

Abstract
Glioblastoma (GBM) progression is associated with metabolic remodeling in both glioma and immune cells, resulting in the use of aerobic glycolysis as the main source energy and biosynthetic molecules. The transcription factor HIF-1α drives this metabolic reorganization. Oxygen levels as well as other factors control the activity of HIF-1α. In addition, the ligand-activated transcription factor AHR modulates tumor-specific immunity and can also participate in metabolic remodeling. AHR activity is regulated by tryptophan derivatives present in the tumor microenvironment. Thus, the tumor microenvironment, signaling via HIF-1α and AHR, regulates the metabolism of gliomas and immune cells, modulating tumor-specific immunity and consequently, tumor growth. Here, we will review the roles of HIF-1α and AHR in cancer and immune cell metabolism in glioblastoma.
Keywords: glioblastoma, cancer, metabolism, AHR, HIF-1α
Metabolic alterations associated with cancer
Metabolism is a complex set of coordinated biochemical reactions that support the bioenergetic needs of the cell and maintain basic life processes such as growth and differentiation. Depending on the availability of nutrients and environmental cues, different metabolic pathways are used by specific cell types to produce energy. Most cells utilize oxidative phosphorylation to obtain the energy needed to support their needs from glucose [1]. Oxidative phosphorylation involving the mitochondrial tricarboxylic acid (TCA)/Krebs cycle, is very efficient in the production of energy in the form of adenosine 5′-triphosphate (ATP). Under hypoxic conditions, glucose participates in the less energetically efficient anaerobic glycolysis [2]. Cancer cells, however, support their metabolism through glycolysis: aerobic glycolysis (the so called Warburg effect) or anaerobic glycolysis under normoxic or hypoxic conditions, respectively [3]. Although aerobic glycolysis produces less ATP than oxidative phosphorylation, this process provides biosynthetic molecules needed to support the proliferation of cancer cells [2]. Therefore, augmented glycolysis is considered a hallmark of cancer metabolism.
The hypoxia-inducible factor (HIF) is a transcription factor that is activated in response to reduced oxygen levels and other environmental changes. HIF plays a central role in sensing environmental cues and coordinating the transcriptional control of metabolic pathways that drive glycolysis [4]. Aryl hydrocarbon receptor (AHR) is a transcription factor that is activated by small molecules provided by the diet, the gut flora, the metabolism, and the environment [5]. In the context of GBM the activity of AHR is controlled by tryptophan derivatives present in the tumor microenvironment. AHR has been shown to cooperate with HIF-1α to control immune responses and the differentiation of type 1 regulatory T cells [6]. AHR also has additional well-established functions in the immune response, controlling the generation of effector and regulatory T cells, while also modulating the innate immune response [7–12]. This review will focus on the roles of HIF-1α and AHR in abnormal metabolic processes that contribute to the pathogenicity of the most aggressive and currently incurable primary brain tumor of a glioma type, glioblastoma.
Glioblastoma
GBM is the highest-grade glioma (Grade IV) according to World Health Organization (WHO) classification and belongs to the most malignant form of brain tumors. It is an incurable disease despite the use of aggressive treatments that include surgery and radiotherapy, usually in combination with chemotherapy [13]. GBM utilizes several mechanisms to achieve fast growth and dissemination in the brain parenchyma, among them are rapid proliferation, invasion, the promotion of angiogenesis and the induction of immunosuppression [14, 15]. Gliomas actively recruit cells of the peripheral immune system by releasing several chemokines, such as CCL2 [16–19]. Once within the tumor environment, immune cells are exposed to immunomodulatory cytokines and factors, such as TGFβ1 [20], resulting in the suppression of tumor-specific immunity. Thus, the recruitment of peripheral immune cells into the tumor suppresses tumor-specific immunity and promotes tumor growth.
Myeloid cells, such as macrophages and microglia, infiltrate the GBM to which they are attracted, at least partly, by the chemokine CCL2 [18, 21–23]. Myeloid cells comprise the predominant immune cell population in GBM tissue; indeed, their abundance correlates with the GBM grade [17, 20, 24]. Notably, within the tumor microenvironment, macrophages acquire a tumorigenic antiinflammatory phenotype [17, 25]. As a result, these glioma-infiltrating macrophages support glioma invasion, angiogenesis and suppress GBM-specific immunity [15, 24, 25]. Other types of immune cells, such as T and B lymphocytes, NK cells and DCs infiltrate gliomas to a lower extent [26]. Some reports demonstrate that a higher degree of infiltration by CD8+ T cells in brain tumors is associated with less aggressive disease while CD4+ Treg infiltration is linked to poor outcome, however the association of other immune cells with disease prognosis should be further investigated [27]. Finally, the molecular mechanisms regulating the immunosuppressive activities of immune cells in GBM and potential therapeutic targets to interfere with this process are mostly unknown.
The malignancy of GBM is manifested by its high proliferation rate, ability to invade the surrounding brain parenchyma and immunosuppression. These aspects of GBM malignancy are supported by the manipulation of a number of biological pathways to exploit not only intracellular tumor resources but also the microenvironment provided by surrounding cells [28]. Augmented glycolysis or Warburg effect [3, 28, 29] and abnormal tryptophan catabolism [30] are hallmarks of GBM. Specifically, Xiong and colleagues observed that a mutated form of a critical component of the TCA cycle, isocitrate dehydrogenase 1 (IDH1), is associated with HIF-1α-mediated carcinogenesis. Their findings provide an important line of evidence that links metabolic dysfunction, through disruption of the IDH1 pathway and increases in HIF-1α activity, to increased transcriptional activity leading to more aggressive glioma growth.
In addition, Michael Platten’s group reported that kynurenine, an endogenous ligand for the transcription factor AHR derived from tryptophan is produced by glioma cells. The group suggested that the kynurenine-AHR pathway contributes to GBM pathology by increasing the growth and motility of tumor cells and suppressing the immune response. Therefore, to understand the role of metabolism in GBM pathology it is important to link specific metabolic signaling pathways to specific cell populations in the tumor microenvironment.
Multiple lines of evidence indicate that the increase in aerobic glycolysis detected in GBM supports the elevated nutrient demands of fast proliferating cancer cells by providing lipid and nucleotide biosynthesis [28]. In addition, this increased glycolysis has important effects on tumor-specific immunity and consequently, tumor pathogenesis [31, 32]. Indeed, the Warburg effect also promotes the production of lactate in anaerobic conditions, which attracts immune cells to the tumor microenvironment where they are imprinted with a tumor suppressing phenotype [33–35]. For example, excessive accumulation of lactic acid in the tumor microenvironment can lead to disruption of the lactic acid gradient between the intracellular space of lymphoid cells and the extracellular milieu [21]. Through this mechanism, T-cells can no longer export intracellular lactic acid efficiently, which leads to a disruption of metabolic processes and a consequent decrease in T-cell function. Lactic acid can activate the IL-23/IL-17 pathway, which is a canonical pro-inflammatory pathway [21]. However, lactic acid acts on tumor associated macrophages to polarize them toward an M2 state, augmenting tumor growth rates through a mechanism contingent on HIF-1α [33]. Therefore, lactic acid is an oncometabolite with important functions in cellular communication in the tumor microenvironment, which can reprogram immune cells, such as T-cells and macrophages, to become pathologic tumor-assisting agents. Moreover, the tumor microenvironment activates HIF-1α and AHR signaling to promote the metabolic reprogramming of immune cells and further modulate anti-tumor immunity [29, 30]. Thus, metabolic adaptation and the changes it imposes on the tumor microenvironment promote GBM survival and propagation by acting both on glioma and immune cells. Although several pathways contribute to GBM pathology, we will focus on the roles of HIF-1α and AHR signaling.
HIF-1α
HIF-1α signaling in cancer cells contributes to tumor progression by promoting angiogenesis, invasion, metastasis and the recruitment of immunosuppressive cells via secreted modulators [36, 37]. One example of a HIF-1α immunosuppressive pathway is through increased expression of inducible nitric oxide synthase (iNOS), which can act on myeloid cells to impair immune cell recruitment to tumors [38]. Similarly, HIF-1α activity regulates the chemotactic properties of immune cell-mediated tumor infiltration by increasing vascular endothelial growth factor (VEGF) production from endothelial cells [39]. VEGF binds to its receptor, neuropilin-1, and attracts Tregs to the tumor site, which is a positive regulator of tumor growth and is therefore associated with worse survival outcomes. Similar relationships were observed for TGF-β, which is secreted by malignant cells to attract Tregs to GBM and prevent the killing of cancer cells [23]. HIF-1α has been identified as an important driver of the Warburg effect in several tumors, including GBM [40–42]. Hypoxia, a condition of reduced oxygen supply in tissue, is a well-characterized trigger of HIF-1α dependent signaling. Under normoxic conditions, prolyl hydroxylase domain-containing proteins (PHD) promote the degradation of HIF-1α, while the HIF-1α inhibitor protein (gene: Hif1an) suppresses its transcriptional activity [42, 43]. However, PHD proteins and the HIF-1α inhibitor are inactivated by hypoxia, resulting in HIF-1α stabilization and the induction of HIF-1α—dependent cellular responses such as promoting angiogenesis, regulating immune cell tumor infiltration, and exerting control over anaerobic and aerobic metabolic processes [37, 43]. HIF-1α is frequently up-regulated in solid malignances, such as GBM, due to the hypoxic conditions that characterize the tumor microenvironment [41]. HIF-1α is also activated by additional stimuli besides low oxygen tension, such as extracellular ATP [41, 44] or lactate [33].
HIF-1α complexes with HIF-1β (also known as the AHR nuclear translocator, ARNT) to regulate the expression of target genes [45]. The HIF-1α/HIF-1β complex translocates to the nucleus to control the expression of genes that contain hypoxia response consensus sequences (HREs) in their regulatory regions [40, 46]. HIF-1α is an important regulator of the expression of glycolytic enzymes under hypoxic conditions [47]. For example, HIF-1α promotes the expression of hexokinase 2 (HK2), an enzyme up-regulated in GBM that plays a critical role in the initiation of glycolysis [48, 49]. HIF-1α also promotes the expression of the pyruvate dehydrogenase kinase 1 (PDK1), which inhibits pyruvate dehydrogenase thereby limiting the entry of pyruvate into the TCA cycle [50]. Thus, HIF-1α increases glycolysis by direct stimulation of key components of this metabolic pathway. Surprisingly, HIF-1α and its close isoform HIF-2α may have opposing activities in cancer depending on the cellular context [51, 52]. Moreover, HIF-1α and HIF-2α are relatively ubiquitous in cells but are regulated differentially. For instance, HIF-1α expression increases under hypoxic conditions while HIF-2α expression is increased with higher oxygen levels [43]. Production of nitric oxide (NO) is dependent on differential expression of HIF-1α and HIF-2α, which are themselves regulated by IFN-γ [43, 53, 54]. NO production controls immune cell migration to the tumor site and high NO levels, caused by HIF-1α, lead to immunosuppressive functions. However, only HIF-2α but not HIF-1α is associated with poor prognosis in glioma [55, 56]. These effects are thought to be mediated at least partially through the inhibition of stem-like features of tumor cells, thereby arresting tumor growth [55].
Additional mechanisms enhance glycolysis in glioma. IDH1, for example, is found to be mutated in 20% of GBM cases, especially in younger patients [57, 58]. IDH1 is a critical component of the TCA through the generation of alpha-ketoglutarate, a metabolite that destabilizes HIF-1α. A unique CpG island methylation pattern was identified in GBM patients bearing IDH1 mutations [59]. Importantly, the mutant form of IDH1 can inhibit the activity of normal IDH1 in a dominant fashion. Moreover, mutated IDH1 increases the levels of HIF-1α by protecting it from degradation [29, 60, 61]. In gliomas that express a mutated form of IDH1 (R132H), HIF-1α transcriptional activity is increased, which leads to worsened glioma outcomes, at least partially through HIF-1α as a result of increased angiogenesis and other HIF-1α dependent processes that occur in hypoxic environments. Similar effects have been described in association with over-expressed microRNAs that correlate with poor survival in GBM. Indeed, miR-148a and miR-31 which involve HIF-1α signaling were shown to regulate glioma growth and angiogenesis [62]. Thus, in addition to hypoxia and other metabolic changes associated with the tumor microenvironment, IDH1 mutations and epigenetic changes accompanying GBM activate HIF-1α, reinforcing the metabolic remodeling that promotes tumorigenesis and glioma pathogenesis.
Interestingly, hypoxia and enhanced Warburg effect ultimately lead to increased lactate production. The accumulation of lactate results in an acidic tumor microenvironment, which induces local inflammation. Lactate also promotes the polarization of macrophages that express immunosuppressive arginase 1 and boost tumor growth through a mechanism mediated by HIF-1α [33]. Attracted immune cells then promote immune suppression, providing an advantage for tumor cell proliferation [34, 35]. Thus, changes in the tumor microenvironment resulting from metabolic alterations attract immune cells and modulate their function, suppressing tumor-specific immunity and facilitating tumor proliferation.
Physiologic conditions different from hypoxia can also activate HIF-1α in immune cells associated with GBM pathogenesis. For example, the activation of CD4+ T helper 17 (Th17), CD4+ type 1 regulatory T cells (Tr1 cells) and CD8+ T cells (cytotoxic T lymphocytes, CTLs) results in HIF-1α stabilization in an oxygen-independent manner [6, 63–66]. In these cells, HIF-1α signaling is thought to reflect the activation of mTORc1 [66, 67]. The stabilization of HIF-1α in these cells leads to increased glycolysis that supports the metabolic requirements of proliferating cells following their stimulation. For example, Finlay et al. showed that the HIF-1 pathway sustains the expression of several genes that control glycolysis and pyruvate metabolism in effector CD8+ T cells [67]. However, it should be noted that although the later stages of the glycolytic process are significantly impaired, the early processes of glycolysis are intact in the context of HIF-1 deficiency in effector CD8+ T cells [67]. Moreover, HIF-1-deficient CD8+ T cells show reduced trafficking and expression of granzymes and perforin, indicating that HIF-1 supports cytotoxic effector responses in CTLs [67]. Supporting the involvement of HIF-1 in activating cytolytic effector molecules, CD8+ T cells cultured under hypoxic conditions up-regulate perforin expression. These findings explain earlier observations demonstrating that CTLs exhibit cytotoxic activity under hypoxia [68]. Collectively, these data suggest that HIF-1α activation may impact the effector function of tumor-specific T cells.
HIF-1α also influences the differentiation of helper T cell lineages that play an important role in GBM pathogenesis [63, 66]. For example, HIF-1α promotes the generation, maintenance and function of Th17 cells [63, 66, 69]. In addition, HIF-1α promotes Foxp3 ubiquitination and its degradation by the proteasome in Th17 cells [63], suppressing Foxp3+ Treg differentiation. Conversely, the expression of HIF-1α is reduced under conditions promoting Foxp3+ Treg differentiation [66]. These findings suggest that aerobic glycolysis promotes Th17 cell differentiation. Indeed, the blockade of glycolysis inhibits Th17 cell generation while stimulating Foxp3+ Treg cell development [66]. Finally, HIF-1α signaling in antigen presenting cells has been shown to modulate their ability to promote the differentiation of Th1 cells and Foxp3+ Tregs [70]. Collectively, these findings suggest that, besides its direct role in cancer cell metabolism, HIF-1α is an important factor in the control of tumor-specific immunity.
In summary, HIF-1α regulation and function in GBM seems to be complex and deserves further investigation. HIF-1α can be activated in glioma and immune cells infiltrating the tumor by multiple factors, including hypoxia, inflammation, ATP levels and IDH mutation state. The exact mechanism that drives HIF-1α function in GBM is not clear and it seems that HIF-1α activity is context and time dependent. By signaling through different cells in glioma microenvironment, HIF-1α can overall promote tumor growth by a number of mechanisms, such as tumor cell invasion, angiogenesis and immune suppression. While a link between HIF-1α and patient’s outcome needs to be established, it is possible that HIF-1α pathologic activity is associated with a specific genetic subtype of GBM. Additional work should be performed to resolve the impact of HIF-1α signaling in different cells of GBM on the net disease outcome.
AHR
AHR has been linked to several forms of cancer, including GBM [30, 71, 72]. In addition, the expression of the AHR repressor (AHRR) is down-regulated due to hyper-methylation in a number of human malignancies [73]. The regulation of AHR expression has not been well described in cancer. NF-kB, dysregulated in cancer, has been shown to activate directly the transcription of AHR in immune cells implying its potential role in the transcriptional regulation of AHR in cancer [74]. AHR acts as a receptor for ligands provided by the diet, gut flora, metabolism and environment. The inactive, cytosolic AHR is part of a protein complex that includes the 90 kDa heat shock protein (HSP90), the c-SRC protein kinase and the AHR—interacting protein Ara9. Ligand binding triggers conformational changes that expose a protein kinase C target site, phosphorylation of which triggers the translocation of the AHR to the nucleus and the dissociation of the AHR/HSP90/c-SRC complex. In the nucleus, AHR partners with ARNT to control the expression of target genes. AHR can also trigger non-genomic signaling pathways independent of its role as a transcription factor [45]. One of these pathways results from the release of c-SRC from the AHR/HSP90/c-SRC complex, enabling c-SRC to then target multiple cellular targets [45].
Recent studies have demonstrated that AHR plays an important role in the interplay between cancer metabolism and tumor-specific immunity. Tryptophan catabolism is increasingly recognized as a metabolic pathway that promotes tumorigenesis through its role in immune suppression [75]. The rate-limiting enzymes tryptophan-2,3-dioxygenase (TDO) and idoleamine-2,3-dioxygenase (IDO), expressed by tumor cells and antigen presenting cells, are thought to drive the increased catabolism of tryptophan in cancer [30, 76]. This metabolic pathway is up-regulated in several tumors including GBM, where it creates an immunosuppressive microenvironment through the depletion of the essential amino acid tryptophan and the generation of immunomodulatory tryptophan metabolites such as kynurenine (Kyn), which suppresses T cell function and induce apoptosis [77]. Importantly, Kyn is an agonist of AHR. Indeed, AHR activation by Kyn promotes Foxp3+ Treg differentiation, supporting earlier reports of a role of AHR in Tregs [9, 78]. Of note, AHR also promotes the differentiation of IL-10 producing Tr1 cells [7, 8], which together with Foxp3+ Tregs have been shown to contribute to tumor-associated immunosuppression [79]. In addition, AHR has also been linked to Th17 differentiation [79, 80]. However, detailed analyses suggest that AHR is more associated with non-pathogenic Th17 cells [80, 81]. Indeed, it has been shown that AHR drives the expression of the ectoenzyme CD39, which together with CD73 promotes the production of immunosuppressive adenosine. CD39-expressing Th17 cells with suppressive function have been associated with tumor immunosuppression [82].
AHR also regulates the function of dendritic cells (DC) [45], modulating their ability to promote the differentiation of effector and regulatory T cells [11, 13, 83]. AHR affects antigen presenting cell function through several mechanisms. AHR induces the production of Kyn and retinoic acid (RA) by DCs, boosting the differentiation of Tregs and interfering with effector T cells [84]. AHR also suppresses NF-kB activation in DCs through a mechanism mediated by SOCS2 [85], consequently interfering with the production of cytokines that promote effector T cell differentiation. In addition, AHR controls the activity of resident cells in the central nervous system with inflammatory function, such as astrocytes and microglia [12], which play important roles in the control of the tumor microenvironment. Collectively, these data suggest that Kyn produced by tryptophan metabolism in GBM triggers AHR-dependent effects in multiple components of the immune system to impair tumor-specific immunity.
Less is known about the direct role of AHR in the metabolism of cancer cells. Opitz et al. recently reported that TDO in glioma cells produce Kyn, which acts in an autocrine manner to activate AHR and increased tumor growth and invasiveness [30] and can potentially activate AHR on tumor-infiltrating immune cells. Thus, AHR signaling could be elevated in both glioma and inflammatory immune cells in GBM. Taken together, these findings suggest that AHR activation by tryptophan metabolites acts on gliomas and immune cells to promote GBM pathogenesis.
Of note, Kyn was proposed as a novel biomarker for meningioma grades as it correlates with cancer pathology, further implicating the role of AHR in brain tumors [86]. Thus, tryptophan metabolites and other tumor-associated AHR ligands may offer useful biomarkers to stratify cancer patients for immunotherapy.
Cross-talk between AHR and HIF-1α
AHR- and HIF-1α-dependent signaling pathways have several points of contact, with important biological consequences. Both AHR and HIF-1α are environmental sensors [87] that dimerize with the same partner (HIF-1β/ARNT) to exert their biological effects [45]. Studies using HIF-1β-deficient T cells demonstrated that HIF-1β/ARNT is needed to sustain glycolysis in CD8+ effector T cells [67]. This evidence, therefore, suggests a functional cross-talk between AHR and HIF-1α signaling pathways. Another common binding partner of HIF-1α and AHR, chaperone HSP90, can provide an additional link between two proteins [88].
It has been recently shown that AHR and HIF-1α cooperate to support the metabolism of Tr1 cells [6]. Interestingly, AHR and HIF-1α act sequentially to orchestrate the metabolic remodeling of lymphocytes. While HIF-1α regulates the early metabolic reprogramming of Tr1 cells, AHR takes over at later time points, inducing HIF-1α degradation. This cross-regulation between AHR and HIF-1α may also operate in other cell types. For example, lipopolysaccharide induces AHR and HIF-1α expression in macrophages, and both pathways have been independently shown to affect the biologic response of these cells [89, 90]. Thus, it is possible that AHR and HIF-1α cooperate to regulate the response of macrophages and other cell populations implicated in GBM pathology. Therefore, by acting both in gliomas and immune cells in the tumor microenvironment, AHR and HIF-1α may synergize to support GBM malignant growth.
Concluding remarks
Hypoxia and enhanced tryptophan metabolism characterize the cancer microenvironment. These metabolic features of the tumor microenvironment induce the activation of HIF-1α and AHR signaling to regulate the metabolic reprograming of glioma and immune cells and to activate specific transcriptional programs that interfere with tumor-specific immunity. The combined effect of these pathways is the amplification of glioma pathogenesis (Fig. 1). A number of questions, however, remain open and need to be addressed by future studies (see Outstanding Questions). For example, it is important to delineate the effects of tryptophan metabolites on different cells comprising the GBM microenvironment, the contribution of AHR and HIF-1α in these effects and their potential as therapeutic targets. The successful results reported in recent checkpoint inhibitor clinical trials highlight the potential of immunotherapeutic approaches for cancer [90–95]. Thus, considering their dual roles as regulators of both glioma and immune cells, HIF-1α and AHR offer new and exciting opportunities for therapeutic intervention.
Figure 1. Factors in the tumor microenvironment drive immunosuppression in GBM.
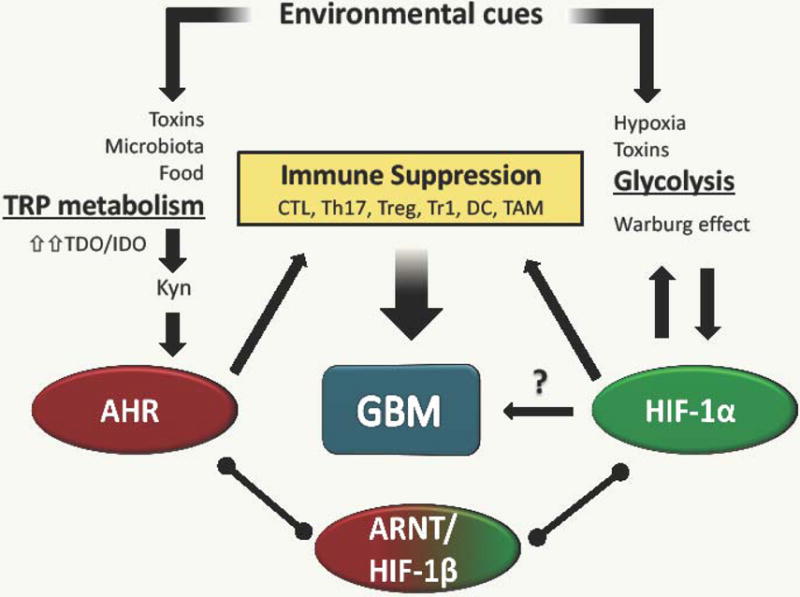
Soluble factors in the periphery and tumor microenvironment can shift adaptive and innate immune cell function to an immunosuppressive state. Microbiota products and onco-metabolites provide AHR agonists that inhibit inflammatory cytokine production by dendritic cells and macrophages, favoring the differentiation of CD4+ Treg cells. Moreover, hypoxic microenvironments favor tumor growth by inducing pro-tumorigenic macrophage polarization.
Outstanding Questions Box.
What is the primary signal that initiates metabolic changes in GBM: low oxygen or kynurenine accumulation?
Is the function of HIF-1α and AHR coordinated in the GBM microenvironment? What is the role of ARNT in this process?
How do AHR and HIF-1α affect immune cells in the tumor microenvironment to support gliomagenesis?
What is the contribution of AHR and HIF-1α signaling in different immune cells (e.g. effector T cells, Tregs and myeloid cells) to GBM pathogenesis?
What is the potential of metabolism as a therapeutic target for GBM?
What other metabolic pathways are involved in gliomagenesis?
Trends Box.
Metabolic remodeling in tumor and immune cells promotes GBM malignancy.
In addition to the well-characterized Warburg effect, tryptophan catabolism contributes to gliomagenesis through a kynurenine/AHR signaling pathway.
HIF-1α and AHR sense metabolic changes in tumor environment and promote glioma progression by signaling in cancer and also, in immune cells such as CD4 and CD8 T lymphocytes and macrophages.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Balaban RS. Regulation of oxidative phosphorylation in the mammalian cell. Am J Physiol. 1990;258(3 Pt 1):C377–89. doi: 10.1152/ajpcell.1990.258.3.C377. [DOI] [PubMed] [Google Scholar]
- 2.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124(3215):269–70. [PubMed] [Google Scholar]
- 4.Semenza GL. Regulation of metabolism by hypoxia-inducible factor 1. Cold Spring Harb Symp Quant Biol. 2011;76:347–53. doi: 10.1101/sqb.2011.76.010678. [DOI] [PubMed] [Google Scholar]
- 5.Murray IA, Patterson AD, Perdew GH. Aryl hydrocarbon receptor ligandsin cancer: friend and foe. Nat Rev Cancer. 2014;14(12):801–14. doi: 10.1038/nrc3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mascanfroni ID, et al. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-alpha. Nat Med. 2015;21(6):638–46. doi: 10.1038/nm.3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Apetoh L, et al. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat Immunol. 2010;11(9):854–61. doi: 10.1038/ni.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mascanfroni ID, et al. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-α. Nat Med. 2015;21(6):638–46. doi: 10.1038/nm.3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quintana FJ, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453(7191):65–71. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- 10.Quintana FJ, et al. Aiolos promotes T(H)17 differentiation by directly silencing Il2 expression. Nature Immunology. 2012;13(8):770–777. doi: 10.1038/ni.2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quintana FJ, et al. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2010;107(48):20768–73. doi: 10.1073/pnas.1009201107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rothhammer V, et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med. 2016;22(6):586–97. doi: 10.1038/nm.4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeste A, et al. Nanoparticle-mediated codelivery of myelin antigen and a tolerogenic small molecule suppresses experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2012;109(28):11270–5. doi: 10.1073/pnas.1120611109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wen PY, Kesari S. Malignant gliomas in adults. The New England journal of medicine. 2008;359(5):492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 15.Charles NA, et al. The brain tumor microenvironment. Glia. 2012;60(3):502–14. doi: 10.1002/glia.21264. [DOI] [PubMed] [Google Scholar]
- 16.Furnari FB, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes & development. 2007;21(21):2683–710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 17.Roggendorf W, Strupp S, Paulus W. Distribution and characterization of microglia/macrophages in human brain tumors. Acta Neuropathol. 1996;92(3):288–93. doi: 10.1007/s004010050520. [DOI] [PubMed] [Google Scholar]
- 18.Platten M, et al. Monocyte chemoattractant protein-1 increases microglial infiltration and aggressiveness of gliomas. Annals of neurology. 2003;54(3):388–92. doi: 10.1002/ana.10679. [DOI] [PubMed] [Google Scholar]
- 19.Okada M, et al. Tumor-associated macrophage/microglia infiltration in human gliomas is correlated with MCP-3, but not MCP-1. International journal of oncology. 2009;34(6):1621–7. doi: 10.3892/ijo_00000292. [DOI] [PubMed] [Google Scholar]
- 20.Sciume G, Santoni A, Bernardini G. Chemokines and glioma: invasion and more. Journal of neuroimmunology. 2010;224(1–2):8–12. doi: 10.1016/j.jneuroim.2010.05.019. [DOI] [PubMed] [Google Scholar]
- 21.Platten M, Wick W, Weller M. Malignant glioma biology: role for TGF-beta in growth, motility, angiogenesis, and immune escape. Microscopy research and technique. 2001;52(4):401–10. doi: 10.1002/1097-0029(20010215)52:4<401::AID-JEMT1025>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 22.Prat E, et al. The human astrocytoma cell line U373MG produces monocyte chemotactic protein (MCP)-1 upon stimulation with beta-amyloid protein. Neurosci Lett. 2000;283(3):177–80. doi: 10.1016/s0304-3940(00)00966-6. [DOI] [PubMed] [Google Scholar]
- 23.Yang I, et al. The role of microglia in central nervous system immunity and glioma immunology. J Clin Neurosci. 2010;17(1):6–10. doi: 10.1016/j.jocn.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mantovani A, et al. Tumor-associated macrophages and the related myeloid-derived suppressor cells as a paradigm of the diversity of macrophage activation. Human immunology. 2009;70(5):325–30. doi: 10.1016/j.humimm.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 25.Laoui D, et al. Mononuclear phagocyte heterogeneity in cancer: different subsets and activation states reaching out at the tumor site. Immunobiology. 2011;216(11):1192–202. doi: 10.1016/j.imbio.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 26.Domingues P, et al. Tumor infiltrating immune cells in gliomas and meningiomas. Brain Behav Immun. 2016;53:1–15. doi: 10.1016/j.bbi.2015.07.019. [DOI] [PubMed] [Google Scholar]
- 27.Platten M, Wick W. Tregs in gliomas - the jury is still out. Neuro Oncol. 2015;17(6):769–70. doi: 10.1093/neuonc/nov034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Agnihotri S, Zadeh G. Metabolic reprogramming in glioblastoma: The influence of cancer metabolism on epigenetics and unanswered questions. Neuro Oncol. 2015 doi: 10.1093/neuonc/nov125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao S, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324(5924):261–5. doi: 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Opitz CA, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478(7368):197–203. doi: 10.1038/nature10491. [DOI] [PubMed] [Google Scholar]
- 31.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 32.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331(6024):1565–70. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 33.Colegio OR, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513(7519):559–63. doi: 10.1038/nature13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fischer K, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109(9):3812–9. doi: 10.1182/blood-2006-07-035972. [DOI] [PubMed] [Google Scholar]
- 35.Shime H, et al. Tumor-secreted lactic acid promotes IL-23/IL-17 proinflammatory pathway. J Immunol. 2008;180(11):7175–83. doi: 10.4049/jimmunol.180.11.7175. [DOI] [PubMed] [Google Scholar]
- 36.Deng B, et al. Intratumor hypoxia promotes immune tolerance by inducing regulatory T cells via TGF-beta1 in gastric cancer. PLoS One. 2013;8(5):e63777. doi: 10.1371/journal.pone.0063777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Palazon A, et al. HIF transcription factors, inflammation, and immunity. Immunity. 2014;41(4):518–28. doi: 10.1016/j.immuni.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peyssonnaux C, et al. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest. 2005;115(7):1806–15. doi: 10.1172/JCI23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hansen W, et al. Neuropilin 1 deficiency on CD4+Foxp3+ regulatory T cells impairs mouse melanoma growth. J Exp Med. 2012;209(11):2001–16. doi: 10.1084/jem.20111497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang GL, et al. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92(12):5510–4. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maynard MA, Ohh M. The role of hypoxia-inducible factors in cancer. Cell Mol Life Sci. 2007;64(16):2170–80. doi: 10.1007/s00018-007-7082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Womeldorff M, Gillespie D, Jensen RL. Hypoxia-inducible factor-1 and associated upstream and downstream proteins in the pathophysiology and management of glioblastoma. Neurosurg Focus. 2014;37(6):E8. doi: 10.3171/2014.9.focus14496. [DOI] [PubMed] [Google Scholar]
- 43.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148(3):399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Amoroso F, et al. The P2X7 receptor is a key modulator of aerobic glycolysis. Cell Death Dis. 2012;3:e370. doi: 10.1038/cddis.2012.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quintana FJ, Sherr DH. Aryl hydrocarbon receptor control of adaptive immunity. Pharmacol Rev. 2013;65(4):1148–61. doi: 10.1124/pr.113.007823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE. 2005;2005(306):re12. doi: 10.1126/stke.3062005re12. [DOI] [PubMed] [Google Scholar]
- 47.Iyer NV, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12(2):149–62. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wolf A, et al. Developmental profile and regulation of the glycolytic enzyme hexokinase 2 in normal brain and glioblastoma multiforme. Neurobiol Dis. 2011;44(1):84–91. doi: 10.1016/j.nbd.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 49.Wolf A, et al. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J Exp Med. 2011;208(2):313–26. doi: 10.1084/jem.20101470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim JW, et al. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3(3):177–85. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 51.Acker T, et al. Genetic evidence for a tumor suppressor role of HIF-2alpha. Cancer Cell. 2005;8(2):131–41. doi: 10.1016/j.ccr.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 52.Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2012;12(1):9–22. doi: 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takeda N, et al. Differential activation and antagonistic function of HIF-{alpha} isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010;24(5):491–501. doi: 10.1101/gad.1881410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Doedens AL, et al. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. 2010;70(19):7465–75. doi: 10.1158/0008-5472.CAN-10-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li Z, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15(6):501–13. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Scrideli CA, et al. Prognostic significance of co-overexpression of the EGFR/IGFBP-2/HIF-2A genes in astrocytomas. J Neurooncol. 2007;83(3):233–9. doi: 10.1007/s11060-007-9328-0. [DOI] [PubMed] [Google Scholar]
- 57.Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cohen AL, Holmen SL, Colman H. IDH1 and IDH2 mutations in gliomas. Curr Neurol Neurosci Rep. 2013;13(5):345. doi: 10.1007/s11910-013-0345-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Noushmehr H, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17(5):510–22. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang G, et al. Mutation of isocitrate dehydrogenase 1 induces glioma cell proliferation via nuclear factor-kappaB activation in a hypoxia-inducible factor 1-alpha dependent manner. Mol Med Rep. 2014;9(5):1799–805. doi: 10.3892/mmr.2014.2052. [DOI] [PubMed] [Google Scholar]
- 61.Dimitrov L, et al. New developments in the pathogenesis and therapeutic targeting of the IDH1 mutation in glioma. Int J Med Sci. 2015;12(3):201–13. doi: 10.7150/ijms.11047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wong HK, et al. The Cancer Genome Atlas Analysis Predicts MicroRNA for Targeting Cancer Growth and Vascularization in Glioblastoma. Mol Ther. 2015;23(7):1234–47. doi: 10.1038/mt.2015.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dang EV, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146(5):772–84. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Doedens AL, et al. Hypoxia-inducible factors enhance the effector responses of CD8(+)T cells to persistent antigen. Nat Immunol. 2013;14(11):1173–82. doi: 10.1038/ni.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nakamura H, et al. TCR engagement increases hypoxia-inducible factor-1 alpha protein synthesis via rapamycin-sensitive pathway under hypoxic conditions in human peripheral T cells. J Immunol. 2005;174(12):7592–9. doi: 10.4049/jimmunol.174.12.7592. [DOI] [PubMed] [Google Scholar]
- 66.Shi LZ, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208(7):1367–76. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Finlay DK, et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med. 2012;209(13):2441–53. doi: 10.1084/jem.20112607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Caldwell CC, et al. Differential effects of physiologically relevant hypoxic conditions on T lymphocyte development and effector functions. J Immunol. 2001;167(11):6140–9. doi: 10.4049/jimmunol.167.11.6140. [DOI] [PubMed] [Google Scholar]
- 69.Kryczek I, et al. Human TH17 cells are long-lived effector memory cells. Sci Transl Med. 2011;3(104):104ra100. doi: 10.1126/scitranslmed.3002949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu G, et al. Dendritic cell SIRT1-HIF1alpha axis programs the differentiation of CD4+ T cells through IL-12 and TGF-beta1. Proc Natl Acad Sci U S A. 2015;112(9):E957–65. doi: 10.1073/pnas.1420419112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gramatzki D, et al. Aryl hydrocarbon receptor inhibition downregulates the TGF-beta/Smad pathway in human glioblastoma cells. Oncogene. 2009;28(28):2593–605. doi: 10.1038/onc.2009.104. [DOI] [PubMed] [Google Scholar]
- 72.Andersson P, et al. A constitutively active dioxin/aryl hydrocarbon receptor induces stomach tumors. Proc Natl Acad Sci U S A. 2002;99(15):9990–5. doi: 10.1073/pnas.152706299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zudaire E, et al. The aryl hydrocarbon receptor repressor is a putative tumor suppressor gene in multiple human cancers. J Clin Invest. 2008;118(2):640–50. doi: 10.1172/JCI30024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vogel CF, et al. Cross-talk between aryl hydrocarbon receptor and the inflammatory response: a role for nuclear factor-kappaB. J Biol Chem. 2014;289(3):1866–75. doi: 10.1074/jbc.M113.505578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res. 2012;72(21):5435–40. doi: 10.1158/0008-5472.CAN-12-0569. [DOI] [PubMed] [Google Scholar]
- 76.Muller AJ, et al. Chronic inflammation that facilitates tumor progression creates local immune suppression by inducing indoleamine 2,3 dioxygenase. Proc Natl Acad Sci U S A. 2008;105(44):17073–8. doi: 10.1073/pnas.0806173105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Opitz CA, et al. Tryptophan degradation in autoimmune diseases. Cell Mol Life Sci. 2007;64(19–20):2542–63. doi: 10.1007/s00018-007-7140-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mezrich JD, et al. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. 2010;185(6):3190–8. doi: 10.4049/jimmunol.0903670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6(4):295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 80.Veldhoen M, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453(7191):106–9. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- 81.Lee Y, et al. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol. 2012;13(10):991–9. doi: 10.1038/ni.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Longhi MS, et al. Characterization of human CD39+ Th17 cells with suppressor activity and modulation in inflammatory bowel disease. PLoS One. 2014;9(2):e87956. doi: 10.1371/journal.pone.0087956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Takenaka MC, Robson S, Quintana FJ. Regulation of the T Cell Response by CD39. Trends Immunol. 2016;37(7):427–39. doi: 10.1016/j.it.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yeste A, et al. Tolerogenic nanoparticles inhibit T cell-mediated autoimmunity through SOCS2. Sci Signal. 2016;9(433):ra61. doi: 10.1126/scisignal.aad0612. [DOI] [PubMed] [Google Scholar]
- 85.Quintana FJ, Yeste A, Mascanfroni ID. Role and therapeutic value of dendritic cells in central nervous system autoimmunity. Cell Death Differ. 2015;22(2):215–24. doi: 10.1038/cdd.2014.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bosnyak E, et al. Molecular imaging correlates of tryptophan metabolism via the kynurenine pathway in human meningiomas. Neuro Oncol. 2015;17(9):1284–92. doi: 10.1093/neuonc/nov098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ramsay G, Cantrell D. Environmental and metabolic sensors that control T cell biology. Front Immunol. 2015;6:99. doi: 10.3389/fimmu.2015.00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gradin K, et al. Functional interference between hypoxia and dioxin signal transduction pathways: competition for recruitment of the Arnt transcription factor. Mol Cell Biol. 1996;16(10):5221–31. doi: 10.1128/mcb.16.10.5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Blouin CC, et al. Hypoxic gene activation by lipopolysaccharide in macrophages: implication of hypoxia-inducible factor 1alpha. Blood. 2004;103(3):1124–30. doi: 10.1182/blood-2003-07-2427. [DOI] [PubMed] [Google Scholar]
- 90.Sekine H, et al. Hypersensitivity of aryl hydrocarbon receptor-deficient mice to lipopolysaccharide-induced septic shock. Mol Cell Biol. 2009;29(24):6391–400. doi: 10.1128/MCB.00337-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Littman DR. Releasing the Brakes on Cancer Immunotherapy. Cell. 2015;162(6):1186–90. doi: 10.1016/j.cell.2015.08.038. [DOI] [PubMed] [Google Scholar]
- 92.Palucka AK, Coussens LM. The Basis of Oncoimmunology. Cell. 2016;164(6):1233–47. doi: 10.1016/j.cell.2016.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Preusser M, et al. Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat Rev Neurol. 2015;11(9):504–14. doi: 10.1038/nrneurol.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Reardon DA, et al. Immunotherapy advances for glioblastoma. Neuro Oncol. 2014;16(11):1441–58. doi: 10.1093/neuonc/nou212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]