrial metabolism, and similarly, the metabolic reprogramming typically associated with nutrient deprivation also occurs during oxidative stress conditions to maintain energy reserve and cell survival.
Oxidative Stress in Aging and Diseases
Numerous studies have set out to test the free radical theory of aging. Surprisingly, lifespan studies of various antioxidant enzymes show that only one of 18 genetic manipulations influences lifespan [60]. On the other hand, new evidence supporting a causal role of oxidative stress in aging continues to emerge from studies in various model systems. In yeast, overexpression of the H2O2-scavenging enzyme Tsa1 extends lifespan [61]. Mice overexpressing glucose-6-phosphate dehydrogenase, a rate-limiting enzyme of the pentose phosphate pathway that generate the reducing agent NADPH, are protected from oxidative damage and exhibit improved healthspan [62]. Repressed NRF2 activity and increased oxidative stress recapitulate Hutchinson-Gilford progeria syndrome (HGPS) defects, while NRF2 activation decreases oxidative stress and reverses cellular HGPS defects [63]. These findings add a surprising twist to the free radical theory of aging and form the foundation to formulate new hypotheses to scrutinize this half-century old theory of aging.
A common approach to test an aging theory is the lifespan analyses. While critically important and informative, these studies do not offer the resolution necessary to tease out the numerous variables that act together to influence the organismal lifespan, which include but are not limited to the rate of aging. It is therefore imperative to dissect the impact of oxidative stress on aging-associated conditions in specific tissues or even cell types. Further, lifespan is influenced by life-threatening diseases, such as cancer. Admittedly, the incidence of cancer increases with aging and these two rival demons are likely to share some common origins, such as oxidative stress [64]. However, recent advances in cancer research also reveal that oxidative stress prevents aspects of cancer development and can complicate the effect of oxidative stress in lifespan [65–69]. Our constantly improving capacity to study physiology and pathology with increasing spatial and temporal resolution usher in a new era for the free radical theory of aging.
Stem Cell Aging and Tissue Degeneration
Adult stem cells or tissue specific stem cells persist throughout the lifespan to maintain and repair tissues. This lifetime commitment requires stem cells to develop robust cellular protective mechanisms to ensure their integrity. Using the hematopoietic system as an example, the cellular ROS level is considerably lower in HSCs than in their differentiated progeny [23]. The low cellular ROS level in HSCs is in part due to reduced ROS production. Adult HSCs mostly stay in a quiescent state, which is associated with low metabolic rate and mitochondrial number [70]. Quiescent HSCs primarily rely on glycolysis for energy production [71, 72]. Compared to mitochondrial oxidative phosphorylation, glycolysis is much less efficient for energy production but is sufficient to support the low energy requirement of quiescent HSCs. This metabolic feature is essential for the maintenance of HSCs, because less ROS are produced [70]. HSCs are also armed with heightened capacity for ROS disposal. Oxidative stress regulators are highly enriched in HSCs and activate robust oxidative stress responses to scavenge ROS. FOXOs tend to be enriched in the nucleus of HSCs but excluded from the nucleus of differentiated progeny [73]. SIRT3 is highly expressed in HSCs but its expression is much repressed in differentiated hematopoietic cells [23].
The transition of HSCs from the quiescent state to proliferation is regulated by a metabolic checkpoint that monitors the health of mitochondria and repairs mitochondrial damage before the cells progress through the restriction point and enter the cell cycle [74, 75]. Mitochondrial damage beyond repair leads to cell death. A tight correlation has been observed between increased ROS levels and HSC proliferation and death in numerous mouse models. Increased ROS production due to aberrant activation of the TSC-mTOR pathway [52, 76] or defective DNA damage response [77, 78], failure to engage the oxidative stress response resulting from defective FOXOs, SIRT3, Nrf2, or thioredoxin-interacting protein (Txnip) [23, 79–82], and dysregulation of ROS signaling, such as SIRT7-mediated mitochondrial unfolded protein response [75, 83] or p38 MAPK signaling [84], all result in loss of HSC quiescence and maintenance, and attrition of HSC regenerative capacity. These studies suggest that ROS act as a signal that dictates the balance between HSC quiescence, proliferation, and survival. The ROS level is increased in HSCs with aging [84], consistent with the role of ROS as a trigger of the functional deterioration of HSC aging. ROS are also essential regulators of neural stem cells [85–88] and intestinal stem cells [89]. Thus, ROS regulation of stem cell fate decision appears to be conserved across tissues.
ROS management differs in HSCs and their progeny. Hematopoietic progenitor cells (HPCs) display increased levels of ROS under physiological conditions. Scavenging ROS from the HPCs prevents their differentiation into mature blood cells, while increasing the ROS level triggers precocious differentiation into mature blood cells, indicating that ROS function as signaling molecules that prime the differentiation of HPCs [90]. How ROS signaling leads to cell fate decision making remains unclear, but different cellular programs are likely to be activated by ROS signaling to render distinct cell fate decisions in HSCs and HPCs (Figure 2).
Figure 2. ROS govern cell fate decision in hematopoietic stem progenitor cells.
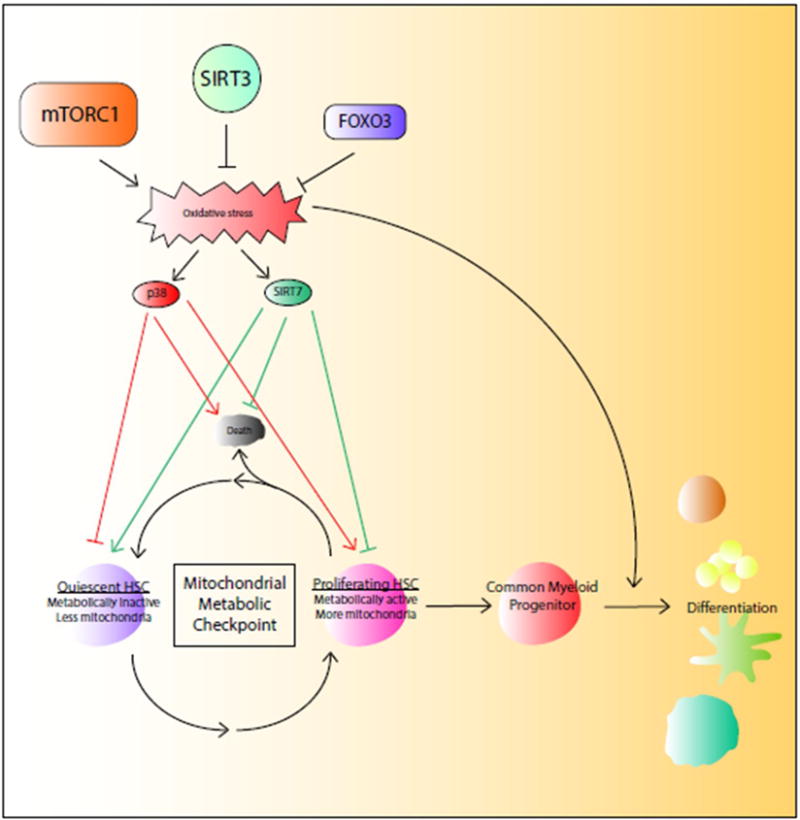
The transition of hematopoietic stem cells (HSCs) from quiescence to proliferation is associated with increased mitochondrial biogenesis and oxidative stress, which is monitored by the mitochondrial metabolic checkpoint that determines the cell fate decision on quiescence, proliferation, or death through SIRT7-mediated mitochondrial unfolded protein response or p38 signaling. ROS prime common myeloid progenitor cells for differentiation.
If ROS are indeed a cause of stem cell aging, it is tempting to ask whether ROS cause aging due to the chronic accumulation of oxidative damage over lifetime or acute effects of high levels of ROS, and whether ROS-induced physiological aging is reversible. The expression of SIRT3 and SIRT7 is reduced in aged HSCs, which may contribute to increased ROS level and defective ROS signaling in aged HSCs [23, 75]. Importantly, overexpression of SIRT3 or SIRT7 improves the regenerative capacity of aged HSCs [23, 75]. These studies suggest that ROS-induced physiological aging is likely to be acute and reversible. It appears that HSC aging is not due to the passive accumulation of cellular damage over the lifetime but to the regulated repression of cellular protective programs, giving hope for targeting the dysregulated cellular protective programs to reverse HSC aging and rejuvenate tissue homeostasis.
Innate Immunity
Host defense is dependent on the burst of oxidative stress in phagocytes as part of the innate immune response during infection. Various pathways are employed by immune cells to produce ROS and facilitate the immediate killing of pathogens. Neutrophils and inflammatory monocytes produce ROS by metabolic enzymes, such as NADPH oxidases and myeloperoxidases [91]. In contrast, killer lymphocytes deliver cytotoxic granules containing granzymes into the invading bacterial strains, where granzymes disrupt the electron transport chain proteins as well as the antioxidant proteins, resulting in increased ROS [92]. It was shown recently that activated macrophages undergo a metabolic reprogramming and the resulting increase in mitochondrial oxidation of succinate and an elevation of mitochondrial membrane potential combine to drive mitochondrial ROS production [93]. Interestingly, pathogens also develop defense mechanisms against the oxidative stress generated by the host immune system [94, 95]. The diverse pathways employed by innate immune cells to produce ROS and various strategies developed by pathogens to defend against oxidative stress are a strong indication that oxidative stress is crucial in supporting innate immunity (Figure 3).
Figure 3. Oxidative stress supports the innate immunity.
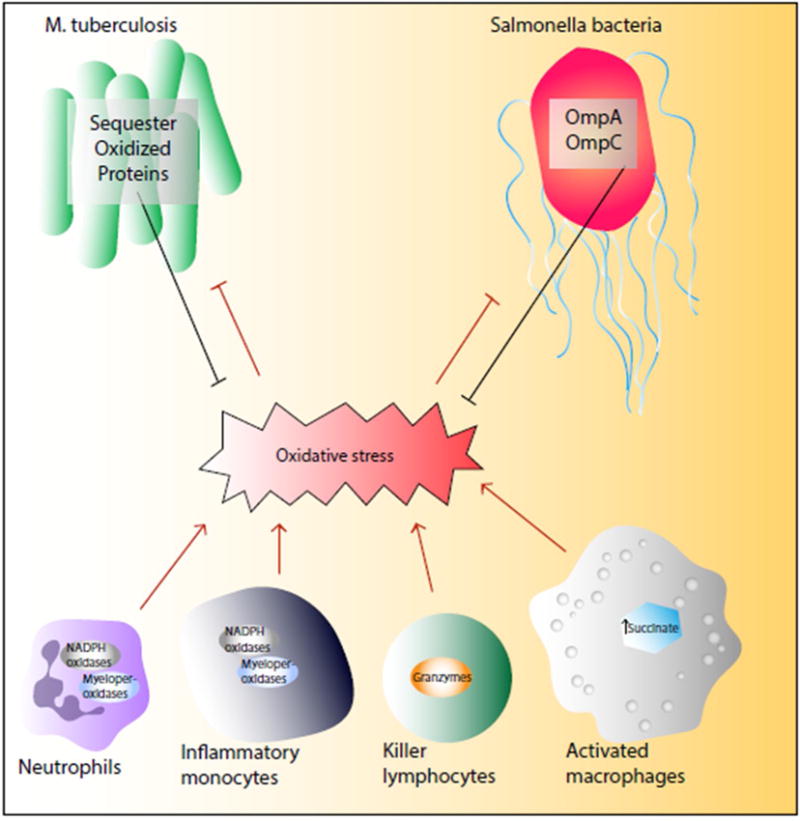
Phagocytes employ different mechanisms of ROS production to facilitate pathogen clearance while pathogens develop strategies to alleviate ROS damage.
Although oxidative stress is necessary in ridding host cells of foreign pathogens, the host may suffer from tissue damage and dysfunction due to over-accumulation of ROS that is characteristic of the inflammatory processes. Mice deficient in NRF2 display enhanced host defense 4 hours after the S. pneumonia challenge, but exhibit increased lung injury and death in 24 hours [96]. Similarly, mice deficient in Negative Regulator of ROS (NRROS), a negative regulator of ROS production in phagocytes, exhibit enhanced bactericidal activity, but develop severe experimental autoimmune encephalomyelitis due to oxidative damage in the central nervous system [97]. Thus, oxidative stress levels in the innate immune cells require a tightly controlled regulation in order to ensure effective host defense while maintaining the tissue integrity.
The findings in stem and progenitor cells as well as innate immunity have challenged the view that ROS are produced as the byproducts of cellular respiration and supported the idea that ROS can act as signaling molecules and support physiological functions. These new findings make us rethink the evolution of ROS production and motivate future studies to explore new roles of ROS in supporting cellular physiology. Along this line, it is likely that the robustness of oxidative stress regulation but not the inhibition of ROS that is optimal for slowing aging.
Cancer
ROS cause DNA damage and genomic instability, a major driving force for tumorigenesis. Thus, ROS have long been thought to play a critical role in tumor initiation. Indeed, SIRT3 KO cells exhibit increased genomic instability and susceptibility to transformation, and SIRT3 KO mice develop spontaneous tumors [98]. Complementary to this traditional view, recent studies have revealed several novel roles ROS play in tumor initiation. SIRT3 deficiency results in ROS stabilization and upregulation of hypoxia-inducible factor-1α (HIF1α) [25], a transcription factor that triggers a metabolic reprogramming characterized by increased glycolysis in the presence of oxygen, a hallmark of tumor cells known as the Warburg effect [99]. This metabolic feature provides tumor cells with the substrates required for biomass generation. Furthermore, ROS alter the cellular signaling events to promote cancer cell proliferation [100]. Finally, heightened ROS levels can result in necrosis, an unprogrammed cell death pathway that triggers the recruitment of macrophages followed by the secretion of inflammatory mediators. The chronic inflammatory state in the tumor niche provides a warm house for malignant transformation [101].
Although oxidative stress has a pro-cancer role at the tumor initiation stage by promoting transformation, proliferation, and establishment of the tumor microenvironment, cancer cells seem to hijack the oxidative stress response to survive (Figure 4). As a result, oxidative stress also plays an anti-cancer role at the development stage by limiting tumor progression and metastasis. In lung cancer cells, the regulatory property of the glycolytic enzyme pyruvate kinase M2 (PKM2) for ROS detoxification is essential for PKM2 to promote tumor progression [65]. The transition into anchorage-independent growth is associated with the induction of isocitrate dehydrogenase-1 (IDH1), which mitigates ROS and promotes anchorage-independent growth [66]. Further, melanoma cells that are capable of distant metastasis undergo metabolic changes during metastasis to increase their capacity to withstand oxidative stress [68], and age-related changes in the tumor microenvironment also affect cancer cell metastasis through the modulation of the oxidative stress response pathways [67]. In line with the importance of redox control of tumor development, inducing heightened levels of oxidative stress in cancer cells has been explored as a potential therapy for certain cancers [69].
Figure 4. Oxidative stress regulation of cancer.
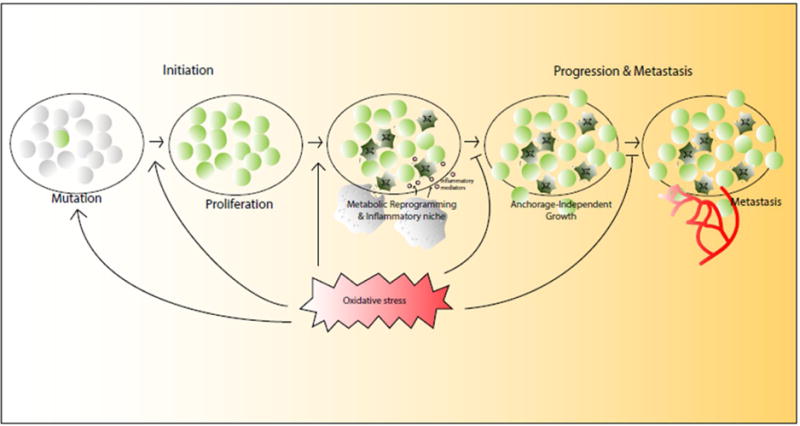
Although oxidative stress has a pro-cancer role at the tumor initiation stage by promoting DNA damage, proliferation, metabolic reprogramming, and the establishment of the tumor microenvironment, cancer cells also hijack the oxidative stress responses to survive, progress, and metastasize.
Conclusion and Perspectives
Recent molecular links between nutrient sensors and the oxidative stress response support the notion that CR triggers an active defense program to reduce oxidative stress. It would be important to determine whether nutrient sensors are required to reduce oxidative stress during CR, and if so, whether they reduce oxidative stress in particular cell types or ubiquitously. Much work is needed to demonstrate that these nutrient sensors indeed exert their physiological functions to impact the aging process and development of diseases at least in part by reducing oxidative stress. In this regard, the studies in SIRT3 are the most advanced and SIRT3 KO mice have been extensively characterized in various cell types and tissues for oxidative stress and tissue functions. Gain-of-function studies would be of particular interest, as this information is relevant for therapeutic implications. Mouse models for other nutrient sensing pathways have been extensively characterized, although oxidative stress has not been analyzed extensively in these mouse models and whether redox regulation contributes to the physiological functions of these nutrient sensing pathways remains largely unknown [54, 102–104].
Compelling evidence supports the new role of ROS as signaling molecules to support physiological functions, in addition to damaging agents. It is also increasingly appreciated the effects of ROS on aging and cancer are more complex than previously thought. Future studies will unravel new biological processes and physiological functions that require ROS as signaling molecules and elucidate whether CR interferes with these processes. Much work is needed to scrutinize the context when ROS promote or prevent aging or cancer. So far, the evidence that ROS promote cancer initiation but suppress cancer progression and metastasis derives from studies of different genetic manipulations in different cancers. Studies of the same genetic manipulations of ROS using diverse cancer models are necessary to crystalize the differential effects of ROS at the various developmental stages of cancers. It is also tempting to ask whether CR has differential effects at different stages of cancer development. This knowledge builds a solid foundation for designing CR mimetics and antioxidants with the maximal health benefits.
Outstanding Questions.
Are nutrient sensors required to reduce oxidative stress during CR? Do nutrient sensors exert their physiological functions at least in part through their redox regulation?
In addition to cell fate decision and innate immunity, what biological processes and physiological functions require ROS as signaling molecules? Does CR interfere with these processes?
In what context do ROS promote or prevent aging or cancer? Does CR have differential effects at different stages of cancer development?
Can small molecule regulators of nutrient sensors function as CR mimetics to extend lifespan and healthspan?
Acknowledgments
Supported by NIH R01 AG040990 (D.C.), R01DK101885 (D.C.), National Institute of Food and Agriculture (D.C.), PackerWentz Endowment (D.C.), Glenn/AFAR Scholarship (H.L.), and James C.Y. Soong Fellowship (H. C.).
References
- 1.McCay CM, Crowell MF. Prolonging the life span. The Scientific Monthly. 1934;39(5):405–414. [Google Scholar]
- 2.Arumugam TV, et al. Age and energy intake interact to modify cell stress pathways and stroke outcome. Ann Neurol. 2010;67(1):41–52. doi: 10.1002/ana.21798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Colman RJ, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325(5937):201–4. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Godar RJ, et al. Repetitive stimulation of autophagy-lysosome machinery by intermittent fasting preconditions the myocardium to ischemia-reperfusion injury. Autophagy. 2015;11(9):1537–60. doi: 10.1080/15548627.2015.1063768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Longo VD, Mattson MP. Fasting: molecular mechanisms and clinical applications. Cell Metab. 2014;19(2):181–92. doi: 10.1016/j.cmet.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weindruch R, Walford RL. Retardation of Aging and Disease by Dietary Restriction. CC Thomas; pringfield, IL: 1988. [Google Scholar]
- 7.Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273(5271):59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen D, et al. Increase in activity during calorie restriction requires Sirt1. Science. 2005;310(5754):1641. doi: 10.1126/science.1118357. [DOI] [PubMed] [Google Scholar]
- 9.Qiu X, et al. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010;12(6):662–7. doi: 10.1016/j.cmet.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 10.Someya S, et al. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143(5):802–12. doi: 10.1016/j.cell.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120(4):483–95. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 12.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11(3):298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 13.Reczek CR, Chandel NS. ROS-dependent signal transduction. Curr Opin Cell Biol. 2015;33:8–13. doi: 10.1016/j.ceb.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chae YC, et al. Landscape of the mitochondrial Hsp90 metabolome in tumours. Nat Commun. 2013;4:2139. doi: 10.1038/ncomms3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liang Q, et al. Bioenergetic and autophagic control by Sirt3 in response to nutrient deprivation in mouse embryonic fibroblasts. Biochem J. 2013;454(2):249–57. doi: 10.1042/BJ20130414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Z, et al. Nutrient deprivation-related OXPHOS/glycolysis interconversion via HIF-1alpha/C-MYC pathway in U251 cells. Tumour Biol. 2016;37(5):6661–71. doi: 10.1007/s13277-015-4479-7. [DOI] [PubMed] [Google Scholar]
- 17.Bruss MD, et al. Calorie restriction increases fatty acid synthesis and whole body fat oxidation rates. Am J Physiol Endocrinol Metab. 2010;298(1):E108–16. doi: 10.1152/ajpendo.00524.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nisoli E, et al. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005;310(5746):314–7. doi: 10.1126/science.1117728. [DOI] [PubMed] [Google Scholar]
- 19.Schwer B, Verdin E. Conserved metabolic regulatory functions of sirtuins. Cell Metab. 2008;7(2):104–12. doi: 10.1016/j.cmet.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 20.Hallows WC, et al. Sirt3 promotes the urea cycle and fatty acid oxidation during dietary restriction. Mol Cell. 2011;41(2):139–49. doi: 10.1016/j.molcel.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi T, et al. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J Biol Chem. 2005;280(14):13560–7. doi: 10.1074/jbc.M414670200. [DOI] [PubMed] [Google Scholar]
- 22.Bell EL, et al. SirT3 suppresses hypoxia inducible factor 1alpha and tumor growth by inhibiting mitochondrial ROS production. Oncogene. 2011;30(26):2986–96. doi: 10.1038/onc.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brown K, et al. SIRT3 reverses aging-associated degeneration. Cell Rep. 2013;3(2):319–27. doi: 10.1016/j.celrep.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen IC, et al. Role of SIRT3 in the regulation of redox balance during oral carcinogenesis. Mol Cancer. 2013;12:68. doi: 10.1186/1476-4598-12-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finley LW, et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell. 2011;19(3):416–28. doi: 10.1016/j.ccr.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirschey MD, et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol Cell. 2011;44(2):177–90. doi: 10.1016/j.molcel.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jang YC, et al. Dietary restriction attenuates age-associated muscle atrophy by lowering oxidative stress in mice even in complete absence of CuZnSOD. Aging Cell. 2012;11(5):770–82. doi: 10.1111/j.1474-9726.2012.00843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jing E, et al. Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc Natl Acad Sci U S A. 2011;108(35):14608–13. doi: 10.1073/pnas.1111308108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Traba J, et al. Fasting and refeeding differentially regulate NLRP3 inflammasome activation in human subjects. J Clin Invest. 2015;125(12):4592–600. doi: 10.1172/JCI83260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu W, et al. Loss of SIRT3 Provides Growth Advantage for B Cell Malignancies. J Biol Chem. 2016;291(7):3268–79. doi: 10.1074/jbc.M115.702076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng A, et al. Mitochondrial SIRT3 Mediates Adaptive Responses of Neurons to Exercise and Metabolic and Excitatory Challenges. Cell Metab. 2016;23(1):128–42. doi: 10.1016/j.cmet.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tao R, et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell. 2010;40(6):893–904. doi: 10.1016/j.molcel.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shin J, Zhang D, Chen D. Reversible acetylation of metabolic enzymes celebration: SIRT2 and p300 join the party. Mol Cell. 2011;43(1):3–5. doi: 10.1016/j.molcel.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 34.Boily G, et al. SirT1 regulates energy metabolism and response to caloric restriction in mice. PLoS One. 2008;3(3):e1759. doi: 10.1371/journal.pone.0001759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herranz D, Serrano M. SIRT1: recent lessons from mouse models. Nat Rev Cancer. 2010;10(12):819–23. doi: 10.1038/nrc2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kume S, et al. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J Clin Invest. 2010;120(4):1043–55. doi: 10.1172/JCI41376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brunet A, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303(5666):2011–5. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 38.Kops GJ, et al. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419(6904):316–21. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- 39.Greer EL, Brunet A. Different dietary restriction regimens extend lifespan by both independent and overlapping genetic pathways in C. elegans. Aging Cell. 2009;8(2):113–27. doi: 10.1111/j.1474-9726.2009.00459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greer EL, et al. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr Biol. 2007;17(19):1646–56. doi: 10.1016/j.cub.2007.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson EC, et al. Altered metabolism and persistent starvation behaviors caused by reduced AMPK function in Drosophila. PLoS One. 2010;5(9) doi: 10.1371/journal.pone.0012799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schulz TJ, et al. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007;6(4):280–93. doi: 10.1016/j.cmet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 43.Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013;493(7432):338–45. doi: 10.1038/nature11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kapahi P, et al. With TOR, less is more: a key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab. 2010;11(6):453–65. doi: 10.1016/j.cmet.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mirzaei H, Suarez JA, Longo VD. Protein and amino acid restriction, aging and disease: from yeast to humans. Trends Endocrinol Metab. 2014;25(11):558–66. doi: 10.1016/j.tem.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Molin M, et al. Life span extension and H(2)O(2) resistance elicited by caloric restriction require the peroxiredoxin Tsa1 in Saccharomyces cerevisiae. Mol Cell. 2011;43(5):823–33. doi: 10.1016/j.molcel.2011.07.027. [DOI] [PubMed] [Google Scholar]
- 47.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13(4):251–62. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Efeyan A, Comb WC, Sabatini DM. Nutrient-sensing mechanisms and pathways. Nature. 2015;517(7534):302–10. doi: 10.1038/nature14190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hinnebusch AG. Translational regulation of GCN4 and the general amino acid control of yeast. Annu Rev Microbiol. 2005;59:407–50. doi: 10.1146/annurev.micro.59.031805.133833. [DOI] [PubMed] [Google Scholar]
- 50.Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485(7400):661–5. doi: 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saito Y, et al. AMPK Protects Leukemia-Initiating Cells in Myeloid Leukemias from Metabolic Stress in the Bone Marrow. Cell Stem Cell. 2015;17(5):585–96. doi: 10.1016/j.stem.2015.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen C, et al. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med. 2008;205(10):2397–408. doi: 10.1084/jem.20081297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ravindran R, et al. The amino acid sensor GCN2 controls gut inflammation by inhibiting inflammasome activation. Nature. 2016;531(7595):523–7. doi: 10.1038/nature17186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morita M, et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013;18(5):698–711. doi: 10.1016/j.cmet.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 56.Chen Y, et al. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011;12(6):534–41. doi: 10.1038/embor.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alexander A, et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci U S A. 2010;107(9):4153–8. doi: 10.1073/pnas.0913860107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hawley SA, et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010;11(6):554–65. doi: 10.1016/j.cmet.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zmijewski JW, et al. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J Biol Chem. 2010;285(43):33154–64. doi: 10.1074/jbc.M110.143685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Perez VI, et al. Is the oxidative stress theory of aging dead? Biochim Biophys Acta. 2009;1790(10):1005–14. doi: 10.1016/j.bbagen.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hanzen S, et al. Lifespan Control by Redox-Dependent Recruitment of Chaperones to Misfolded Proteins. Cell. 2016;166(1):140–51. doi: 10.1016/j.cell.2016.05.006. [DOI] [PubMed] [Google Scholar]
- 62.Nobrega-Pereira S, et al. G6PD protects from oxidative damage and improves healthspan in mice. Nat Commun. 2016;7:10894. doi: 10.1038/ncomms10894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kubben N, et al. Repression of the Antioxidant NRF2 Pathway in Premature Aging. Cell. 2016;165(6):1361–74. doi: 10.1016/j.cell.2016.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Campisi J. Cancer and ageing: rival demons? Nat Rev Cancer. 2003;3(5):339–49. doi: 10.1038/nrc1073. [DOI] [PubMed] [Google Scholar]
- 65.Anastasiou D, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334(6060):1278–83. doi: 10.1126/science.1211485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jiang L, et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature. 2016;532(7598):255–8. doi: 10.1038/nature17393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kaur A, et al. sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nature. 2016;532(7598):250–4. doi: 10.1038/nature17392. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 68.Piskounova E, et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527(7577):186–91. doi: 10.1038/nature15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yun J, et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science. 2015;350(6266):1391–6. doi: 10.1126/science.aaa5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Folmes CD, et al. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell. 2012;11(5):596–606. doi: 10.1016/j.stem.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Takubo K, et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell. 2010;7(3):391–402. doi: 10.1016/j.stem.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 72.Takubo K, et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell. 2013;12(1):49–61. doi: 10.1016/j.stem.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Naka K, et al. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature. 2010;463(7281):676–80. doi: 10.1038/nature08734. [DOI] [PubMed] [Google Scholar]
- 74.Mohrin M, Chen D. The mitochondrial metabolic checkpoint and aging of hematopoietic stem cells. Curr Opin Hematol. 2016;23(4):318–24. doi: 10.1097/MOH.0000000000000244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mohrin M, et al. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science. 2015;347(6228):1374–7. doi: 10.1126/science.aaa2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Juntilla MM, et al. AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood. 2010;115(20):4030–8. doi: 10.1182/blood-2009-09-241000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ito K, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431(7011):997–1002. doi: 10.1038/nature02989. [DOI] [PubMed] [Google Scholar]
- 78.Maryanovich M, et al. The ATM-BID pathway regulates quiescence and survival of haematopoietic stem cells. Nat Cell Biol. 2012;14(5):535–41. doi: 10.1038/ncb2468. [DOI] [PubMed] [Google Scholar]
- 79.Jung H, et al. TXNIP maintains the hematopoietic cell pool by switching the function of p53 under oxidative stress. Cell Metab. 2013;18(1):75–85. doi: 10.1016/j.cmet.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 80.Miyamoto K, et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell. 2007;1(1):101–12. doi: 10.1016/j.stem.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 81.Tothova Z, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128(2):325–39. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 82.Tsai JJ, et al. Nrf2 regulates haematopoietic stem cell function. Nat Cell Biol. 2013;15(3):309–16. doi: 10.1038/ncb2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shin J, et al. SIRT7 represses Myc activity to suppress ER stress and prevent fatty liver disease. Cell Rep. 2013;5(3):654–65. doi: 10.1016/j.celrep.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ito K, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med. 2006;12(4):446–51. doi: 10.1038/nm1388. [DOI] [PubMed] [Google Scholar]
- 85.Paik JH, et al. FoxOs cooperatively regulate diverse pathways governing neural stem cell homeostasis. Cell Stem Cell. 2009;5(5):540–53. doi: 10.1016/j.stem.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Renault VM, et al. FoxO3 regulates neural stem cell homeostasis. Cell Stem Cell. 2009;5(5):527–39. doi: 10.1016/j.stem.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Webb AE, et al. FOXO3 shares common targets with ASCL1 genome-wide and inhibits ASCL1-dependent neurogenesis. Cell Rep. 2013;4(3):477–91. doi: 10.1016/j.celrep.2013.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yeo H, et al. FoxO3 coordinates metabolic pathways to maintain redox balance in neural stem cells. EMBO J. 2013;32(19):2589–602. doi: 10.1038/emboj.2013.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hochmuth C, et al. Redox regulation by Keap1 and Nrf2 controls intestinal stem cell proliferation in Drosophila. Cell Stem Cell. 2011;8(2):188–99. doi: 10.1016/j.stem.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Owusu-Ansah E, Banerjee U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature. 2009;461(7263):537–41. doi: 10.1038/nature08313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Burton NA, et al. Disparate impact of oxidative host defenses determines the fate of Salmonella during systemic infection in mice. Cell Host Microbe. 2014;15(1):72–83. doi: 10.1016/j.chom.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 92.Walch M, et al. Cytotoxic cells kill intracellular bacteria through granulysin-mediated delivery of granzymes. Cell. 2014;157(6):1309–23. doi: 10.1016/j.cell.2014.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mills EL, et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell. 2016;167(2):457–470 e13. doi: 10.1016/j.cell.2016.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.van der Heijden J, et al. Salmonella Rapidly Regulates Membrane Permeability To Survive Oxidative Stress. MBio. 2016;7(4) doi: 10.1128/mBio.01238-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vaubourgeix J, et al. Stressed mycobacteria use the chaperone ClpB to sequester irreversibly oxidized proteins asymmetrically within and between cells. Cell Host Microbe. 2015;17(2):178–90. doi: 10.1016/j.chom.2014.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gomez JC, et al. Nrf2 Modulates Host Defense during Streptococcus pneumoniae Pneumonia in Mice. J Immunol. 2016;197(7):2864–79. doi: 10.4049/jimmunol.1600043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Noubade R, et al. NRROS negatively regulates reactive oxygen species during host defence and autoimmunity. Nature. 2014;509(7499):235–9. doi: 10.1038/nature13152. [DOI] [PubMed] [Google Scholar]
- 98.Kim HS, et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell. 2010;17(1):41–52. doi: 10.1016/j.ccr.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sebastián C, et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell. 2012;151(6):1185–99. doi: 10.1016/j.cell.2012.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tonks NK. Redox redux: revisiting PTPs and the control of cell signaling. Cell. 2005;121(5):667–70. doi: 10.1016/j.cell.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 101.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Castilho BA, et al. Keeping the eIF2 alpha kinase Gcn2 in check. Biochim Biophys Acta. 2014;1843(9):1948–68. doi: 10.1016/j.bbamcr.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 103.Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460(7255):587–91. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Viollet B, et al. AMPK: Lessons from transgenic and knockout animals. Front Biosci (Landmark Ed) 2009;14:19–44. doi: 10.2741/3229. [DOI] [PMC free article] [PubMed] [Google Scholar]