

Abstract
Reactive oxygen species (ROS) are harnessed by the cell for signaling at the same time as being detrimental to cellular components such as DNA. The genome and transcriptome contain instructions that can alter cellular processes when oxidized. The guanine (G) heterocycle in the nucleotide pool, DNA, or RNA is the base most prone to oxidation. The oxidatively-derived products of G consistently observed in high yields from hydroxyl radical, carbonate radical, or singlet oxygen oxidations under conditions modeling the cellular reducing environment are discussed. The major G base oxidation products are 8-oxo-7,8-dihydroguanine (OG), 5-carboxamido-5-formamido-2-iminohydantoin (2Ih), spiroiminodihydantoin (Sp), and 5-guanidinohydantoin (Gh). The yields of these products show dependency on the oxidant and the reaction context that includes nucleoside, single-stranded DNA (ssDNA), double-stranded DNA (dsDNA), and G-quadruplex DNA (G4-DNA) structures. Upon formation of these products in cells, they are recognized by the DNA glycosylases in the base excision repair (BER) pathway. This review focuses on initiation of BER by the mammalian Nei-like1-3 (NEIL1-3) glycosylases for removal of 2Ih, Sp, and Gh. The unique ability of the human NEILs to initiate removal of the hydantoins in ssDNA, bulge-DNA, bubble-DNA, dsDNA, and G4-DNA is outlined. Additionally, when Gh exists in a G4 DNA found in a gene promoter, NEIL-mediated repair is modulated by the plasticity of the G4-DNA structure provided by additional G-runs flanking the sequence. On the basis of these observations and cellular studies from the literature, the interplay between DNA oxidation and BER to alter gene expression is discussed.
Keywords: 8-oxo-7, 8-dihydroguanine; 5-carboxamido-5-formamido-2-iminohydantoin; spiroiminodihydantoin; 5-guanidinohydantoin; NEIL DNA glycosylase; G-quadruplex
Graphical abstract
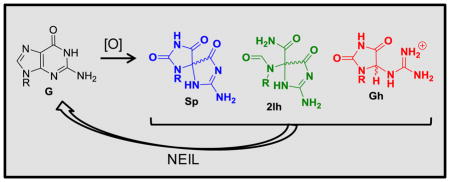
1. Introduction1
Reactive oxygen species (ROS) are electron deficient and readily oxidize most biomolecules. At low concentrations, the ROS hydrogen peroxide (H2O2) and peroxynitrite (ONOO−) are signaling agents harnessed by the cell to guide biological processes [1, 2]. In contrast, when the concentration of H2O2 or ONOO− surpasses their signaling threshold, they promote oxidation of biomolecules, such as proteins, lipids, and particularly, nucleic acids [1, 3]. High levels of ROS released under control in phagocytes provide protection from invading bacteria and viruses [4]; however, uncontrolled and high levels of ROS breakdown cellular components leading to cancer, neurological disorders, and cardiovascular disease, to name a few [5–7]. Oxidation of the genome represents a critical location in which oxidatively-derived damage under high levels of ROS can lead to these diseases.
The guanine heterocycle (G) is the most electron rich of the four bases, and as a consequence, it has the lowest reduction potential resulting in G nucleotides as major sites of ROS-mediated oxidations [8]. Scientists have unearthed detailed information concerning the ROS-dependent pathways for oxidizing G leading to many products of varying stability [9–27]. On the basis of this information, the G oxidation products can be placed into groups dependent on whether the oxidation occurs at the sugar or heterocyclic base [12]. A product observed in nearly all studies conducted under biologically relevant reaction conditions is the base oxidation product 8-oxo-7,8-dihydroguanine (OG; Scheme 1) [9–25]. Furthermore, OG is found in cell extracts and has been identified as a biomarker for monitoring the extent of cellular oxidative stress [28]. Lastly, OG is dramatically more sensitive toward oxidation than is G [29] yielding the hydantoin products spiroiminodihydantoin (Sp) and 5-guanidinohydantoin (Gh) [30–32], and these products also result from direct 4-electron oxidation of G [33, 34]. The present review will briefly summarize the reaction conditions leading to OG, Sp, and Gh. Additionally, work to establish the possible importance of 5-carboxamido-5-formamido-2-iminohydantoin (2Ih) in cellular oxidations will be described. A detailed analysis of the mechanisms and intermediates leading to these products will not be provided and can be found in other reviews [9–24].
Scheme 1.

Overview of G oxidation products and the extent to which each was oxidized leading to their formation. The products inside the dashed boxes are the main focus of this review because they are the best substrates for the hNEIL glycosylases (2Ih, Sp, or Gh) or a key precursor to these products (OG).
The picture of a static B-form helical genome that simply holds the cellular blueprint has emerged as incomplete. Genomes are rich in regulatory chemical modifications, and DNA adopts other secondary structures beyond B-form helices. Mounting evidence supports hairpin [35], A-form [36], Z-form [37], triple helix [38], Holliday junction [39], i-motif [40], and G-quadruplex (G4) [41] structures having the capability to form in genomes. Therefore, this review will cover the current state of our knowledge regarding how some of these various structural contexts impact G oxidation, leading to a discussion of knowledge gaps in the literature on this topic.
Oxidatively-derived damage to DNA, if left unrepaired, would be detrimental to cellular survival because the distorted blueprint would be incapable of providing proper instructions to the cell. To maintain a complete set of instructions, biology has evolved a DNA repair system to fix damaged nucleotides. The importance of DNA repair mechanisms was highlighted in 2015 when Tomas Lindahl, Paul Modrich, and Aziz Sancar shared the Nobel Prize in Chemistry for their work on this topic. With regard to repair of G-base oxidation products, both the base excision repair (BER) and nucleotide excision repair (NER) pathways are invoked [42–45]. This review will focus on BER repair of G-base oxidation products by the three members of the NEIL DNA glycosylase family, ending with a focus on repair in structural contexts beyond B-form helices. The literature summary presented will guide a discussion on recent cellular studies hypothesizing how G oxidation and its repair may serve as a signaling pathway in cells, beyond constituting unwanted damage that must be removed and corrected.
2. ROS formed in the cellular context
Oxidative phosphorylation is a metabolic pathway for generation of energy that occurs in the mitochondria of eukaryotic organisms. This metabolic pathway represents a controlled redox chain for reduction of the strong oxidant O2 by four electrons to H2O, while generating a proton gradient to synthesize ATP. Under ideal conditions, O2 is reduced to H2O; however, partial reduction of O2 yielding superoxide (O2•−) occurs in a ~0.1–2% yield depending on the cellular state [46]. The O2•− formed exists in low picomolar concentrations and has a ~1 μs half-life [1]. The major pathway for removal of O2•− in cells is conversion to H2O2 by superoxide dismutase (SOD, Fig. 1A Reaction 1) [1]. The H2O2 formed upon dismutation has a cellular half-life of ~10 μs due to its removal by catalases and peroxidases [1]. The concentration of H2O2 is ~200 nM under normal conditions that increases to low micromolar concentrations under oxidative stress [1]. The half-life for H2O2 is sufficiently long to allow intracellular and intercellular diffusion because it readily passes through membranes. Neither O2•− (Ered = 0.94 V vs. NHE) nor H2O2 (Ered = 0.32 V vs. NHE) are strong oxidants, although they both lead to generation of HO• (Ered = 2.31 V vs. NHE), a potent one-electron oxidant (Fig. 1B) [1]. Generation of HO• from H2O2 generally occurs by the iron- or copper-mediated Fenton reactions (Fig. 1A Reaction 2; only an example with iron is provided) [12]. In cells, iron and copper are tightly regulated and guarded from reaction under normal physiological states, but they become deregulated under conditions of oxidative stress [47]. The half-life of HO• is 10−9 s resulting in a low cellular abundance of ~10−15 molar [1]. Importantly, HO• indiscriminately oxidizes biomolecules near its site of formation. Additional enzymatic mechanisms for O2•− and H2O2 production in cells include phagocytic oxidases, NADP/H oxidases, amine oxidases, peroxisomal oxidases cyclooxygenases, lipid oxidases, and xanthine oxidase, as examples [1]. Lastly, ionizing radiation causes radiolysis of water to yield HO• (Fig. 1A Reaction 3) [48]. These pathways represent the major sources of O2•− leading to H2O2 and HO• in cells.
Figure 1.

Mechanisms for ROS formation, redox potentials for nucleosides, ROS, and reductants. (A) Reaction schemes for generation of ROS. (B) Plot of the redox potentials for nucleosides, reductants (Asc = ascorbate, NAC = N-acetylcysteine, GSH = glutathione), and various ROS.
Another reaction channel of O2•− is addition to NO in a diffusion-controlled process to yield ONOO− (Fig. 1A, Reaction 4),and this reaction proceeds faster than dismutation of O2•− by SOD [3]. Cellular ONOO− has a half-life of ~1 ms and can be produced in mid-micromolar amounts per minute [49]. The pKa for ONOO− is ~6.8, allowing both the acid and conjugate base to be present at physiological pH [49]. This is important because ONOOH decomposes in a proton-catalyzed pathway to yield HO• and •NO2 in ~30% yield (Fig. 1A Reaction 5) [49]. The •NO2 (Ered = 1.04 V vs. NHE) formed is a much weaker oxidant than HO• (Fig. 1B) [50]. On the other hand, ONOO− rapidly reacts with intracellular CO2 to yield nitrosoperoxycarbonate (ONOOCO2−) that undergoes homolytic cleavage of the O-O bond to yield carbonate radical (CO3•−) and •NO2 in ~35% yield (Fig. 1A, Reaction 6) [49]. The CO3•− (Ered = 1.59 V vs. NHE) so formed is a much stronger one-electron oxidant than •NO2 (Fig. 1B) [51]. Each time ONOOH or ONOOCO2− decomposes, it yields a stronger ROS (i.e., HO• or CO3•−) and a weaker reactive nitrogen species (RNS; i.e., •NO2). Yields of ONOO− are maximal when NO and O2•− are actively synthesized in the cell, for instance, during inflammation [3].
Molecular oxygen is a diradical that is kinetically inert in the biological context due to a quantum mechanical spin restriction [52]. The restriction on O2 can be removed upon excitation of an electron to yield 1O2, more specifically, 1ΔgO2 (referred to as 1O2 in the remainder of the text), with a half-life in H2O of 1–3 μs that decreases by ~2-fold in cellular systems due to rapid oxidation of biomolecules (Fig. 1A, Reaction 7) [52]. Intracellularly, 1O2 can be generated by photoexcitation of a chromophore (i.e., sensitizer) followed by energy transfer to O2 in a process known as Type II photochemistry [52]. Chromophores found within cells and capable of Type II photochemistry include flavins, porphyrins, cytochromes, and 4-thiouridine, and this list extends to a number of drugs and photosensitizers used in photodynamic therapies (e.g., tetracycline, quinine, haematoporphyrin, chlorins, etc.) [52]. Another pathway for cellular generation of 1O2 is via the Russell reaction that occurs when two alkylperoxyl radicals react with one another and decompose to yield 1O2 (Fig. 1A, Reaction 8). Leukocytes generate 1O2 enzymatically [53]. In biology, 1O2 is a potent oxidant that affects two- and four-electron oxidation of G.
Oxidative stress from ROS usually occurs from the intermediates O2•− or H2O2 that react and decompose to yield the one-electron oxidants HO• or CO3•−. Additionally, electronic excitation of 3O2 can yield 1O2. In general, production of ROS in cells is a rare event, tightly controlled by a number of mechanisms, for example, the enzymes SOD and catalase as well as small-molecule antioxidants. The latter reductants include glutathione (GSH) and ascorbate (Asc) for deactivation of ROS to protect cells [54]. The oxidations of G described next focus on the roles that Asc or the GSH mimic N-acetylcysteine (NAC) provide in defining the observed oxidation products.
3. Products resulting from oxidation of G
The G heterocycle has a reduction potential of 1.29 V vs. NHE, a value lowest amongst the four standard DNA bases (Fig. 1B) [8]. Consequently, G is the most labile toward ROS-mediated oxidation in the genome. Numerous studies have reported exposure of G in many contexts to the ROS mentioned above leading to products resulting from base and sugar oxidation. Oxidation of the sugar on G nucleotides leads to strand breaks that can be lethal if not repaired. Sugar oxidation in DNA has been reviewed by Dedon [23] and Tullius and coworker [55], and therefore, these products will not be further described; additionally, sugar oxidation products are not established substrates for BER initiated by the NEIL glycosylases. Experiments to identify G-base oxidation products found the yields depend on the nature of the ROS, the reaction conditions, and the structural context in which the guanine is located [9–25]. The G heterocycle is susceptible to two-, four-, and six-electron oxidations (Scheme 1) [11]. Oxidation of G can occur in a stepwise, one-electron fashion or a direct four-electron oxidation.
A base damage product of G observed during some oxidations that is not formally an oxidized product is 2,6-diamino-4-hydroxy-5-formamidopyrimidine (Fapy-G) resulting from formal hydrolysis of the imidazole ring of G (Scheme 1) [19, 26, 27]. Formation of Fapy-G is promoted under anaerobic oxidative conditions. Upon two-electron oxidation of the G heterocycle, the products OG or 2Ih have been identified, while four-electron oxidation of G yields Sp, Gh, 2,5-diaminoimidazolone (Iz) and its hydrolysis product 2,2,4-triamino-2H-oxazol-5-one (Z) (Scheme 1). The hydantoin compounds Sp and Gh more typically result from oxidation of OG as an intermediate (Scheme 1), because OG has a much lower reduction potential than G (OG = 0.74 V and G = 1.29 V, both vs. NHE) (Fig. 1B) [8, 29]. The 0.55 V difference in potential between G and OG translates to a ΔG = −12.7 kcal/mol favorability in OG oxidation. The hydantoins can also form directly from G (Scheme 1), as described below. Recently, our laboratory demonstrated that Gh is in a pH-dependent equilibrium with its constitutional isomer iminoallantoin (Ia, Scheme 1) [56], supporting a past hypothesis of ours [32]. Lastly, six-electron oxidation of G yields oxidized 5-guanidinohydantoin (Ghox, Scheme 1) that has limited stability and decomposes through several intermediates to yield a 2′-deoxyribosyl-urea lesion [16]. The formation of Ghox during one-electron oxidation reactions results from Gh having a low standard reduction potential of ~1.0 V vs. NHE (Fig. 1B) [32]. Further oxidation of Ia to Iaox also can occur (Scheme 1) [32]. Because oxidations are a rare event, it is anticipated that six-electron oxidation of G would be very uncommon in cells, and the appearance of such products in model reactions results from over oxidation of the samples; therefore, these six-electron oxidation products will not be described further.
The oxidation products Sp, Gh, Ia, and 2Ih have been the most challenging to detect chromatographically because they are very hydrophilic, causing them to be easily overlooked. Furthermore, Sp, Gh, Ia, and 2Ih nucleosides are generated as pairs of diastereomers, and each of them elutes from an HPLC column as two peaks [33, 56–60]. Additionally, the pH-dependent isomerization between Gh and Ia dramatically impacts the elution time of these isomers and must be considered during product analysis [56]. The following text describes the products of G oxidation observed when each of the cellular ROS was the oxidant with a focus on how the reaction context and the presence of physiologically relevant amounts of reductant impacts the products observed.
4. Key products, reaction sites, and context effects when G is oxidized by HO•
The most potent ROS, HO•, is generated in its free form via radiolysis of water by γ-rays or X-rays, as well as by the iron-mediated Fenton reaction [48, 61]. In contrast, the copper-mediated Fenton reaction most likely yields a metal-bound HO• impacting its reactivity [62]. Early product profiles found free, HO•-mediated oxidations of G yielded OG and Fapy-G (Scheme 2) [63–66], in which OG was observed under aerobic conditions and Fapy-G was observed under anaerobic, reducing conditions. An additional mechanism for the formation of OG by HO• is oxidation of a 5′ or 3′ pyrimidine adjacent to a G nucleotide to yield a pyrimidine peroxyl radical that degrades to yield OG and a damaged pyrimidine tandem lesion [67–69]. Recent work in our laboratory using HPLC methods that allowed a more complete analysis of the products found 2Ih as the major product during HO• oxidations in the nucleoside context [62, 70]. The HO• was generated by the iron- or copper-mediated Fenton reaction, as well as via X-ray radiolysis of water under aerobic reactions in the presence of physiological amounts of Asc (1–3 mM) or NAC (1–5 mM; Scheme 2). In our studies and others [65, 66, 70, 71], Fapy-G was only observed in the complete absence of O2. Thorough reviews on Fapy-G formation have been written by Dizdaroglu [27], Greenberg [19], and Tudek [26], thus, we guide the reader to these resources for more information regarding Fapy-G. Lastly, minor products observed in the G oxidations by HO• were Z and Iz (Scheme 1) [62, 70, 72].
Scheme 2.

HO•-mediated oxidation of the G heterocycle. The dominate pathways are shown with bold reaction arrows. The compounds in boxes are the focus of this review because they have been identified as the best substrates for the hNEIL glycosylases in the BER pathway.
The OG formed during these oxidations is very stable, but it is susceptible to further oxidation to yield the hydantoins Sp or Gh (Fig. 1B and Scheme 2) [30–32, 50, 73–77]. In nucleoside studies, Sp is formed during HO•-mediated oxidations, an observation consistent across many laboratories [62, 70, 78, 79]. In vitro oxidation of G nucleoside aids in identification of how cellular ROS would impact nucleotide and nucleoside pools. These studies support 2Ih and OG as the major HO•-generated G oxidation products to be found in cellular nucleotide pools, along with lower yields of the OG oxidation product Sp (Fig. 2).
Figure 2.

Context dependency of major G oxidation pathways and products. The contexts studied in DNA include nucleoside, ssDNA, dsDNA, and G4 structures and the products shown are those appearing during oxidations conducted in the presence of physiological amounts of reductant. In several cases, OG is an initial product that undergoes subsequent oxidation to Sp as indicated by straight arrows, or Sp is directly formed from oxidation of G bypassing OG as indicated by no arrow between OG and Sp.
Mechanistically, HO• can add to the G heterocycle at either the C5 or C8 carbon and, upon transfer of a proton and electron, yield C5 or C8 hydroxy derivatives of G, respectively (5-HO-G or 8-HO-G, Scheme 2). Next, 5-HO-G reacts via a [1,2] acyl shift followed by hydrolytic ring opening of the sugar bound 5-membered ring to yield 2Ih. The mechanism of direct formation of 2Ih from G was proposed by our laboratory [62, 70] and by Meunier, Pratviel, and coworkers [11, 80]. The 2Ih structure was determined by multi-dimensional NMR in the Ball laboratory [81]. As for 8-HO-G, a tautomerization to the thermodynamically stable 8-oxo-7,8-dihydroguanine (OG) occurs [82]. These two mechanisms best explain 2Ih and OG as the major HO• oxidation products of G (Scheme 2). The mechanism for Fapy-G formation was proposed to occur from reduction of the 8-HO-G• intermediate followed by opening of the imidazole ring under anaerobic conditions (Scheme 2).
The nucleoside studies provide a solid footing for extending oxidation product analysis to contexts of increasing complexity, such as single-stranded DNA (ssDNA) and double-stranded DNA (dsDNA). Product analysis in these contexts occurs by oxidation of small oligomers with a single reactive site followed by direct mass spectrometric analysis [72], or the DNA is digested with nucleases and a phosphatase to liberate the nucleosides for analysis by HPLC coupled to a mass spectrometer (LC-MS) [83]. One concern with nuclease digestion is whether the efficiency of digesting damaged nucleosides from DNA is great enough to allow quantification of each lesion. This limitation requires a good understanding of the nuclease digestion kinetics for the lesions [84, 85]. An additional method to liberate DNA bases from the polymer utilizes a harsh chemical hydrolysis procedure [27]. This approach has been demonstrated to significantly over estimate the oxidation product yields due to artifactual oxidation of the G nucleosides during the derivatization steps [86–88]. The yields of OG and Fapy-G have been demonstrated to be highly over estimated by GC-MS methods [89, 90]. Consequently, the European Standards Committee on Oxidative DNA Damage (ESCODD) established protocols that verified the use of enzymatic methods followed by LC-MS/MS, or in the unique case of OG, LC with an electrochemical detector (ECD) [28], as the best approaches for quantification of lesions from DNA. The analytical methods and concerns with artifactual formation of lesions during the derivatization steps for GC-MS have been reviewed and will not be further discussed [89, 90].
When HO•-mediated oxidation of G was studied in ssDNA and dsDNA contexts, the products modestly changed relative to the nucleoside studies (Fig. 2). The early analyses with GC-MS methods identified OG and Fapy-G as major products of the oxidation with the same O2 and reducing agent dependency in their yields [64], as previously discussed in the nucleoside context. In contrast, LC-MS or LC-ECD methods observed OG to dominate under aerobic conditions [63, 65]. Studies in the Cadet laboratory utilizing LC-MS/MS to quantify OG and Fapy-G in radiation exposed cells found similar amounts of the two products at low radiation flux and greater amounts of Fapy-G at very high radiation exposure [91, 92]. When product analysis was more complete after nuclease digestion, 2Ih and OG were found to be the major two-electron oxidation products observed from iron- and copper-Fenton chemistry, as well as from X-ray radiolysis of water under aerobic reducing conditions [62, 70]. In the DNA contexts, OG can be further oxidized to Sp and Gh, in which the Sp yield is maximal in ssDNA and the Gh yield is maximal in dsDNA (Scheme 2) [62, 70, 77]. Product analysis from these contexts revealed Z and Iz in low overall yields in our studies and others [62, 70, 93]. In addition to these base-oxidation products, lower yields of sugar oxidation occur under aerobic, reducing conditions [62, 70]. This supports a previous observation that reducing agents, such as Asc or NAC at physiologically relevant concentrations, repair oxidized sugars before they can further react to cause strand breaks [94]. Lastly, HO•-mediated oxidations in DNA contexts have revealed cross-links between oxidized G and neighboring nucleotides with the yields varying depending on the conditions [9, 95, 96]. A study from our laboratory oxidized the G4 that folds from the human telomere repeat sequence (5′-TTAGGG-3′)n with HO• generated by the copper-Fenton reaction with NAC present under aerobic conditions [97]. In the telomeric G4 context, 2Ih, OG, and Sp were the products observed. These studies support the conclusion that 2Ih and OG would be the major products resulting from G oxidation by HO• in all DNA contexts and further oxidation of OG would yield lower amounts of Sp in ssDNA and G4 DNA, or Gh in dsDNA (Fig. 2).
Hydroxyl radical is a potent and short-lived oxidant that shows very little discrimination in the site of oxidation. This is best exemplified in the work conducted in ssDNA demonstrating nearly equal oxidation at all G nucleotides by HO• generated with iron-Fenton chemistry or water radiolysis [70, 98]. Closer inspection of HO•-mediated G oxidation in dsDNA shows a very modest preference for oxidation of the 5′ G in the sequence context 5′-GG-3′ [99]. The preference for one-electron oxidation of the 5′ most G in 5′-GG-3′ sequence contexts results from a decrease in the ionization potential of the 5′ G due to π stacking in the dsDNA context, first described by Saito, et al. [100]. In DNA duplexes, π stacking allows oxidation at a remote site to induce chemistry at a distance because electron holes migrate through the duplex. When oxidation occurs by this pathway, G oxidation occurs at the 5′ G in 5′-GG-3′ or 5′-GGG-3′ sequence contexts, a property of DNA experimentally well validated by the Barton and Schuster laboratories [101, 102]. The modest preference for 5′ G oxidation with HO• is likely due to the fact that the principal reaction of HO• is addition to C5 or C8 of G rather than abstraction of an electron. The formation of guanine radical cation (G•+) has been invoked by a mechanism in which HO• adds and then HO− (or its conjugate acid, H2O) departs to generate G•+ allowing hole migration to a reactive G site [103]. When HO• was generated by copper-mediated Fenton chemistry, nearly equal reactivity at all Gs in ssDNA, dsDNA, nucleosomal, and G4 contexts was observed (Fig. 2) [62, 97, 104]. This observation was attributed to indiscriminate copper coordination with N7 of G eliminating any sequence effects on the oxidation. For this reason, Fenton oxidation of DNA is modulated by the nature of the redox-active transition metal. Lastly, HO• oxidation of G runs in model nucleosomes produced the greatest extent of reactivity at G runs directed outward, away from protection by the nucleosome core proteins [105], demonstrating histone proteins can protect Gs from oxidation. Thus, HO• readily oxidizes most G nucleotides with a slight preference for 5′ Gs in 5′-GG-3′ sequence contexts; however, this is dependent on how HO• was generated.
5. Key products, reaction sites, and context effects when G is oxidized by CO3•− and one-electron oxidants
Details obtained from the HO•-mediated oxidation studies guide a discussion of how G oxidation differs when CO3•− is the oxidant. The dominant pathway of CO3•−-mediated oxidation occurs on the G heterocycle to yield OG when a low radical flux is used [106]. This oxidation occurs via one-electron abstraction from G by the radical to yield a G•+ that rapidly deprotonates (pKa ~3.9) to the neutral radical (G•) [107]. The neutral radical couples with other radicals such as O2•−, or with CO3•− at either the C5 or C8 carbon (Scheme 3) [108]. At present, regardless of the radical that couples, the products are generally the same, therefore, only chemistry with O2•− will be described because it applies to one-electron oxidants other than CO3•−. Radical addition of O2•− at C8 plus addition of a proton yields 8-HOO-G that in the presence of reductant is reduced to 8-HO-G followed by tautomerization to OG (Scheme 3). When O2•− (+H+) addition occurs at C5, the hydroperoxide 5-HOO-G is formed and can be reduced to 5-HO-G to yield 2Ih (Scheme 3). Additionally, 5-HOO-G can yield Iz and Z if reduction of the peroxide does not occur (Scheme 3). In the dsDNA context, one-electron oxidation of G yields G•+. The dsDNA results in G•+ persisting because the acidic proton is trapped in the Watson-Crick base pair with C modulating the reactivity of the radical intermediate. In dsDNA, G•+ hydrates at C8 followed by a second oxidation and proton loss to yield OG (Scheme 3). An additional mechanism for G•+ formation in dsDNA is via one-electron oxidation by HO• that has been experimentally observed and computationally studied (Scheme 3) [109, 110]. These pathways have been supported computationally by work in the Schlegel laboratory [111, 112]. In summary, OG is the major product formed and 2Ih can form in a similar yield under the few conditions studied so far with physiologically relevant amounts of reductant present.
Scheme 3.

Proposed one-electron oxidation mechanisms of the G heterocycle. The key products repaired by hNEIL1 resulting from this mechanism are identified by the dashed boxes. RSH = cellular reductant such as glutathione.
The OG thus formed is subject to a second two-electron oxidation to yield Sp and Gh (Scheme 3) [73, 76]. The oxidation sequence from G to OG to Sp and Gh dominates CO3•− oxidations in the nucleoside, ssDNA, dsDNA, and G4 contexts (Fig. 2) [51, 97, 113]. The main difference observed in the context dependency of product formation resides in the relative yields of Sp and Gh. The product Sp dominates in nucleoside, ssDNA, and G4 contexts, while the Gh yield is maximal in dsDNA contexts (Fig. 2). A recent estimate placed the base reaction dominating over sugar oxidation by >2000-fold supporting the base oxidation chemistry described as the mode of CO3•− oxidation of G [114].
When OG is formed in dsDNA, polymerase bypass of a template OG results in insertion of either A or C nucleotides opposite the damaged site [115]. This is a consequence of the energetic preference of OG for the syn configuration allowing Hoogsteen base pairing between OG and A. In our laboratory, dsDNAs with either an OG base paired with an A or C were oxidized with CO3•− to find OG:C exclusively yields Gh while OG:A yields Sp and Gh in a 1:3 ratio, respectively [73]. The Geacintov and Shafirovich laboratories uncovered a characteristic cross-link formed during CO3•− oxidations between G and T in the DNA sequence contexts 5′-GCT-3′ and 5′-TCG-3′ (underlined nucleotides form the cross-link) [116]. In cells, CO3•− is generated in tandem with the weaker one-electron oxidant •NO2. The •NO2 formed is incapable of oxidizing G nucleotides, but it can radical couple with oxidized G to yield 8-nitroguanine or 5-guanidino-4-nitroimidazole [50]. These products will not be further discussed as their kinetic properties with respect to DNA BER repair have not been extensively evaluated. The •NO2 formed can also oxidize OG nucleotides to yield Sp and Gh [50]. Lastly, oxidation of G by a high flux of CO3•− can yield 2Ih in ssDNA; however, 2Ih formation in other DNA contexts post CO3•− oxidation has yet to be determined (Fig. 2) [106, 117]. Characteristic G oxidation products resulting from CO3•− oxidations include OG, Sp, and Gh, depending on the context of the reaction, and as more studies are conducted, 2Ih might be revealed as a major product in future studies.
In the context of dsDNA, CO3•− oxidizes G with a strong sequence dependency on the reaction rate. Oxidation of G is biased toward 5′ Gs in the sequence context 5′-GG-3′ and 5′-GGG-3′ (bold = reactive Gs), similar to that observed for HO•; however, the bias is much stronger for CO3•− (Fig. 2) [118]. The greater bias to oxidation at 5′ Gs in G runs results from CO3•− favoring reaction channels that yield G•+ allowing electron hole migration through the DNA π stack. Additionally, CO3•−-mediated oxidation of nucleosomal DNA with G runs indicated 5′ G oxidation in G runs regardless of whether the Gs were protected by the nucleosomal proteins, a contrasting observation in comparison to HO•-mediated oxidation [105]. Again, this supports oxidation of DNA at a distance followed by electron hole migration to the most reactive 5′ G of a G run. When OG is in dsDNA, there does not appear to be a strong sequence dependency in its further oxidation by CO3•− [119]. In the G4 that folds from the human telomere repeat sequence (5′-TTAGGG-3′)n, the 5′ most G of 5′-GGG-3′ runs is susceptible to CO3•− oxidation, while the middle and 3′ Gs are protected [97]. Additionally, the G4 that folds in the VEGF promoter yields oxidation at G nucleotides in loop positions in addition to the 5′ most G of the G-quadruplex core-forming G runs (Fig. 2) [113]. Using these studies, a hypothesis of likely locations and products for CO3•−-mediated oxidation of G can be formulated. In ssDNA regions of replication and transcription bubbles, G can be oxidized without sequence preference to OG and Sp, while in dsDNA oxidation of G will occur at 5′ Gs in G runs to yield OG and preferentially Gh. Oxidation of G4 folds will yield damage at loop Gs and the 5′ most G of core G runs to yield OG and Sp (Fig. 2). Because long-range electron transfer can occur when CO3•− is the oxidant, both open and compact regions of the genome could be subject to this form of damage.
6. Key products, reaction sites, and context effects when G is oxidized by 1O2
The path to understanding G oxidation by 1O2 has led to many challenges in determination of the products observed and their mechanisms of formation [120, 121]. The mechanism by which G is oxidized via 1O2 remains a topic of study in the literature [122–124]. To study 1O2 in the laboratory, its photochemical generation with a Type II photosensitizer is the easiest [52]. One drawback with these chemical sensitizers is they do not generate a pure source of 1O2--there is always some amount of Type I photochemistry (i.e., electron transfer from G to yield radical intermediates and O2•−) [52]. The Foote laboratory conducted oxidations with a photooxidant and monitored the intermediates and products by 1D- and 2D-NMR to propose the leading mechanism [125]. On the basis of the NMR results, 1O2 initiates oxidation of G via a [4+2] cycloaddition across the imidazole ring to yield a 4,8-endoperoxide intermediate, a result that was supported by isotopic labeling studies conducted in our laboratory (Scheme 4) [33, 125]. The 4,8-endoperoxide rearranges to the 8-HOO-G intermediate that is reduced to yield OG, or collapses to yield an electrophilic-quininoid intermediate (OGox) [33, 34, 97, 126]. Next, OGox adds water at C5 to furnish 5-HO-OG that undergoes a [1,2] acyl migration to yield Sp or hydrates at C6 followed by elimination of CO2 to yield Gh (Scheme 4). The hydantoin yields are dependent on the pH and reaction context (Scheme 4) [33, 34, 124, 126]. Lastly, OG oxidation by 1O2 yields Sp, Gh, Iz, Z, cyanuric acid, and Ghox, and like G oxidation, the products observed show dependency on the reaction conditions and context [127–129]. From these studies, OG, Sp, and Gh are the products observed in nucleosides under modeled reducing conditions found in cells, and they are the only substrates studied with the hNEIL glycosylases; therefore, they will remain the focus of this review.
Scheme 4.

Proposed mechanism for oxidation of the G heterocycle by 1O2. The key products repaired by the hNEILs are within the dashed boxes.
Rose Bengal is a high quantum yield Type II photosensitizer with a minimal amount of Type I photochemical side reactions [52]. Utility of Rose Bengal to generate 1O2 readily oxidizes G nucleotides in the ssDNA context and poorly oxidizes G nucleotides in the dsDNA context (Fig. 2) [97, 130]. This observation is consistent with the mechanism of oxidation because π stacking around G in dsDNA sterically blocks 1O2 from undergoing [4+2] cycloaddition, resulting in poor reactivity. The G oxidation observed in dsDNA under 1O2 conditions must result from Type I photochemistry and not 1O2, a concept described in the literature [130]. Lastly, G4 contexts show G oxidation on all G nucleotides in the exterior tetrads [97]. To mitigate the inconsistent chemistry with photooxidants, Cadet and coworkers synthesized a naphthalene endoperoxide that generates a clean source of 1O2 upon thermal decomposition [131]. First, they used the endoperoxide to oxidize calf thymus DNA with 1O2 and found OG as a product [132]; however, this study did not look for hydantoin products (i.e., Gh or Sp). In a second set of studies, they used the endoperoxide to understand 1O2 oxidation of cellular DNA, in which OG was the observed product [133, 134]. In the second set of studies the analytical method could detect Sp (i.e., 4-hydroxy-8-oxo-4,8-dihydro-2′-deoxyguanosine, the structure of Sp was incorrectly assigned by this laboratory when these studies were conducted and has since been shown to be the spirocyclic compound [30, 31, 135, 136]) but it was not found. Cellular DNA resides in many structural contexts impacting the G oxidation observed, in which the most reactive contexts are ssDNA and G4-DNA that might be the locations the OG was formed in cells by 1O2.
7. Additional contexts and reaction partners of G studied under oxidative conditions
Inside cells, G nucleotides exist in other contexts such as A-form duplexes, Z-form duplexes, Holliday junctions, i-motifs, and hairpins. Like B-form DNA duplexes, A-form and Z-form duplexes allow electron hole migration to affect initiation of oxidation at one site followed by chemical modification of G at a distance [101]. Interestingly, G oxidation products resulting from such chemistry in A- and Z-duplex forms have not been evaluated by nuclease digestion followed with LC-MS analysis. Hairpin DNA has the potential to place G in duplex and single-stranded contexts, in which oxidation by the ROS mentioned should yield the same products in each of these contexts as described; however, these studies have not been systematically undertaken. The key difference is likely to be the relative reactivity of G in these contexts favoring single-stranded oxidations by >100-fold [73]. The core of i-motif folds is comprised of C+:C base pairs built from runs of contiguous Cs in a sequence that occurs in complementary strands to G4 sequences [137]. Oxidation of G in the G4 context is expected to dominate in this situation, although i-motif folds can place Gs in highly reactive loop positions to bias the strand reactivity when these structures are present. This effect on G oxidation has yet to be evaluated.
Finally, G oxidation in RNA represents a frontier in which systematic studies to evaluate reactivity, reaction sites, and products have not been as thoroughly conducted as those in DNA. Studies to monitor cellular excretion of OG from DNA or RNA observe up to ~3-fold more OG in RNA than DNA supporting high levels of RNA oxidation in vivo [138]. Reports from our laboratory [139, 140], Pratviel and coworkers [141], and Thorp and coworkers [142] have described RNA oxidation in various contexts. The recent work by Pratviel and coworkers analyzed G oxidation by a manganese porphyrin complex and KHSO5 in model anticodon tRNA stem-loops to identify the oxidation product Sp [141]. We found 1O2 oxidation of G in ssRNA can yield Sp or Gh in a pH-dependent reaction [140], and a nickel macrocycle to activate the oxidant KHSO5 yields 2Ih [140]. Because RNA adopts many different structural motifs (i.e., pseudoknots, bulges, etc.), studies in these contexts may present new patterns of reactivity not observed in DNA. The products will likely remain the same; however, this has not been thoroughly studied.
The products described to this point result from radical G intermediates reacting with either, O2•− CO3•−, or electrophilic G oxidation intermediates reacting with H2O as a nucleophile. Genomic DNA intimately interacts with many other possible reaction partners, such as polyamines and protein side chains. The polyamines spermine and spermidine exist at millimolar concentrations in cells and closely interact with genomic DNA [143]. The primary amines in the polyamines and lysine residues in proteins such as histones are excellent nucleophiles for reacting with electrophilic G oxidation intermediates [144–150]. When primary amines form adducts with G or OG during oxidation, products are formed that must be repaired [45]. This is the focus of other reviews in this special issue, and therefore, we guide the reader to these reviews for more information on repair of amine adducts to DNA.
8. Products of G oxidation are mutagenic on the basis of cellular studies
The reports described so far examined G oxidation in vitro. A number of studies have quantified G oxidation products from cellular sources after nuclease digestion using HPLC coupled to a tandem mass spectrometer [83]. In early work, OG was the principal product identified, and its concentration generally showed a strong positive correlation with the extent of oxidative stress [83, 151]. In light of more exhaustive in vitro studies like the ones described above, a deeper inspection of genomic DNA for lesions beyond OG was conducted. Sp was first found in prokaryotic cells exposed to chromate with a dose dependency in Sp formation [152]. Next, Sp and Gh were detected in livers and colons of mice with chronic inflammation, and they both showed a correlation with disease progression [151]. The hydantoins were found at concentrations ~100-fold lower than OG in the mouse studies. Ia was not detected, but the analytical conditions were not conducive to finding it. Importantly, if the nuclease digestion to detect the lesions is performed at high pH, the conversion of Gh to Ia leads to a loss of Gh without concomitant detection of Ia. When livers of diabetic rats were analyzed they found OG and Z to show a positive correlation with the initial extent of the disease [153]. This study found Z to exist ~40-fold lower than OG with a modest correlation to diabetes. Because OG is always observed in higher concentrations, it is regarded as a good indicator for monitoring cellular oxidative stress. In light of recent in vitro oxidations of G to yield 2Ih in greater levels than OG [62, 154], 2Ih may exist in cells at high levels; however, in cellulo studies to detect 2Ih have yet to be reported
Lesions resulting from G oxidation in DNA are sources of mutations when polymerases bypass them in template strands. As stated above, OG can base pair with A via its Hoogsteen face, allowing polymerases to insert either dATP or dCTP opposite a template OG [115]. The ratio of dATP vs. dCTP inserted opposite OG is polymerase dependent with ratios of the two nucleotides around 1:1 for pol δ and pol η and can favor dATP insertion by nearly 200-fold for pol α [115, 155]. If repair does not occur [43], a second round of polymerase bypass through an OG:A base pair causes insertion of dTTP opposite A, leading to a T in the position previously occupied by G (i.e., G→T transversion). The potential for OG to cause G→T transversion mutations has been verified in vivo by the restriction endonuclease and post-labeling (REAP) assay [156]. Transversions induced by OG were observed in a 3% yield, supporting the modest mutagenic potential of OG.
When Sp or Gh resides in the template strand, insertion of dATP or dGTP is favored, ultimately causing G→T or G→C transversion mutations [56, 157, 158], respectively. Studies to inspect Sp and Gh mutational profiles by the REAP assay verified their ability to cause transversion mutations in ~100% yield [156, 159, 160]. The results indicated that the Sp base stereochemistry impacted the ratio of G→T and G→C transversions. The REAP assay with Gh identified a strong preference for G→C transversion that is hard to chemically rationalize by the base pairing potential of Gh. Recently, we verified a previous hypothesis of ours [32] that Gh can isomerize to iminoallantoin (Ia), and demonstrated that iminoallantoin base pairs with dGTP ultimately causing G→C transversions [56]. Whether isomerization of Gh to Ia occurs in cells to induce the G→C transversions observed in the REAP assay is not currently known.
When 2Ih is in the template strand, dATP or dGTP are inserted opposite the lesion by a polymerase in vitro with a preference for dGTP [154]. Verification of 2Ih causing mutations in cellulo by the REAP assay consistent with the in vitro studies has yet to be conducted. Lastly, a templating Z lesion when bypassed by a polymerase allows insertion of mainly dATP leading to G→T transversion mutations [161]. Cellular studies using the REAP assay verified the in vitro results in which Z preferentially induces G→T transversions ~95% of the time [159]. Besides OG, the other G-derived lesions are nearly ~100% mutagenic causing G→T or G→C transversion mutations. These mutational profiles exist at high frequency in cancers established to result from oxidative stress [162]. Yet, the actual source of these mutations remains unknown. To evade these potential mutational pathways resulting from G oxidation products, a DNA repair system comprised of BER and NER pathways exists.
The relative reactivity of the nucleotide pool toward oxidation can be >6000-fold more prevalent than oxidations in the genome [73]. Oxidations in these contexts would yield the nucleotide triphosphates of OG, Sp, and 2Ih, on the basis of the discussion above. A nucleotide pool cleansing pathway exists to prevent these triphosphates from interfering with polymerase extension [163]. Evaluation of the ability for polymerases to accept oxidized triphosphates have found they prefer canonical nucleotides by >10,000-fold. For example, pol γ inserts dGTP ~10,000-fold more than dOGTP [164]; however, when dOGTP is incorporated it is only opposite the expected A or C nucleotides, identical to studies looking at OG in template strands. Similarly, Klenow Fragment lacking 3′→5′ exonuclease activity favors dGTP insertion by ~64,000-fold and ~185,000-fold more than (S)-dSpTP and (R)-dSpTP, respectively [165]. However, in the dSpTP studies, the researchers only observed insertion opposite C nucleotides that is inconsistent with polymerase bypass of Sp diastereomers in a template strand, in which dATP or dCTP are inserted opposite the lesion site [157]. This curious inconsistency may result from impure samples because the dSpTP studied was synthesized from dGTP that would easily insert opposite C in a template strand. When the selectivity for the lesion triphosphate is >100,000-fold less than dGTP a miniscule amount of starting material would impact the results. Studies with d2IhTP have yet to be conducted.
9. Three Neil DNA glycosylases in BER remove hydantoin substrates from DNA
The BER pathway has primarily evolved to remove damaged nucleotides from the genome to evade mutations at these sites [44, 166]. Initiation of BER occurs with a DNA glycosylase catalyzing cleavage of the glycosidic bond to remove the damaged base from DNA (Fig. 3A). Each glycosylase can then either release the abasic site (AP) product for further downstream processing by the phosphodiesterase apurinic/apyrimidinic endonuclease (APE1), or further process the AP by catalyzing elimination of a phosphate ester group (Fig. 3A). The former are considered monofunctional enzymes. In the latter case, the glycosylase has a lyase function to cause β or β,δ elimination of the sugar to yield a strand break and is said to be bifunctional. Those bifunctional glycosylases that only induce β elimination of the sugar fragment require APE1 to finish the process, while enzymes that catalyze β,δ elimination do not require APE1, and instead use polynucleotide kinase (PNK) to finish processing the damaged site. Next, polymerase β (POLβ) inserts the correct nucleotide followed by ligation via LIGIII of the nicked strand to complete the repair process (Fig. 3A). POLβ also has lyase capabilities to aid in removal of sugar fragments to yield a nicked site for its polymerase activity. When OG is in the genome, it is removed by members of the GO repair system that has been the topic of reviews by David [166, 167] and Wallace [44, 168], and many more in this special issue. When Sp and Gh are present in DNA they are predominantly extracted by endonuclease VIII (Nei) in bacteria and endonuclease VIII-like 1 (Neil1), Neil2, or Neil3 in mammalian cells, and the NEIL1, NEIL2, or NEIL3 homologs in humans (NEIL1 has been referred to as hNEIL1 in the literature). During establishment of the nomenclature of the Neils, they have been referred to as hFPG [169], hNEI [170], and NEH1 [171], ultimately the name NEIL (Nei-Like) was suggested by the Human Genome Organization. NEIL1 and 2 are bifunctional glycosylases with β,δ elimination activity through the action of an N-terminal proline [169, 171–173], and NEIL3 is mainly a monofunctional glycosylase via an N-terminal valine residue [174, 175]. The remainder of this review will focus on DNA glycosylases of the BER pathway processing the hydantoin substrates Sp or Gh and the single report on 2Ih in many different structural contexts.
Figure 3.

BER mechanism and relative reaction rates for NEIL1 removal of DNA lesions. (A) Outline of the BER pathway (dRP = 2′-deoxyribosephosphate). (B) Comparison of edited NEIL1 reaction rates vs. DNA lesions relative to Tg:A (arrow). The inset is for unedited NEIL1 reaction rates relative to Tg:G (arrow). (C) Edited NEIL1 reaction rates toward the diastereomers of Sp and Gh vs. their base pairing partner. (D) Context-dependent reaction rates for edited NEIL1 relative to the ssDNA context (arrow).
The Wallace laboratory first identified E. coli Nei (EcoNei) [176] and found this protein to have sequence and structural similarity to formamidopyrimidine DNA glycosylase (Fpg) that was discovered in Tomas Lindahl’s laboratory [177]. After sequencing of the human genome was completed, in silico analysis allowed the Wallace, Mitra, and Seeberg laboratories to identify, clone, and initially characterize three Fpg/Nei homologs in humans [169–171, 178]. Mouse, plant, fungal, and mimiviral Neil homologs have also been identified and characterized [172, 173, 179]. X-Ray crystal structures for EcoNei [180], NEIL1 [181, 182], a viral NEIL1 ortholog [179], mouse Neil3 [183], and a viral ortholog of NEIL2/3 [184] have been determined. At present, a structure for NEIL2 or an ortholog of this protein has yet to be solved. Detailed reviews on the structures of the Neil glycosylases by Wallace, Doublié, and coworkers exist and should be consulted for structural insights into these glycosylases [168, 185, 186].
In an attempt for completeness, recent studies have found Sp and Gh are also substrates for NER. This was reported by our laboratory in collaboration with the David laboratory utilizing the NER complex UvrABC found in bacteria [45]. Additionally, Shafirovich, Geacintov, and coworkers found Sp and Gh were good substrates for mammalian NER [42]. The details of these results will not be further discussed in this review.
10. The hydantoins are substrates for EcFpg, EcNei, and NEIL1
The story of hydantoins as substrates for BER started with a report by our laboratory in collaboration with the David laboratory in 2000 [187]. The initial study used an 18-mer duplex in which Sp or Gh were recognized and cleaved by the bifunctional DNA glycosylase E. coli Fpg (EcFpg). EcFpg utilizes an N-terminal proline to catalyze base removal and β,δ-elimination reactions to yield a strand break. This work led to a follow-up paper with greater details about the repair process in a 30-mer duplex leading to many key observations [188]. Diastereomeric mixtures of each hydantoin were studied opposite all four canonical nucleotides to determine whether Sp or Gh were removed by EcFpg as efficiently as the signature substrate OG:C. When Sp or Gh were base paired with A they were operated on by EcFpg more efficiently than an OG:C base pair, and when the hydantoins were opposite C, T, or G they were nearly as good substrates as OG:C. One key point in comparisons between the 18-mer and 30-mer duplex studies is that the duplex length is critical to capturing a true snapshot of the removal kinetics for highly disruptive lesions like the hydantoins in dsDNA. The longer duplexes provided a better context for glycosylase binding and catalysis of lesion removal. In a recent study from our laboratory, we found that EcFpg could catalyze excision of each diastereomer of 2Ih opposite A, C, T, or G in a 15-mer duplex with ~50% of the efficiency of removal of Sp from the same duplex [154]. Moreover, the removal efficiency showed little bias for the R or S stereochemistry of 2Ih. A 15-mer strand was studied because synthesis of the 2Ih diastereomers was only achievable in this short system. These results demonstrate that the hydantoins Sp, Gh, and 2Ih are all substrates for EcFpg and pave the way for analysis with EcNei and the NEILs.
In 2001, our laboratory in collaboration with the Mitra laboratory demonstrated that the Sp and Gh diastereomers were substrates for EcoNei [189]. EcoNei catalyzes hydrolysis of the glycosidic bond and β,δ-elimination of the sugar via an N-terminal proline as for EcFpg. The enzymatic analysis was conducted with mixtures of the Sp or Gh diastereomers opposite A, C, or G in an 18-mer duplex context. For Sp or Gh, the best kinetic rate removal by EcoNei was observed when the hydantoins were base paired with G. This concept was then extended by the Sugden laboratory to the mouse Neil1 and Neil2 glycosylases removing the hydantoins [190]. They found Neil1 was catalytically competent on Sp or Gh in ssDNA and dsDNA. Further, Neil1 cleaved Sp when it was base paired with any of the four canonical nucleotides. Neil2 processed Sp and Gh in ssDNA while the activity in dsDNA was only observed on Sp substrates, and this will be further discussed below. Prior to these studies, EcoNei, NEIL1, and NEIL2 were only thought to function on oxidized pyrimidines such as thymine glycol (Tg), 5-hydroxy-2′-deoxyuridine (5-HOU), 5-hydroxy-2′-deoxycytidine (5-HOC), 5,6-dihydro-2′-deoxyuridine (DHU), 5,6-dihydro-2′-deoxythymidine (DHT), as well as the purine lesions Fapy-G and 4,6-diamino-5-formamidopyrimidine (Fapy-A), and lastly with very poor activity on the OG:C or OG:A base pairs [169, 171, 173, 176, 191]. The wide substrate scope observed for EcNei, NEIL1, and NEIL2 led to questions concerning which substrates are the best. The oxidized pyrimidines were already established to be repaired by endonuclease III (i.e., Nth) in bacteria or its homolog NTH1 in humans [168], and OG is repaired by the elaborate GO repair system [44, 167]. Thus, questions of why such an overlap in substrates of repair by these BER DNA glycosylases began to emerge.
An approach to find the best substrates for the NEILs is to conduct detailed kinetic analysis for identification of those that are acted upon with the best rates. Such studies were conducted in our laboratory in collaboration with the David laboratory, the Dalhus laboratory, and the Ide laboratory for NEIL1 [154, 192–194]. To date, extensive kinetic analysis for NEIL2 or NEIL3 vs. substrate identity has yet to be reported. Before discussing the results, four points must be made. (1) The data presented in Fig. 3B are all relative to the repair rate for Tg:A (5R diastereomer of Tg), a substrate analyzed in most studies except the one with 2Ih from our laboratory. The 2Ih study made a comparison to Sp allowing a comparison into the rest of the data. This comparison minimizes inter-laboratory bias based on differing reaction conditions. (2) When the Sp diastereomers were originally studied, the absolute configuration for each was still in question [59, 60], and therefore, they were referred to as Sp1 and Sp2 in the original publications. At present, we know that in oligomers Sp1 = (S)-Sp and Sp2 = (R)-Sp [57], and therefore, the individual Sp diastereomers will be referred to by their absolute configurations in the present discussion. (3) The Gh diastereomers readily interconvert [33], and they are consequently studied as a mixture. (4) The initial results are all with respect to the edited form of NEIL1 [195], a topic discussed later.
The relative kinetic rate comparison for NEIL1 acting on the following substrates was made: (R)-Sp:C, (S)-Sp:C, Gh:C, (R)-2Ih:C, (S)-2Ih:C, 5-HOC:G, 5-HOU:G, DHU:G, abasic site (AP):A, Tg:G, Tg:A, 2,6-diamino-4-hydroxy-5-N-methylformamidopyrimidine (meFapyG):C, urea:A, DHT:A, 5-formyl-2′-deoxyuridine (fU):A, OG:C, and 5-hydroxymethyl-2′-deoxyuridine (hmU):A [154, 192–194]. Inspection of the relative kinetic rate comparisons immediately identifies the hydantoin diastereomers Sp, Gh, and 2Ih as the most preferred substrates for edited NEIL1 (Fig. 3B). NEIL1 favors the Sp diastereomers by >100-fold and Gh or 2Ih by >80-fold over the lesions Tg:A, AP:A, meFapyG:C, DHT:A, and OG:C (Fig. 3B). NEIL1 even favors Sp, Gh, and 2Ih over the 5-hydroxypyrimidines, 5-HOU and 5-HOC, by ~1.5- to 4-fold. The 5-hydroxypyrimidines were the first discovered NEIL1 substrates [169, 173, 178], followed later by studies of the hydantoins from G oxidation [154, 190, 192, 196]. The relative repair rate for meFapyG:C in dsDNA is ~150-fold less efficient than the Sp diastereomers base paired with C, supporting the favorability of NEIL1 for recognition of the hydantoins (Fig. 3B).
Next, the base pairing impact for NEIL1 reaction on (R)-Sp, (S)-Sp, and Gh was studied and a strong base pair dependency in the rates was found (Fig. 3C). For the Sp diastereomers, the base pair dependency decreased from T>G>C>A, and for Gh the rate decreased from T>G>A~C, a result consistent with the report from Sugden’s laboratory [190]. The comparisons of Sp or Gh vs. other lesions were made with C opposite the lesion that is kinetically slower; comparisons to G opposite would have led to an even larger relative favorability for NEIL1 acting upon the hydantoins.
The studies in the Burrows/David collaboration also looked into the impact Sp stereochemistry has on the reaction rate and found the S diastereomer was preferred for removal by NEIL1 (Figs. 3B and 3C) [192]. Interestingly, this experimental observation was supported by a previous computational prediction by the Broyde laboratory [197]. In their calculations, (S)-Sp provided better hydrogen bonding contacts with NEIL1 via methionine 80, an interaction that was found to be repulsive for (R)-Sp leading to an enlargement of the binding site. These observations provide a fascinating case study in which the stereochemical dependency in an enzymatic reaction was readily predicted by computational modeling.
The coding potential of mRNA can change via RNA editing to modify nucleotides in the sequence [198]. One mechanism for RNA editing is via the actions of adenosine deaminases acting on RNA (ADAR) for conversion of an A to I (inosine) nucleotide [199]. An intriguing observation by the David and Beal laboratories was that NEIL1 undergoes editing at the 242nd codon [195]. The genomic, or unedited, sequence for this codon is AAA and codes for a lysine, while the AIA edited sequence codes for an arginine. This led the team to study the repair kinetics as a function of editing state to identify a profound change in the repair rates for Sp, Gh, and Tg [195]. The unedited version of NEIL1 shows a much stronger preference for Tg:G (~30x) that was not observed in the edited version of the previous studies (Fig. 3B). In the case of Gh or Sp, the unedited version of NEIL1 showed decreased activity compared to the edited version by 0.4 and 0.5 (ratio = unedited/edited), respectively. When the unedited NEIL1 rates were compared relative to Tg:G, the hydantoins (S)-Sp:G and Gh:G were kinetically favored by ~1.5-fold (Fig. 3B inset); this is in contrast to the >80-fold favorability of the hydantoins for edited NEIL1. A recent x-ray crystal structure of a Tg-containing DNA housed inside the active site of edited vs. unedited NEIL1 found interactions between the lysine or arginine at position 242 and Tg [182]. This observation verifies that the editing state of NEIL1 can impact recognition of Tg, and whether this also impacts NEIL1 interactions with Sp or Gh remains an open question. Lastly, in the work of Beal and David they discuss that ADAR expression leading to RNA editing is stimulated by a pathway upregulated during inflammatory stress [195]. This suggests the possibility that cellular stress induces RNA editing of the hNEIL1 mRNA to tailor the substrate preference for the protein.
Hydantoins could exist in many different contexts in the genome. Evidence in the literature supports the idea that hydantoins in template strands are extruded during polymerase bypass leading to frameshift mutations [192]. These mutation types could result in Sp or Gh to exist in a bulge structure. As stated above, G4 contexts are highly susceptible to oxidation yielding Sp. Lastly, during transcription or replication a bubble structure is formed in which Sp or Gh might stop polymerase progression and need to be repaired (i.e., transcription-coupled or replication-coupled repair) [166, 168]. Therefore, Sp and Gh have a high potential to exist in contexts within the genome other than dsDNA. Moreover, NEIL1 and NEIL2 were recognized early on to bind and remove damaged nucleotides in contexts other than dsDNA [172, 200]. The context dependency of NEIL1 removal of each Sp diastereomer and Gh was evaluated in ssDNA, bulge DNA, bubble DNA, and the human telomere G4 (G4 hTelo) by our laboratory in collaboration with the David and Wallace laboratories [196, 201, 202]. The kinetic rate data for the Sp diastereomers and Gh in each context studied are plotted relative to the rates observed in ssDNA (Fig. 3D). Also, the hydantoins were studied in many positions within the G4 hTelo context; the data currently discussed are within the sequence 5′-TAGGGTTAGXGTTAGGGTTAGGGTT-3′, in which X = (S)-Sp, (R)-Sp, or Gh in solutions containing KCl [201].
The context-dependent comparison of NEIL1 repair of the Sp isomers and Gh demonstrates these hydantoins are favorably repaired in dsDNA and the G4 hTelo sequence (Fig. 3D). Furthermore, NEIL1 glycosylase repair rates for Sp and Gh in dsDNA and the G4 hTelo sequence are >400-fold more efficient than repair initiation in ssDNA. Hydantoin removal by hNEIL1 in the bulge and bubble structures is similar to ssDNA with the exception of Gh in the bubble structure (Fig. 3D, 87x more efficient than the ssDNA context). Thus, NEIL1 may have evolved to operate on its superior substrates, Sp and Gh, in dsDNA and G4 DNA contexts (Figs. 3B and 3D).
Because NEIL1 cleaves lesions from hTelo G4 sequences, we then wondered if NEIL1 was operational on other G4s such as those found in gene promoters. The vascular endothelial growth factor-A (VEGF) promoter G4 was selected as a case study [201]. The VEGF G4 sequence is comprised of five G runs, in which the four runs on the 5′ end of the sequence were demonstrated to adopt a parallel-stranded G4 [203]. When Gh was synthesized at a site occupying a loop position or a core position in the four G-track sequence and folded in KCl, NEIL1 was not capable of initiating removal of Gh. This led us to incorporate the 5th G run and repeat the repair assay, and to our surprise, NEIL1 efficiently removed Gh from the 5-track VEGF sequence [113]. We determined when 5 G-runs are present, the damage-containing segment can be extruded and replaced by the fifth track to establish a G4 fold with a long loop that is acted upon by NEIL1 (Fig. 4). Therefore, the plasticity of G4 motifs provides a structure switching mechanism to regulate NEIL1 activity and possibly substrate recognition and binding as well.
Figure 4.

NEIL initiated repair in the VEGF G4 requires the 5th G track to allow a structural transition to a repair-competent fold.
11. The hydantions are substrates for NEIL2 and NEIL3
Early on it was recognized that NEIL2 was operational on 5-HOC in ssDNA and bubble DNA [200], which was followed by the observation of NEIL2 processing Gh and Sp in ssDNA [190]. More recently, NEIL2 was shown to be ineffective at removing the Sp isomers or Gh from the hTelo G4 in comparison to NEIL1 and NEIL3 [201]. In contrast, NEIL2 provided similar reactivity toward Gh in the five G track VEGF sequence when compared to NEIL1, in which Gh was extruded into a large loop to maintain G4 folding (Fig. 4) [113]. This final observation is consistent with NEIL2 favorably removing lesions from ssDNA or large bubble DNA structures [190, 200]. Detailed kinetic analysis for NEIL2 acting on Sp and Gh has not been achieved, preventing comparisons as was provided for NEIL1 above. Kinetic studies for NEIL2 operating on Tg:A, fU, hmU, urea, 5-HOU, 5-HOC, OG, and meFapyG were conducted and the enzyme was found to be generally much slower than NEIL1 when acting on the same substrates in the same contexts [194].
Expression and purification of functional NEIL3 was the most challenging of the NEILs [175]. The Wallace laboratory first demonstrated the mouse ortholog of NEIL3, MmuNeil3, was a functional glycosylase [204]. Additionally, MmuNeil3 is a large glycosylase when expressed in the wild-type state (606 amino acids) and is prone to aggregation, leading to the construction of a truncated mutant of MmuNeil3 (MmuNeil3Δ324) possessing the glycosylase domain. In their studies, MmuNeil3 and MmuNeil3Δ324 were tested on Tg, OG, 5-HOU, 5-HOC, DHT, DHU, Sp, Gh, meFapyG, and AP substrates in ssDNA and dsDNA contexts (Figs. 5A and 5B). The main conclusions were that MmuNeil3 and MmuNeil3Δ324 were much more active on lesions in ssDNA than in dsDNA. In ssDNA and dsDNA, diastereomeric mixtures of Sp and Gh were found to be the best substrates out of all lesions tested. The hydantoins were found to be even better substrates than meFapyG (Fig. 5A). A recent X-ray crystal structure by the Doublié laboratory explains why MmuNeil3 favors ssDNA substrates [183]. In the structure, MmuNeil3 lacks two of the three void-filling residues responsible for binding the complementary strand, and it also harbors negatively charged residues causing an electrostatic repulsion of the complementary strand. A second distinct characteristic of MmuNeil3/NEIL3 is that the active site nucleophile is an N-terminal valine (Val-2), in contrast to the N-terminal proline (Pro-2) functioning as the active site nucleophile in EcFpg, NEIL1, and NEIL2. These differences help address why MmuNeil3/NEIL3 is mainly a monofunctional glycosylase with poor lyase activity with favorable activity on ssDNA [174, 175].
Figure 5.

Context and substrate preference for MmuNeil3 and cellular properties of the NEIL glycosylases. (A) Efficiency of removal for various substrates by MmuNeil3 and MmuNeil3Δ324 in dsDNA contexts. (B) Efficiency of removal for various substrates by MmuNeil3 and MmuNeil3Δ324 in ssDNA contexts. The graphs in parts A and B are adapted from the original publication on this work [204]. (C) Table to compare the NEILs on the basis of their substrate preference, reaction context preference, protein interaction partners, and cell cycle of maximal expression. The graphs in part A are adapted from the original publication [204].
The observation that NEIL3 prefers substrates in ssDNA over dsDNA contexts led to questions of whether this glycosylase could function in other contexts, such as G4-DNA. To address this question, the hydantoins were synthesized in hTelo G4 sequences and subjected to NEIL3 repair [201, 202]. When the hTelo sequences were studied in KCl solutions with the individual Sp diastereomers or Gh, it was found that NEIL3 was quite proficient at removing hydantoins in G4-DNA relative to dsDNA [201]. The stereochemistry of Sp did not impact the reaction kinetics in G4-DNA, as was observed for NEIL1. In a set of position-dependent studies in the hTelo G4 context, the lesion location was found to be the major factor impacting the reaction rate. Next, NEIL3 activity on promoter G4s was evaluated in the VEGF promoter G4 to find nearly identical results as observed with NEIL1 and NEIL2 (Fig. 4) [113]. When repair was attempted in the 4-track VEGF G4 no reaction was observed; in contrast, when the 5-track VEGF G4 was studied, Gh was efficiently removed (Fig. 4). The dynamics of the G4 provided by the 5th G run regulates NEIL3 activity and may serve additional cellular functions. In summary, NEIL3 is mainly a monofunctional glycosylase that acts on hydantoins in ssDNA and G4-DNA; however, in G4-DNA the lesion needs to be a located in a relatively large loop. A recent publication found NEIL3 can repair DNA interstrand cross-links; thus, expanding the possible roles NEIL3 has in cells [205].
12. The biology of the NEILs and hydantoins
The NEIL glycosylases were found to possess distinct properties with respect to substrate selection and the impact of reaction context on their activity. For instance, NEIL1 readily removes Sp and Gh from dsDNA and all G4-DNA contexts, NEIL2 has a preference for Sp and Gh in ssDNA and promoter G4s, and NEIL3 operates on Sp and Gh in ssDNA and all G4-DNA contexts (Fig. 5C). In cells where would NEIL activity on these hydantoins and each context be utilized?
The glycosylase NEIL1 has strong cell cycle dependency; its expression peaks during the S phase [171], and it is found widely distributed in many cell types [206] and mitochondria [207]. Further, NEIL1 was shown to interact during replication with the proteins proliferating cell nuclear antigen (PCNA) [208], replication protein A (RPA) [209], flap endonuclease 1 (FEN1) [210], and Werner syndrome protein (WRN) (Fig. 5C) [211]. Additional NEIL1 interactions occur with the DNA repair proteins PNK, POLβ, XRCC1, and LIGIII [212], via the C-terminal domain (Fig. 5C) [213]. Furthermore, NEIL1 deficiency slows replication fork progression in oxidatively stressed cells [214]. These observations support NEIL1 acting as a “cowcatcher” to identify DNA damage and initiate repair in front of the DNA replication complex conducting replication-coupled BER repair (Fig. 5C).
A surprising interaction is one with WRN helicase that preferentially unravels G4s, especially those in gene promoters and the first intron [215, 216]. Potentially, NEIL1 may function with WRN for repair of Sp or Gh with a preference for G4 DNA. Additionally, WRN and FEN1 have both been demonstrated to localize to telomeric regions [217, 218], which may recruit NEIL1 to the telomeres for maintenance during oxidative stress. These possible roles for NEIL1 to repair Sp and Gh in G-rich regions, such as G4s in telomeres and gene promoters, are anticipated to position NEIL1 for removal of G oxidation products at sites possibly most prone to oxidation.
Stimulation of NEIL1 occurs by the 9-1-1 complex [219], WRN [220], and Y-box protein 1 (YB-1) (Fig. 5C) [221], while NEIL1 stimulates the activity of OGG1 on DNA containing OG [222]. These proteins activate transcription or the stress response, leading to the possibility that NEIL1 plays a more important role in the cell beyond removal of damaged bases. One possibility is that NEIL1 guides the efforts of stimulating proteins to alter protein expression levels during oxidative stress. Moreover, whether the NEIL1 mRNA has been edited or not impacts the affinity of the NEIL1 protein for DNA damage substrates that will provide another layer of control. Future cell studies will aid in understanding if NEIL1 has a signaling function in cells.
For NEIL2, its expression levels are independent of cell cycle [178], and it is distributed throughout many cell types, as well as being present in mitochondria [223]. Protein-protein interactions with NEIL2 have been identified with RNA pol II and the transcriptional regulator heterogeneous nuclear ribonucleoprotein-U (hnRNP-U) [224]. So far, YB-1 is the only protein recognized to stimulate NEIL2 glycosylase activity (Fig. 5C) [225]. Moreover, NEIL2 knocked out cells accumulated more DNA damage in active compared to silent genes [224]. These observations have led to the conclusion that NEIL2 is involved in transcription-coupled BER (Fig. 5C). During transcription NEIL2 can actively repair Sp and Gh in ssDNA and promoter G4-DNA contexts.
The biology of NEIL3 has been reviewed by Wallace and coworkers [185], and the present discussion will focus on possible genomic contexts and roles that Sp and Gh could have when recognized by NEIL3. Expression of NEIL3 is highly dependent on cell type, and so far, it has not been observed in the mitochondria. NEIL3 has been detected in human thymus and testis [169, 226], and mouse Neil3 has also been additionally observed in the spleen, bone marrow, and the brain, and its expression levels vary as a function of development, particularly in the brain [227]. NEIL3 is overexpressed in malignant melanomas [227]. NEIL3 is also a required host factor for HIV replication [228]. Further, Neil3 was found to promote neurogenesis induced by hypoxia-ischemia [229], to regulate lipid metabolism preventing atherosclerosis [230], and to function during development prior to implantation [227]. Knockdown of Neil3 and Ogg1 in mice decreases embryonic neural stem cell differentiation [231]. The expression of NEIL3 is induced during S phase by the Ras-dependent ERK-MAP kinase pathway and peaks during G2 phase, and expression can be repressed by the DREAM complex (Fig. 5C) [232]. The only established protein interaction to NEIL3 is with RPA (Fig. 5C) [169]. Lastly, Neil3-deficient mice do not present with a phenotype indicative of genome instability [229]. These observations paint a very broad and poorly defined mechanism by which NEIL3 is functional in cells, but it does appear to be involved in cellular regulation, particularly in proliferating cells.
The biochemical studies described above found that NEIL3 accepts Sp and Gh as the best possible substrates in ssDNA, G4-DNA in telomeres, and G4-DNA in promoters, while it has very poor affinity for lesions in dsDNA contexts (Fig. 5C). One possibility is that NEIL3 is involved in replication-coupled repair, consistent with induction during S phase and interacting with RPA (Fig. 5C), although a role in replication-coupled repair does not fit with the observations of NEIL3 functioning in cell cycle processes. Therefore, we have proposed in tandem with the Wallace laboratory that NEIL3 may function as a transcriptional regulator when binding to DNA lesions in promoter G-quadruplexes [113, 202]. Many examples of genes regulated by promoter G4s have been observed [41], visualization of G4s is maximal in cells during S phase (i.e., during replication) [233], and this is a stage when DNA is most susceptible to oxidation at G nucleotides, particularly in G4s. Interestingly, HIV replication is associated with G4 formation and facilitated by oxidative stress [234]. Thus, oxidation of a promoter G4 induces G oxidation and in the case of four G-track G4s NEIL3 does not operate as demonstrated with the VEGF 4-track promoter sequence. However, if its 5th domain is present a structural switch occurs allowing NEIL3 to bind and initiate lesion removal (Fig. 3E). The concept of coupling BER and gene induction has recently emerged in other systems [235]. Utilization of NEIL3 as a “transcription factor” for recognition of the oxidative modifications Sp or Gh to induce gene induction ascribes a regulatory function to NEIL3 and the hydantoins (Fig. 5C). Is it possible that Sp or Gh are epigenetic-like DNA modifications and NEIL3 is the reader protein to alter cellular phenotype? This mechanism would begin to address why NEIL3 does not fit the mold of a traditional BER glycosylase. Future studies will aid in addressing these questions.
Highlights.
Guanine oxidation in the presence of cellular reductants yields OG, 2Ih, Sp, and Gh as products
Hydantoins and OG are formed in nucleoside, single-, double-, and G-quadruplex contexts
Sp, Gh, and 2Ih are the best substrates for the NEIL DNA glycosylases in base excision repair
Hydantoins in single-, double-, bulge-, bubble-, and G-quadruplexes are NEIL substrates
NEIL3 removal of Gh from promoter G-quadruplexes may be a cellular signaling pathway
Acknowledgments
We are greatly appreciative of our wonderful collaborators in our own laboratory and others, including Prof. Sheila David (University of California-Davis) and Prof. Susan Wallace (University of Vermont). Our work on this topic was funded by the National Cancer Institute (R01 CA090689).
Footnotes
Abbreviations: AP, abasic site; APE1, apurinic/apyrimidinic endonuclease; Asc, ascorbate; BER, base excision repair; DHT, 5,6-dihydro-2′-deoxythymidine; DHU, 5,6-dihydro-2′-deoxyuridine; dsDNA, double-stranded DNA; ESCODD, European Standards Committee on Oxidative DNA Damage; Fapy-A, 4,6-diamino-5-formamidopyrimidine; Fapy-G, 2,6-diamino-4-hydroxy-5-formamidopyrimidine; FEN1, flap endonuclease 1; Fpg and EcFpg, formamidopyrimidine DNA glycosylase; G, guanine; G•+, guanine radical cation; G•, guanine radical; G4, G-quadruplex; Gh, 5-guanidinohydantoin; Ghox, oxidized 5-guanidinohydantoin; hmU, 5-hydroxymethyl-2′-deoxyuridine; GSH, glutathione; hnRNP-U, heterogeneous nuclear ribonucleoprotein-U; 5-HOC, 5-hydroxy-2′-deoxycytidine; 5-HO-G, 5-hydroxy-guanine; 5-HOO-G, 5-hydroperoxy-guanine; 8-HOO-G, 8-hydroperoxy-guanine; 8-HO-G, 8-hydroxy-guanine; 5-HOU, 5-hydroxy-2′-deoxyuridine; hTelo, human telomere sequence; Ia, iminoallantoin; Iaox, oxidized iminoallantoin; I, inosine; 2Ih, 5-carboxamido-5-formamido-2-iminohydantoin; Iz, 2,5-diaminoimidazolone; LCMS, liquid chromatography mass spectrometer; meFapyG; 2,6-diamino-4-hydroxy-5-N-methylformamidopyrimdine; NAC, N-acetylcysteine; Nei and EcoNei, endonuclease VIII; Neil1 and NEIL1, endonuclease VIII like-1; Neil2 and NEIL2, endonuclease VIII like-2, Neil3 and NEIL3, endonuclease VIII like-3; NER, nucleotide excision repair; Nth, endonuclease III; OG, 8-oxo-7,8-dihydroguanine; OGox, oxidized 8-oxo-7,8-dihydroguanine; PCNA, proliferating cell nuclear antigen; PNK, polynucleotide kinase; POLβ, polymerase beta; REAP, restriction endonuclease and post-labeling; RPA, replication protein A; ROS, reactive oxygen species; ssDNA, single-stranded DNA; Sp, spiroiminodihydantoin; Tg, thymine glycol; VEGF, vascular endothelial growth factor; WRN, Werner syndrome protein; YB-1, Y-box protein 1; Z, 2,2,4-triamino-2H-oxazol-5-one
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Giorgio M, Trinei M, Migliaccio E, Pelicci PG. Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nat Rev Mol Cell Biol. 2007;8:722–728. doi: 10.1038/nrm2240. [DOI] [PubMed] [Google Scholar]
- 2.Speckmann B, Steinbrenner H, Grune T, Klotz LO. Peroxynitrite: From interception to signaling. Arch Biochem Biophys. 2016;595:153–160. doi: 10.1016/j.abb.2015.06.022. [DOI] [PubMed] [Google Scholar]
- 3.Dedon PC, Tannenbaum SR. Reactive nitrogen species in the chemical biology of inflammation. Arch Biochem Biophys. 2004;423:12–22. doi: 10.1016/j.abb.2003.12.017. [DOI] [PubMed] [Google Scholar]
- 4.Knoops B, Argyropoulou V, Becker S, Ferte L, Kuznetsova O. Multiple roles of peroxiredoxins in inflammation. Mol Cells. 2016;39:60–64. doi: 10.14348/molcells.2016.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lonkar P, Dedon PC. Reactive species and DNA damage in chronic inflammation: reconciling chemical mechanisms and biological fates. Int J Cancer. 2011;128:1999–2009. doi: 10.1002/ijc.25815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kurian GA, Rajagopal R, Vedantham S, Rajesh M. The role of oxidative stress in myocardial ischemia and reperfusion injury and remodeling: Revisited. Oxid Med Cell Longev. 2016;2016:1656450. doi: 10.1155/2016/1656450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Markesbery WR. The role of oxidative stress in Alzheimer disease. Arch Neurology. 1999;56:1449–1452. doi: 10.1001/archneur.56.12.1449. [DOI] [PubMed] [Google Scholar]
- 8.Steenken S, Jovanovic SV. How easily oxidizable is DNA? One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution. J Am Chem Soc. 1997;119:617–618. [Google Scholar]
- 9.Cadet J, Wagner JR, Shafirovich V, Geacintov NE. One-electron oxidation reactions of purine and pyrimidine bases in cellular DNA. Int J Radiat Biol. 2014;90:423–432. doi: 10.3109/09553002.2013.877176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cadet J, Douki T, Ravanat JL. Oxidatively generated damage to cellular DNA by UVB and UVA radiation. Photochem Photobiol. 2015;91:140–155. doi: 10.1111/php.12368. [DOI] [PubMed] [Google Scholar]
- 11.Pratviel G, Meunier B. Guanine oxidation: one- and two-electron reactions. Chem Eur J. 2006;12:6018–6030. doi: 10.1002/chem.200600539. [DOI] [PubMed] [Google Scholar]
- 12.Burrows CJ, Muller JG. Oxidative nucleobase modifications leading to strand scission. Chem Rev. 1998;98:1109–1152. doi: 10.1021/cr960421s. [DOI] [PubMed] [Google Scholar]
- 13.Delaney S, Jarem DA, Volle CB, Yennie CJ. Chemical and biological consequences of oxidatively damaged guanine in DNA. Free Radical Res. 2012;46:420–441. doi: 10.3109/10715762.2011.653968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cadet J, Douki T, Ravanat J-L. Oxidatively generated base damage to cellular DNA. Free Radical Biol Med. 2010;49:9–21. doi: 10.1016/j.freeradbiomed.2010.03.025. [DOI] [PubMed] [Google Scholar]
- 15.Jena NR, Mishra PC. Formation of ring-opened and rearranged products of guanine: mechanisms and biological significance. Free Radic Biol Med. 2012;53:81–94. doi: 10.1016/j.freeradbiomed.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 16.Neeley WL, Essigmann JM. Mechanisms of formation, genotoxicity, and mutation of guanine oxidation products. Chem Res Toxicol. 2006;19:491–505. doi: 10.1021/tx0600043. [DOI] [PubMed] [Google Scholar]
- 17.Gimisis T, Cismas C. Isolation, characterization, and independent synthesis of guanine oxidation products. Eur J Org Chem. 2006;2006:1351–1378. [Google Scholar]
- 18.Chatgilialoglu C, Ferreri C, Terzidis MA. Purine 5′,8-cyclonucleoside lesions: chemistry and biology. Chem Soc Rev. 2011;40:1368–1382. doi: 10.1039/c0cs00061b. [DOI] [PubMed] [Google Scholar]
- 19.Greenberg MM. The formamidopyrimidines: purine lesions formed in competition with 8-oxopurines from oxidative stress. Acc Chem Res. 2012;45:588–597. doi: 10.1021/ar2002182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gates K. An overview of chemical processes that damage cellular DNA: spontaneous hydrolysis, alkylation, and reactions with radicals. Chem Res Toxicol. 2009;22:1747–1760. doi: 10.1021/tx900242k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y. Bulky DNA lesions induced by reactive oxygen species. Chem Res Toxicol. 2008;21:276–281. doi: 10.1021/tx700411g. [DOI] [PubMed] [Google Scholar]
- 22.Tretyakova N, Villalta PW, Kotapati S. Mass spectrometry of structurally modified DNA. Chem Rev. 2013;113:2395–2436. doi: 10.1021/cr300391r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dedon PC. The chemical toxicology of 2-deoxyribose oxidation in DNA. Chem Res Toxicol. 2008;21:206–219. doi: 10.1021/tx700283c. [DOI] [PubMed] [Google Scholar]
- 24.Nikitaki Z, Hellweg CE, Georgakilas AG, Ravanat JL. Stress-induced DNA damage biomarkers: applications and limitations. Front Chem. 2015;3:35. doi: 10.3389/fchem.2015.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakabeppu Y, Sakumi K, Sakamoto K, Tsuchimoto D, Tsuzuki T, Nakatsu Y. Mutagenesis and carcinogenesis caused by the oxidation of nucleic acids. Biol Chem. 2006;387:373–379. doi: 10.1515/BC.2006.050. [DOI] [PubMed] [Google Scholar]
- 26.Tudek B. Imidazole ring-opened DNA purines and their biological significance. J Biochem Mol Biol. 2003;36:12–19. doi: 10.5483/bmbrep.2003.36.1.012. [DOI] [PubMed] [Google Scholar]
- 27.Dizdaroglu M. Oxidatively induced DNA damage and its repair in cancer. Mutat Res Rev Mutat Res. 2015;763:212–245. doi: 10.1016/j.mrrev.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 28.Gedik CM, Collins A. Establishing the background level of base oxidation in human lymphocyte DNA: results of an interlaboratory validation study. FASEB J. 2005;19:82–84. doi: 10.1096/fj.04-1767fje. [DOI] [PubMed] [Google Scholar]
- 29.Steenken S, Jovanovic SV, Bietti M, Bernhard K. The trap depth (in DNA) of 8-oxo-7,8-dihydro-2′deoxyguanosine as derived from electron-transfer equilibria in aqueous solution. J Am Chem Soc. 2000;122:2373–2374. [Google Scholar]
- 30.Luo W, Muller JG, Rachlin EM, Burrows CJ. Characterization of spiroiminodihydantoin as a product of one-electron oxidation of 8-oxo-7,8-dihydroguanosine. Org Lett. 2000;2:613–616. doi: 10.1021/ol9913643. [DOI] [PubMed] [Google Scholar]
- 31.Niles JC, Wishnok JS, Tannenbaum SR. Spiroiminodihydantoin is the major product of the 8-oxo-7,8-dihydroguanosine reaction with peroxynitrite in the presence of thiols and guanosine photooxidation by methylene blue. Org Lett. 2001;3:963–966. [PubMed] [Google Scholar]
- 32.Luo W, Muller JG, Rachlin EM, Burrows CJ. Characterization of hydantoin products from one-electron oxidation of 8-oxo-7,8-dihydroguanosine in a nucleoside model. Chem Res Toxicol. 2001;14:927–938. doi: 10.1021/tx010072j. [DOI] [PubMed] [Google Scholar]
- 33.Ye Y, Muller JG, Luo W, Mayne CL, Shallop AJ, Jones RA, Burrows CJ. Formation of 13C-, 15N-, and 18O-labeled guanidinohydantoin from guanosine oxidation with singlet oxygen. Implications for structure and mechanism. J Am Chem Soc. 2003;125:13926–13927. doi: 10.1021/ja0378660. [DOI] [PubMed] [Google Scholar]
- 34.Ravanat J-L, Martinez GR, Medeiros MHG, Di Mascio P, Cadet J. Singlet oxygen oxidation of 2′-deoxyguanosine. Formation and mechanistic insights. Tetrahedron. 2006;62:10709–10715. [Google Scholar]
- 35.Avila Figueroa A, Delaney S. Mechanistic studies of hairpin to duplex conversion for trinucleotide repeat sequences. J Biol Chem. 2010;285:14648–14657. doi: 10.1074/jbc.M109.061853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nekludova L, Pabo CO. Distinctive DNA conformation with enlarged major groove is found in Zn-finger-DNA and other protein-DNA complexes. Proc Natl Acad Sci U S A. 1994;91:6948–6952. doi: 10.1073/pnas.91.15.6948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang G, Vasquez KM. Z-DNA, an active element in the genome. Front Biosci. 2007;12:4424–4438. doi: 10.2741/2399. [DOI] [PubMed] [Google Scholar]
- 38.Buske FA, Bauer DC, Mattick JS, Bailey TL. Triplexator: detecting nucleic acid triple helices in genomic and transcriptomic data. Genome Res. 2012;22:1372–1381. doi: 10.1101/gr.130237.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sarbajna S, West SC. Holliday junction processing enzymes as guardians of genome stability. Trends Biochem Sci. 2014;39:409–419. doi: 10.1016/j.tibs.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 40.Kendrick S, Kang HJ, Alam MP, Madathil MM, Agrawal P, Gokhale V, Yang D, Hecht SM, Hurley LH. The dynamic character of the BCL2 promoter i-motif provides a mechanism for modulation of gene expression by compounds that bind selectively to the alternative DNA hairpin structure. J Am Chem Soc. 2014;136:4161–4171. doi: 10.1021/ja410934b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Balasubramanian S, Hurley LH, Neidle S. Targeting G-quadruplexes in gene promoters: A novel anticancer strategy? Nat Rev Drug Discov. 2011;10:261–275. doi: 10.1038/nrd3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shafirovich V, Kropachev K, Anderson T, Liu Z, Kolbanovskiy M, Martin BD, Sugden K, Shim Y, Chen X, Min JH, Geacintov NE. Base and nucleotide excision repair of oxidatively generated guanine lesions in DNA. J Biol Chem. 2016;291:5309–5319. doi: 10.1074/jbc.M115.693218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.David S, O’Shea V, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wallace SS. Base excision repair: A critical player in many games. DNA Repair. 2014;19:14–26. doi: 10.1016/j.dnarep.2014.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McKibbin PL, Fleming AM, Towheed MA, Van Houten B, Burrows CJ, David SS. Repair of hydantoin lesions and their amine adducts in DNA by base and nucleotide excision repair. J Am Chem Soc. 2013;135:13851–13861. doi: 10.1021/ja4059469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kozlowski H, Janicka-Klos A, Brasun J, Gaggelli E, Valensin D, Valensin G. Copper, iron, and zinc ions homeostasis and their role in neurodegenerative disorders (metal uptake, transport, distribution and regulation) Coord Chem Rev. 2009;253:2665–2685. [Google Scholar]
- 48.Reisz JA, Bansal N, Qian J, Zhao W, Furdui CM. Effects of ionizing radiation on biological molecules-mechanisms of damage and emerging methods of detection. Antioxid Redox Signal. 2014;21:260–292. doi: 10.1089/ars.2013.5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Szabo C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 50.Misiaszek R, Crean C, Geacintov NE, Shafirovich V. Combination of nitrogen dioxide radicals with 8-oxo-7,8-dihydroguanine and guanine radicals in DNA: Oxidation and nitration end-products. J Am Chem Soc. 2005;127:2191–2200. doi: 10.1021/ja044390r. [DOI] [PubMed] [Google Scholar]
- 51.Joffe A, Geacintov NE, Shafirovich V. DNA lesions derived from the site selective oxidation of guanine by carbonate radical anions. Chem Res Toxicol. 2003;16:1528–1538. doi: 10.1021/tx034142t. [DOI] [PubMed] [Google Scholar]
- 52.Ogilby PR. Singlet oxygen: there is indeed something new under the sun. Chem Soc Rev. 2010;39:3181–3209. doi: 10.1039/b926014p. [DOI] [PubMed] [Google Scholar]
- 53.Onyango AN. Endogenous generation of singlet oxygen and ozone in human and animal tissues: Mechanisms, biological significance, and influence of dietary components. Oxid Med Cell Longev. 2016;2016:2398573. doi: 10.1155/2016/2398573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kurutas EB. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: current state. Nutr J. 2016;15:71. doi: 10.1186/s12937-016-0186-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pogozelski WK, Tullius TD. Oxidative strand scission of nucleic acids: Routes initiated by hydrogen abstraction from the sugar moiety. Chem Rev. 1998;98:1089–1108. doi: 10.1021/cr960437i. [DOI] [PubMed] [Google Scholar]
- 56.Zhu J, Fleming AM, Orendt AM, Burrows CJ. pH-Dependent equilibrium between 5-guanidinohydantoin and iminoallantoin affects nucleotide insertion opposite the DNA lesion. J Org Chem. 2016;81:351–359. doi: 10.1021/acs.joc.5b02180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fleming AM, Orendt AM, He Y, Zhu J, Dukor RK, Burrows CJ. Reconciliation of chemical, enzymatic, spectroscopic and computational data to assign the absolute configuration of the DNA base lesion spiroiminodihydantoin. J Am Chem Soc. 2013;135:18191–18204. doi: 10.1021/ja409254z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fleming AM, Alshykhly O, Orendt AM, Burrows CJ. Computational studies of electronic circular dichroism spectra predict absolute configuration assignments for the guanine oxidation product 5-carboxamido-5-formamido-2-iminohydantoin. Tetrahedron Lett. 2015;56:3191–3196. doi: 10.1016/j.tetlet.2014.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Karwowski B, Dupeyrat F, Bardet M, Ravanat JL, Krajewski P, Cadet J. Nuclear magnetic resonance studies of the 4R and 4S diastereomers of spiroiminodihydantoin 2′-deoxyribonucleosides: absolute configuration and conformational features. Chem Res Toxicol. 2006;19:1357–1365. doi: 10.1021/tx060088f. [DOI] [PubMed] [Google Scholar]
- 60.Ding S, Jia L, Durandin A, Crean C, Kolbanovskiy A, Shafirovich V, Broyde S, Geacintov NE. Absolute configurations of spiroiminodihydantoin and allantoin stereoisomers: Comparison of computed and measured electronic circular dichroism spectra. Chem Res Toxicol. 2009;22:1189–1193. doi: 10.1021/tx900107q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lloyd RV, Hanna PM, Mason RP. The origin of the hydroxyl radical oxygen in the Fenton reaction. Free Radic Biol Med. 1997;22:885–888. doi: 10.1016/s0891-5849(96)00432-7. [DOI] [PubMed] [Google Scholar]
- 62.Fleming AM, Muller JG, Ji I, Burrows CJ. Characterization of 2′-deoxyguanosine oxidation products observed in the Fenton-like system Cu(II)/H2O2/reductant in nucleoside and oligodeoxynucleotide contexts. Org Biomol Chem. 2011;9:3338–3348. doi: 10.1039/c1ob05112a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Douki T, Martini R, Ravanat JL, Turesky RJ, Cadet J. Measurement of 2,6-diamino-4-hydroxy-5-formamidopyrimidine and 8-oxo-7,8-dihydroguanine in isolated DNA exposed to gamma radiation in aqueous solution. Carcinogenesis. 1997;18:2385–2391. doi: 10.1093/carcin/18.12.2385. [DOI] [PubMed] [Google Scholar]
- 64.Aruoma OI, Halliwell B, Dizdaroglu M. Iron ion-dependent modification of bases in DNA by the superoxide radical-generating system hypoxanthine/xanthine oxidase. J Biol Chem. 1989;264:13024–13028. [PubMed] [Google Scholar]
- 65.Frelon S, Douki T, Favier A, Cadet J. Comparative study of base damage induced by gamma radiation and Fenton reaction in isolated DNA. J Chem Soc, Perkin Trans 1. 2002:2866–2870. [Google Scholar]
- 66.Berger M, Cadet J. Isolation and characterization of the radiation-induced degradation products of 2′-deoxyguanosine in oxygen-free aqueous solutions. Z Naturforsch, B: Chem Sci. 1985;40:1519–1531. [Google Scholar]
- 67.Bergeron F, Auvre R, Radicella JP, Ravanat J-L. HO radicals induce an unexpected high proportion of tandem base lesions refractory to repair by DNA glycosylases. Proc Natl Acad Sci USA. 2010;107:5528–5533. doi: 10.1073/pnas.1000193107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Box HC, Freund HG, Budzinski EE, Wallace JC, MacCubbin AE. Free radical-induced double base lesions. Radiat Res. 1995;141:91–94. [PubMed] [Google Scholar]
- 69.Bourdat A-Gl, Douki T, Frelon S, Gasparutto D, Cadet J. Tandem base lesions are generated by hydroxyl radical within isolated DNA in aerated aqueous solution. J Am Chem Soc. 2000;122:4549–4556. [Google Scholar]
- 70.Alshykhly OR, Fleming AM, Burrows CJ. 5-Carboxamido-5-formamido-2-iminohydantoin, in addition to 8-oxo-7,8-dihydroguanine, is the major product of the iron-Fenton or X-ray radiation-induced oxidation of guanine under aerobic reducing conditions in nucleoside and DNA contexts. J Org Chem. 2015;80:6996–7007. doi: 10.1021/acs.joc.5b00689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Crespo-Hernandez CE, Arce R. Formamidopyrimidines as major products in the low- and high-intensity UV irradiation of guanine derivatives. J Photochem Photobiol B. 2004;73:167–175. doi: 10.1016/j.jphotobiol.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 72.Lim KS, Cui L, Taghizadeh K, Wishnok JS, Chan W, DeMott MS, Babu IR, Tannenbaum SR, Dedon PC. In situ analysis of 8-oxo-7,8-dihydro-2′-deoxyguanosine oxidation reveals sequence- and agent-specific damage spectra. J Am Chem Soc. 2012;134:18053–18064. doi: 10.1021/ja307525h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fleming AM, Muller JG, Dlouhy AC, Burrows CJ. Context effects in the oxidation of 8-oxo-7,8-dihydro-2′-deoxyguanosine to hydantoin products: Electrostatics, base stacking, and base pairing. J Am Chem Soc. 2012;134:15091–15102. doi: 10.1021/ja306077b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ye Y, Muller JG, Burrows CJ. Synthesis and charactorization of the oxidized dGTP lesions spiroiminodihydantoin-2′-deoxynucleoside-5′-triphosphate and guanidinohydantoin-2′-deoxynucleoside-5′-triphosphate. J Org Chem. 2006;71:2181–2184. doi: 10.1021/jo052484t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Niles JC, Wishnok JS, Tannenbaum SR. Spiroiminodihydantoin and guanidinohydantoin are the dominant products of 8-oxoguanosine oxidation at low fluxes of peroxynitrite: Mechanistic studies with 18O. Chem Res Toxicol. 2004;17:1510–1519. doi: 10.1021/tx0400048. [DOI] [PubMed] [Google Scholar]
- 76.Crean C, Geacintov NE, Shafirovich V. Oxidation of guanine and 8-oxo-7,8-dihydroguanine by carbonate radical anions: Insight from oxygen-18 labeling experiments. Angew Chem, Int Ed. 2005;44:5057–5060. doi: 10.1002/anie.200500991. [DOI] [PubMed] [Google Scholar]
- 77.White B, Smyth MR, Stuart JD, Rusling JF. Oscillating formation of 8-oxoguanine during DNA oxidation. J Am Chem Soc. 2003;125:6604–6605. doi: 10.1021/ja0343252. [DOI] [PubMed] [Google Scholar]
- 78.White B, Tarun MC, Gathergood N, Rusling JF, Smyth MR. Oxidised guanidinohydantoin (Ghox) and spiroiminodihydantoin (Sp) are major products of iron- and copper-mediated 8-oxo-7,8-dihydroguanine and 8-oxo-7,8-dihydro-2′-deoxyguanosine oxidation. Mol Biosyst. 2005;1:373–381. doi: 10.1039/b511756a. [DOI] [PubMed] [Google Scholar]
- 79.Cui L, Ye W, Prestwich EG, Wishnok JS, Taghizadeh K, Dedon PC, Tannenbaum SR. Comparative analysis of four oxidized guanine lesions from reactions of DNA with peroxynitrite, singlet oxygen, and gamma-radiation. Chem Res Toxicol. 2013;26:195–202. doi: 10.1021/tx300294d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lapi A, Pratviel G, Meunier B. Guanine oxidation in double-stranded DNA by MnTMPyP/KHSO5: at least three independent reaction pathways. Met-Based Drugs. 2001;8:47–56. doi: 10.1155/MBD.2001.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ye W, Sangaiah R, Degen DE, Gold A, Jayaraj K, Koshlap KM, Boysen G, Williams J, Tomer KB, Mocanu V, Dicheva N, Parker CE, Schaaper RM, Ball LM. Iminohydantoin lesion induced in DNA by peracids and other epoxidizing oxidants. J Am Chem Soc. 2009;131:6114–6123. doi: 10.1021/ja8090752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Culp SJ, Cho BP, Kadlubar FF, Evans FE. Structural and conformational analyses of 8-hydroxy-2′-deoxyguanosine. Chem Res Toxicol. 1989;2:416–422. doi: 10.1021/tx00012a010. [DOI] [PubMed] [Google Scholar]
- 83.Taghizadeh K, McFaline JL, Pang B, Sullivan M, Dong M, Plummer E, Dedon PC. Quantification of DNA damage products resulting from deamination, oxidation and reaction with products of lipid peroxidation by liquid chromatography isotope dilution tandem mass spectrometry. Nat Protocols. 2008;3:1287–1298. doi: 10.1038/nprot.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen X, Fleming AM, Muller JG, Burrows CJ. Endonuclease and exonuclease activities on oligodeoxynucleotides containing spiroiminodihydantoin depend on the sequence context and the lesion stereochemistry. New J Chem. 2013;37:3440–3449. doi: 10.1039/C3NJ00418J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yu H, Venkatarangan L, Wishnok JS, Tannenbaum SR. Quantitation of four guanine oxidation products from reaction of DNA with varying doses of peroxynitrite. Chem Res Toxicol. 2005;18:1849–1857. doi: 10.1021/tx050146h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ravanat JL, Turesky RJ, Gremaud E, Trudel LJ, Stadler RH. Determination of 8-oxoguanine in DNA by gas chromatography--mass spectrometry and HPLC--electrochemical detection: overestimation of the background level of the oxidized base by the gas chromatography--mass spectrometry assay. Chem Res Toxicol. 1995;8:1039–1045. doi: 10.1021/tx00050a007. [DOI] [PubMed] [Google Scholar]
- 87.Collins A. Measurement of DNA oxidation in human cells by chromatographic and enzymic methods. Free Radical Biol Med. 2003;34:1089–1099. doi: 10.1016/s0891-5849(03)00041-8. [DOI] [PubMed] [Google Scholar]
- 88.Ravanat JL, Guicherd P, Tuce Z, Cadet J. Simultaneous determination of five oxidative DNA lesions in human urine. Chem Res Toxicol. 1999;12:802–808. doi: 10.1021/tx980194k. [DOI] [PubMed] [Google Scholar]
- 89.Cadet J, Douki T, Ravanat J-L, Wagner JR. Measurement of oxidatively generated base damage to nucleic acids in cells: facts and artifacts. Bioanal Rev. 2012;4:55–74. [Google Scholar]
- 90.Cadet J, Douki T, Ravanat JL. Artifacts associated with the measurement of oxidized DNA bases. Environ Health Perspect. 1997;105:1034–1039. doi: 10.1289/ehp.105-1470384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pouget JP, Frelon S, Ravanat JL, Testard I, Odin F, Cadet J. Formation of modified DNA bases in cells iexposed either to gamma radiation or to high-LET particles. Radiat Res. 2002;157:589–595. doi: 10.1667/0033-7587(2002)157[0589:fomdbi]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 92.Frelon S, Douki T, Ravanat J-L, Pouget J-P, Tornabene C, Cadet J. High-performance liquid chromatography-tandem mass spectrometry measurement of radiation-induced base damage to isolated and cellular DNA. Chem Res Toxicol. 2000;13:1002–1010. doi: 10.1021/tx000085h. [DOI] [PubMed] [Google Scholar]
- 93.Frelon S, Douki T, Favier A, Cadet J. Hydroxyl radical is not the main reactive species involved in the degradation of DNA bases by copper in the presence of hydrogen peroxide. Chem Res Toxicol. 2003;16:191–197. doi: 10.1021/tx025650q. [DOI] [PubMed] [Google Scholar]
- 94.O’Neill P. Pulse radiolytic study of the interaction of thiols and ascorbate with OH adducts of dGMP and dG: implications for DNA repair processes. Radiat Res. 1983;96:198–210. [PubMed] [Google Scholar]
- 95.Patrzyc HB, Dawidzik JB, Budzinski EE, Freund HG, Wilton JH, Box HC. Covalently linked tandem lesions in DNA. Radiat Res. 2012;178:538–542. doi: 10.1667/RR2915.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hong H, Cao H, Wang Y. Identification and quantification of a guanine-thymine intrastrand cross-link lesion induced by Cu(II)/H2O2/ascorbate. Chem Res Toxicol. 2006;19:614–621. doi: 10.1021/tx060025x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fleming AM, Burrows CJ. G-Quadruplex folds of the human telomere sequence alter the site reactivity and reaction pathway of guanine oxidation compared to duplex DNA. Chem Res Toxicol. 2013;26:593–607. doi: 10.1021/tx400028y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Terzidis MA, Ferreri C, Chatgilialoglu C. Radiation-induced formation of purine lesions in single and double stranded DNA: revised quantification. Front Chem. 2015;3 doi: 10.3389/fchem.2015.00018. doi.org/10.3389/fchem.2015.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Margolin Y, Shafirovich V, Geacintov NE, DeMott MS, Dedon PC. DNA sequence context as a determinant of the quantity and chemistry of guanine oxidation produced by hydroxyl radicals and one-electron oxidants. J Biol Chem. 2008;283:35569–35578. doi: 10.1074/jbc.M806809200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Saito I, Takayama M, Sugiyama H, Nakatani K, Tsuchida A, Yamamoto M. Photoinduced DNA cleavage via electron transfer: demonstration that guanine residues located 5′ to guanine are the most electron-donating sites. J Am Chem Soc. 1995;117:6406–6407. [Google Scholar]
- 101.Genereux JC, Barton JK. Mechanisms for DNA charge transport. Chem Rev. 2010;110:1642–1662. doi: 10.1021/cr900228f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schuster GB. Long-range charge transfer in DNA: Transient structural distortions control the distance dependence. Acc Chem Res. 2000;33:253–260. doi: 10.1021/ar980059z. [DOI] [PubMed] [Google Scholar]
- 103.Steenken S. Purine bases, nucleosides, and nucleotides: aqueous solution redox chemistry and transformation reactions of their radical cations and e− and OH adducts. Chem Rev. 1989;89:503–520. [Google Scholar]
- 104.Liang Q, Dedon PC. Cu(II)/H2O2-induced DNA damage is enhanced by packaging of DNA as a nucleosome. Chem Res Toxicol. 2001;14:416–422. doi: 10.1021/tx0002278. [DOI] [PubMed] [Google Scholar]
- 105.Liu Y, Liu Z, Geacintov NE, Shafirovich V. Proton-coupled hole hopping in nucleosomal and free DNA initiated by site-specific hole injection. Phys Chem Chem Phys. 2012;14:7400–7410. doi: 10.1039/c2cp40759k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rokhlenko Y, Geacintov NE, Shafirovich V. Lifetimes and reaction pathways of guanine radical cations and neutral guanine radicals in an oligonucleotide in aqueous solutions. J Am Chem Soc. 2012;134:4955–4962. doi: 10.1021/ja212186w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Candeias LP, Steenken S. Structure and acid-base properties of one-electron-oxidized deoxyguanosine, guanosine, and 1-methylguanosine. J Am Chem Soc. 1989;111:1094–1099. [Google Scholar]
- 108.Misiaszek R, Crean C, Joffe A, Geacintov NE, Shafirovich V. Oxidative DNA Damage Associated with Combination of Guanine and Superoxide Radicals and Repair Mechanisms via Radical Trapping. J Biol Chem. 2004;279:32106–32115. doi: 10.1074/jbc.M313904200. [DOI] [PubMed] [Google Scholar]
- 109.Chryssostomos C, Mila DA, Maurizio G, Panagiotis K, Quinto GM. A reevaluation of the ambident reactivity of the guanine moiety towards hydroxyl radicals. Angew Chem, Int Ed. 2009;48:2214–2217. doi: 10.1002/anie.200805372. [DOI] [PubMed] [Google Scholar]
- 110.Kumar A, Pottiboyina V, Sevilla MD. Hydroxyl radical (OH radical) reaction with guanine in an aqueous environment: A DFT study. J Phys Chem B. 2011;115:15129–15137. doi: 10.1021/jp208841q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Munk BH, Burrows CJ, Schlegel HB. An exploration of mechanisms for the transformation of 8-oxoguanine to guanidinohydantoin and spiroiminodihydantoin by density functional theory. J Am Chem Soc. 2008;130:5245–5256. doi: 10.1021/ja7104448. [DOI] [PubMed] [Google Scholar]
- 112.Thapa B, Munk BH, Burrows CJ, Schlegel HB. Computational Study of the Radical Mediated Mechanism of the Formation of C8, C5 and C4 Guanine:Lysine Adducts in Presence of the Benzophenone Photosensitizer. Chem Res Toxicol. 2016 doi: 10.1021/acs.chemrestox.1026b00057. [DOI] [PubMed] [Google Scholar]
- 113.Fleming AM, Zhou J, Wallace SS, Burrows CJ. A role for the fifth G-track in G-quadruplex forming oncogene promoter sequences during oxidative stress: Do these “spare tires” have an evolved function? ACS Cent Sci. 2015;1:226–233. doi: 10.1021/acscentsci.5b00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Roginskaya M, Moore TJ, Ampadu-Boateng D, Razskazovskiy Y. Efficacy and site specificity of hydrogen abstraction from DNA 2-deoxyribose by carbonate radicals. Free Rad Res. 2015;49:1431–1437. doi: 10.3109/10715762.2015.1081187. [DOI] [PubMed] [Google Scholar]
- 115.Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349:431–434. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 116.Crean C, Uvaydov Y, Geacintov NE, Shafirovich V. Oxidation of single-stranded oligonucleotides by carbonate radical anions: generating intrastrand cross-links between guanine and thymine bases separated by cytosines. Nucleic Acids Res. 2008;36:742–755. doi: 10.1093/nar/gkm1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wolna AH, Fleming AM, Burrows CJ. Single-molecule detection of a guanine(C8)-thymine(N3) cross-link using ion channel recording. J Phys Org Chem. 2014;27:247–251. doi: 10.1002/poc.3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Margolin Y, Cloutier JF, Shafirovich V, Geacintov NE, Dedon PC. Paradoxical hotspots for guanine oxidation by a chemical mediator of inflammation. Nat Chem Biol. 2006;2:365–366. doi: 10.1038/nchembio796. [DOI] [PubMed] [Google Scholar]
- 119.Lim KS, Taghizadeh K, Wishnok JS, Babu IR, Shafirovich V, Geacintov NE, Dedon PC. Sequence-dependent variation in the reactivity of 8-oxo-7,8-dihydro-2′-deoxyguanosine toward oxidation. Chem Res Toxicol. 2012;25:366–373. doi: 10.1021/tx200422g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cadet J, Decarroz C, Wang SY, Midden WR. Mechanisms and products of photosensitized degradation of nucleic acids and related model compounds. Israel J Chem. 1983;23:420–429. [Google Scholar]
- 121.Kang P, Foote CS. Formation of transient intermediates in low-temperature photosensitized oxidation of an 8-(13)C-guanosine derivative. J Am Chem Soc. 2002;124:4865–4873. doi: 10.1021/ja012038x. [DOI] [PubMed] [Google Scholar]
- 122.Dumont E, Gruber R, Bignon E, Morell C, Aranda J, Ravanat JL, Tunon I. Singlet oxygen attack on guanine: Reactivity and structural signature within the B-DNA helix. Chemistry. 2016;22:12358–12362. doi: 10.1002/chem.201601287. [DOI] [PubMed] [Google Scholar]
- 123.Dumont E, Graber R, Bignon E, Morell C, Moreau Y, Monari A, Ravanat J-L. Probing the reactivity of singlet oxygen with purines. Nucleic Acids Res. 2016;44:56–62. doi: 10.1093/nar/gkv1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lu W, Liu J. Capturing transient endoperoxide in the singlet oxygen oxidation of guanine. Chemistry. 2016;22:3127–3138. doi: 10.1002/chem.201504140. [DOI] [PubMed] [Google Scholar]
- 125.Sheu C, Foote CS. Endoperoxide formation in a guanosine derivative. J Am Chem Soc. 1993;115:10446–10447. [Google Scholar]
- 126.Peres PS, Valerio A, Cadena SMSC, Winnischofer SMB, Scalfo AC, Di Mascio P, Martinez GR. Glutathione modifies the oxidation products of 2′-deoxyguanosine by singlet molecular oxygen. Arch Biochem Biophys. 2015;586:33–44. doi: 10.1016/j.abb.2015.09.020. [DOI] [PubMed] [Google Scholar]
- 127.Raoul S, Cadet J. Photosensitized reaction of 8-oxo-7,8-dihydro-2′-deoxyguanosine: Identification of 1-(2-deoxy-β-d-erythro-pentofuranosyl)cyanuric acid as the major singlet oxygen oxidation product. J Am Chem Soc. 1996;118:1892–1898. [Google Scholar]
- 128.McCallum JEB, Kuniyoshi CY, Foote CS. Characterization of 5-hydroxy-8-oxo-7,8-dihydroguanosine in the photosensitized oxidation of 8-oxo-7,8-dihydroguanosine and its rearrangement to spiroiminodihydantoin. J Am Chem Soc. 2004;126:16777–16782. doi: 10.1021/ja030678p. [DOI] [PubMed] [Google Scholar]
- 129.Duarte V, Gasparutto D, Yamaguchi LF, Ravanat J-L, Martinez GR, Medeiros MHG, Di Mascio P, Cadet J. Oxaluric acid as the major product of singlet oxygen-mediated oxidation of 8-oxo-7,8-dihydroguanine in DNA. J Am Chem Soc. 2000;122:12622–12628. [Google Scholar]
- 130.Hickerson RP, Prat F, Muller JG, Foote CS, Burrows CJ. Sequence and stacking dependence of 8-oxoguanine oxidation: Comparison of one-electron vs singlet oxygen mechanisms. J Am Chem Soc. 1999;121:9423–9428. [Google Scholar]
- 131.Martinez GR, Ravanat J-L, Medeiros MHG, Cadet J, Di Mascio P. Synthesis of a naphthalene endoperoxide as a source of 18O-labeled singlet oxygen for mechanistic studies. J Am Chem Soc. 2000;122:10212–10213. [Google Scholar]
- 132.Ravanat J-L, Saint-Pierre C, Di Mascio P, Martinez GR, Medeiros MHG, Cadet J. Damage to isolated DNA mediated by singlet oxygen. Helv Chim Acta. 2001;84:3702–3709. [Google Scholar]
- 133.Cadet J, Ravanat JL, Martinez GR, Medeiros MH, Di Mascio P. Singlet oxygen oxidation of isolated and cellular DNA: product formation and mechanistic insights. Photochem Photobiol. 2006;82:1219–1225. doi: 10.1562/2006-06-09-IR-914. [DOI] [PubMed] [Google Scholar]
- 134.Ravanat J-L, Di Mascio P, Martinez GR, Medeiros MHG, Cadet J. Singlet oxygen induces oxidation of cellular DNA. J Biol Chem. 2000;275:40601–40604. doi: 10.1074/jbc.M006681200. [DOI] [PubMed] [Google Scholar]
- 135.Buchko GW, Cadet J, Berger M, Revanat J-L. Photooxidation of d(TpG) by phthalocyanines riboflavin Isolation and characterization of dinucleoside monophosphates containing the 4R* and 4S* d iastereoisomers of 4,8-di hydro-4-hydroxy-8-oxo-2′-deoxyguanosine. Nucleic Acids Res. 1992;20:4847–4851. doi: 10.1093/nar/20.18.4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Adam W, Arnold MA, Grune M, Nau WM, Pischel U, Saha-Moller CR. Spiroiminodihydantoin Is a major product in the photooxidation of 2′-deoxyguanosine by the triplet states and oxyl radicals generated from hydroxyacetophenone photolysis and dioxetane thermolysis. Org Lett. 2002;4:537–540. doi: 10.1021/ol017138m. [DOI] [PubMed] [Google Scholar]
- 137.Kendrick S, Hurley LH. The role of G-quadruplex/i-motif secondary structures as cis-acting regulatory elements. Pure Appl Chem. 2010;82:1609–1621. doi: 10.1351/PAC-CON-09-09-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Poulsen HE, Specht E, Broedbaek K, Henriksen T, Ellervik C, Mandrup-Poulsen T, Tonnesen M, Nielsen PE, Andersen HU, Weimann A. RNA modifications by oxidation: a novel disease mechanism? Free Radic Biol Med. 2012;52:1353–1361. doi: 10.1016/j.freeradbiomed.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 139.Hickerson RP, Watkins-Sims CD, Burrows CJ, Atkins JF, Gesteland RF, Felden B. A nickel complex cleaves uridine in folded RNA structures: application to E. coli tmRNA and related engineered molecules. J Mol Biol. 1998;279:577–587. doi: 10.1006/jmbi.1998.1813. [DOI] [PubMed] [Google Scholar]
- 140.Fleming AM, Alshykhly O, Zhu J, Muller JG, Burrows CJ. Rates of chemical cleavage of DNA and RNA oligomers containing guanine oxidation products. Chem Res Toxicol. 2015;28:1292–1300. doi: 10.1021/acs.chemrestox.5b00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tomaszewska-Antczak A, Guga P, Nawrot B, Pratviel G. Guanosine in a single stranded region of anticodon stem-loop tRNA models is prone to oxidatively generated damage resulting in dehydroguanidinohydantoin and spiroiminodihydantoin lesions. Chemistry. 2015;21:6381–6385. doi: 10.1002/chem.201406409. [DOI] [PubMed] [Google Scholar]
- 142.Ciftan SA, Theil EC, Holden Thorp H. Oxidation of guanines in the iron-responsive element RNA: similar structures from chemical modification and recent NMR studies. Chem Biol. 1998;5:679–687. doi: 10.1016/s1074-5521(98)90661-5. [DOI] [PubMed] [Google Scholar]
- 143.Igarashi K, Kashiwagi K. Polyamines: mysterious modulators of cellular functions. Biochem Biophys Res Commun. 2000;271:559–564. doi: 10.1006/bbrc.2000.2601. [DOI] [PubMed] [Google Scholar]
- 144.Hosford ME, Muller JG, Burrows CJ. Spermine participates in oxidative damage of guanosine and 8-oxoguanosine leading to deoxyribosylurea formation. J Am Chem Soc. 2004;126:9540–9541. doi: 10.1021/ja047981q. [DOI] [PubMed] [Google Scholar]
- 145.Xu X, Muller JG, Ye Y, Burrows CJ. DNA-protein cross-links between guanine and lysine depend on the mechanism of oxidation for formation of C5 vs C8 guanosine adducts. J Am Chem Soc. 2008;130:703–709. doi: 10.1021/ja077102a. [DOI] [PubMed] [Google Scholar]
- 146.Silerme S, Bobyk L, Taverna-Porro M, Cuier C, Saint-Pierre C, Ravanat JL. DNA-polyamine cross-links generated upon one electron oxidation of DNA. Chem Res Toxicol. 2014;27:1011–1018. doi: 10.1021/tx500063d. [DOI] [PubMed] [Google Scholar]
- 147.Uvaydov Y, Geacintov NE, Shafirovich V. Generation of guanine-amino acid cross-links by a free radical combination mechanism. Phys Chem Chem Phys. 2014;16:11729–11736. doi: 10.1039/c4cp00675e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Solivio MJ, Joy TJ, Sallans L, Merino EJ. Copper generated reactive oxygen leads to formation of lysine-DNA adducts. J Inorg Biochem. 2010;104:1000–1005. [PubMed] [Google Scholar]
- 149.Kurbanyan K, Nguyen KL, To P, Rivas EV, Lueras AMK, Kosinski C, Steryo M, Gonzalez A, Mah DA, Stemp EDA. DNA-protein cross-linking via guanine oxidation: Dependence upon protein and photosensitizer. Biochemistry. 2003;42:10269–10281. doi: 10.1021/bi020713p. [DOI] [PubMed] [Google Scholar]
- 150.Perrier S, Hau J, Gasparutto D, Cadet J, Favier A, Ravanat JL. Characterization of lysine-guanine cross-links upon one-electron oxidation of a guanine-containing oligonucleotide in the presence of a trilysine peptide. J Am Chem Soc. 2006;128:5703–5710. doi: 10.1021/ja057656i. [DOI] [PubMed] [Google Scholar]
- 151.Mangerich A, Knutson CG, Parry NM, Muthupalani S, Ye W, Prestwich E, Cui L, McFaline JL, Mobley M, Ge Z, Taghizadeh K, Wishnok JS, Wogan GN, Fox JG, Tannenbaum SR, Dedon PC. Infection-induced colitis in mice causes dynamic and tissue-specific changes in stress response and DNA damage leading to colon cancer. Proc Natl Acad Sci USA. 2012;109:E1820–E1829. doi: 10.1073/pnas.1207829109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hailer MK, Slade PG, Martin BD, Sugden KD. Nei deficient Escherichia coli are sensitive to chromate and accumulate the oxidized guanine lesion spiroiminodihydantoin. Chem Res Toxicol. 2005;18:1378–1383. doi: 10.1021/tx0501379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Matter B, Malejka-Giganti D, Csallany AS, Tretyakova N. Quantitative analysis of the oxidative DNA lesion, 2,2-diamino-4-(2-deoxy-β-D-erythro-pentofuranosyl)amino]-5(2H)-oxazolone (oxazolone), in vitro and in vivo by isotope dilution-capillary HPLC-ESI-MS/MS. Nucleic Acids Res. 2006;34:5449–5460. doi: 10.1093/nar/gkl596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Alshykhly OR, Fleming AM, Burrows CJ. Guanine oxidation product 5-carboxamido-5-formamido-2-iminohydantoin induces mutations when bypassed by DNA polymerases and is a substrate for base excision repair. Chem Res Toxicol. 2015;28:1861–1871. doi: 10.1021/acs.chemrestox.5b00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.McCulloch SD, Kokoska RJ, Garg P, Burgers PM, Kunkel TA. The efficiency and fidelity of 8-oxo-guanine bypass by DNA polymerases δ and η. Nucleic Acids Res. 2009;37:2830–2840. doi: 10.1093/nar/gkp103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Henderson PT, Delaney JC, Muller JG, Neeley WL, Tannenbaum SR, Burrows CJ, Essigmann JM. The hydantoin lesions formed from oxidation of 7,8-dihydro-8-oxoguanine are potent sources of replication errors in vivo. Biochemistry. 2003;42:9257–9262. doi: 10.1021/bi0347252. [DOI] [PubMed] [Google Scholar]
- 157.Kornyushyna O, Berges AM, Muller JG, Burrows CJ. In vitro nucleotide misinsertion opposite the oxidized guanosine lesions spiroiminodihydantoin and guanidinohydantoin and DNA synthesis past the lesions using Escherichia coli DNA polymerase I (Klenow Fragment) Biochemistry. 2002;41:15304–15314. doi: 10.1021/bi0264925. [DOI] [PubMed] [Google Scholar]
- 158.Kornyushyna O, Burrows CJ. Effect of the oxidized guanosine lesions spiroiminodihydantoin and guanidinohydantoin on proofreading by Escherichia coli DNA polymerase I (Klenow Fragment) in different sequence contexts. Biochemistry. 2003;42:13008–13018. doi: 10.1021/bi0350755. [DOI] [PubMed] [Google Scholar]
- 159.Delaney S, Delaney JC, Essigmann JM. Chemical-biological fingerprinting: probing the properties of DNA lesions formed by peroxynitrite. Chem Res Toxicol. 2007;20:1718–1729. doi: 10.1021/tx700273u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Neeley WL, Delaney S, Alekseyev YO, Jarosz DF, Delaney JC, Walker GC, Essigmann JM. DNA Polymerase V allows bypass of toxic guanine oxidation products in vivo. J Biol Chem. 2007;282:12741–12748. doi: 10.1074/jbc.M700575200. [DOI] [PubMed] [Google Scholar]
- 161.Duarte V, Gasparutto D, Jaquinod M, Cadet J. In vitro DNA synthesis opposite oxazolone and repair of this DNA damage using modified oligonucleotides. Nucleic Acids Res. 2000;28:1555–1563. doi: 10.1093/nar/28.7.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Pfeifer G, Besaratinia A. Mutational spectra of human cancer. Human Genetics. 2009;125:493–506. doi: 10.1007/s00439-009-0657-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nakabeppu Y, Oka S, Sheng Z, Tsuchimoto D, Sakumi K. Programmed cell death triggered by nucleotide pool damage and its prevention by MutT homolog-1 (MTH1) with oxidized purine nucleoside triphosphatase. Mutat Res. 2010;703:51–58. doi: 10.1016/j.mrgentox.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 164.Hanes JW, Thal DM, Johnson KA. Incorporation and replication of 8-oxo-deoxyguanosine by the human mitochondrial DNA polymerase. J Biol Chem. 2006;281:36241–36248. doi: 10.1074/jbc.M607965200. [DOI] [PubMed] [Google Scholar]
- 165.Huang J, Yennie CJ, Delaney S. Klenow fragment discriminates against the incorporation of the hyperoxidized dGTP lesion spiroiminodihydantoin into DNA. Chem Res Toxicol. 2015;28:2325–2333. doi: 10.1021/acs.chemrestox.5b00330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.David SS, Williams SD. Chemistry of glycosylases and endonucleases involved in base-excision repair. Chem Rev. 1998;98:1221–1262. doi: 10.1021/cr980321h. [DOI] [PubMed] [Google Scholar]
- 167.David SS, O’Shea VL, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wallace SS. DNA glycosylases search for and remove oxidized DNA bases. Environ Mol Mutagen. 2013;54:691–704. doi: 10.1002/em.21820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Morland I, Rolseth V, Luna L, Rognes T, Bjoras M, Seeberg E. Human DNA glycosylases of the bacterial Fpg/MutM superfamily: an alternative pathway for the repair of 8-oxoguanine and other oxidation products in DNA. Nucleic Acids Res. 2002;30:4926–4936. doi: 10.1093/nar/gkf618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Bandaru V, Sunkara S, Wallace SS, Bond JP. A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair (Amst) 2002;1:517–529. doi: 10.1016/s1568-7864(02)00036-8. [DOI] [PubMed] [Google Scholar]
- 171.Hazra TK, Izumi T, Boldogh I, Imhoff B, Kow YW, Jaruga P, Dizdaroglu M, Mitra S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc Natl Acad Sci U S A. 2002;99:3523–3528. doi: 10.1073/pnas.062053799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Takao M, Kanno S, Kobayashi K, Zhang QM, Yonei S, van der Horst GT, Yasui A. A back-up glycosylase in Nth1 knock-out mice is a functional Nei (endonuclease VIII) homologue. J Biol Chem. 2002;277:42205–42213. doi: 10.1074/jbc.M206884200. [DOI] [PubMed] [Google Scholar]
- 173.Wallace SS, Bandaru V, Kathe SD, Bond JP. The enigma of endonuclease VIII. DNA Repair (Amst) 2003;2:441–453. doi: 10.1016/s1568-7864(02)00182-9. [DOI] [PubMed] [Google Scholar]
- 174.Krokeide SZ, Laerdahl JK, Salah M, Luna L, Cederkvist FH, Fleming AM, Burrows CJ, Dalhus B, Bjoras M. Human NEIL3 is mainly a monofunctional DNA glycosylase removing spiroimindiohydantoin and guanidinohydantoin. DNA Repair. 2013;12:1159–1164. doi: 10.1016/j.dnarep.2013.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Liu M, Bandaru V, Holmes A, Averill AM, Cannan W, Wallace SS. Expression and purification of active mouse and human NEIL3 proteins. Protein Expr Purif. 2012;84:130–139. doi: 10.1016/j.pep.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Melamede RJ, Hatahet Z, Kow YW, Ide H, Wallace SS. Isolation and characterization of endonuclease VIII from. Escherichia coli Biochemistry. 1994;33:1255–1264. doi: 10.1021/bi00171a028. [DOI] [PubMed] [Google Scholar]
- 177.Chetsanga CJ, Lindahl T. Release of 7-methylguanine residues whose imidazole rings have been opened from damaged DNA by a DNA glycosylase from. Escherichia coli Nucleic Acids Res. 1979;6:3673–3684. doi: 10.1093/nar/6.11.3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Hazra TK, Kow YW, Hatahet Z, Imhoff B, Boldogh I, Mokkapati SK, Mitra S, Izumi T. Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J Biol Chem. 2002;277:30417–30420. doi: 10.1074/jbc.C200355200. [DOI] [PubMed] [Google Scholar]
- 179.Imamura K, Wallace SS, Doublie S. Structural characterization of a viral NEIL1 ortholog unliganded and bound to abasic site-containing DNA. J Biol Chem. 2009;284:26174–26183. doi: 10.1074/jbc.M109.021907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zharkov DO, Golan G, Gilboa R, Fernandes AS, Gerchman SE, Kycia JH, Rieger RA, Grollman AP, Shoham G. Structural analysis of an Escherichia coli endonuclease VIII covalent reaction intermediate. EMBO J. 2002;21:789–800. doi: 10.1093/emboj/21.4.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Doublie S, Bandaru V, Bond JP, Wallace SS. The crystal structure of human endonuclease VIII-like 1 (NEIL1) reveals a zincless finger motif required for glycosylase activity. Proc Natl Acad Sci U S A. 2004;101:10284–10289. doi: 10.1073/pnas.0402051101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zhu C, Lu L, Zhang J, Yue Z, Song J, Zong S, Liu M, Stovicek O, Gao YQ, Yi C. Tautomerization-dependent recognition and excision of oxidation damage in base-excision DNA repair. Proc Natl Acad Sci U S A. 2016;113:7792–7797. doi: 10.1073/pnas.1604591113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Liu M, Imamura K, Averill AM, Wallace SS, Doublie S. Structural characterization of a mouse ortholog of human NEIL3 with a marked preference for single-stranded DNA. Structure. 2013;21:247–256. doi: 10.1016/j.str.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Prakash A, Eckenroth BE, Averill AM, Imamura K, Wallace SS, Doublie S. Structural investigation of a viral ortholog of human NEIL2/3 DNA glycosylases. DNA Repair (Amst) 2013;12:1062–1071. doi: 10.1016/j.dnarep.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Liu M, Doublie S, Wallace SS. Neil3, the final frontier for the DNA glycosylases that recognize oxidative damage. Mutat Res. 2013;743–744:4–11. doi: 10.1016/j.mrfmmm.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Prakash A, Doublie S, Wallace SS. The Fpg/Nei family of DNA glycosylases: substrates, structures, and search for damage. Prog Mol Biol Transl Sci. 2012;110:71–91. doi: 10.1016/B978-0-12-387665-2.00004-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Leipold MD, Muller JG, Burrows CJ, David SS. Removal of hydantoin products of 8-oxoguanine oxidation by the Escherichia coli DNA repair enzyme, FPG. Biochemistry. 2000;39:14984–14992. doi: 10.1021/bi0017982. [DOI] [PubMed] [Google Scholar]
- 188.Krishnamurthy N, Muller JG, Burrows CJ, David SS. Unusual structural features of hydantoin lesions translate into efficient recognition by Escherichia coli Fpg. Biochemistry. 2007;46:9355–9365. doi: 10.1021/bi602459v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Hazra TK, Muller JG, Manuel RC, Burrows CJ, Lloyd RS, Mitra S. Repair of hydantoins, one electron oxidation product of 8-oxoguanine, by DNA glycosylases of Escherichia coli. Nucleic Acids Res. 2001;29:1967–1974. doi: 10.1093/nar/29.9.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hailer MK, Slade PG, Martin BD, Rosenquist TA, Sugden KD. Recognition of the oxidized lesions spiroiminodihydantoin and guanidinohydantoin in DNA by the mammalian base excision repair glycosylases NEIL1 and NEIL2. DNA Repair (Amst) 2005;4:41–50. doi: 10.1016/j.dnarep.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 191.Rosenquist TA, Zaika E, Fernandes AS, Zharkov DO, Miller H, Grollman AP. The novel DNA glycosylase, NEIL1, protects mammalian cells from radiation-mediated cell death. DNA Repair (Amst) 2003;2:581–591. doi: 10.1016/s1568-7864(03)00025-9. [DOI] [PubMed] [Google Scholar]
- 192.Krishnamurthy N, Zhao X, Burrows CJ, David SS. Superior removal of hydantoin lesions relative to other oxidized bases by the human DNA glycosylase hNEIL1. Biochemistry. 2008;47:7137–7146. doi: 10.1021/bi800160s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Vik ES, Alseth I, Forsbring M, Helle IH, Morland I, Luna L, Bjoras M, Dalhus B. Biochemical mapping of human NEIL1 DNA glycosylase and AP lyase activities. DNA Repair (Amst) 2012;11:766–773. doi: 10.1016/j.dnarep.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 194.Katafuchi A, Nakano T, Masaoka A, Terato H, Iwai S, Hanaoka F, Ide H. Differential specificity of human and Escherichia coli endonuclease III and VIII homologues for oxidative base lesions. J Biol Chem. 2004;279:14464–14471. doi: 10.1074/jbc.M400393200. [DOI] [PubMed] [Google Scholar]
- 195.Yeo J, Goodman RA, Schirle NT, David SS, Beal PA. RNA editing changes the lesion specificity for the DNA repair enzyme NEIL1. Proc Natl Acad Sci U S A. 2010;107:20715–20719. doi: 10.1073/pnas.1009231107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Zhao X, Krishnamurthy N, Burrows CJ, David SS. Mutation versus Repair: NEIL1 removal of hydantoin lesions in single-stranded, bulge, bubble, and duplex DNA contexts. Biochemistry. 2010;49:1658–1666. doi: 10.1021/bi901852q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Jia L, Shafirovich V, Geacintov NE, Broyde S. Lesion specificity in the base excision repair enzyme hNeil1: Modeling and dynamics studies. Biochemistry. 2007;46:5305–5314. doi: 10.1021/bi062269m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Licht K, Jantsch MF. Rapid and dynamic transcriptome regulation by RNA editing and RNA modifications. J Cell Biol. 2016;213:15–22. doi: 10.1083/jcb.201511041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Pullirsch D, Jantsch MF. Proteome diversification by adenosine to inosine RNA editing. RNA Biol. 2010;7:205–212. doi: 10.4161/rna.7.2.11286. [DOI] [PubMed] [Google Scholar]
- 200.Dou H, Mitra S, Hazra TK. Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J Biol Chem. 2003;278:49679–49684. doi: 10.1074/jbc.M308658200. [DOI] [PubMed] [Google Scholar]
- 201.Zhou J, Fleming AM, Averill AM, Burrows CJ, Wallace SS. The NEIL glycosylases remove oxidized guanine lesions from telomeric and promoter quadruplex DNA structures. Nucleic Acids Res. 2015;43:4039–4054. doi: 10.1093/nar/gkv252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Zhou J, Liu M, Fleming AM, Burrows CJ, Wallace SS. Neil3 and NEIL1 DNA glycosylases remove oxidative damages from quadruplex DNA and exhibit preferences for lesions in the telomeric sequence context. J Biol Chem. 2013;288:27263–27272. doi: 10.1074/jbc.M113.479055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Agrawal P, Hatzakis E, Guo K, Carver M, Yang D. Solution structure of the major G-quadruplex formed in the human VEGF promoter in K+: insights into loop interactions of the parallel G-quadruplexes. Nucleic Acids Res. 2013;41:10584–10592. doi: 10.1093/nar/gkt784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Liu M, Bandaru V, Bond JP, Jaruga P, Zhao X, Christov PP, Burrows CJ, Rizzo CJ, Dizdaroglu M, Wallace SS. The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc Natl Acad Sci U S A. 2010;107:4925–4930. doi: 10.1073/pnas.0908307107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Semlow DR, Zhang J, Budzowska M, Drohat AC, Walter JC. Replication-dependent unhooking of DNA interstrand cross-links by the NEIL3 glycosylase. Cell. 2016;167:498–511. doi: 10.1016/j.cell.2016.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Yamamoto R, Yamamoto M, Kusaka H, Masatsugu H, Matsuyama S, Tajima T, Ide H, Kubo K. NEIL1 mRNA splicing variants are expressed in normal mouse organs. J Radiat Res. 2012;53:234–241. doi: 10.1269/jrr.11029. [DOI] [PubMed] [Google Scholar]
- 207.Hu J, de Souza-Pinto NC, Haraguchi K, Hogue BA, Jaruga P, Greenberg MM, Dizdaroglu M, Bohr VA. Repair of formamidopyrimidines in DNA involves different glycosylases: role of the OGG1, NTH1, and NEIL1 enzymes. J Biol Chem. 2005;280:40544–40551. doi: 10.1074/jbc.M508772200. [DOI] [PubMed] [Google Scholar]
- 208.Dou H, Theriot CA, Das A, Hegde ML, Matsumoto Y, Boldogh I, Hazra TK, Bhakat KK, Mitra S. Interaction of the human DNA glycosylase NEIL1 with proliferating cell nuclear antigen. The potential for replication-associated repair of oxidized bases in mammalian genomes. J Biol Chem. 2008;283:3130–3140. doi: 10.1074/jbc.M709186200. [DOI] [PubMed] [Google Scholar]
- 209.Theriot CA, Hegde ML, Hazra TK, Mitra S. RPA physically interacts with the human DNA glycosylase NEIL1 to regulate excision of oxidative DNA base damage in primer-template structures. DNA Repair (Amst) 2010;9:643–652. doi: 10.1016/j.dnarep.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Hegde ML, Theriot CA, Das A, Hegde PM, Guo Z, Gary RK, Hazra TK, Shen B, Mitra S. Physical and functional interaction between human oxidized base-specific DNA glycosylase NEIL1 and flap endonuclease 1. J Biol Chem. 2008;283:27028–27037. doi: 10.1074/jbc.M802712200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Das A, Boldogh I, Lee JW, Harrigan JA, Hegde ML, Piotrowski J, de Souza Pinto N, Ramos W, Greenberg MM, Hazra TK, Mitra S, Bohr VA. The human Werner syndrome protein stimulates repair of oxidative DNA base damage by the DNA glycosylase NEIL1. J Biol Chem. 2007;282:26591–26602. doi: 10.1074/jbc.M703343200. [DOI] [PubMed] [Google Scholar]
- 212.Wiederhold L, Leppard JB, Kedar P, Karimi-Busheri F, Rasouli-Nia A, Weinfeld M, Tomkinson AE, Izumi T, Prasad R, Wilson SH, Mitra S, Hazra TK. AP endonuclease-independent DNA base excision repair in human cells. Mol Cell. 2004;15:209–220. doi: 10.1016/j.molcel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 213.Hegde PM, Dutta A, Sengupta S, Mitra J, Adhikari S, Tomkinson AE, Li GM, Boldogh I, Hazra TK, Mitra S, Hegde ML. The C-terminal domain (CTD) of human DNA glycosylase NEIL1 is required for forming BERosome repair complex with DNA replication proteins at the replicating genome: DOMINANT NEGATIVE FUNCTION OF THE CTD. J Biol Chem. 2015;290:20919–20933. doi: 10.1074/jbc.M115.642918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Hegde ML, Hegde PM, Bellot LJ, Mandal SM, Hazra TK, Li GM, Boldogh I, Tomkinson AE, Mitra S. Prereplicative repair of oxidized bases in the human genome is mediated by NEIL1 DNA glycosylase together with replication proteins. Proc Natl Acad Sci U S A. 2013;110:E3090–E3099. doi: 10.1073/pnas.1304231110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Mohaghegh P, Karow JK, Brosh RM, Jr, Bohr VA, Hickson ID. The Bloom’s and Werner’s syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res. 2001;29:2843–2849. doi: 10.1093/nar/29.13.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Tang W, Robles AI, Beyer RP, Gray LT, Nguyen GH, Oshima J, Maizels N, Harris CC, Monnat RJ., Jr The Werner syndrome RECQ helicase targets G4 DNA in human cells to modulate transcription. Hum Mol Genet. 2016 doi: 10.1093/hmg/ddw1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Vallur AC, Maizels N. Distinct activities of exonuclease 1 and flap endonuclease 1 at telomeric G4 DNA. PLoS ONE. 2009;5:e8908. doi: 10.1371/journal.pone.0008908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Ghosh A, Rossi ML, Aulds J, Croteau D, Bohr VA. Telomeric D-loops containing 8-oxo-2′-deoxyguanosine are preferred substrates for Werner and Bloom syndrome helicases and are bound by POT1. J Biol Chem. 2009;284:31074–31084. doi: 10.1074/jbc.M109.027532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Guan X, Bai H, Shi G, Theriot CA, Hazra TK, Mitra S, Lu AL. The human checkpoint sensor Rad9-Rad1-Hus1 interacts with and stimulates NEIL1 glycosylase. Nucleic Acids Res. 2007;35:2463–2472. doi: 10.1093/nar/gkm075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Popuri V, Croteau DL, Bohr VA. Substrate specific stimulation of NEIL1 by WRN but not the other human RecQ helicases. DNA Repair (Amst) 2010;9:636–642. doi: 10.1016/j.dnarep.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Alemasova EE, Moor NA, Naumenko KN, Kutuzov MM, Sukhanova MV, Pestryakov PE, Lavrik OI. Y-box-binding protein 1 as a non-canonical factor of base excision repair. Biochim Biophys Acta. 2016;1864:1631–1640. doi: 10.1016/j.bbapap.2016.08.012. [DOI] [PubMed] [Google Scholar]
- 222.Mokkapati SK, Wiederhold L, Hazra TK, Mitra S. Stimulation of DNA glycosylase activity of OGG1 by NEIL1: functional collaboration between two human DNA glycosylases. Biochemistry. 2004;43:11596–11604. doi: 10.1021/bi049097i. [DOI] [PubMed] [Google Scholar]
- 223.Mandal SM, Hegde ML, Chatterjee A, Hegde PM, Szczesny B, Banerjee D, Boldogh I, Gao R, Falkenberg M, Gustafsson CM, Sarkar PS, Hazra TK. Role of human DNA glycosylase Nei-like 2 (NEIL2) and single strand break repair protein polynucleotide kinase 3′-phosphatase in maintenance of mitochondrial genome. J Biol Chem. 2012;287:2819–2829. doi: 10.1074/jbc.M111.272179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Banerjee D, Mandal SM, Das A, Hegde ML, Das S, Bhakat KK, Boldogh I, Sarkar PS, Mitra S, Hazra TK. Preferential repair of oxidized base damage in the transcribed genes of mammalian cells. J Biol Chem. 2011;286:6006–6016. doi: 10.1074/jbc.M110.198796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Das S, Chattopadhyay R, Bhakat KK, Boldogh I, Kohno K, Prasad R, Wilson SH, Hazra TK. Stimulation of NEIL2-mediated oxidized base excision repair via YB-1 interaction during oxidative stress. J Biol Chem. 2007;282:28474–28484. doi: 10.1074/jbc.M704672200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Torisu K, Tsuchimoto D, Ohnishi Y, Nakabeppu Y. Hematopoietic tissue-specific expression of mouse Neil3 for endonuclease VIII-like protein. J Biochem Biophys Methods. 2005;138:763–772. doi: 10.1093/jb/mvi168. [DOI] [PubMed] [Google Scholar]
- 227.Hildrestrand GA, Neurauter CG, Diep DB, Castellanos CG, Krauss S, Bjoras M, Luna L. Expression patterns of Neil3 during embryonic brain development and neoplasia. BMC Neurosci. 2009;10:45. doi: 10.1186/1471-2202-10-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Zhou H, Xu M, Huang Q, Gates AT, Zhang XD, Castle JC, Stec E, Ferrer M, Strulovici B, Hazuda DJ, Espeseth AS. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe. 2008;4:495–504. doi: 10.1016/j.chom.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 229.Sejersted Y, Hildrestrand GA, Kunke D, Rolseth V, Krokeide SZ, Neurauter CG, Suganthan R, Atneosen-Asegg M, Fleming AM, Saugstad OD, Burrows CJ, Luna L, Bjoras M. Endonuclease VIII-like 3 (Neil3) DNA glycosylase promotes neurogenesis induced by hypoxia-ischemia. Proc Natl Acad Sci USA. 2011;108:18802–18807. doi: 10.1073/pnas.1106880108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Skarpengland T, Holm S, Scheffler K, Gregersen I, Dahl TB, Suganthan R, Segers FM, Ostlie I, Otten JJ, Luna L, Ketelhuth DF, Lundberg AM, Neurauter CG, Hildrestrand G, Skjelland M, Bjorndal B, Svardal AM, Iversen PO, Hedin U, Nygard S, Olstad OK, Krohg-Sorensen K, Slupphaug G, Eide L, Kusnierczyk A, Folkersen L, Ueland T, Berge RK, Hansson GK, Biessen EA, Halvorsen B, Bjoras M, Aukrust P. Neil3-dependent base excision repair regulates lipid metabolism and prevents atherosclerosis in Apoe-deficient mice. Sci Rep. 2016;6:28337. doi: 10.1038/srep28337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Reis A, Hermanson O. The DNA glycosylases OGG1 and NEIL3 influence differentiation potential, proliferation, and senescence-associated signs in neural stem cells. Biochem Biophys Res Commun. 2012;423:621–626. doi: 10.1016/j.bbrc.2012.04.125. [DOI] [PubMed] [Google Scholar]
- 232.Neurauter CG, Luna L, Bjoras M. Release from quiescence stimulates the expression of human NEIL3 under the control of the Ras dependent ERK-MAP kinase pathway. DNA Repair (Amst) 2012;11:401–409. doi: 10.1016/j.dnarep.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 233.Biffi G, Tannahill D, McCafferty J, Balasubramanian S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat Chem. 2013;5:182–186. doi: 10.1038/nchem.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Amrane S, Kerkour A, Bedrat A, Vialet B, Andreola ML, Mergny JL. Topology of a DNA G-quadruplex structure formed in the HIV-1 promoter: a potential target for anti-HIV drug development. J Am Chem Soc. 2014;136:5249–5252. doi: 10.1021/ja501500c. [DOI] [PubMed] [Google Scholar]
- 235.Fong YW, Cattoglio C, Tjian R. The intertwined roles of transcription and repair proteins. Mol Cell. 2013;52:291–302. doi: 10.1016/j.molcel.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]