

Abstract
Glucocorticoids are steroid hormones that regulate diverse cellular functions and are essential to facilitate normal physiology. Yet, stress-induced levels of glucocorticoids result in several pathologies including profound reproductive dysfunction. Compelling new evidence indicates glucocorticoids are crucial to the establishment and maintenance of reproductive function. The fertility promoting or inhibiting activity of glucocorticoids depends on timing, dose, and glucocorticoid-responsiveness within a given tissue, which is mediated by the glucocorticoid receptor. The glucocorticoid receptor gene and protein are subject to cellular processing, contributing to signaling diversity and providing a mechanism by which both physiological and stress-induced levels of glucocorticoids function in a cell-specific function. Understanding how glucocorticoids regulate fertility and infertility may lead to novel approaches to the regulation of reproductive function.
Keywords: glucocorticoids, fertility, HPA axis, uterus, ovary, testis
INTRODUCTION
Glucocorticoids are essential for stress adaptation through regulation of metabolic activity, behavior, and reproduction, in which trade-offs are made between immediate survival and future offspring. Stress-induced levels of glucocorticoids mediate this process through direct and indirect actions on the hypothalamic-pituitary-gonadal axis (HPG). Interestingly, the presenting complaint of the first Cushing’s syndrome (see glossary) patient in 1912 was secondary amenorrhea. However, the molecular mechanisms by which high levels of glucocorticoids suppress fertility are complex and have yet to be fully discovered. Moreover, basal glucocorticoid levels are critically important for the establishment and maintenance of fertility, as evidenced in patients with Addison’s disease that present with premature ovarian failure and oligospermia [1, 2]. Our current understanding of glucocorticoid signaling in the context of reproductive physiology is limited. Notably, a balance between low and high levels of glucocorticoids differentiates between fertility and infertility and elucidating the molecular mechanisms governing this shift is a potential new direction for future research.
Circulating levels of glucocorticoids are a function of the activation status of the hypothalamic-pituitary-adrenal (HPA) axis (Box 1). The biological effects of glucocorticoids are determined by serum concentrations in concert with relative expression of the 11β-hydroxysteroid dehydrogenase isoenzymes (11β-HSD) and the glucocorticoid receptor (gene: NR3C1; protein: GR), a ligand-dependent transcription factor (Box 2). The transcriptional activity of GR demonstrates profound diversity, stimulating or suppressing the expression of 10–20% of all genes in the human genome (Box 3) [3]. Identifying which genes are targets of GR regulation in reproductive tissues will provide a basis for understanding the mechanisms by which glucocorticoids regulate reproduction and a framework for the complex regulatory networks. Establishing which signaling networks are associated with stress-induced infertility will ultimately provide new avenues for therapy. Here, we review recent findings of glucocorticoid action in the reproductive system, summarizing historical data and highlighting new insights where research is ongoing.
Box 1. Glucocorticoid Production and Bioavailability.
Glucocorticoids are steroid hormones synthesized and released by the adrenal cortex under the regulation of the HPA axis. The production of glucocorticoids by the HPA axis is pulsatile, demonstrating both circadian and ultradian rhythms. Corticotrophin-releasing hormone (CRH) and arginine vasopressin are secreted from the parvicellular neurons of the hypothalamus into the pituitary portal circulation, which stimulates adrenocorticotropic hormone (ACTH) release from the anterior pituitary gland. ACTH acts on the adrenal gland to induce steroidogenesis and the production of glucocorticoids. Endocrine feedback loops exist where glucocorticoids inhibit CRH expression and secretion and ACTH output [119–121]. The rhythmic secretion of glucocorticoids is critical to the maintenance of physiological homeostasis, but acute exposures to stress transiently induce HPA activity and glucocorticoid release.
Approximately 95% of secreted glucocorticoids circulate bound to cortisol-binding globulin (CBG) or albumin, and therefore, expression of these binding proteins determines the level of biologically free glucocorticoids [122, 123]. The circulating half-life of glucocorticoids is variable (66–120 minutes) but glucocorticoids bound to CBG have a longer half-life compared to unbound glucocorticoids [124, 125]. Unbound glucocorticoids are highly lipophilic and able to cross the plasma membrane through simple diffusion. In cells which express the multi-drug resistance p-glycoprotein, entry of glucocorticoids to the intracellular compartment can be restricted by active transport out of the cell [126]. Within the cell, interconversion of glucocorticoids to the active (cortisol and corticosterone) or inactive (cortisone and 11-dehydrocorticosterone) forms by 11β-HSD I and II controls ligand availability to GR [127]. Collectively, the biological activity of glucocorticoids is a balance between synthesis and secretion, diffusion and transport, and metabolism and clearance.
Box 2. The Glucocorticoid Receptor: Structure and Regulation.
The molecular response to glucocorticoids is mediated by intracellular GR. GR is a member of the nuclear receptor superfamily of ligand-dependent transcription factors [128]. All members of this superfamily share a similar domain structure: an N-terminal transactivation (AF1) domain, a central DNA-binding domain (DBD), and a C-terminal ligand-binding domain (LBD) (Figure I). In addition to comprising the ligand-binding pocket, the LBD also contains sequences important for receptor dimerization and nuclear localization, and a second transactivation domain (AF2), which mediates ligand-dependent interactions with coregulators. The DBD and the LBD are separated by a small flexible hinge region, which plays a role in ligand-binding mediated conformational changes.
Human GR is the product of one gene (NR3C1) consisting of nine exons. Exon 1 forms the 5′-untranslated region of human GR, and exon 2 encodes the NTD, which is poorly conserved and therefore the most variable domain among nuclear receptors. The DBD, the most conserved region in the nuclear receptor family, is comprised of exons 3 and 4, and exons 5–9 encode the hinge region and LBD. Alternative splicing of the NR3C1 gene primarily from the C’-terminal end produces the human GRα, GRβ, GRγ, GR-A, and GR-P transcriptional isoforms [129–132]. Research has mainly focused on glucocorticoid signaling through GRα and GRβ, which are identical proteins through amino acid 727 that diverge in their C-terminal exon 9. GRβ lacks sequences that encode helices 11 and 12 of the LBD and is therefore, unable to bind glucocorticoids [133]. GRβ was thought to function primarily as a dominant negative regulator of GRα, but genome-wide transcriptional analysis of overexpressed human GRβ indicates it maintains inherent transcriptional activity [130, 134, 135]. The sequence of GR-γdiffersfromGR-α by the inclusion of an arginine residue between exons 3 and 4 of the DBD from the use of an alternative intronic splice donor site [132]. This single amino acid insertion demonstrates the functional importance of this region, as the transcriptional activity of GR-γ is significantly reduced [136, 137]. Interestingly, differences in the DBD of GR-γ alter DNA binding sequence specificity and result in a unique transcriptome [138, 139]. The GR-A and GR-P isoforms contain incomplete LBDs and relatively little is known about their functions in vivo [131, 140]. Each GR transcriptional splice variant is capable of generating additional isoforms through alternative translation initiation mechanisms [141]. In exon 2, eight highly conserved AUG start codons generate the GR-A, -B, -C1, -C2, -C3, -D1, -D2, and -D3 isoforms. These translational isoforms have distinct tissue distribution and subcellular localization, regulate unique genes, and differentially regulate the cellular response to glucocorticoids [142–144].
Box 3. Glucocorticoid Receptor Signaling.
Once in the cytoplasm, glucocorticoids bind GR resulting in a conformational change, un-masking of the nuclear localization signals, and subsequent nuclear translocation of the glucocorticoid-GR complex. Classically, glucocorticoids were thought to act exclusively through the genomic actions of activated GR. Recent evidence indicates glucocorticoids can also act through signaling cascades secondary to the conformational change of the glucocorticoid-GR complex or through extranuclear glucocorticoid receptors, though the identity of the membrane-associated GR remains unknown [145–147]. In the nucleus, ligand-bound GR mediates the genomic actions of GR by regulating gene transcription in direct and indirect mechanisms. Direct transcriptional regulation requires binding of GR homodimer complexes to well-defined GREs. However, motif analysis of GR chromatin immunoprecipitation sequencing (ChIP-seq) data sets evaluating GR-bound regions indicates <42% of binding peaks contain a high-stringency motif match [148]. This suggests that GRE binding is not a requirement for DNA association. Alternatively, the glucocorticoid-GR complex can directly bind distinct negative GREs (nGRE) to inhibit gene transcription [149]. GR association with DNA can also occur through mechanisms of indirect gene regulation by GR that involve interactions with other DNA-bound transcription factors (protein:protein tethering) or through cooperative binding with other transcription factors at neighboring sites (composite regulation). DNA- or complex-bound GR recruits cofactors which facilitate converting the signal into transcriptional activity via RNA polymerases or by regulating paused elongation complexes [150].
GR is constitutively expressed in almost all cells, but the actions of glucocorticoids demonstrate tissue- and cell-type specific diversity. The glucocorticoid response is determined by many factors including the availability of glucocorticoids and the sensitivity to glucocorticoids in a target tissue. The relative level of GR and the relative expression of the isoforms can determine the response to glucocorticoids in a given tissue or cell [151]. GR is subject to various post-translational modifications, which can mediate subcellular trafficking, promoter specificity, cofactor interaction, receptor stability, and turnover [152]. At specific gene loci, the response to glucocorticoids is regulated by the recruitment of cofactors, which can act in varying mechanisms, including: remodeling the chromatin, facilitating the assembly of transcriptional machinery, or modifying histones or other components of the transcription factor complex [153–156]. These coregulators of GR function maintain tissue-specific expression [157, 158]. The unique transcriptional profiles of GR also rely on distal enhancers that loop to the promoter of target genes and open regions of chromatin, which are highly cell-type specific [159, 160]. Individual glucocorticoid sensitivity has been associated with single nucleotide polymorphisms in NR3C1 that alter protein stability, ligand binding affinity, transactivating capacity, and interactions with co-activators or co-repressors [161]. Polymorphisms in the genes that encode the 11-β- hydroxysteroid dehydrogenase isoenzymes have been identified and may also contribute to glucocorticoid sensitivity [162–164].
Glucocorticoid actions in the hypothalamus and pituitary: recent developments
The hypothalamus and pituitary preside over reproductive physiology by acting as a central terminal for the input and output of endocrine signals [4]. Gonadotropin-releasing hormone (GnRH) is synthesized and secreted by the hypothalamus, which then crosses through the hypophyseal portal system into the anterior pituitary. In the pituitary, GnRH induces the synthesis and release of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) from the gonadotropes, which trigger testosterone release from the testis and estradiol and progesterone release from the ovaries. A feedback loop exists by which testosterone inhibits the secretion of GnRH and therefore FSH and LH production. In females, ovarian estradiol also exerts negative feedback on the hypothalamus and pituitary. However, during the late follicular phase of the menstrual cycle, rising levels of estradiol from the follicle triggers a switch from negative to positive feedback on the hypothalamus resulting in increased GnRH secretion. During the luteal phase of the menstrual cycle, rising levels of progesterone negatively feedback to suppress activity in the hypothalamus and pituitary. At the late luteal phase the levels of estradiol and progesterone decline, allowing gonadotrophins to again rise and triggering the beginning of a new cycle.
Increased glucocorticoid exposure, either by stress or exogenous treatment, leads to profound reproductive dysfunction through well-described effects in the hypothalamus and pituitary [5–7]. Glucocorticoids modulate the HPG axis by directly inhibiting the release of GnRH from the hypothalamus and the synthesis and release of gonadotropins from the pituitary. However, the signaling pathways by which this occurs are still under investigation and are further complicated by the recent discovery of new neuropeptides, such as kisspeptin (KISS1) and gonadotropin-inhibitory hormone (GnIH). These neuropeptides have opposing effects on GnRH release from the hypothalamus and are responsive to high levels of glucocorticoids. KISS1 exerts stimulatory effects on GnRH secretion through its cognate receptor KISS-1R (also known as g-protein coupled receptor 54; GPR54) that is co-expressed in GnRH neurons. KISS1 neurons in the anteroventral periventricular nucleus and periventricular nucleus continuum of the preoptic area of the hypothalamus express GR, suggesting that glucocorticoids can directly act in these neurons [8]. In mice, corticosterone treatment diminished hypothalamic expression of KISS1 during the estradiol-induced LH surge and decreased the activation of KISS1 neurons [9]. Impairment of KISS1 neurons represents a newly discovered mechanism by which glucocorticoids are able to suppress the HPG axis. GnIH (mammalian orthologue Rfamide-related peptide-3 (RFRP3)) neurons inhibit the activity of GnRH neurons and KISS1 neurons [10]. Acute (audiovisual imposition of a predator, hypoglycemia, or lipopolysaccharide challenge) and chronic stress (synthetically-induced plasma cortisol levels) in sheep increase the function of GnIH neurons and increased contacts with GnRH neurons [11]. Immobilization stress-induced up-regulation of GnIH led to inhibition of LH release from the pituitary [12]. Adrenalectomy blocked induction of GnIH, suggesting that glucocorticoids released in response to stress mediated this effect. NR3C1 is coexpressed in GnIH neurons, and corticosterone treatment in quail and a rat hypothalamic neuronal cell line increased GnIH expression through glucocorticoid response elements (GREs) in the GnIH promotor [13]. Regulation of GnRH expression and release by KISS1 and GnIH represent two novel mechanisms by which glucocorticoids indirectly modulate reproductive functions of the hypothalamus and pituitary.
Glucocorticoids regulate the function of organs in the reproductive tract
Ovary
In addition to the effects on ovarian cyclicity mediated through the hypothalamus and pituitary, glucocorticoids impact ovarian physiology through regulating the functions of granulosa cells, oocytes, cumulus cells, and luteal cells [9]. Glucocorticoids differentially induce and repress steroidogenesis in the ovary. In rat preovulatory granulosa cells, dexamethasone increased production of progesterone by increasing the expression of caspase-3 and the steroidogenic proteins steroidogenic acute regulatory protein (StAR) and cytochrome P450 cholesterol side chain cleavage enzyme [14]. Glucocorticoids (cortisol and dexamethasone) have also been shown to decrease LH-induced StAR protein levels and progesterone production in rat and human granulosa cells [15, 16]. These differences may reflect differences in dose or stage of follicular development. Cortisol treatment or elevated glucocorticoid levels due to restraint stress in mice impairs oocyte development potential, when evaluated ex vivo [17]. Isolated mural granulosa cells demonstrated increased apoptosis in response to elevated cortisol, which was associated with higher levels of Fas ligand, and cumulus cells were sensitized to undergo apoptosis following serum-starved culture conditions. Glucocorticoid exposure is also associated with decreased mRNA expression of the NR3C1 and ovarian growth factors insulin-like growth factor-1 (IGF-1) and brain-derived neurotrophic factor (BDNF) in mural granulosa cells. Interestingly, culturing of in vitro fertilized bovine oocytes increased blastocyst development rates, indicating that the response of oocytes to glucocorticoids may be context dependent [18]. Moreover, what is known regarding the function of endogenous glucocorticoids in ovarian physiology stems largely from exposure studies and may not represent direct functions that occur at physiological levels in the ovary.
Uterus, placenta, and labor
Glucocorticoid regulation of uterine biology has historically been described in terms of the antagonistic effects glucocorticoids on estrogen action and only recently been explored as an independent regulator of uterine function. Dexamethasone blocks estrogen-induced uterine growth and proliferation in mice, reducing the number of implantation sites when administered prior to estradiol-induced implantation [19, 20]. Gene ontology analysis of dexamethasone and estradiol regulated genes in the mouse uterus indicates that cell cycle and embryonic development are among the top regulated networks [21]. Dexamethasone administration in mice also uniquely regulates the expression of genes whose functions are related to cellular development, growth and proliferation, and signaling. In the neonatal mouse uterus, dexamethasone inhibits epithelial cell proliferation through both GR-dependent and -independent mechanisms [22]. Decidualized human endometrial cells also demonstrate a unique GR-dependent transcriptome enriched for Krüppel-associated box domain containing zinc-finger proteins, a family of transcriptional repressors [23]. Interestingly, GR signaling is responsible for repressing global expression of trimethylated H3K9 levels during stromal cell differentiation, suggesting that GR can both directly regulate gene expression and globally alter transcription through regulation of histone modifications in human uterine cells.
The placenta is developmentally plastic and therefore responsive to multiple stimuli, including altered glucocorticoid levels. Dexamethasone administered during early placentation inhibits proliferation, migration, and invasion of trophoblasts, induces placental oxidative damage, and manifests as reduced placental and fetal size [24]. Maternal administration of glucocorticoids reduces placental weight and is associated with reduced expression of proliferative markers and increased expression of apoptotic factors [25, 26]. In animal models of maternal prenatal stress, increased glucocorticoid levels are correlated with intrauterine growth restriction. Fetal growth restriction mediated through excess endogenous corticosterone or synthetic dexamethasone exposure results in altered placental transporter function and glucose transport and reduced expression of pro-angiogenic factors [27, 28]. Glucocorticoid exposure at the maternal-fetal interface is in part regulated through the expression of the 11β-HSD isoenzymes in discrete maternal and fetal compartments [29]. Dysregulated expression of the 11β-HSD isoenzymes by glucocorticoid excess further potentiates the adverse effects of high levels of glucocorticoid on placental function. Interestingly, expression of the NR3C1 translational isoforms varies between term and preterm human placenta and with prenatal glucocorticoid exposure, suggesting a mechanism by which changes to the GR protein profile may also mediate the response to increased glucocorticoids [30].
Studies conducted on animals and pregnant woman have indicated the importance of glucocorticoid signaling for the initiation of labor [31, 32]. In sheep, glucocorticoids drive the shift to an estrogen-primed contractile myometrium through the induction of the placental enzyme cytochrome P450 17αhydoxylase (P450c17), adrenal dehydroepiandrosterone sulfate (DHEAS) in humans [31]. Once the myometrium has achieved an estrogen-primed state, prostaglandins critically stimulate myometrial contractions, induce cervical ripening, and trigger fetal membrane rupture. Glucocorticoids contribute to the synthesis of prostaglandins by stimulating the production of cyclooxygenase-2 (COX-2) in human amnion cells and ovine placental trophoblast cells [33, 34]. During parturition, cortisol also contributes to the rupture of fetal membranes by inducing prostaglandins and inducing apoptosis in the amnion epithelial cells [35]. Intravenous dexamethasone improved the Bishop score of the cervix and reduced the length of time between labor induction and active labor in women. However, synthetic glucocorticoids administered preterm for fetal lung maturation do not induce labor, which may denote differential responses dependent on the extent of myometrial priming.
Fetal development in utero
It is well appreciated that the in utero environment directly contributes to physiological disorders that manifest postpartum (DOHaD hypothesis). Fetal exposure to high levels of glucocorticoids is believed to be a major contributor to early-life programming of adult onset disease. Data supporting the role of stress and elevated glucocorticoids in fetal programming has been documented in many species and thoroughly reviewed [36–41]. Interestingly programming of the HPA axis in response to prenatal stressors demonstrates sex-specific differences [42]. A systematic review of human studies concluded that the placenta of female offspring altered permeability to maternal glucocorticoids through regulation of the 11β-HSD enzymes in response to maternal stress [42]. Maternal exposure to dexamethasone in rats results in sexually dimorphic behavior in offspring, associated with structural changes to neuronal populations [43, 44]. In mice, the cardiovascular and renal renin-angiotensin-aldosterone systems also respond to elevated prenatal corticosterone in a sexually-dimorphic manner, potentially through altering adrenal function [45– 47]. Epigenetic factors may also play a key role in the glucocorticoid-driven effects of fetal programming, including altered methylation status of GR following neonatal dexamethasone exposure [48]. It is important to recognize the timing of exposure when considering the correlation between animal models and human fetuses. Development in the prenatal and early postnatal rodent is more similar to the first two trimesters of human pregnancy, and therefore may not accurately model the response in humans during the third trimester. Synthetic analogs of glucocorticoids are routinely administered during the third trimester to women at risk of early or late preterm delivery, therefore elucidating the long-term effects of late term glucocorticoid exposure is more accurately evaluated in a human population than a rodent model.
Testis
Both physiological and stress-induced levels of glucocorticoids also mediate testicular functions. Adrenalectomy in male rats demonstrated that homeostatic levels of adrenal hormones, including glucocorticoids, are required to maintain steroidogenesis, spermatogenesis, and sperm maturation [49]. Testosterone production increased following adrenalectomy in rats, which was prevented by glucocorticoid replacement [50]. Decreased spermatid number following adrenalectomy was rescued by concurrent dexamethasone treatment, indicating that adrenally produced hormones are responsible for maintaining sperm production. Interestingly, glucocorticoid replacement exacerbated morphological defects evident in the seminiferous tubules of adrenalectomized males, which may reflect differences between the endogenous and synthetic hormone. Alternatively, other endocrine hormones produced by the adrenal gland such as mineralocorticoids and epinephrine could be responsible for the maintenance of testicular morphology or morphology within the seminiferous epithelium could be sensitive to glucocorticoid dose. High levels of circulating glucocorticoids, related to stress, Cushing’s disease, or exogenous hormone treatment, suppress male fertility through repressing the expression of steroidogenic enzymes, inducing testicular oxidative stress, reducing the testicular response to gonadotropins, and prompting apoptosis of Leydig and germ cells [5]. Elevated glucocorticoids may also control Leydig cell number through the induction of cell cycle arrest, as was demonstrated following dexamethasone treatment in the R2C rat Leydig tumor cells in vitro [51]. Understanding the physiological range of adrenal steroids which supports male fertility is critical in patients who undergo adrenalectomy, have Addison’s disease or Cushing’s syndrome.
Mouse models for uncovering GR actions
Whole-body GR-null mice exhibit perinatal lethality due to defects in lung maturation and subsequent respiratory failure, which precludes the study of reproductive function in these mice [52]. Investigators have made use of tissue transplant studies from embryonic GR-null mice into syngeneic hosts, but this method is limited by the types of tissues that can be transplanted and endpoints which can be measured. The advent of Cre-lox technology allowed researchers to investigate the cell autonomous versus indirect effects of GR in tissues that critically support reproductive function. However, models which have investigated the direct actions of glucocorticoids are limited. Currently in the reproductive system, the tissue- or cell-type specific functions of GR have been evaluated in the uterus, Sertoli cells, mammary epithelial cells, and prostate epithelial cells [53–56].
Conditional ablation of uterine NR3C1 demonstrated the direct actions of glucocorticoids in early pregnancy, indicating that glucocorticoid signaling is critical to establishing uterine receptivity and the subsequent endometrial remodeling that occurs during decidualization. [53]. The immune response likely plays a key role in establishing endometrial receptivity [57], and genes related to the inflammatory response and immune cell trafficking were substantially dysregulated in the absence of uterine GR [53]. Thus, GR can locally coordinate the immune response in early pregnancy, thereby contributing to endometrial receptivity. Moreover, the expression of genes known to contribute to proper implantation was altered when NR3C1 was conditionally ablated from the uterus, suggesting that uterine glucocorticoid signaling may be necessary for the molecular framework that decides uterine receptivity. Glucocorticoid signaling in the uterus may also be required for the cell-fate decision of stromal cells during decidualization, although the direct gene targets of GR that regulate cell proliferation or apoptosis in the uterus have yet to be determined. Deletion of NR3C1 in the Sertoli cells through the use of anti-Mullerian hormone (AMH) Cre also produced a profound phenotype, where GR was determined to be required for Sertoli cell maintenance [54]. Although male Sertoli cell-specific NR3C1 knockout mice are fertile, the numbers of Sertoli cells and stage-specific spermatocytes/spermatids were significantly reduced. The Sertoli cell NR3C1-knockout model also revealed that glucocorticoid signaling in Sertoli cells mediates testicular steroidogenesis and critically maintains endocrine feedback pathways. The uterine and Sertoli cell transgenic mouse models have proven that glucocorticoids play a homeostatic role in regulating fertility and also provide a direct link between the stress response and fertility.
Prior to the creation of a tissue-specific model of glucocorticoid action in mammary epithelial cells, researchers utilized mice harboring a deletion in the NR3C1 gene (GRdim), which was reported to prevent GR transcriptional regulation through direct DNA-binding, and tissue-transplant techniques from GR-deficient mice [58, 59]. In the viable GRdim mice, mammary gland development was impaired due to reduced ductal epithelial cell proliferation in virgin mice, although mammary gland differentiation and milk production during pregnancy was normal [59]. The GRdim mutant maintains transcriptional regulation through protein:protein interactions, and direct transactivation by the GRdim mutant has been described for some genes, indicating that the mechanisms of GR action in the mammary gland remain poorly understood in this model [60]. Embryonic mammary buds from mice with a disrupted second exon of NR3C1 were transplanted into the cleared mammary fat pad of three-week-old recipient mice to circumvent perinatal lethality and study the role of GR in mammary gland development [52, 58]. These transplants displayed abnormal ductal morphogenesis in virgin mice but no defects in development during pregnancy, lactation, or involution. However, the exon 2 hypomorph yields a ligand-responsive truncated GR fragment, which suggests studies with this model were unable to fully appreciate the functions of GR [61]. Transgenic mice with a mammary epithelial cell-specific deletion of NR3C1 late in pregnancy exhibited defects in lobuloalveolar development due to reduced cell proliferation, although expression of milk proteins and milk secretion was not affected [55]. Interestingly, none of the in vivo models of disrupted glucocorticoid signaling in the mammary gland demonstrated defects in milk protein production compared to previous experiments which determined that glucocorticoids are essential to the transcription of milk protein genes and support milk secretion [62]. The differences in observed phenotypes may reflect the limits of current transgenic models, and may be resolved with targeted deletion of NR3C1 in other mammary cell types.
The prostate expresses GR and the direct actions of glucocorticoid signaling in prostate physiology are not well understood. A prostate epithelial-specific NR3C1 knockout was created using Probasin Cre to determine the cell-specific role of glucocorticoids in the prostate [56]. Deletion of prostate epithelial NR3C1 demonstrated that glucocorticoid signaling in epithelial cells is not required for prostate development or for the prostate’s morphological response to the presence or absence of androgens. This model showed that glucocorticoid-mediated prostate epithelial cell hyperplasia results from paracrine actions of GR originating in the stroma and not direct signaling within the epithelial cells. These studies suggest that the actions of glucocorticoids vary by prostate cell-type, which likely will be important for interpreting results from human prostate cell lines in vitro and understanding the role of GR in prostate cancer.
Changes to the bioavailability of glucocorticoids have been modeled through the creation of whole-body and tissue-specific 11β-HSD I and II knockout mice and rats [63–67]. These models have provided insights into the consequence of intrauterine glucocorticoid excess during pregnancy. In the mouse, 11β-HSDII is expressed in the stromal cells of the endometrium and the labyrinthine zone of the placenta, the primary site of maternal-fetal exchange where maternal blood flows over embryo-derived trophoblasts [68]. Therefore, impaired expression of 11β-HSD II exposes both the placenta and the fetus to locally high levels of glucocorticoids. 11β-HSD II null mice weigh less at birth, which was correlated with reduced fetal capillary development accompanied by lower placental expression of the angiogenic factors vascular endothelial growth factor A (Vegfa) and peroxisome proliferator-activated receptor gamma (Ppary), decreased expression of glucose transporter 3 (Slc2a3; GLUT3), reduced placental transport of glucose to the offspring, and smaller placentas [69]. The free transfer of maternal glucocorticoids through the placenta in the absence of 11β-HSD II indicates that glucocorticoid overexposure is detrimental to placentation and placental function. The role for physiological levels of cortisol has not been determined.
It is important to note that tissue-specific deletion of NR3C1 has been accomplished through the use of several unique floxed contructs which excise different exons of NR3C1. For instance, exon 3 floxed mice were used to create the Sertoli cell, mammary epithelial cell, and prostate epithelial cell knockout mice, and the uterine knockout model utilized mice with exon 3 and 4 floxed [70, 71]. Exon 2 floxed mice have been generated by two groups, though this model has not been utilized to develop tissue-specific knockouts of GR in the reproductive system [72, 73]. Zebrafish have also been utilized to study the actions of GR. Reported models include the use of morpholinos and a transgenic model harboring a point mutation in the DNA binding region [74, 75]. In both zebrafish models, disruption to GR signaling alters embryo development and adult physiology, although assessment of fertility was not reported. Zebrafish offer a promising alternative model for studying glucocorticoid signaling in the reproductive tract [76]. New gene editing techniques will expand our understanding of glucocorticoid signaling through the ability to insert, delete, or replace DNA at precise regions of the genome and enable researchers to use other experimental animal models (Box 4).
Box 4. Insights from non-classical models.
In most vertebrates studied, the exon length and amino acid sequence of the NR3C1 gene is well conserved and the HPA response to a physiological stressor is intact [165]. Therefore, studies describing the impact of stress on reproductive fitness may provide insight into the evolutionarily conserved mechanisms linking fertility to glucocorticoid signaling. Fertility in both male and female cheetahs is sensitive to the physiological stress that accompanies captivity. Living arrangements on-exhibit compared to off-exhibit increased glucocorticoid concentrations, reduced total mobile sperm numbers in males, and decreased ovarian cyclicity in females [166, 167]. Heat stress leads to infertility in dairy cows by inhibiting follicular development. Specifically, heat stress dramatically alters gene expression, reduces estrogen synthesis, and induces apoptosis in the follicle supporting granulosa cells [168]. In male zebra finches, fasting-increased levels of glucocorticoids were associated with decreased testicular parameters, including lower testosterone and expression of steroidogenic enzymes, in the absence of hypothalamic input, indicating that glucocorticoids integrate the cues from stress directly in the testis in this species [169]. As seen in rodent models, a single exposure to dexamethasone decreased serum testosterone and expression of genes involved in cholesterol synthesis and steroidogenesis in the stallion testis [170]. Dexamethasone exposure in breeder roosters decreased testosterone, sperm motility, and sperm viability [171]. Further evidence for a conserved mechanism of glucocorticoid regulation of steroidogenesis was demonstrated in the three-spot wrasse, Halichoeres trimaculatus [172]. In this species, prolonged exposure to cortisol induced female to male sex change and repression of plasma estradiol. Deciphering the mechanisms by which stress impacts fertility in other species is not only important to understanding the origin of infertility in humans but also essential to alleviating the financial consequences of infertility in agricultural species.
Reproductive immunology: immuno-modulatory actions of glucocorticoids
Endometrial receptivity and successful pregnancy require immune adaptations that precede embryo implantation and allow the semi-allogenic fetus to be tolerated (Figure 1A). During the menstrual cycle and increasingly in early pregnancy, immune cells are recruited to and activated in the endometrial compartment in a tightly regulated manner [77–79]. Conditional ablation of NR3C1 in the mouse uterus determined that glucocorticoid signaling in the endometrium is essential for appropriate immune cell recruitment [53]. Studies that utilize exogenous glucocorticoid administration have also demonstrated effects on immune cell number and function in the uterus [57]. Importantly, a variety of immune cells are present in the uterus that work to create the dynamic immune response essential for implantation and tolerance. Disturbances to this immune balance likely contribute to the pathogenesis of miscarriage, preeclampsia, or preterm labor. Direct actions on immune cells and indirection actions through signaling networks in the endometrium represent two mechanisms by which glucocorticoids can regulate the uterine immune system. Uterine natural killer (uNK) cells, stemming from resident and recruited populations, are the predominant immune cell population in the uterus at implantation representing approximately 70% of all leucocytes [80, 81]. During early pregnancy, uNK cells regulate trophoblast invasion and the vascular changes that support placental development. uNK cells express GR, and uNK cell-mediated cytotoxicity is sensitive cortisol treatment [82, 83]. Prednisolone treatment in women with recurrent miscarriage is associated with reduced numbers of uNK cells, although a link between the glucocorticoid-mediated reduction in uNK cell numbers and pregnancy success was not demonstrated [84]. Moreover, uNK cells differ in function from circulating natural killer cells and the specific effects of glucocorticoids on each of these populations have not been investigated.
Figure 1.
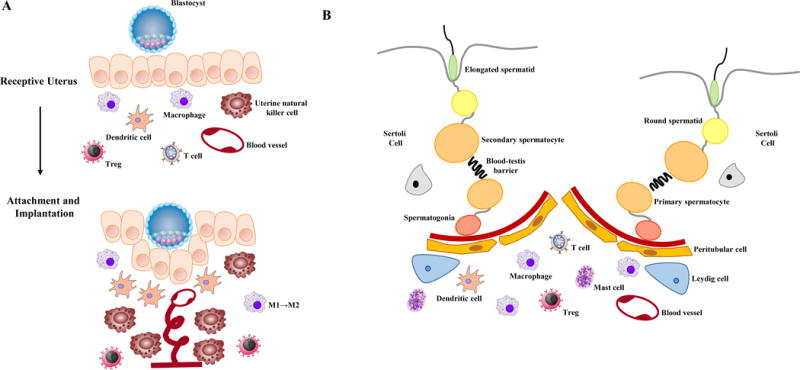
Infiltrating monocytes that develop into macrophages and uterine resident myeloid cells constitute the second largest population of immune cells in the uterus during early pregnancy [85]. Macrophages exist as two distinct subpopulations, an Ml “proinflammatory” phenotype and an M2 phenotype linked to tissue remodeling, immunosuppression, and angiogenesis [86]. Endometrial macrophages are skewed toward the M1 phenotype during the peri-implantation period and polarized toward the M2 phenotype post-implantation [87, 88]. Human endometrial macrophages express GR during the secretory phase, corresponding to the period of uterine receptivity and implantation, menstrual phase, and at term, although the function of glucocorticoid signaling in macrophages is not well understood [89, 90]. Macrophage-specific NR3C1 knockout mice have been generated using LysM Cre recombinase, although no associated infertility has been reported [91].
Dendritic cells control the innate adaptive immune response, but mouse studies suggest they also contribute to uterine receptivity. Selective ablation of CD11c+ dendritic cells in the murine uterus caused implantation failure, impaired stromal cell decidualization, and altered angiogenesis, indicating that dendritic cells are important for the initiation of pregnancy [92]. Dendritic cells are antigen presenting cells and play a key role in determining tolerance or rejection of the blastocyst. Regulatory T cells are critical to maintaining tolerance at the maternal-fetal interface and are generated from the CD4+ T cell through antigen presentation in the thymus or in the uterus. Depletion of regulatory T cells in mice leads to pregnancy loss and fetal resorption [93]. The relative abundance of another derivative of the CD4+ T cell, Th17 cells, has been associated with miscarriage, although the importance of this immune cell type in normal pregnancy has not been investigated [94]. Moreover, the physiological effects of glucocorticoids on dendritic cells, regulatory T cells, or Th17 cells in the endometrium have not been studied. Dendritic cell-specific NR3C1 knockout mice have been utilized in models of sepsis and arthritis but have not been evaluated for functions relating to fertility [95, 96]. T cell-specific ablation of NR3C1 has been reported, although Cre recombinase activity occurs early in T-cell development and prevents discriminating the contribution of glucocorticoid signaling among T cell subtypes [91].
Much like the uterus during pregnancy, immune cells present in the testis mediate the process of spermatogenesis through a delicate balance of inflammation and immunotolerance (Figure 1B) [97]. The testis is an immunoprivileged site by virtue of the blood-testis barrier, systemic immune tolerance and local immunosuppression, which protects the immunogenic germ cells from provoking an immune response [98]. However, effective local innate immunity is required to prevent bacterial or viral infections in the testis. Cells of both the innate and adaptive immune response reside within the interstitial space of the testis and are able to interact with the steroidogenic Leydig cells and the seminiferous tubules where spermatogenesis occurs. Under physiological conditions, subsets of dendritic cells and regulatory T cells mediate tolerance to germ cells. Under inflammatory conditions, however, proinflammatory cytokines produced by macrophages, dendritic cells, and effector T cells disrupt the blood-testis barrier and induce apoptosis of germ cells. To date, there are no studies in humans or mouse models which assess the impact of glucocorticoids on immune cells in the testis. Endocrine regulation of the testicular immune system by androgens suggests that glucocorticoids may also have indirect effects on immune cell functions.
In the peripheral immune system glucocorticoids demonstrate a variety of direct actions on immune cell development and function, which may influence the relative pool of immune cells able to be recruited to the reproductive tract [99]. Glucocorticoids prevent the generation of immature dendritic cells from monocytes and inhibit differentiation of immature dendritic cells into mature dendritic cell in vitro [100, 101]. High serum glucocorticoid levels induced by psychoactive drug administration were shown to associate with rapid accumulation of mature natural killer cells in the peripheral circulation in vivo [102]. Glucocorticoids also induce robust apoptosis of T and B cells, mature dendritic cells, basophils, and eosinophils [103]. Glucocorticoids also regulate the immune system by impacting the function of immune cells. For example, glucocorticoids inhibit the transcription of pro-inflammatory cytokines and chemokines in macrophages and drive macrophages toward the M2 phenotype [57]. Dexamethasone can also induce components of the inflammasome to sensitize the innate immune response in macrophages [104]. The nature of the response to glucocorticoids in the immune system is dependent on physiological or stress-induced levels of hormone and the duration of the stimulus, suggesting that the immune response governing reproduction is regulated in part through physiological glucocorticoid signaling and sensitive to hormone fluctuations.
Concluding Remarks
Stress-induced activation of the HPA axis or administration of exogenous glucocorticoids produces adverse effects on male and female fertility and negatively impacts in utero development. In models of adrenal insufficiency, the absence of glucocorticoids is also associated with reduced fertility and disturbances to fetal growth. However, basal physiological concentrations of glucocorticoids are supportive to all stages of reproduction, indicating dose is critically important to outcome. The timing of glucocorticoid exposure may also dictate whether glucocorticoid signaling promotes or inhibits fertility. The tipping point between supportive and adverse effects is not clear (Key Figure). For example, exogenous glucocorticoids are clinically utilized during in vitro fertilization (IVF) and in patients with recurrent miscarriage with the rationale that the immunomodulatory actions of glucocorticoids improve the intra-uterine environment [105, 106]. Following conception, women may receive glucocorticoids during pregnancy as treatment for asthma, autoimmune diseases, or adrenal insufficiency or for the management of preterm labor [107–109]. Glucocorticoid use during preterm labor critically improves neonatal outcomes through enhanced fetal development. However, exposure to exogenous or stress-induced levels of glucocorticoids during the window of receptivity or during fetal development may attenuate the maternal immune system required to establish pregnancy and alter fetal development in utero. Evidence from animal studies demonstrates that prenatal exposure to synthetic or stress-induced levels of glucocorticoids leads to reduced fetal growth and dysfunction of the cardiovascular, metabolic, endocrine, nervous, and reproductive systems in adults [26]. The effects of overexposure are dependent on dose and type of glucocorticoid and determined by gestational age at exposure. In humans, prenatal glucocorticoid exposure is associated with higher blood pressure and insulin levels and mild behavioral defects, although it is difficult to separate the effects of glucocorticoid exposure from the impact of premature birth [110]. Given the clear benefits to fetal survival, prenatal glucocorticoids are part of the standard management for women who present with preterm labor. However, the additional benefit of repeated treatment or the long-term consequences of exposure during early pregnancy remain uncertain and call for further investigation (see Outstanding Questions).
Figure 2, Key Figure. Impact of glucocorticoids on reproductive physiology.
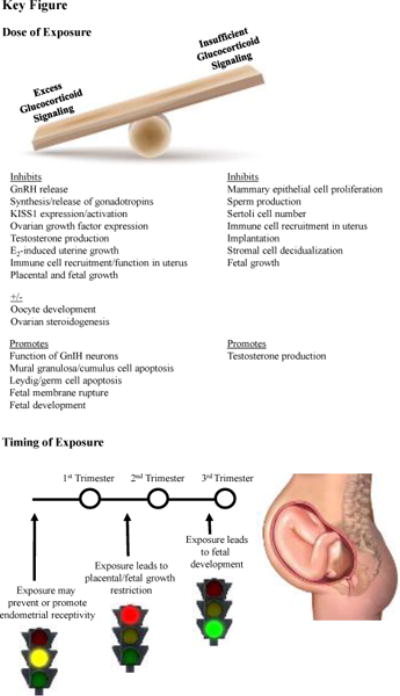
The impact of glucocorticoids on reproductive physiology is dependent on dose and timing. Both excess and insufficient levels of glucocorticoids result in reproductive dysfunction, though occurring through differing pathophysiology. The timing of exposure also dictates the relative benefit or harm of glucocorticoid exposure. The example is provided in which early exposure to high levels of glucocorticoids may inhibit endometrial receptivity, although physiological levels of glucocorticoids may be required for this process. Excess glucocorticoid exposure during early is associated with reduced placental weight and fetal size. However, glucocorticoid signaling in the third trimester is required for fetal development. Timing of exposure also plays a role in fetal programming of postnatal development, with more profound influence occurring as a result of elevated glucocorticoids during the first and second trimester.
Other deficits in our current understanding of glucocorticoid functions in the reproductive system stem from a lack of knowledge regarding the sites of direct GR action and the cross-talk of glucocorticoids with other nuclear receptors. Glucocorticoids and estrogens have demonstrated antagonistic actions in the uterus [19, 20]. The presence of estradiol with dexamethasone alters GR recruitment to target genes in immortalized uterine cells and is dependent on estrogen receptor-α (ERα) [111]. It is not clear whether this molecular antagonism is a result of direct interference with GR-DNA interactions, and the biological significance of GR-ER cross-talk is not well understood. Glucocorticoids bind with a low affinity to PR, and similarly progesterone weakly binds GR [112, 113]. The abortifacient drug RU486 acts as both an anti-progestin and antiglucocorticoid, which prevents discriminating between the GR- and PR-mediated effects in the uterus [114]. Cells in culture have provided a genetically tractable model to dissect the relative contribution of GR and PR, and propose cross-talk between progesterone and GR in cells of the reproductive tract [115–118]. New techniques utilizing cell-specific knockdown or targeted genome editing in vivo will help clarify the specific roles of glucocorticoids in normal reproductive physiology and stress-induced infertility. By identifying the GR target genes in tissues of the reproductive tract and framing that data in reference to the published ChIP-seq databases, we will begin to provide a picture of the regulatory networks that orchestrate fertility. Additionally, future studies should assess the contribution of the NR3C1 isoforms, post-translational modifications, and single nucleotide polymorphisms in the heterogeneity of stress responses among individuals. In the placenta, GR isoform expression varied between women who delivered with no complications at term and those who delivered preterm [30]. Context-specific expression provides insight in the molecular mechanisms by which stress impacts individuals uniquely. This knowledge is increasingly important to identify new avenues of treatment for infertility and potential new mechanisms to enhance fertility in agricultural species.
Outstanding Questions Box.
The ovarian hormones and their receptors regulate reproductive functions prior to and during pregnancy. Many in vitro studies have demonstrated dynamic molecular interplay in GR and ER binding, and glucocorticoids and estrogen regulate many common genes. What role does GR play in mediating estrogen responses, if any?
Glucocorticoid sensitivity in the reproductive system may also be mediated by the relative expression of the 11-β-hydroxysteroid dehydrogenase isoenzymes and MR. Do glucocorticoids signal through MR in the normal physiology of reproduction or the pathologies of adverse events? Could 11-βHSD be a therapeutic target in women?
What are the signaling pathways regulated by glucocorticoids during parturition in humans and could our knowledge of these improve pregnancy outcomes?
Antenatal corticosteroid administration for fetal maturation is critical for survival in preterm delivery. Recent updates have shown that antenatal corticosteroids may be beneficial in late preterm birth (34– <37 weeks) and should now be considered in treatment. What is the magnitude of benefits or risk of long-term effects in this population?
How is glucocorticoid signaling in reproduction impacted by environmental exposures? Are there tissue-specific effects or long-term consequences? The stress response to environmental compounds may represent a new adaptive mechanism to incorporate exposures into reproductive fitness.
Trends Box.
Glucocorticoids exhibit both central and peripheral regulation of the reproductive axis.
Within reproductive organs, basal glucocorticoids contribute to reproductive function, while stress-induced levels induce suppression of reproductive function.
Transgenic animal models have aided in the discovery of the tissue-specific functions of glucocorticoids. However, studies in reproductive tissues are needed.
Immune cells play an active role in male and female fertility, and administration of glucocorticoids or stress may disrupt the immune system resulting in reproductive dysfunction.
Acknowledgments
We thank Dr. Robert Oakley and Dr. Francesco for their critical reading of the manuscript. Supported in part by the Intramural Research Program of the National Institutes of Health (NIH)/NIEHS and by the R00 Grant ES022983 from the NIH (SW) and an Albert McKern Scholar Award (SW).
Glossary
- 11β-hydroxysteroid dehydrogenase isoenzymes (11β-HSD)
enzymes which catalyze the interconversion of active glucocorticoids (11-oxoreductase activity) and their inactive metabolites (11-β-dehydrogenase activity). The type I isoenzyme demonstrates both 11-β-dehydrogenase and 11-oxoreductase activities and the type II isoenzyme has only 11-β-dehydrogenase activity.
- Addison’s disease
severe or total deficiency of the hormones made in the adrenal cortex. Also known as primary adrenal insufficiency or hypocortisolism.
- Adrenalectomized
surgical removal of the adrenal glands (bi- or unilateral)
- Bishop score
a pre-labor cervix score used to predict the response to labor induction or chance of spontaneous labor. A low Bishop score is associated with a greater probability that induction of labor will fail
- Blood-testis barrier (BTB)
a physical barrier within the seminiferous tubules formed by tight junctions, adherens junctions, and desmosome junctions between adjacent Sertoli cells. The BTB forms an immunological barrier that segregates the basal and adluminal compartments
- Chromatin Immunoprecipitation-sequencing (ChIP-seq)
a method to identify genome-wide DNA binding sites for proteins. Proteins crosslinked to chromatin are isolated by antibody immunoprecipitation. Enriched DNA binding sites are sequenced and mapped to a reference genome. Sequencing identifies location and quantity of sites bound by a protein
- Cre-lox technology
system for creating tissue-specific or inducible knockout mouse models by catalyzing the recombination of two loxP recognition (floxed) sites introduced into the gene of interest with tissue-specific/inducible expression of the cre enzyme
- Cushing’s syndrome
a disorder resulting from prolonged exposure to elevated levels of the endogenous glucocorticoid cortisol. Cushing’s syndrome often results from exogenous glucocorticoid use, but can also be caused by pituitary adenomas and adrenal tumors
- DOHaD (Developmental Origins of Health and Disease) hypothesis
theory, previously known as the Fetal Origins Hypothesis, based on David Barker’s analysis of epidemiological studies in the early 1900’s that states that environmental factors encountered by a developmentally plastic organism early in life influence health and disease outcomes throughout adulthood
- Glucocorticoid response element (GRE)
a short palindromic sequence of DNA consisting of two 6-bp sequences separated by a three nucleotide spacer which GR recognizes and binds to directly regulate transcription. 5′ RGRACAnnnTGTYCY3′ R= purine (A or G) Y= pyrimidine (C or T), n= any nucleotide
- Inflammasome
a multiprotein intracellular complex that activates caspase enzymes leading to the processing and secretion of pro-inflammatory cytokines in response to pathogenic microorganisms and sterile stressors
- Negative glucocorticoid response element (nGRE)
related DNA sequences bound by GR distinct to the transrepression activities of glucocorticoids. The nGRE motif is tolerable to single base-pair mutations and contains a 0–2 base pair spacer between inverted repeated motifs
- Post-translational modification
chemical modifications to a protein, including phosphorylation, glycosylation, ubiquitination, nitrosylation, methylation, acetylation, lipidation, and proteolysis, which increase the functional diversity of the proteome. Post-translational modifications can occur at many stages in the life of a protein and can also be reversible
- Single nucleotide polymorphism (SNP)
a DNA sequence variation at a single nucleotide that is appreciable within a population. The variation may be a substitution, deletion, or insertion. SNPs are found within genes, in non-coding regions of DNA, and in the intergenic regions. When occurring within a gene, the SNP may lead to an amino acid substitution. SNPs can be associated with traits or diseases or have no known function
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Erichsen MM, et al. Sexuality and fertility in women with Addison’s disease. The Journal of clinical endocrinology and metabolism. 2010;95:4354–4360. doi: 10.1210/jc.2010-0445. [DOI] [PubMed] [Google Scholar]
- 2.Kowal BF, et al. Addison’s disease presenting as male infertility. Fertility and sterility. 2006;85:1059, e1051–1054. doi: 10.1016/j.fertnstert.2005.09.056. [DOI] [PubMed] [Google Scholar]
- 3.Oakley RH, Cidlowski JA. Cellular processing of the glucocorticoid receptor gene and protein: new mechanisms for generating tissue-specific actions of glucocorticoids. The Journal of biological chemistry. 2011;286:3177–3184. doi: 10.1074/jbc.R110.179325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dagklis T, et al. Common features and differences of the hypothalamic-pituitary-gonadal axis in male and female. Gynecological endocrinology : the official journal of the International Society of Gynecological Endocrinology. 2015;31:14–17. doi: 10.3109/09513590.2014.959917. [DOI] [PubMed] [Google Scholar]
- 5.Whirledge S, Cidlowski JA. A role for glucocorticoids in stress-impaired reproduction: beyond the hypothalamus and pituitary. Endocrinology. 2013;154:4450–4468. doi: 10.1210/en.2013-1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Breen KM, Mellon PL. Influence of stress-induced intermediates on gonadotropin gene expression in gonadotrope cells. Molecular and cellular endocrinology. 2014;385:71–77. doi: 10.1016/j.mce.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geraghty AC, Kaufer D. Glucocorticoid Regulation of Reproduction. Advances in experimental medicine and biology. 2015;872:253–278. doi: 10.1007/978-1-4939-2895-8_11. [DOI] [PubMed] [Google Scholar]
- 8.Takumi K, et al. Immunohistochemical analysis of the colocalization of corticotropin-releasing hormone receptor and glucocorticoid receptor in kisspeptin neurons in the hypothalamus of female rats. Neuroscience letters. 2012;531:40–45. doi: 10.1016/j.neulet.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 9.Luo E, et al. Corticosterone Blocks Ovarian Cyclicity and the LH Surge via Decreased Kisspeptin Neuron Activation in Female Mice. Endocrinology. 2016;157:1187–1199. doi: 10.1210/en.2015-1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ubuka T, et al. Identification of human GnIH homologs, RFRP-1 and RFRP-3, and the cognate receptor, GPR147 in the human hypothalamic pituitary axis. PloS one. 2009;4:e8400. doi: 10.1371/journal.pone.0008400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clarke IJ, et al. Stress Increases Gonadotropin Inhibitory Hormone Cell Activity and Input to GnRH Cells in Ewes. Endocrinology. 2016;157:4339–4350. doi: 10.1210/en.2016-1513. [DOI] [PubMed] [Google Scholar]
- 12.Kirby ED, et al. Stress increases putative gonadotropin inhibitory hormone and decreases luteinizing hormone in male rats. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:11324–11329. doi: 10.1073/pnas.0901176106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Son YL, et al. Molecular basis for the activation of gonadotropin-inhibitory hormone gene transcription by corticosterone. Endocrinology. 2014;155:1817–1826. doi: 10.1210/en.2013-2076. [DOI] [PubMed] [Google Scholar]
- 14.Yuan XH, et al. Dexamethasone altered steroidogenesis and changed redox status of granulosa cells. Endocrine. 2014;47:639–647. doi: 10.1007/s12020-014-0250-x. [DOI] [PubMed] [Google Scholar]
- 15.Huang TJ, Shirley Li P. Dexamethasone inhibits luteinizing hormone-induced synthesis of steroidogenic acute regulatory protein in cultured rat preovulatory follicles. Biology of reproduction. 2001;64:163–170. doi: 10.1095/biolreprod64.1.163. [DOI] [PubMed] [Google Scholar]
- 16.Michael AE, et al. Direct inhibition of ovarian steroidogenesis by cortisol and the modulatory role of 11 beta-hydroxysteroid dehydrogenase. Clinical endocrinology. 1993;38:641–644. doi: 10.1111/j.1365-2265.1993.tb02147.x. [DOI] [PubMed] [Google Scholar]
- 17.Yuan HJ, et al. Glucocorticoids impair oocyte developmental potential by triggering apoptosis of ovarian cells via activating the Fas system. Scientific reports. 2016;6:24036. doi: 10.1038/srep24036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.da Costa NN, et al. Effect of cortisol on bovine oocyte maturation and embryo development in vitro. Theriogenology. 2016;85:323–329. doi: 10.1016/j.theriogenology.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 19.Rhen T, et al. Dexamethasone blocks the rapid biological effects of 17beta-estradiol in the rat uterus without antagonizing its global genomic actions. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2003;17:1849–1870. doi: 10.1096/fj.02-1099com. [DOI] [PubMed] [Google Scholar]
- 20.Johnson DC, Dey SK. Role of histamine in implantation: dexamethasone inhibits estradiol-induced implantation in the rat. Biology of reproduction. 1980;22:1136–1141. doi: 10.1093/biolreprod/22.5.1136. [DOI] [PubMed] [Google Scholar]
- 21.Whirledge S, et al. Global gene expression analysis in human uterine epithelial cells defines new targets of glucocorticoid and estradiol antagonism. Biology of reproduction. 2013;89:66. doi: 10.1095/biolreprod.113.111054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nanjappa MK, et al. Maximal Dexamethasone Inhibition of Luminal Epithelial Proliferation Involves Progesterone Receptor (PR)- and Non-PR-Mediated Mechanisms in Neonatal Mouse Uterus. Biology of reproduction. 2015;92:122. doi: 10.1095/biolreprod.114.123463. [DOI] [PubMed] [Google Scholar]
- 23.Kuroda K, et al. Induction of 11beta-HSD 1 and activation of distinct mineralocorticoid receptor- and glucocorticoid receptor-dependent gene networks in decidualizing human endometrial stromal cells. Molecular endocrinology. 2013;27:192–202. doi: 10.1210/me.2012-1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang D, et al. Glucocorticoid exposure in early placentation induces preeclampsia in rats via interfering trophoblast development. General and comparative endocrinology. 2016;225:61–70. doi: 10.1016/j.ygcen.2015.09.019. [DOI] [PubMed] [Google Scholar]
- 25.Braun T, et al. Early dexamethasone treatment induces placental apoptosis in sheep. Reproductive sciences. 2015;22:47–59. doi: 10.1177/1933719114542028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fowden AL, Forhead AJ. Glucocorticoids as regulatory signals during intrauterine development. Experimental physiology. 2015;100:1477–1487. doi: 10.1113/EP085212. [DOI] [PubMed] [Google Scholar]
- 27.Vaughan OR, et al. Corticosterone alters materno-fetal glucose partitioning and insulin signalling in pregnant mice. The Journal of physiology. 2015;593:1307–1321. doi: 10.1113/jphysiol.2014.287177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ozmen A, et al. Glucocorticoid exposure altered angiogenic factor expression via Akt/mTOR pathway in rat placenta. Annals of anatomy = Anatomischer Anzeiger : official organ of the Anatomische Gesellschaft. 2015;198:34–40. doi: 10.1016/j.aanat.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 29.Yang Q, et al. Compartmentalized localization of 11beta-HSD 1 and 2 at the feto-maternal interface in the first trimester of human pregnancy. Placenta. 2016;46:63–71. doi: 10.1016/j.placenta.2016.08.079. [DOI] [PubMed] [Google Scholar]
- 30.Saif Z, et al. Expression of eight glucocorticoid receptor isoforms in the human preterm placenta vary with fetal sex and birthweight. Placenta. 2015;36:723–730. doi: 10.1016/j.placenta.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li XQ, et al. Roles of glucocorticoids in human parturition: a controversial fact? Placenta. 2014;35:291–296. doi: 10.1016/j.placenta.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 32.Laloha F, et al. Effect of Intravenous Dexamethasone on Preparing the Cervix and Labor Induction. Acta medica Iranica. 2015;53:568–572. [PubMed] [Google Scholar]
- 33.Wang W, et al. Phosphorylation of STAT3 mediates the induction of cyclooxygenase-2 by cortisol in the human amnion at parturition. Science signaling. 2015;8:ra106. doi: 10.1126/scisignal.aac6151. [DOI] [PubMed] [Google Scholar]
- 34.Challis JRG, et al. Endocrine and paracrine regulation of birth at term and preterm. Endocrine reviews. 2000;21:514–550. doi: 10.1210/edrv.21.5.0407. [DOI] [PubMed] [Google Scholar]
- 35.Wang W, et al. Induction of Amnion Epithelial Apoptosis by Cortisol via tPA/Plasmin System. Endocrinology. 2016;157:4487–4498. doi: 10.1210/en.2016-1464. [DOI] [PubMed] [Google Scholar]
- 36.Gopalakrishnan GS, et al. Programming of adult cardiovascular function after early maternal undernutrition in sheep. American journal of physiology. Regulatory, integrative and comparative physiology. 2004;287:R12–20. doi: 10.1152/ajpregu.00687.2003. [DOI] [PubMed] [Google Scholar]
- 37.De Blasio MJ, et al. Maternal exposure to dexamethasone or cortisol in early pregnancy differentially alters insulin secretion and glucose homeostasis in adult male sheep offspring. American journal of physiology. Endocrinology and metabolism. 2007;293:E75–82. doi: 10.1152/ajpendo.00689.2006. [DOI] [PubMed] [Google Scholar]
- 38.Slotkin TA, et al. Programming of brainstem serotonin transporter development by prenatal glucocorticoids. Brain research. Developmental brain research. 1996;93:155–161. doi: 10.1016/0165-3806(96)00027-2. [DOI] [PubMed] [Google Scholar]
- 39.Pechnick RN, et al. Developmental exposure to corticosterone: behavioral changes and differential effects on leukemia inhibitory factor (LIF). and corticotropin-releasing hormone (CRH). gene expression in the mouse. Psychopharmacology. 2006;185:76–83. doi: 10.1007/s00213-005-0258-2. [DOI] [PubMed] [Google Scholar]
- 40.Fowden AL, et al. Glucocorticoid programming of intrauterine development. Domestic animal endocrinology. 2016;56(Suppl):S121–132. doi: 10.1016/j.domaniend.2016.02.014. [DOI] [PubMed] [Google Scholar]
- 41.Holmes MC, et al. Fetal programming of adult behaviour by stress and glucocorticoids. Psychoneuroendocrinology. 2015;61:9. [Google Scholar]
- 42.Carpenter T, et al. Sex differences in early-life programming of the hypothalamic-pituitary-adrenal axis in humans suggest increased vulnerability in females: a systematic review. Journal of developmental origins of health and disease. 2017:1–12. doi: 10.1017/S204017441600074X. [DOI] [PubMed] [Google Scholar]
- 43.Gillies GE, et al. Enduring, Sexually Dimorphic Impact of In Utero Exposure to Elevated Levels of Glucocorticoids on Midbrain Dopaminergic Populations. Brain sciences. 2016;7 doi: 10.3390/brainsci7010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hiroi R, et al. Sex-dependent programming effects of prenatal glucocorticoid treatment on the developing serotonin system and stress-related behaviors in adulthood. Neuroscience. 2016;320:43–56. doi: 10.1016/j.neuroscience.2016.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cuffe JS, et al. Maternal corticosterone exposure in the mouse programs sex-specific renal adaptations in the renin-angiotensin-aldosterone system in 6-month offspring. Physiological reports. 2016;4 doi: 10.14814/phy2.12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O’Sullivan L, et al. Excess prenatal corticosterone exposure results in albuminuria, sex-specific hypotension, and altered heart rate responses to restraint stress in aged adult mice. American journal of physiology. Renal physiology. 2015;308:F1065–1073. doi: 10.1152/ajprenal.00676.2014. [DOI] [PubMed] [Google Scholar]
- 47.Cuffe JS, et al. Prenatal corticosterone exposure programs sex-specific adrenal adaptations in mouse offspring. The Journal of endocrinology. 2017;232:37–48. doi: 10.1530/JOE-16-0417. [DOI] [PubMed] [Google Scholar]
- 48.Sun Y, et al. Prenatal Dexamethasone Exposure Increases the Susceptibility to Autoimmunity in Offspring Rats by Epigenetic Programing of Glucocorticoid Receptor. BioMed research international. 2016;2016:9409452. doi: 10.1155/2016/9409452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Silva EJ, et al. Impact of adrenalectomy and dexamethasone treatment on testicular morphology and sperm parameters in rats: insights into the adrenal control of male reproduction. Andrology. 2014;2:835–846. doi: 10.1111/j.2047-2927.2014.00228.x. [DOI] [PubMed] [Google Scholar]
- 50.Gao HB, et al. Suppression of endogenous corticosterone levels in vivo increases the steroidogenic capacity of purified rat Leydig cells in vitro. Endocrinology. 1996;137:1714–1718. doi: 10.1210/endo.137.5.8612506. [DOI] [PubMed] [Google Scholar]
- 51.Panza S, et al. Glucocorticoid Receptor as a Potential Target to Decrease Aromatase Expression and Inhibit Leydig Tumor Growth. The American journal of pathology. 2016;186:1328–1339. doi: 10.1016/j.ajpath.2015.12.024. [DOI] [PubMed] [Google Scholar]
- 52.Cole TJ, et al. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes & development. 1995;9:1608–1621. doi: 10.1101/gad.9.13.1608. [DOI] [PubMed] [Google Scholar]
- 53.Whirledge SD, et al. Uterine glucocorticoid receptors are critical for fertility in mice through control of embryo implantation and decidualization. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:15166–15171. doi: 10.1073/pnas.1508056112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hazra R, et al. In vivo actions of the Sertoli cell glucocorticoid receptor. Endocrinology. 2014;155:1120–1130. doi: 10.1210/en.2013-1940. [DOI] [PubMed] [Google Scholar]
- 55.Wintermantel TM, et al. The epithelial glucocorticoid receptor is required for the normal timing of cell proliferation during mammary lobuloalveolar development but is dispensable for milk production. Molecular endocrinology. 2005;19:340–349. doi: 10.1210/me.2004-0068. [DOI] [PubMed] [Google Scholar]
- 56.Zhao B, et al. Glucocorticoid receptor in prostate epithelia is not required for corticosteroid-induced epithelial hyperproliferation in the mouse prostate. The Prostate. 2014;74:1068–1078. doi: 10.1002/pros.22825. [DOI] [PubMed] [Google Scholar]
- 57.Robertson SA, et al. Corticosteroid therapy in assisted reproduction - immune suppression is a faulty premise. Human reproduction. 2016;31:2164–2173. doi: 10.1093/humrep/dew186. [DOI] [PubMed] [Google Scholar]
- 58.Kingsley-Kallesen M, et al. The mineralocorticoid receptor may compensate for the loss of the glucocorticoid receptor at specific stages of mammary gland development. Molecular endocrinology. 2002;16:2008–2018. doi: 10.1210/me.2002-0103. [DOI] [PubMed] [Google Scholar]
- 59.Reichardt HM, et al. Mammary gland development and lactation are controlled by different glucocorticoid receptor activities. European journal of endocrinology. 2001;145:519–527. doi: 10.1530/eje.0.1450519. [DOI] [PubMed] [Google Scholar]
- 60.Jewell CM, et al. Complex human glucocorticoid receptor dim mutations define glucocorticoid induced apoptotic resistance in bone cells. Molecular endocrinology. 2012;26:244–256. doi: 10.1210/me.2011-1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mittelstadt PR, Ashwell JD. Disruption of glucocorticoid receptor exon 2 yields a ligand-responsive C-terminal fragment that regulates gene expression. Molecular endocrinology. 2003;17:1534–1542. doi: 10.1210/me.2002-0429. [DOI] [PubMed] [Google Scholar]
- 62.Casey TM, Plaut K. The role of glucocorticoids in secretory activation and milk secretion, a historical perspective. Journal of mammary gland biology and neoplasia. 2007;12:293–304. doi: 10.1007/s10911-007-9055-3. [DOI] [PubMed] [Google Scholar]
- 63.Kotelevtsev Y, et al. 11beta-hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:14924–14929. doi: 10.1073/pnas.94.26.14924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kotelevtsev Y, et al. Hypertension in mice lacking 11beta-hydroxysteroid dehydrogenase type 2. The Journal of clinical investigation. 1999;103:683–689. doi: 10.1172/JCI4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lavery GG, et al. Lack of significant metabolic abnormalities in mice with liver-specific disruption of 11beta-hydroxysteroid dehydrogenase type 1. Endocrinology. 2012;153:3236–3248. doi: 10.1210/en.2012-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wyrwoll C, et al. Fetal brain 11beta-hydroxysteroid dehydrogenase type 2 selectively determines programming of adult depressive-like behaviors and cognitive function, but not anxiety behaviors in male mice. Psychoneuroendocrinology. 2015;59:59–70. doi: 10.1016/j.psyneuen.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mullins LJ, et al. Mineralocorticoid excess or glucocorticoid insufficiency: renal and metabolic phenotypes in a rat Hsd11b2 knockout model. Hypertension. 2015;66:e20. doi: 10.1161/HYP.0000000000000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thompson A, et al. Spatial and temporal patterns of expression of 11beta-hydroxysteroid dehydrogenase types 1 and 2 messenger RNA and glucocorticoid receptor protein in the murine placenta and uterus during late pregnancy. Biology of reproduction. 2002;67:1708–1718. doi: 10.1095/biolreprod.102.005488. [DOI] [PubMed] [Google Scholar]
- 69.Wyrwoll CS, et al. Altered placental function of 11beta-hydroxysteroid dehydrogenase 2 knockout mice. Endocrinology. 2009;150:1287–1293. doi: 10.1210/en.2008-1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tronche F, et al. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nature genetics. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- 71.Oakley RH, et al. Essential role of stress hormone signaling in cardiomyocytes for the prevention of heart disease. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:17035–17040. doi: 10.1073/pnas.1302546110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jeanneteau FD, et al. BDNF and glucocorticoids regulate corticotrophin-releasing hormone (CRH). homeostasis in the hypothalamus. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:1305–1310. doi: 10.1073/pnas.1114122109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brewer JA, et al. T-cell glucocorticoid receptor is required to suppress COX-2-mediated lethal immune activation. Nature medicine. 2003;9:1318–1322. doi: 10.1038/nm895. [DOI] [PubMed] [Google Scholar]
- 74.Pikulkaew S, et al. The knockdown of maternal glucocorticoid receptor mRNA alters embryo development in zebrafish. Developmental dynamics : an official publication of the American Association of Anatomists. 2011;240:874–889. doi: 10.1002/dvdy.22586. [DOI] [PubMed] [Google Scholar]
- 75.Griffiths BB, et al. A zebrafish model of glucocorticoid resistance shows serotonergic modulation of the stress response. Frontiers in behavioral neuroscience. 2012;6:68. doi: 10.3389/fnbeh.2012.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hoo JY, et al. Zebrafish: A Versatile Animal Model for Fertility Research. BioMed research international. 2016;2016:9732780. doi: 10.1155/2016/9732780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang J, et al. To serve and to protect: the role of decidual innate immune cells on human pregnancy. Cell and tissue research. 2016;363:249–265. doi: 10.1007/s00441-015-2315-4. [DOI] [PubMed] [Google Scholar]
- 78.Figueiredo AS, Schumacher A. The T helper type 17/regulatory T cell paradigm in pregnancy. Immunology. 2016;148:13–21. doi: 10.1111/imm.12595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vinketova K, et al. Human Decidual Stromal Cells as a Component of the Implantation Niche and a Modulator of Maternal Immunity. Journal of pregnancy. 2016;2016:8689436. doi: 10.1155/2016/8689436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mori M, et al. The decidua-the maternal bed embracing the embryo-maintains the pregnancy. Seminars in immunopathology. 2016;38:635–649. doi: 10.1007/s00281-016-0574-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bartmann C, et al. Quantification of the predominant immune cell populations in decidua throughout human pregnancy. American journal of reproductive immunology. 2014;71:109–119. doi: 10.1111/aji.12185. [DOI] [PubMed] [Google Scholar]
- 82.Henderson TA, et al. Steroid receptor expression in uterine natural killer cells. The Journal of clinical endocrinology and metabolism. 2003;88:440–449. doi: 10.1210/jc.2002-021174. [DOI] [PubMed] [Google Scholar]
- 83.Chen Y, et al. Mifepristone increases the cytotoxicity of uterine natural killer cells by acting as a glucocorticoid antagonist via ERK activation. PloS one. 2012;7:e36413. doi: 10.1371/journal.pone.0036413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Quenby S, et al. Prednisolone reduces preconceptual endometrial natural killer cells in women with recurrent miscarriage. Fertility and sterility. 2005;84:980–984. doi: 10.1016/j.fertnstert.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 85.Trundley A, et al. Methods for isolation of cells from the human fetal-maternal interface. Methods in molecular medicine. 2006;122:109–122. doi: 10.1385/1-59259-989-3:109. [DOI] [PubMed] [Google Scholar]
- 86.Porta C, et al. Molecular and epigenetic basis of macrophage polarized activation. Seminars in immunology. 2015;27:237–248. doi: 10.1016/j.smim.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 87.Gustafsson C, et al. Gene expression profiling of human decidual macrophages: evidence for immunosuppressive phenotype. PloS one. 2008;3:e2078. doi: 10.1371/journal.pone.0002078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Heikkinen J, et al. Phenotypic characterization of human decidual macrophages. Clinical and experimental immunology. 2003;131:498–505. doi: 10.1046/j.1365-2249.2003.02092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Thiruchelvam U, et al. Cortisol regulates the paracrine action of macrophages by inducing vasoactive gene expression in endometrial cells. Journal of leukocyte biology. 2016;99:1165–1171. doi: 10.1189/jlb.5A0215-061RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Horton JS, et al. Relaxin modulates proinflammatory cytokine secretion from human decidual macrophages. Biology of reproduction. 2011;85:788–797. doi: 10.1095/biolreprod.110.089201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tuckermann JP, et al. Macrophages and neutrophils are the targets for immune suppression by glucocorticoids in contact allergy. The Journal of clinical investigation. 2007;117:1381–1390. doi: 10.1172/JCI28034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Plaks V, et al. Uterine DCs are crucial for decidua formation during embryo implantation in mice. The Journal of clinical investigation. 2008;118:3954–3965. doi: 10.1172/JCI36682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Aluvihare VR, et al. Regulatory T cells mediate maternal tolerance to the fetus. Nature immunology. 2004;5:266–271. doi: 10.1038/ni1037. [DOI] [PubMed] [Google Scholar]
- 94.Wang WJ, et al. Increased prevalence of T helper 17 (Th17). cells in peripheral blood and decidua in unexplained recurrent spontaneous abortion patients. Journal of reproductive immunology. 2010;84:164–170. doi: 10.1016/j.jri.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 95.Li CC, et al. Suppression of Dendritic Cell-Derived IL-12 by Endogenous Glucocorticoids Is Protective in LPS-Induced Sepsis. PLoS biology. 2015;13:e1002269. doi: 10.1371/journal.pbio.1002269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Baschant U, et al. Glucocorticoid therapy of antigen-induced arthritis depends on the dimerized glucocorticoid receptor in T cells. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:19317–19322. doi: 10.1073/pnas.1105857108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Perez CV, et al. Dual role of immune cells in the testis: Protective or pathogenic for germ cells? Spermatogenesis. 2013;3:e23870. doi: 10.4161/spmg.23870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhao S, et al. Testicular defense systems: immune privilege and innate immunity. Cellular & molecular immunology. 2014;11:428–437. doi: 10.1038/cmi.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Busillo JM, Cidlowski JA. The five Rs of glucocorticoid action during inflammation: ready, reinforce, repress, resolve, and restore. Trends in endocrinology and metabolism: TEM. 2013;24:109–119. doi: 10.1016/j.tem.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Woltman AM, et al. The effect of calcineurin inhibitors and corticosteroids on the differentiation of human dendritic cells. European journal of immunology. 2000;30:1807–1812. doi: 10.1002/1521-4141(200007)30:7<1807::AID-IMMU1807>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 101.Matasic R, et al. Dexamethasone inhibits dendritic cell maturation by redirecting differentiation of a subset of cells. Journal of leukocyte biology. 1999;66:909–914. doi: 10.1002/jlb.66.6.909. [DOI] [PubMed] [Google Scholar]
- 102.Bigler MB, et al. Stress-Induced In Vivo Recruitment of Human Cytotoxic Natural Killer Cells Favors Subsets with Distinct Receptor Profiles and Associates with Increased Epinephrine Levels. PloS one. 2015;10:e0145635. doi: 10.1371/journal.pone.0145635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cruz-Topete D, Cidlowski JA. One hormone, two actions: anti- and pro-inflammatory effects of glucocorticoids. Neuroimmunomodulation. 2015;22:20–32. doi: 10.1159/000362724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Busillo JM, et al. Glucocorticoids sensitize the innate immune system through regulation of the NLRP3 inflammasome. The Journal of biological chemistry. 2011;286:38703–38713. doi: 10.1074/jbc.M111.275370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Polak de Fried E, et al. Improvement of clinical pregnancy rate and implantation rate of in- vitro fertilization-embryo transfer patients by using methylprednisone. Human reproduction. 1993;8:393–395. doi: 10.1093/oxfordjournals.humrep.a138058. [DOI] [PubMed] [Google Scholar]
- 106.Quenby S, et al. Successful pregnancy outcome following 19 consecutive miscarriages: case report. Human reproduction. 2003;18:2562–2564. doi: 10.1093/humrep/deg502. [DOI] [PubMed] [Google Scholar]
- 107.Breton MC, et al. Risk of perinatal mortality associated with inhaled corticosteroid use for the treatment of asthma during pregnancy. The Journal of allergy and clinical immunology. 2010;126:772–777 e772. doi: 10.1016/j.jaci.2010.08.018. [DOI] [PubMed] [Google Scholar]
- 108.Ponticelli C, Moroni G. Immunosuppression in pregnant women with systemic lupus erythematosus. Expert review of clinical immunology. 2015;11:549–552. doi: 10.1586/1744666X.2015.1033404. [DOI] [PubMed] [Google Scholar]
- 109.Msan AK, et al. Use of antenatal corticosteroids in the management of preterm delivery. American journal of perinatology. 2015;32:417–426. doi: 10.1055/s-0034-1395476. [DOI] [PubMed] [Google Scholar]
- 110.Harris A, Seckl J. Glucocorticoids, prenatal stress and the programming of disease. Hormones and behavior. 2011;59:279–289. doi: 10.1016/j.yhbeh.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 111.Whirledge S, Cidlowski JA. Estradiol antagonism of glucocorticoid-induced GILZ expression in human uterine epithelial cells and murine uterus. Endocrinology. 2013;154:499–510. doi: 10.1210/en.2012-1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Haslam SZ, et al. An empirical basis for the competition by dexamethasone to progesterone receptors as estimated with the synthetic progestin R5020. Journal of receptor research. 1981;2:435–451. doi: 10.3109/107998981809038877. [DOI] [PubMed] [Google Scholar]
- 113.Attardi BJ, et al. Comparison of progesterone and glucocorticoid receptor binding and stimulation of gene expression by progesterone, 17-alpha hydroxyprogesterone caproate, and related progestins. American journal of obstetrics and gynecology. 2007;197:599, e591–597. doi: 10.1016/j.ajog.2007.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schreiber JR, et al. Binding of the anti-progestin RU-486 to rat ovary steroid receptors. Contraception. 1983;28:77–85. doi: 10.1016/s0010-7824(83)80008-0. [DOI] [PubMed] [Google Scholar]
- 115.Lei K, et al. Progesterone acts via the nuclear glucocorticoid receptor to suppress IL-1beta-induced COX-2 expression in human term myometrial cells. PloS one. 2012;7:e50167. doi: 10.1371/journal.pone.0050167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Leo JC, et al. Glucocorticoid and mineralocorticoid cross-talk with progesterone receptor to induce focal adhesion and growth inhibition in breast cancer cells. Endocrinology. 2004;145:1314–1321. doi: 10.1210/en.2003-0732. [DOI] [PubMed] [Google Scholar]
- 117.Louw-du Toit R, et al. Medroxyprogesterone acetate differentially regulates interleukin (IL)- 12 and IL-10 in a human ectocervical epithelial cell line in a glucocorticoid receptor (GR)-dependent manner. The Journal of biological chemistry. 2014;289:31136–31149. doi: 10.1074/jbc.M114.587311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lei K, et al. Progesterone and the Repression of Myometrial Inflammation: The Roles of MKP- 1 and the AP-1 System. Molecular endocrinology. 2015;29:1454–1467. doi: 10.1210/me.2015-1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sawchenko PE. Evidence for a local site of action for glucocorticoids in inhibiting CRF and vasopressin expression in the paraventricular nucleus. Brain research. 1987;403:213–223. doi: 10.1016/0006-8993(87)90058-8. [DOI] [PubMed] [Google Scholar]
- 120.Plotsky PM, et al. Inhibition of immunoreactive corticotropin-releasing factor secretion into the hypophysial-portal circulation by delayed glucocorticoid feedback. Endocrinology. 1986;119:1126–1130. doi: 10.1210/endo-119-3-1126. [DOI] [PubMed] [Google Scholar]
- 121.Aguilera G. Regulation of pituitary ACTH secretion during chronic stress. Frontiers in neuroendocrinology. 1994;15:321–350. doi: 10.1006/frne.1994.1013. [DOI] [PubMed] [Google Scholar]
- 122.Daughaday WH. Binding of corticosteroids by plasma proteins. II. Paper electrophoresis and equilibrium paper electrophoresis. The Journal of clinical investigation. 1956;35:1434–1438. doi: 10.1172/JCI103401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Baker ME. Albumin, steroid hormones and the origin of vertebrates. The Journal of endocrinology. 2002;175:121–127. doi: 10.1677/joe.0.1750121. [DOI] [PubMed] [Google Scholar]
- 124.Weitzman ED, et al. Twenty-four hour pattern of the episodic secretion of cortisol in normal subjects. The Journal of clinical endocrinology and metabolism. 1971;33:14–22. doi: 10.1210/jcem-33-1-14. [DOI] [PubMed] [Google Scholar]
- 125.Wood PJ, et al. Evidence for the low dose dexamethasone suppression test to screen for Cushing’s syndrome–recommendations for a protocol for biochemistry laboratories. Annals of clinical biochemistry. 1997;34(Pt 3):222–229. doi: 10.1177/000456329703400302. [DOI] [PubMed] [Google Scholar]
- 126.Pariante CM. The role of multi-drug resistance p-glycoprotein in glucocorticoid function: studies in animals and relevance in humans. European journal of pharmacology. 2008;583:263–271. doi: 10.1016/j.ejphar.2007.11.067. [DOI] [PubMed] [Google Scholar]
- 127.Woods C, Tomlinson JW. The Dehydrogenase Hypothesis. Advances in experimental medicine and biology. 2015;872:353–380. doi: 10.1007/978-1-4939-2895-8_16. [DOI] [PubMed] [Google Scholar]
- 128.Wira C, Munck A. Specific glucocorticoid receptors in thymus cells. Localization in the nucleus and extraction of the cortisol-receptor complex. The Journal of biological chemistry. 1970;245:3436–3438. [PubMed] [Google Scholar]
- 129.Bamberger CM, et al. Glucocorticoid receptor beta, a potential endogenous inhibitor of glucocorticoid action in humans. The Journal of clinical investigation. 1995;95:2435–2441. doi: 10.1172/JCI117943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Oakley RH, et al. The human glucocorticoid receptor beta isoform. Expression, biochemical properties, and putative function. The Journal of biological chemistry. 1996;271:9550–9559. doi: 10.1074/jbc.271.16.9550. [DOI] [PubMed] [Google Scholar]
- 131.Moalli PA, et al. Alternatively spliced glucocorticoid receptor messenger RNAs in glucocorticoid-resistant human multiple myeloma cells. Cancer research. 1993;53:3877–3879. [PubMed] [Google Scholar]
- 132.Rivers C, et al. Insertion of an amino acid in the DNA-binding domain of the glucocorticoid receptor as a result of alternative splicing. The Journal of clinical endocrinology and metabolism. 1999;84:4283–4286. doi: 10.1210/jcem.84.11.6235. [DOI] [PubMed] [Google Scholar]
- 133.Hollenberg SM, et al. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature. 1985;318:635–641. doi: 10.1038/318635a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kino T, et al. Glucocorticoid receptor (GR). beta has intrinsic, GRalpha-independent transcriptional activity. Biochemical and biophysical research communications. 2009;381:671–675. doi: 10.1016/j.bbrc.2009.02.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.He B, et al. Human Glucocorticoid Receptor beta Regulates Gluconeogenesis and Inflammation in Mouse Liver. Molecular and cellular biology. 2015;36:714–730. doi: 10.1128/MCB.00908-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ray DW, et al. Glucocorticoid receptor structure and function in glucocorticoid-resistant small cell lung carcinoma cells. Cancer research. 1996;56:3276–3280. [PubMed] [Google Scholar]
- 137.Meijsing SH, et al. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009;324:407–410. doi: 10.1126/science.1164265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Thomas-Chollier M, et al. A naturally occurring insertion of a single amino acid rewires transcriptional regulation by glucocorticoid receptor isoforms. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:17826–17831. doi: 10.1073/pnas.1316235110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Morgan DJ, et al. Glucocorticoid receptor isoforms direct distinct mitochondrial programs to regulate ATP production. Scientific reports. 2016;6:26419. doi: 10.1038/srep26419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gaitan D, et al. Glucocorticoid receptor structure and function in an adrenocorticotropin-secreting small cell lung cancer. Molecular endocrinology. 1995;9:1193–1201. doi: 10.1210/mend.9.9.7491111. [DOI] [PubMed] [Google Scholar]
- 141.Lu NZ, Cidlowski JA. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Molecular cell. 2005;18:331–342. doi: 10.1016/j.molcel.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 142.Lu NZ, et al. Selective regulation of bone cell apoptosis by translational isoforms of the glucocorticoid receptor. Molecular and cellular biology. 2007;27:7143–7160. doi: 10.1128/MCB.00253-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nehme A, et al. Glucocorticoids with different chemical structures but similar glucocorticoid receptor potency regulate subsets of common and unique genes in human trabecular meshwork cells. BMC medical genomics. 2009;2:58. doi: 10.1186/1755-8794-2-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gross KL, et al. Glucocorticoid receptor alpha isoform-selective regulation of antiapoptotic genes in osteosarcoma cells: a new mechanism for glucocorticoid resistance. Molecular endocrinology. 2011;25:1087–1099. doi: 10.1210/me.2010-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Song IH, Buttgereit F. Non-genomic glucocorticoid effects to provide the basis for new drug developments. Molecular and cellular endocrinology. 2006;246:142–146. doi: 10.1016/j.mce.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 146.Nahar J, et al. Rapid Nongenomic Glucocorticoid Actions in Male Mouse Hypothalamic Neuroendocrine Cells Are Dependent on the Nuclear Glucocorticoid Receptor. Endocrinology. 2015;156:2831–2842. doi: 10.1210/en.2015-1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nahar J, et al. Further evidence for a membrane receptor that binds glucocorticoids in the rodent hypothalamus. Steroids. 2016;114:33–40. doi: 10.1016/j.steroids.2016.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Starick SR, et al. ChIP-exo signal associated with DNA-binding motifs provides insight into the genomic binding of the glucocorticoid receptor and cooperating transcription factors. Genome research. 2015;25:825–835. doi: 10.1101/gr.185157.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Surjit M, et al. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell. 2011;145:224–241. doi: 10.1016/j.cell.2011.03.027. [DOI] [PubMed] [Google Scholar]
- 150.Gupte R, et al. Glucocorticoid receptor represses proinflammatory genes at distinct steps of the transcription cycle. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:14616–14621. doi: 10.1073/pnas.1309898110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lu NZ, Cidlowski JA. Glucocorticoid receptor isoforms generate transcription specificity. Trends in cell biology. 2006;16:301–307. doi: 10.1016/j.tcb.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 152.Ramamoorthy S, Cidlowski JA. Corticosteroids: Mechanisms of Action in Health and Disease. Rheumatic diseases clinics of North America. 2016;42:15–31, vii. doi: 10.1016/j.rdc.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Barnes PJ. Histone deacetylase-2 and airway disease. Therapeutic advances in respiratory disease. 2009;3:235–243. doi: 10.1177/1753465809348648. [DOI] [PubMed] [Google Scholar]
- 154.Trotter KW, et al. Glucocorticoid Receptor Transcriptional Activation via the BRG1-Dependent Recruitment of TOP2beta and Ku70/86. Molecular and cellular biology. 2015;35:2799–2817. doi: 10.1128/MCB.00230-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Grontved L, et al. C/EBP maintains chromatin accessibility in liver and facilitates glucocorticoid receptor recruitment to steroid response elements. The EMBO journal. 2013;32:1568–1583. doi: 10.1038/emboj.2013.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Jones CL, et al. Loss of TBL1XR1 disrupts glucocorticoid receptor recruitment to chromatin and results in glucocorticoid resistance in a B-lymphoblastic leukemia model. The Journal of biological chemistry. 2014;289:20502–20515. doi: 10.1074/jbc.M114.569889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Goi C, et al. Cell-type and transcription factor specific enrichment of transcriptional cofactor motifs in ENCODE ChIP-seq data. BMC genomics. 2013;14(Suppl 5):S2. doi: 10.1186/1471-2164-14-S5-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Smith CL, O’Malley BW. Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocrine reviews. 2004;25:45–71. doi: 10.1210/er.2003-0023. [DOI] [PubMed] [Google Scholar]
- 159.John S, et al. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nature genetics. 2011;43:264–268. doi: 10.1038/ng.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.John S, et al. Interaction of the glucocorticoid receptor with the chromatin landscape. Molecular cell. 2008;29:611–624. doi: 10.1016/j.molcel.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 161.Koper JW, et al. Glucocorticoid receptor polymorphisms and haplotypes and their expression in health and disease. Steroids. 2014;92:62–73. doi: 10.1016/j.steroids.2014.07.015. [DOI] [PubMed] [Google Scholar]
- 162.Konstantakou P, et al. Dysregulation of 11beta-hydroxysteroid dehydrogenases: implications during pregnancy and beyond. The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal Obstet. 2016:1–10. doi: 10.3109/14767058.2016.1171308. [DOI] [PubMed] [Google Scholar]
- 163.Lutz SZ, et al. Genetic Variation in the 11beta-hydroxysteroid-dehydrogenase 1 Gene determines NAFLD and Visceral Obesity. The Journal of clinical endocrinology and metabolism. 2016 doi: 10.1210/jc.2016-2498. jc20162498. [DOI] [PubMed] [Google Scholar]
- 164.Ragnarsson O, et al. Common genetic variants in the glucocorticoid receptor and the 11beta-hydroxysteroid dehydrogenase type 1 genes influence long-term cognitive impairments in patients with Cushing’s syndrome in remission. The Journal of clinical endocrinology and metabolism. 2014;99:E1803–1807. doi: 10.1210/jc.2014-1906. [DOI] [PubMed] [Google Scholar]
- 165.Stolte EH, et al. Evolution of glucocorticoid receptors with different glucocorticoid sensitivity. The Journal of endocrinology. 2006;190:17–28. doi: 10.1677/joe.1.06703. [DOI] [PubMed] [Google Scholar]
- 166.Wells A, et al. The stress response to environmental change in captive cheetahs (Acinonyx jubatus) Journal of zoo and wildlife medicine : official publication of the American Association of Zoo Veterinarians. 2004;35:8–14. doi: 10.1638/02-084. [DOI] [PubMed] [Google Scholar]
- 167.Koester DC, et al. Motile Sperm Output by Male Cheetahs (Acinonyx jubatus). Managed Ex Situ Is Influenced by Public Exposure and Number of Care-Givers. PloS one. 2015;10:e0135847. doi: 10.1371/journal.pone.0135847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Li L, et al. The effect of heat stress on gene expression, synthesis of steroids, and apoptosis in bovine granulosa cells. Cell stress & chaperones. 2016;21:467–475. doi: 10.1007/s12192-016-0673-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lynn SE, et al. Food, stress, and circulating testosterone: Cue integration by the testes, not the brain, in male zebra finches (Taeniopygia guttata) General and comparative endocrinology. 2015;215:1–9. doi: 10.1016/j.ygcen.2015.03.010. [DOI] [PubMed] [Google Scholar]
- 170.Ing NH, et al. Dexamethasone acutely down-regulates genes involved in steroidogenesis in stallion testes. The Journal of steroid biochemistry and molecular biology. 2014;143:451–459. doi: 10.1016/j.jsbmb.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 171.Min Y, et al. Vitamin C and vitamin E supplementation alleviates oxidative stress induced by dexamethasone and improves fertility of breeder roosters. Animal reproduction science. 2016;171:1–6. doi: 10.1016/j.anireprosci.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 172.Nozu R, Nakamura M. Cortisol administration induces sex change from ovary to testis in the protogynous Wrasse, Halichoeres trimaculatus. Sexual development : genetics, molecular biology, evolution, endocrinology, embryology, and pathology of sex determination and differentiation. 2015;9:118–124. doi: 10.1159/000373902. [DOI] [PubMed] [Google Scholar]