

Abstract
Twist transcription factors, members of the basic helix-loop-helix family, play crucial roles in mesoderm development in all animals. Humans have two paralogous genes, TWIST1 and TWIST2, and mutations in each gene have been identified in specific craniofacial disorders. Here, we describe a new clinical entity, Sweeney-Cox syndrome, associated with distinct de novo amino acid substitutions (p.Glu117Val and p.Glu117Gly) at a highly conserved glutamic acid residue located in the basic DNA binding domain of TWIST1, in two subjects with frontonasal dysplasia and additional malformations. Although about one hundred different TWIST1 mutations have been reported in patients with the dominant haploinsufficiency Saethre-Chotzen syndrome (typically associated with craniosynostosis), substitutions uniquely affecting the Glu117 codon were not observed previously. Recently, subjects with Barber-Say and Ablepharon-Macrostomia syndromes were found to harbor heterozygous missense substitutions in the paralogous glutamic acid residue in TWIST2 (p.Glu75Ala, p.Glu75Gln and p.Glu75Lys). To study systematically the effects of these substitutions in individual cells of the developing mesoderm, we engineered all five disease-associated alleles into the equivalent Glu29 residue encoded by hlh-8, the single Twist homolog present in Caenorhabditis elegans. This allelic series revealed that different substitutions exhibit graded severity, in terms of both gene expression and cellular phenotype, which we incorporate into a model explaining the various human disease phenotypes. The genetic analysis favors a predominantly dominant-negative mechanism for the action of amino acid substitutions at this highly conserved glutamic acid residue and illustrates the value of systematic mutagenesis of C. elegans for focused investigation of human disease processes.
Introduction
Twist transcription factors, which are tissue-specific members of the basic helix-loop-helix (bHLH) family, play important roles in development and are highly conserved across metazoans (1). A single homolog exists in Drosophila (twist) and in Caenorhabditis elegans (hlh-8), whereas in humans and other mammals there are two paralogous genes, TWIST1 and TWIST2 (2–4). Mutations in both paralogs have been described in humans, causing distinct diseases. In the case of TWIST2, biallelic loss-of-function mutations lead to Setleis syndrome or Type III focal facial dermal dysplasia [MIM: 227260] (5). In contrast, specific heterozygous point mutations, mostly involving a single amino acid, were recently described in two different syndromes that are distinct from Setleis syndrome. Patients with Ablepharon-Macrostomia syndrome (AMS [MIM: 200110]) and Barber-Say syndrome (BSS [MIM: 209885]) have missense substitutions in a conserved glutamic acid in the basic DNA binding domain of TWIST2; p.Glu75Lys substitutions are associated with AMS and p.Glu75Ala or p.Glu75Gln are associated with BSS (6). These distinct syndromes are characterized by unique phenotypes (e.g. sparse hair and absent or reduced eyelids in AMS and excess hair and bulbous nose in BSS) while also sharing overlapping features (e.g. large mouth and small ears) (7). The mutant proteins are proposed to act dominantly by altering the degree of DNA binding as well as the identity of the genomic sites bound by the mutant proteins compared with wild-type (WT), as measured by ChIP-seq (6).
Studies in C. elegans are relevant to interpreting these human genetic findings, because the mutation exactly equivalent to the TWIST2 p.Glu75Lys (AMS) was previously analyzed in detail in this species (8). Null mutations of hlh-8 lead to egg-laying defective (Egl) and constipated (Con) animals, owing to lack of proper differentiation of the muscles required to lay embryos and eliminate waste (9). Null hlh-8 mutations are recessive, since heterozygous animals are indistinguishable from WT. In hlh-8, the residue in the basic DNA-binding domain equivalent to TWIST2-Glu75 is Glu29. hlh-8 mutants homozygous for the Glu29Lys substitution are also Egl and Con, and Glu29Lys heterozygotes have Egl defects but are not Con (8). Further, Glu29Lys homozygotes have a more severe phenotype (earlier defect in development) compared with null mutants, and Glu29Lys heterozygotes, similar to null homozygotes, do not express the target gene, egl-15, which is an FGF receptor (FGFR) homolog. Since the mutant HLH-8 Glu29Lys could still bind DNA in vitro, it was proposed to represent a dominant-negative protein that interfered with some aspects of WT HLH-8 function (8).
Dominant mutations of the paralogous gene TWIST1 are also associated with a human disorder, Saethre-Chotzen syndrome (SCS [MIM: 101400]). SCS is characterized by premature fusion of cranial sutures (craniosynostosis; usually involving the coronal sutures), with additional cranial features including eyelid ptosis, blocked naso-lacrimal ducts, deviated nasal septum and small auricles with prominent helical crus, and minor limb anomalies comprising cutaneous syndactyly of the digits and broad or bifid halluces (10). Approximately 100 different heterozygous TWIST1 mutations have been described in individuals affected with SCS, including whole gene deletions, nonsense or frameshift mutations distributed throughout the open reading frame, and missense substitutions, which are almost entirely localized to the bHLH region (10–13). To date, all TWIST1 mutations are proposed to cause loss-of-function in the encoded protein, leading to haploinsufficiency with a range of phenotypic severity that does not appear to correlate with the specific genotype (10).
We describe here two individuals who have specific heterozygous missense substitutions (p.Glu117Val and p.Glu117Gly) located at the glutamic acid residue in the basic DNA binding domain of TWIST1, exactly equivalent to that altered in TWIST2 of AMS and BSS patients. In contrast to the general lack of genotype–phenotype correlation for TWIST1 mutations seen previously, the individuals with mutations at TWIST1-Glu117 exhibit a radically different craniofacial phenotype compared with SCS, for which we propose the name Sweeney-Cox syndrome (SwCoS; referring to the clinical geneticists who first identified these individuals). Analogous to the contrasting phenotypes associated with TWIST2 mutations in AMS and BSS compared with Setleis syndrome, the difference in phenotype associated with these specific SwCoS substitutions suggests that they exert their influence through a dominant-negative mechanism rather than the haploinsufficiency observed in SCS.
To dissect further these genotype–phenotype correlations in a model system, we engineered both TWIST1 and all three TWIST2 disease-associated glutamic acid substitutions into the DNA binding domain of the HLH-8 protein. By exploiting the well-characterized phenotypes of C. elegans hlh-8 mutants and the ability to examine development in individual cells, we achieved an extensive analysis of the resulting phenotypes. The various hlh-8 mutants have distinct phenotypes and can be placed in an allelic series of increasing severity.
Results
Recognition of a novel clinical entity, SwCoS
We initially identified Subject 1, a boy with a striking pattern of facial dysmorphism affecting the eyes, nose, mouth, ears and hairline (Fig. 1A and B). This was associated with additional congenital abnormalities involving the hands (long fingers with relatively short distal phalanges held in fixed flexion and 2/3/4 syndactyly bilaterally), bilateral talipes equinovarus, bilateral undescended testes and imperforate anus. Currently aged 6 years, he has a moderate learning disability and speech delay with a bilateral 60 dB conductive hearing loss attributed to narrow external ear canals. A full case report is provided in the Materials and Methods section.
Figure 1.

Craniofacial appearance of individuals with p.Glu117 substitutions in TWIST1. (A–C) Subject 1 with de novo TWIST1 c.350A > T variant, leading to the substitution p.Glu117Val. In (A, B) (aged 4 months), note the hypertelorism, prominent forehead, upper eyelid colobomas, deficient bony orbits with pseudoproptosis, hypoplastic alae nasi, short columella and philtrum and small low-set dysplastic cupped ears. (C) CT of skull aged 4 months. Note severely hypoplastic frontal bones separated by patent metopic suture (arrowhead), massive enlargement of anterior fontanelle (bounded by arrows), patent coronal and sagittal sutures, hypertelorism and hypoplasia of zygomatic bones, mandible and maxilla. (D–F) Subject 2 with a de novo TWIST1 c.350A > G variant, leading to the substitution p.Glu117Gly (D aged 6 days, E and F aged 15 weeks, postoperative tarsorraphies). Note that this individual is more severely affected but with a similar combination of hair configuration in widow’s peak, hypertelorism, upper eyelid colobomas, very broad nasal bridge with a broad and flattened nasal tip and a low columella and small crumpled ears.
Further investigations of Subject 1 included a computed tomographic scan of the head at the age of 4 months, which revealed multiple abnormalities (Fig. 1C). Cranial and spinal magnetic resonance imaging (MRI) scans, chromosomal microarray and TCOF1 testing yielded normal findings. In an attempt to obtain a diagnosis, the clinical features were presented at a national Dysmorphology meeting (London, 2013). A match was proposed with Subject 2 based on several overlapping phenotypic features, including the hypertelorism, bilateral upper eyelid colobomas, configuration of nose and mouth and crumpled ears (Fig. 1D–F, see Materials and Methods for case report on Subject 2). Given the recognizable pattern of features and the identification of a shared pathogenic basis described in this study, we propose to name the new disorder present in Subjects 1 and 2 Sweeney-Cox syndrome (SwCoS), based on the surnames of the clinicians who recognized the common features present in their patients.
Mutations of the TWIST1 Glu117 codon in SwCoS
In view of the sporadic presentation in Subject 1, we performed parent/child trio-based exome sequencing as part of a wider diagnostic study of craniosynostosis (14). We hypothesized that the disease could have arisen either by de novo mutation or by inheritance of rare recessive alleles; functional consequences of non-synonymous substitutions were scored according to a six-point scale (15). Among rare variants potentially associated with altered function, there were two de novo mutations (in TWIST1 and IL12RB1), two biallelic variants in autosomal genes (in PLCB4 and DNAH1) and one hemizygous (X-linked) variant (CFAP47) (Supplementary Material, Table S1). Given the previously described association with craniofacial phenotypes, we initially considered the variants in TWIST1 (SCS) and PLCB4 (auriculocondylar syndrome) most likely to be causative. However, as neither of the predicted amino acid substitutions in PLCB4 localized to the two documented disease hotspot regions in auriculocondylar syndrome (16,17), we focused our analysis on TWIST1.
To investigate a possible role for the TWIST1 variant (c.350A > T; p.Glu117Val) in pathogenesis, we first used dideoxy-sequencing to confirm the presence of the heterozygous mutation in Subject 1 and absence from both his parents (Fig. 2A). The predicted substitution localizes to the DNA-binding region of the TWIST1 protein; although multiple amino acid substitutions of this region (including the adjacent residues Arg116 and Arg118) were already reported in patients with SCS (Fig. 2B), no substitution at the Glu117 residue has been described previously. Following the identification of Subject 2 as a potential phenotypic match to Subject 1, we performed dideoxy-sequencing of the TWIST1 gene in Subject 2 and found, remarkably, a different heterozygous substitution (c.350A > G; p.Glu117Gly) at the identical nucleotide to Subject 1 (Fig. 2A). Sequencing of parental TWIST1 genes of Subject 2 was normal (not shown), indicating that this mutation had also arisen de novo. Given the radically different phenotype present in Subjects 1 and 2 (including absence of any documented craniosynostosis) compared with SCS, we hypothesized that the mechanism by which substitutions at TWIST1–Glu117 lead to disease must be different and highly specific, compared with previously described TWIST1 mutations. Notably, the Glu117 residue in TWIST1 is equivalent to Glu75 in TWIST2, in which specific substitutions were previously described in AMS and BSS (see Introduction); however, different amino acid substitutions were encountered in the two genes [to Val and Gly in TWIST1 compared with Lys (AMS), Ala and Gln (BSS) in TWIST2, see Fig. 2B]. To investigate the comparative genetics and phenotypic consequences of these different substitutions, we generated an allelic series at the corresponding Glu29 in C. elegans hlh-8.
Figure 2.
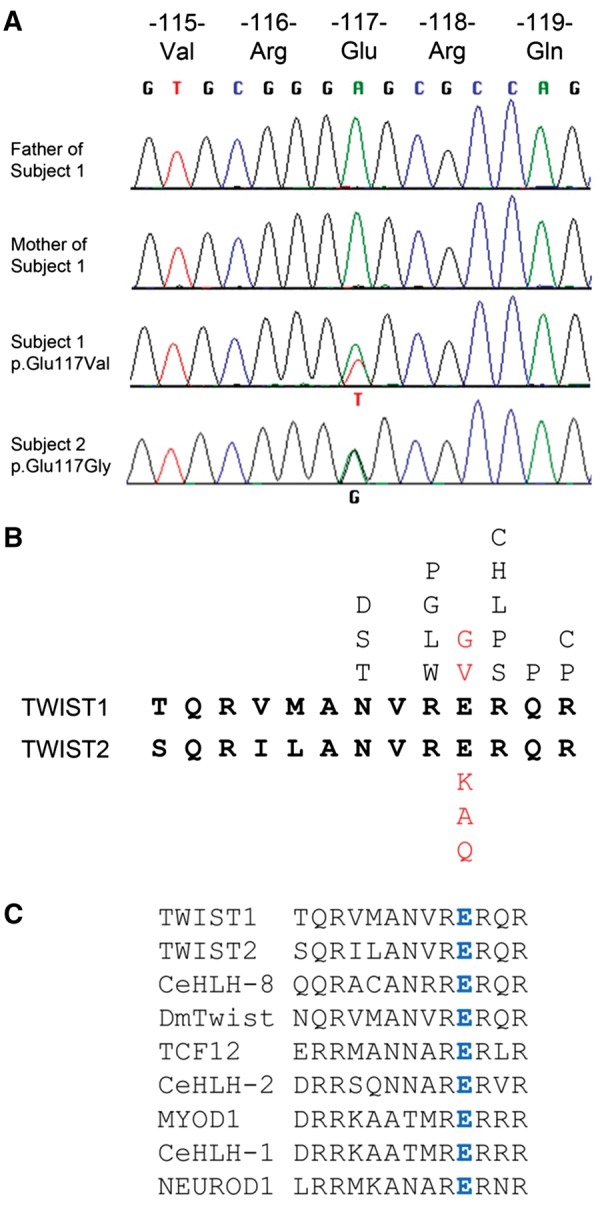
Identification of TWIST1 mutations altering the Glu117 codon. (A) Dideoxy-sequencing traces of TWIST1 in Subject 1 and unaffected parents (above) and Subject 2 (below). The heterozygous substitutions A > T and A > G are indicated in the two probands. (B) Amino acid sequences (bold black lettering) of basic DNA binding domains of human TWIST1 and TWIST2. Above the TWIST1 sequence are shown the positions of amino acid substitutions observed in SCS (unbolded black lettering; public data available from Human Gene Mutation Database, plus an unpublished c.347G > C (p.Arg116Pro) mutation identified by the Oxford Genetics Laboratories) and SwCoS (red lettering). Below the TWIST2 sequence are shown the positions of amino acid substitutions observed in AMS and BSS (red lettering). (C) Amino acid sequence alignment of basic domains from various bHLH proteins; the glutamic acid (blue in all the sequences) is invariant throughout various bHLH human homologs as well as in D. melanogaster (Dm) Twist and C. elegans (Ce) bHLH proteins.
Modeling Twist-related disease mutations in C. elegans
We used CRISPR/Cas9 genome-editing technology to substitute the conserved glutamic acid residue in the basic region encoded by hlh-8 (Supplementary Material, Fig. S1) (18). As previously noted, this residue is invariant between human and C. elegans TWIST homologs, as well as in most other members of the bHLH family (Fig. 2C). We constructed individual strains that contained the disease-associated alleles found in SwCoS (Glu29Val and Glu29Gly), BSS (Glu29Ala and Glu29Gln) and AMS (Glu29Lys) patients, which we collectively referred to as Glu29† mutants. As a control, we generated a 4 bp deletion (fs2728) before Glu29 (deleting all of codon 27 and the first nucleotide of codon 28, which causes a frameshift mutation), essentially representing a null allele of hlh-8. The phenotypes of the fs2728 animals, as reported below, were the same as those of a previously analyzed null mutant with a large deletion in the hlh-8 locus (9). We also constructed a Glu29Asp substitution to ask whether conserving the charge of this residue would be phenotypically silent. Finally, for comparison, we made hlh-8 mutations modeling three SCS alleles that have amino acid substitutions in the conserved Arg116 of human TWIST1 (Fig. 2B, C, Arg28Leu, Arg28Gly, Arg28Trp) (19–21), referred to collectively as Arg28† mutants. We analyzed a variety of phenotypes in these mutant animals (summarized in Fig. 3; Figs 4–6, Supplementary Material, Figs S2–S4).
Figure 3.

Phenotypes of C. elegans hlh-8 CRISPR/Cas9 alleles. (A) Table indicates all of the phenotypes examined for the disease-associated Glu29† and Arg28† alleles and the related control strains. In experiments for which the data in the subsequent figures are summarized (starting with the row ‘Early M lineage’ and all subsequent rows), the + and – marks indicate the percent of the population with the WT phenotype [e.g. for the Early M lineage—two cells in the dorsal/ventral planes or robust expression of the reporter genes (with bright or medium GFP signal): +++, 80–100%; ++, 60–80%; +, 40–60%; +/–, 20–40%; –, 0–20%]; nd, not determined. (B–G) Micrographs representing the major phenotypes in the mutant animals (E–G) compared with WT (B–D). (B, E) The central region of adult hermaphrodites showing the egg-laying defective (Egl) phenotype with two representative embryos (white arrowheads) that are in an early stage of embryogenesis in WT (B) compared with embryos that are just prior to hatching in Egl mutants (E). WT hermaphrodites already would have laid these later stage embryos. (C, F) The central region of L4 larvae showing the constipated (Con) phenotype with the intestinal lumen (white arrow) that is expanded in the constipated mutant (F) compared with the narrow lumen in the WT animal (C). (D, G) The posterior region of adult hermaphrodites showing the deformed anal region (Dar) phenotype with the anal opening (open arrow) surrounded by a smooth, tapered tail in WT (D) and a wide, deformed tail in the mutant (G). In (A), n values for Egl, Con, and development time are all >30 animals per genotype. Subsequent n values and data can be found in the following figures: Average Brood Size (Supplementary Material, Fig. S3), Early M lineage (Fig. 4A), Late M lineage (two sex myoblasts) (Fig. 4B), Late M lineage (SM descendants) (Fig. 4C), arg-1 VM expression (Fig. 5G), arg-1 Ent expression (Fig. 5G), egl-15 expression (Fig. 5J), E29†/+ nonRet (Fig. 6A) Note that the nonRet phenotype in heterozygotes (E29†/+) was analyzed in a egl-15::gfp sensitized background., E29†/+ arg-1 VMs (Fig. 6C), E29†/+ arg-1 Ents (Supplementary Material, Fig. S4), E29†/+ egl-15 (Fig. 6B). Ent refers to the enteric muscles collectively: intestinal muscles (Ints), anal sphincter (Sph) and anal depressor (Dep) (see Supplementary Material, Figs S4, S5).
Figure 4.

hlh-8 homozygous Glu29† mutant animals display distinct defects in the M (postembryonic mesodermal) lineage. (A) Animals were scored for the first division of the M cell in L1 larvae—in WT animals, one daughter cell is on the dorsal side and one is on the ventral side of the animal (D/V). Some mutant animals have both cells on the dorsal (D) or ventral (V) side. (B) L3 larvae were scored for the number of sex myoblasts (SMs), the range being from 1 to 6 or more SMs (6+). (C) L4 larvae at the same stage of vulval development were scored for the presence of SM descendants (SMd). When the SMs divide, the resulting cells are reduced in size such that they are approximately a quarter of the size of WT SM cells. Animals that have six or more of these small cells (indicative of at least three SM divisions) are considered to have SMd (blue bars). Animals that still contained the large blast cells at this stage that would be either undivided or underdivided are also indicated (red bars). Some animals exhibit both phenotypes so the two categories are >100% (e.g. fs2728); an animal with four SMs may have many smaller SMd cells and still have a few large undivided SMs. Here and in subsequent figures, the n values of animals examined with each genotype are indicated in parentheses. Error bars on each graph are standard error of the proportion.
Figure 5.

hlh-8 homozygous Glu29† mutant animals disrupt target gene expression. GFP reporters [arg-1::gfp (A–F) and egl-15::gfp (H, I)] were crossed into the Glu29† mutants and the expression patterns were scored for each cell type: head mesodermal cell (HMC), vulval muscles (VMs), intestinal muscles (Ints), anal sphincter (Sph) and anal depressor (Dep or mu anal). For the egl-15::gfp expression, animals were categorized as having bright green (I), dim green or no green VMs (see images in Fig. 6). The percentage of the animals in the population with each expression pattern is graphed (G, J). A–F and H–I are modified from (46). White arrow in (F) indicates the anal opening. Error bars on each graph are standard error of the proportion.
Figure 6.

hlh-8 Glu29†/+ heterozygotes retain embryos and are distinctly dominant for target gene expression in the vulval muscles. (A) Heterozygous animals were scored for the number of pharyngeal GFP-expressing embryos in the uterus indicating the hermaphrodite is retaining embryos. Since the pharynx develops during a stage of embryogenesis after embryos are typically laid on plates and no longer present in the uterus, hermaphrodites with green embryos in utero are retaining embryos (Ret phenotype). (B, C) Heterozygous animals were examined for vulval muscle expression of egl-15::gfp (B) and arg-1::gfp (C). Animals were scored as having bright (very bright gfp expression, comparable to the pharyngeal gfp expression in the heterozygous animals), medium (clearly defined vulval muscles but not overly bright), dim (can discern outline of the vulval muscles but can barely see the expression) or no GFP expression. (Images of representative egl-15::gfp expression taken with the same exposure time (50 ms) for each category are shown beneath (B and C). Error bars on each graph are standard error of the proportion.
All Glu29† homozygotes have Egl defective phenotypes
We found that all Glu29† mutants yielded animals that, when homozygous, were highly penetrant for an Egl phenotype, similar to that observed for previously identified hlh-8 alleles (8,9). Quantification of the population of animals with the Egl phenotype is a good metric of how defective the vulval muscles are in these hlh-8 mutants. In the case of three mutants (Glu29Gly, Glu29Ala and Glu29Gln), 2–3% of animals are capable of laying a few embryos during their first day of adulthood before their embryos hatch internally (referred to as a ‘bag-of-worms’ or ‘Bag’ phenotype; compare Fig. 3B and E). However, all animals bag by the second day, indicating that none of the mutant animals make fully functional egg-laying muscles. The Arg28† homozygotes differed from the Glu29† and fs2728 null animals in that they were less consistently Egl (99% of the population was Egl for Arg28Trp, 70% for Arg28Gly and 0% for Arg28Leu; Fig. 3A). In the remaining population for each Arg28† genotype that could lay embryos, the hermaphrodites had an embryo retention phenotype (Ret) in which late stage embryos built up in the uterus, but were eventually laid, indicating that these mutants possess partially functional egg-laying muscles and have a weaker phenotype than the Glu29† mutants.
The Glu29† homozygotes display distinct defects in defecation
HLH-8 plays a critical role in the differentiation of the non-striated enteric muscles (the anal depressor, anal sphincter and intestinal muscles) that control defecation; these muscles develop during embryogenesis (Supplementary Material, Fig. S5). Similar to previously studied hlh-8 alleles (8,9), many of the hlh-8 CRISPR/Cas9 mutants, in the homozygous state, were Con indicating a defect in proper functioning of these enteric muscles. Quantification of the Con phenotype is a good metric of how defective the enteric muscles are in these hlh-8 mutants. This phenotype is easily observed because of the expanded intestinal lumen in animals of all ages, but especially in larvae before the production of embryos (compare Fig. 3C and F). This phenotype was highly penetrant in the fs2728, Glu29Asp, Glu29Val and Glu29Lys animals (Fig. 3A). The Con phenotype was more variable in Glu29Ala and Glu29Gly mutants with 19–25% of the population showing the phenotype, and only mildly penetrant in the Glu29Gln strains, in which <5% of the animals were Con (Fig. 3A). We examined the well-characterized and reproducible muscle contractions of the defecation cycle (22) in animals of each Glu29† genotype and found that fs2728, Glu29Asp, Glu29Val and Glu29Lys animals were extremely defective for the final step of defecation that is controlled by the enteric muscles (expulsion). Even among the Glu29Gly, Glu29Ala and Glu29Gln mutants, animals that did not appear visibly Con did not expel waste at the WT rate (Supplementary Material, Fig. S2). Therefore, all of the mutants had some defecation defect but some were much more severe than others. We also observed a deformed hermaphrodite tail in animals exhibiting the Con phenotype (Fig. 3G) associated with all the amino acid substitutions as well as the fs2728 null mutants, similar to the deformed anal region (Dar) phenotype (23) previously described for nematodes bearing bacterial infections. This Dar phenotype appears to be recessive in all the Glu29† mutants. In contrast, none of the Arg28† homozygous mutants were visibly Con or Dar (Fig. 3A), indicating a lack of defects in enteric muscle development.
The Glu29† homozygotes display distinct defects in development time and brood size
Another measure of the severity of the various Glu29† mutants is development time (Fig. 3A). Other Con mutants have been observed to develop more slowly (22). The most severe hlh-8 alleles have a life cycle extended by two days while the alleles that are only weakly Con have a normal life cycle of three days at 20 °C (Fig. 3A). In addition, all the mutants had a smaller brood size compared with WT animals, with mutants in which a larger percentage of the population were Con (e.g. fs2728, Glu29Asp, Glu29Val and Glu29Lys) having a lower average brood size than animals with a smaller fraction of Con animals (Glu29Gly, Glu29Ala, Glu29Gln and the Arg28† mutants) (Fig. 3A, Supplementary Material, Fig. S3). Overall, the differences in average brood size likely indicate how well animals can lay embryos (Egl and Ret phenotypes) and how much space they have in their uterus (the expanded intestinal lumen of Con animals restricts the size of the uterus). Thus, the most severe mutants (as measured by the percent of the population that is Con) display the smallest broods.
Characterization of phenotypes of the postembryonic mesodermal lineage
To determine whether there were differences in the cellular phenotypes leading to the Egl phenotype of the Glu29† homozygotes, we examined the lineage from which the egg-laying muscles are derived (Supplementary Material, Fig. S5). In C. elegans, a cell termed the M mesoblast is born in the embryo and divides throughout larval development to form all the non-gonadal postembryonic mesoderm (24); HLH-8 plays a critical role at several points in the M lineage (9). To follow M lineage development, we used a reporter gene containing the hlh-8 promoter region fused to GFP (hlh-8::gfp) (3). All the Glu29† variants were either generated in, or crossed into, the hlh-8::gfp transgenic background. To determine whether these hlh-8 variants were perturbing the M lineage and the patterning of its descendants, mutants expressing this transgene were examined throughout larval development (Fig. 4). These assays provide separate metrics for the effects of the mutant alleles in a lineage where HLH-8 plays several distinct roles—regulating cell division planes, the development of the correct number of blast cells, and cell proliferation prior to differentiation into vulval muscles.
The first division of the M mesoblast in L1 larvae, in which HLH-8 plays a critical role, occurs in the dorsal–ventral (D–V) plane (Fig. 4A). In WT animals, this first division of M is D–V 100% of the time whereas hlh-8 mutants often have much lower percentages. For the fs2728 mutants, the D–V division plane only occurred ∼50% of the time, whereas in the Glu29Asp, Glu29Val, Glu29Gly, Glu29Ala and Glu29Gln mutants, the M mesoblast divided along the proper plane >60% of the time. The Glu29Lys mutant displayed the most severe phenotype with the M mesoblast dividing in the proper plane only ∼30% of the time (Figs 3A, 4A).
HLH-8 also regulates the number of sex myoblasts (SMs) that arise from the mesoblast lineage. In WT animals, normally two SMs are born in the posterior and migrate to the center of the animal where they proliferate and differentiate into eight vulval and eight uterine muscles (24). We examined the number of SMs generated from the M lineage in the mutants. All the Glu29† homozygotes displayed a variable number of SMs, with Glu29Gly and Glu29Gln having two SMs in ∼60%, Glu29Val and Glu29Ala in ∼50%, and fs2728, Glu29Asp and Glu29Lys in ∼30% or less of the population (Figs 3A, 4B). For the rest of each mutant population, the number of SMs varied from one to six (Fig. 4B).
Animals homozygous for the Glu29Lys mutation had previously been shown to manifest arrested development of the M lineage at an earlier stage, with under-divided or undivided SMs (an SM proliferation defect), compared with hlh-8 null alleles in which the SMs proliferated but did not differentiate, which would be a later phenotype in SM development (8). Both Glu29Lys and null mutants are Egl since they lack functioning egg-laying muscles, yet their underlying cellular defects are distinct. We consider the SM proliferation defect indicative that the mutant Glu29Lys protein may be interfering with the SM divisions and wanted to examine whether the Glu29† disease alleles exhibited this phenotype. We examined animals at the same stage of development (based on the maturity of the developing vulva in L4 stage animals) and determined whether the SMs had proliferated [as evidenced by the small sex myoblast descendant cells (SMd) versus the larger SMs] (Fig. 4C). As compared with WT animals that had evidence of proliferation (SMd) in >90% of the animals, the null fs2728 allele had SMd in >80% of the animals. All of the Glu29† mutants had <60% of the animals exhibit SMd, with the Glu29Lys allele being the most defective with only a few percent of the animals showing SMd (Figs 3A, 4C). Despite the observation that the Glu29† homozygotes had one to six SMs, it was clear which SMs had divided and which had not by the size of the cell. Not all the SMs behaved the same within a given animal; some may divide to generate SMd cells and others may under-divide or not divide at all. Of the five disease-related alleles, Glu29Lys was most defective for the phenotype of lack of SM divisions and the other four disease alleles were not nearly as defective in proliferation.
HLH-8 target gene expression is variably affected
To further distinguish the phenotypes at the molecular level, all homozygous Glu29† mutants were crossed into arg-1::gfp and egl-15::gfp transgenic strains driving a green fluorescent protein (GFP) reporter. ARG-1 is a Delta-related transmembrane signal ligand of the Notch receptor and is expressed in the vulval muscles, the enteric muscles, and a head mesodermal cell (HMC) (25). egl-15 encodes an FGF receptor and null mutations result in Egl animals because the SMs fail to migrate properly to the vulval region (26). Both these genes are direct HLH-8 targets (3,27), so measuring their expression provides a good metric of how defective these hlh-8 alleles are for activating target gene expression in vivo. We scored the expression of these two transgenes in homozygotes (Figs 3A, 5). For arg-1::gfp, the fs2728, Glu29Asp, Glu29Val and Glu29Lys mutants did not express GFP in any cells whereas Glu29Gly, Glu29Ala, Glu29Gln expressed arg-1::gfp in the HMC and enteric muscles and a few Glu29Gly animals expressed it in the vulval muscles (Figs 3A, 5A–G). In contrast, nearly all of the homozygous animals, except for a few Glu29Gly and Glu29Gln animals, failed to express egl-15::gfp in the vulval muscles (or the precursor cells) compared with WT animals (Figs 3A, 5H–J). Overall, these phenotypes indicated that Glu29Gly, Glu29Ala and Glu29Gln were the weakest alleles for transcriptional activation as they retained the ability to activate tissue-specific transcription of arg-1 (only in the HMC and enteric muscles) and to a much lesser extent Glu29Gly and Glu29Gln were able to activate expression of egl-15.
The Glu29† HLH-8 mutants act semi-dominantly
Previous studies of TWIST2 mutations causing AMS and BSS concluded that these variants might be inducing disease owing to either a dominant gain-of-function or dominant-negative activity (6). To determine whether the hlh-8 alleles were behaving dominantly, heterozygous Glu29†/+ animals were generated and assayed for the Egl and Con phenotypes and tissue-specific expression of GFP target gene reporters. The Con phenotypes appear to be recessive traits, as heterozygous animals of all mutant genotypes were not visibly Con (data not shown). Similarly the heterozygous animals were also all able to lay embryos and thus were not Egl (data not shown). To evaluate whether there were any subtle defects in the vulval muscles leading to a slower rate of egg laying, heterozygous hermaphrodites were assayed for their retention of embryos inside the uterus (Ret phenotype). Egl animals fail to lay any embryos and the progeny hatch internally and devour the mother to escape. Ret animals retain embryos, become bloated, but do lay embryos onto the growth medium. We found that the Glu29Lys/+ heterozygotes were visibly Ret but not as severe as homozygotes; their embryos do not hatch internally. Many of the other Glu29†/+ heterozygotes were difficult to score for embryo retention, whereas fs2728/+ heterozygotes did not retain embryos (data not shown).
To further test the possibility that these Glu29† mutants might have some semi-dominant character, we tested them in a sensitized background where the phenotype might be more pronounced. Glu29†/+ heterozygotes in an egl-15::gfp background displayed a more pronounced embryo retention phenotype than in a background without a transgene, suggesting some defects in vulval muscle function (Fig. 6A). We interpret the enhanced embryo retention defect as resulting from the multiple egl-15 promoter copies that are present within the integrated egl-15::gfp transgene titrating out functional HLH-8 in the heterozygotes, to reveal the defect in the mutants. Notably, the fs2728 animals did not exhibit this defect and behaved as a recessive allele with a WT phenotype (nonRet) in this assay (Figs 3A, 6A) making it likely that the phenotype is not strictly due to the reduced dose of HLH-8 available. The results from this experiment are consistent with the Glu29† mutants acting as dominant-negative alleles.
To further address the dominant nature of these hlh-8 alleles, we examined GFP expression in heterozygous animals crossed with reporter lines. Glu29†/+; arg-1::gfp and Glu29†/+; egl-15::gfp animals displayed reduced expression for these markers in the vulval muscles (Fig. 6B, C). In examining egl-15::gfp expression in heterozygotes, the fs2728/+ animals resembled the WT population, confirming the recessive nature of this allele. In contrast the Glu29†/+ animals exhibited a range of defects with Glu29Lys/+ the most defective with nearly 100% of the population not expressing the reporter at all; Glu29Gln/+ and Glu29Ala/+ animals were also quite defective with 60% of the population barely expressing the GFP (Figs 3A, 6B). Reduced expression of arg-1::gfp was less dramatic when comparing the Glu29†/+ and fs2728/+ heterozygotes (Figs 3A, 6C). The only mutant that appeared to have major expression defects was Glu29Gln/+. For both reporters, the Glu29Val/+ animals were closest to WT in their expression patterns, when comparing different Glu29† mutants (Figs 3A, 6B, C). All of the heterozygous Glu29†/+ animals expressed arg-1::gfp similar to WT in the enteric muscles (Fig. 3A, Supplementary Material, Fig. S4). The results from these two target genes in heterozygous mutants indicate that Glu29Gln, Glu29Ala and Glu29Lys are the most likely to represent dominant-negative proteins that interfere with the ability of WT HLH-8 to promote transcription of egl-15.
Discussion
We describe a new clinical disorder, SwCoS, which results from heterozygous amino acid substitutions in the TWIST1 DNA binding domain (p.Glu117Val or p.Glu117Gly). The phenotype of SwCoS differs radically from previously described TWIST1 mutations associated with SCS and craniosynostosis, indicating a distinct pathophysiological mechanism for SwCoS. The striking facial dysostosis in both subjects, with hypertelorism, deficiency of the eyelids and facial bones, cleft palate/velopharyngeal insufficiency, and low set, cupped ears (Fig. 1) is reminiscent of multiple ribosome- and spliceosomopathies such as Treacher-Collins syndrome type 1 ([MIM: 154500]; mutation in TCOF1) and acrofacial dysostosis, Cincinnati type ([MIM: 616462]; mutation in POLR1A). In both these latter cases, analysis of mouse or zebrafish models suggests that tissue deficiency arises through p53-dependent apoptosis of the neural crest cell progenitor population (28–31). Further, Twist1 was shown to be essential in mice for survival of the cranial neural tube partly through directing forebrain neural crest fate (32). SCS phenotypes do not include gross deficiencies in neural crest development, and the mutations associated with SCS are haploinsufficient. These observations suggest a possible dominant-negative mechanism for SwCoS pathogenesis, in which the effective dose of WT TWIST1 would be below the level required for normal maintenance of neural crest, which was still largely unaffected in SCS (Fig. 7).
Figure 7.

Clinical phenotypes associated with TWIST1 and TWIST2 disease alleles. The clinical manifestations of altered TWIST1 (left) and TWIST2 (right) function fall on a continuum of decreasing total protein activity. In all cases, except for the mouse knockout (TWIST1) and Setleis syndrome (TWIST2), phenotypes are associated with heterozygous mutations. Since SCS alleles behave dominantly (due to haploinsufficiency), missense alleles such as Arg116† are grouped with deletion alleles at ∼50% protein activity. For TWIST2 heterozygotes, 50% protein activity yields a normal phenotype. Relative protein activities for substitutions at Glu117 (TWIST1) or Glu75 (TWIST2) are based on the experimental observations in heterozygous worms presented in Figure 6 and summarized in Figure 3. See Discussion for further explanation.
To investigate comprehensively the molecular mechanism of SwCoS alleles and those previously described in BSS and AMS at the equivalent position in TWIST2 (6), we generated an allelic series of six different Glu29† amino acid substitutions in the DNA-binding domain of the sole C. elegans Twist homolog, hlh-8. We also generated three Arg28† alleles at the adjacent codon based on reported SCS-associated mutations. All alleles were generated in an isogenic background to reduce other sources of phenotypic variability. Three broad conclusions are possible from examining the homozygous Glu29† mutants. First, the phenotypic consequences of Glu29† alleles differ in their severity, showing the most variability in enteric muscle phenotypes; however, the Glu29† mutants consistently exhibited more severe phenotypes than the neighboring Arg28† mutants (Fig. 3A). To further distinguish the most severe Arg28† mutant (Arg28Trp) from the Glu29† mutants, we determined that Arg28Trp animals could express egl-15::gfp (data not shown) indicating that these mutants are less severe than all of the Glu29† mutants. These observations correlate with the unique clinical features present in Subjects 1 and 2, and reinforce that TWIST1-Glu117 mutations are causative of SwCoS. Second, these Glu29† alleles show quantitative differences in the magnitude of their homozygous loss-of-function effects. For example, whereas some alleles (Glu29Val, Glu29Lys) were similar to the null in that they did not drive expression of the HLH-8 target gene arg-1 in any of the assayed tissues, for other alleles (Glu29Gly, Glu29Ala and Glu29Gln), expression of arg-1 was observed in a proportion of animals (74–100% in intestinal muscles and 3–37% in anal depressor muscles; Figs 3A, 5G), indicating that these alleles retain residual function. Third, the details of these quantitative differences vary, depending on the phenotypic readout. While all were penetrant for an Egl phenotype, only some (Glu29Val and Glu29Lys) were fully penetrant in affecting the enteric muscles to reveal a Con phenotype. Hence the mutants behave genetically as separation-of-function alleles: all the Glu29† variants perturbed egg-laying capabilities, presumably due to defects in vulval muscle function, yet the Glu29Gly, Glu29Ala and Glu29Gln variants were only mildly Con.
The analysis of homozygous worms demonstrated the pathogenicity of every Glu29† allele; however, homozygous Glu29Val worms consistently exhibited a more severe phenotype than Glu29Gly worms (Fig. 3A), whereas Subject 2 with the Glu117Gly substitution has a more severe phenotype than Subject 1 with Glu117Val (Fig. 1). Considering the clinical situation in which human mutations manifest in the heterozygous state, we assayed several phenotypes and markers in C. elegans to ask whether hlh-8 mutations behaved dominantly and more closely mimicked the clinical situation when heterozygous. The fs2728 mutant served as the null control, and in all assays, fs2728/+ heterozygotes behaved similar to WT animals (Figs 3A, 6), suggesting that this allele is recessive (similar to human TWIST2), and not haploinsufficient (as occurs in human TWIST1). In contrast, in embryo retention and target gene expression assays, the Glu29Lys/+ (‘AMS’) heterozygotes behaved in a semi-dominant manner, as originally reported by Corsi et al. (8). The other Glu29†/+ heterozygotes also behaved semi-dominantly in these assays (except in the case of hermaphrodite Con phenotypes), although not as strongly as the Glu29Lys/+ heterozygotes. Importantly, however, the relative magnitude of effects of different alleles in the heterozygous and homozygous states was not always consistent: notably, Glu29Val, which appeared more strongly loss-of-function in the homozygous state than Glu29Gly, Glu29Ala or Glu29Gln (with respect to the Con phenotype and arg-1 expression; Figs 3A, 5G, Supplementary Material, Fig. S2), deviated the least from WT in arg-1 and egl-15 expression in the heterozygous state (Figs 3A, 6B, C).
Comparing these observations to the human genetic situation suggests that the heterozygous C. elegans phenotypes provide a more accurate reflection of relative allele strength in the patients, compared with the homozygotes. Noting that phenotypes of individuals who are heterozygous for TWIST1 or TWIST2 mutations fall on a continuum (Fig. 7), with null mutations leading to milder phenotypes (SCS or Setleis syndrome carriers, respectively) and mutations at Glu117 (TWIST1) or Glu75 (TWIST2) leading to more severe phenotypes (SwCoS or AMS/BSS, respectively), we speculate that the more severe C. elegans alleles in the heterozygous state (Glu29Lys, Glu29Ala and Glu29Gln) may be lethal as heterozygous substitutions at TWIST1-Glu117 in humans (red stipples, Fig. 7 left). Conversely TWIST2-Glu75Gly and Glu75Val might be milder or subclinical, explaining why they have not been observed in AMS and BSS (green stipples, Fig. 7 right). However, the number of human mutations on which this model is based is currently few, so further clinical observations will be required to confirm or refute this model. An additional factor likely to affect the relative pattern of substitutions in human TWIST1 and TWIST2 is the different sequence context of the respective Glu117 (GAG) codon. In TWIST2 but not TWIST1, this is immediately preceded by a C nucleotide to generate a hypermutable CpG dinucleotide (33), predicting the preferential mutation of GAG to AAG (Lys), which is indeed the most frequently observed mutation in TWIST2 (6).
In summary, we conclude that the pathophysiological actions of this allelic series in C. elegans work in more than one dimension, and that in the heterozygous state, a dominant-negative (antimorphic) action is more consistent with our observations than a gain-of-function (neomorphic) mechanism. This conclusion mirrors that based on analysis of the clinical phenotype presented earlier. However, one discrepant observation on the Glu29Lys allele, as noted previously (8), is that it is almost 100% penetrant for lack of SM cell divisions to generate the 16 SM descendants (SMd, Fig. 4C). The neomorphic nature of the E29K mutant suggests that it may be interfering with, or poisoning, another bHLH partner required for SM divisions with which it normally does not interact. This observation suggests that besides the dominant negative nature of most of these hlh-8 alleles, some may be acting as neomorphic alleles and interfering with genetic pathways normally not associated with HLH-8 regulation. The neomorphic versus antimorphic nature of these alleles can be tested in the future by determining whether or not over-expression of WT HLH-8 suppresses the observed phenotypes.
The highly conserved glutamic acid residue that is the focus of this work plays a critical role in the bHLH transcription factor’s ability to bind DNA, as it is responsible for the sequence-specific contacts of adjacent bases (CA) that constitute the symmetrical E box binding motif CANNTG (34). Given this crucial role, it is reasonable to expect that DNA binding would simply be disrupted by any amino acid substitution at this position. In fact, due to the strong conservation of this Glu residue throughout evolution (Fig. 2C), one might expect it could not readily be substituted to other amino acids and retain WT function at all. Indeed, mutating the orthologous glutamic acid in the human bHLH protein MAX to any other amino acid caused a drop in affinity to near-baseline levels of target gene binding, leading to the conclusion that Glu10 in MAX is absolutely required for sequence-specific binding (34). Moreover, ChIP-seq data previously showed that glutamic acid substitutions associated with AMS and BSS led to a decrease in affinity and sequence-specificity in TWIST2 DNA-binding (6).
Given these prior observations, we expected to observe a reduction or absence of HLH-8 target gene expression in the homozygous Glu29† mutants. Yet, we saw quite good expression in the enteric muscles with the Glu29Ala, Glu29Gly and Glu29Gln mutants, suggesting some residual DNA-binding activity. As further evidence to support the possibility of residual DNA binding of TWIST-Glu29† substitutions in vivo, we note that the detailed study of MAX did systematically document the capacity of substitutions at the equivalent Glu10 position to bind different permutations of the canonical hexanucleotide motif (34), and that HLH-8 Glu29Lys was previously demonstrated to bind DNA in vitro, both as a homodimer and as a heterodimer with its bHLH partner HLH-2, whereas an adjacent substitution, Arg28Trp, did not bind nearly as well (8). Similarly, a recurrent Glu47Lys substitution at the equivalent position of the human bHLH protein TCF3 (E47), occurring in agammaglobulinemia, was able to maintain DNA binding as a heterodimer with MYOD (35). Overall, we conclude that the Glu29† amino acid substitutions do not sterically prevent interaction with HLH-8 target gene promoters and probably sequester WT protein in non-productive heterodimers comprising either HLH-2/HLH-8-Glu29† or HLH-8/HLH-8-Glu29†. Future work to investigate the mechanistic details could include DNA binding assays allied to measurements of sub-cellular localization and protein levels in each of the affected tissues.
Given the roles that Twist homologs and many of their downstream targets play both in craniofacial development in mammals and in the M mesoblast lineage and patterning in C. elegans, the whole genetic pathway appears to be conserved through a long span of evolutionary time. This conservation suggests that the study of homologous disease-specific alleles in C. elegans is likely to shed light on the mechanism by which the equivalent variants cause disease in humans, since the cellular mechanisms are conserved even if the tissue-specific phenotypes are distinct. The homologous phenotypes or phenologs (36,37) of SCS, SwCoS, AMS, BSS and Setleis syndrome in humans are the Egl and Con phenotypes in C. elegans caused by defects in the vulval and enteric muscles, respectively. Vulval muscles, where HLH-8 heterodimer function seems to predominate (38), are more sensitive to the Glu29† perturbations than enteric muscles where HLH-8 homodimers play an important role (38). The observation that all Glu29† homozygous variants in this study were associated with an Egl phenotype suggests that HLH-8 heterodimer function is more compromised than the homodimer function. The stronger Glu29† variants are associated with the Con phenotype, which is likely due to defects in enteric muscle function, where HLH-8 homodimer function may also be compromised for those alleles.
Altogether, our genome-editing studies in C. elegans show that mutations homologous to human disease mutations can easily be generated, provided that the disease variant is in a conserved residue. The C. elegans system may prove useful for the analysis of variants of unknown significance; the system is sensitive enough to detect functional perturbations of hlh-8 variants. Looking to the future, the ability to generate homologous mutations in C. elegans allows us to examine these phenotypes in greater detail. Here, we performed morphological, cellular, and molecular phenotyping to determine the semi-dominant nature of some of the hlh-8 variants. To further understand the mechanisms by which a particular variant perturbs the egg-laying and defecation system of C. elegans, we can use genetic suppressor screens to find intragenic and extragenic mutations that restore function to these processes. We anticipate that such screens will identify genes that influence the DNA affinity of these variants or the heterodimer/homodimer balance in these animals. Additionally, for example, by screening for target gene expression (e.g. using a single copy genome-edited egl-15 promoter driving gfp expression), we may uncover cis-acting promoter mutations that restore transcriptional activation to the egl-15 promoter in a Glu29† mutant background. Such studies should shed further light on the molecular mechanism by which these variants disrupt development and cause disease.
Materials and Methods
Patient histories
Subject 1, a male, was the third child born at term to healthy, unrelated white European parents after an uneventful pregnancy; the 20-week ultrasound scan was reported as normal. At birth he weighed 3.94 kg and multiple congenital abnormalities were immediately evident. The evolving facial dysmorphism included hypertelorism, a prominent metopic ridge, upper eyelid colobomas, deficient bony orbits with pseudoproptosis, small ocular globes, hypoplastic alae nasi, short columella, short philtrum, high-arched palate, mild micrognathia and small low-set dysplastic cupped ears with overfolded helices and upturned lobes. He had flattening of the occiput, a broad neck and narrow shoulders; additional physical features were long fingers with relatively short distal phalanges held in fixed flexion and 2/3/4 syndactyly bilaterally, bilateral talipes equinovarus, bilateral undescended testes, imperforate anus and generalized hirsutism with low hairline.
Cranial and abdominal ultrasound scans performed in the neonatal period were normal. There are no concerns about his vision and electrodiagnostic tests were normal. Skeletal survey at 4 months of age showed short clavicles, unusual scapulae with convex medial margins, normal phalanges (which in the feet were broad relative to the metatarsals), a wide anterior fontanelle with wide coronal sutures and normal lambdoid sutures. The long bones were normal. CT of the head at 4 months confirmed the skull changes with additional findings of very small frontal bones separated by a patent metopic suture and hypoplastic midface. Cranial and spinal MRI scans, array comparative genomic hybridization (aCGH) and TCOF1 testing were normal.
A primary anoplasty was undertaken in the first week of life. He had a pharyngoplasty for non-cleft velopharyngeal insufficiency aged 2 years and surgery to correct the undescended testes. He has a Statement of Special Educational Needs with moderate learning disability, and moderate speech delay associated with a bilateral 60 dB hearing loss caused by narrow external ear canals, which is managed with a bone conducting hearing aid.
Subject 2 was the second girl born to healthy, unrelated white European parents. The pregnancy was complicated by polyhydramnios; ultrasound scanning revealed edema of the scalp and bridge of the nose but further investigation was declined. Onset of labor was at 37+5 weeks and birthweight was 2.16 kg (2nd centile), length 44 cm (0.3rd to 2nd centile) and head circumference 32.5 cm (10–25th centile). Multiple dysmorphic features were evident at birth. The skull was brachycephalic with a widely open metopic suture extending from the anterior fontanelle to the nasal bridge, associated with a widow’s peak. There was marked hypertelorism, bilateral upper eyelid colobomas with disrupted eyebrows and both globes almost fully exposed, and fullness of the soft tissues in the glabellar region. The nasal bridge was wide and the tip of the nose was broad with a low columella. The mouth was small with a full top lip, a midline cleft palate and choanal atresia. The pinnae were low-set, small, fleshy and crumpled. The thumbs appeared proximally placed, the fingers were held in a flexed position and were tapering, with small nails. Digits 2–5 had mild skin syndactyly in both hands and feet. Patches of hair were present in an abnormal distribution on the back and also below the knees bilaterally, persisting in tufts around the ankles to 6 months of age. Investigations in the neonatal period showed patent ductus arteriosus and patent foramen ovale on echocardiography (both resolved without surgery), absent spleen on ultrasound, and widely patent metopic suture, small cerebellum and facial nerves on cranial MRI. Array CGH and testing of the FREM1 gene were normal.
A tracheostomy was required from birth. Gastroesophageal reflux required fundoplication and a gastrostomy. Despite surgical repair of the eyelids, corneal exposure and scarring resulted in right-sided phthisis, with some vision (able to see large objects) retained on the left.
On further assessment aged 2 years, she remained proportionately small (length 79 cm, 2nd centile; weight 7.98 kg, <0.4th centile). Her hearing was normal. A limited CT scan of the head at the age of 3 years 2 months showed a thickened frontal bone, patency of the basal parts of the coronal and lambdoid sutures, and unilateral fusion of the occipito-mastoid suture on the right. Development has been globally delayed; by 2–3 years of age there was evidence of receptive language skills (responding to verbal requests) and an ability to sing along to familiar tunes. She walked at 4 years.
Identification of TWIST1 mutations
Three micrograms of genomic DNA from Subject 1 and both parents was captured using Sure Select v5 (Agilent) and sequenced on the Illumina HiSeq2000 platform. Exome sequence reads were aligned to the GRCh38 reference genome with Bowtie 2 (39) and artefacts (unmapped sequences, duplicate PCR products and likely pseudogene sequences) removed using custom Perl scripts. Variants were called using SAMtools v0.1.19 (40) and annotated using ANNOVAR (41). Variants based on ≥5 reads were filtered out if the minor allele frequency was >0.003 in either the Exome Variant Server, 1000 Genomes or Exome Aggregation Consortium. Remaining variants were examined for segregation in accordance with the sporadic phenotype observed in Subject 1 (de novo mutation; autosomal or X-linked recessive; Supplementary Material, Table S1). TWIST1 variants in Subjects 1 and 2 were analyzed by dideoxy-sequencing according to established clinical diagnostic protocols; permission to confirm biological relationships in the family of Subject 2 could not be obtained. Clinical and molecular data from Subject 1 were briefly summarized in a previous publication (Family 18 in (14)).
Generation of hlh-8 alleles by CRISPR/Cas9
CRISPR/Cas9 technology was used to generate the seven different alleles of hlh-8 at residue E29 and the three different alleles at residue R28 (18). One to two nucleotide changes were made to the Glu29 codon of hlh-8 to create the desired alleles (Glu29Ala: gaa → gct, Glu29Gln: gaa → caa, Glu29Gly: gaa → ggt, Glu29Val: gaa → gtt, Glu29Lys: gaa → aaa, Glu29Asp: gaa → gat). A single silent nucleotide mutation was also made in the PAM site (cgg → cgt; codon 28) to prevent re-cutting of the target sequence by Cas9 (42). Lastly, silent mutations were made 6 nt downstream of the Glu29 codon to create a BsiWI restriction site (aggacc → cgtacg) that facilitated screening for successful CRISPR edits (Supplementary Material, Fig. S1). A similar strategy was used to change the Arg28 codon (Arg28Gly: cgg → ggc, Arg28Leu: cgg → cta, Arg28Trp: cgg → tgg), which also disrupted the PAM site. Note that the PAM site was not destroyed in the Arg28Trp mutant.
To generate the Glu29† mutants, N2 (WT), and AK55 (hlh-8::gfp ayIs7 IV; arg-1::gfp ccIs4444 II) (3,25) young adult animals were injected with pDD162 (43) containing the hlh-8 sgRNA sequence, which was GAGAGCATGTGCCAACAGAC. To address potential ‘off target’ effects, analysis of this single sgRNA at http://crispr.mit.edu/was performed. It scored 98 of a possible 100 within the hlh-8 gene. There were eleven off-target sites, seven of which were in genes. Their scores ranged from 0.1 to 0.3 and none of the seven genes have been implicated in egg-laying muscle or enteric muscle function according to WormBase (www.wormbase.org) and published studies. The hlh-8 gRNA containing plasmid (50 ng/μL) was injected along with repair oligonucleotides with the sequence GTTCGAAAGAA TGAAGTGGA AAATGTGC AGCAGA GAGCATGTGC CAACA GA CGTNNN CG TC AA C G TACGAA Gg taa a tgttcattcaatt gtttt tg ag agtt tcgggttgtttaatat att ttg (30 ng/μL). The bold letters indicate the BsiWI site. The repair oligonucleotides for the six alleles were identical except for the Glu29 codon changes indicated by the underlined Ns in the sequence above. The italicized ‘T’ prior to the Ns is the silent mutation made to prevent recutting at the PAM site. The capitalized bases indicate exon 1 while the lower case bases represent intron 1 of hlh-8. We first injected animals with a mix of all six repair oligonucleotides totaling 30 ng/μL, then removed appropriate repair oligonucleotides from subsequent injection mixes as we screened and obtained different alleles. Using the co-conversion method, the worms were also co-injected with pDD162 containing a dpy-10 sgRNA sequence and dpy-10 (cn64) repair oligonucleotide (44).
At least ten hermaphrodites were injected with each editing mix. Single injected worms were placed on individual plates and transferred to new plates daily for 2 days. F1 rollers (indicating successful editing of a marker gene—i.e. co-conversion) were singled to new plates and allowed to lay embryos for one day. After laying F2 embryos, the F1 rollers were lysed and analyzed by PCR and restriction digest with BsiWI (Supplementary Material, Fig. S1). For the positive F1 rollers, we were able to obtain F2 worms homozygous for the hlh-8 mutations by picking individuals that had a tail defect phenotype or retained embryos in their uterus. The desired mutations for all of the alleles were confirmed by sequencing (Macrogen USA, Rockville, MD, USA and Eurofins MWG Operon, Huntsville, AL, USA). We were able to obtain the Glu29Lys, Glu29Asp, Glu29Gln and Glu29Gly (in both N2 and AK55 using this strategy). The Glu29Ala and fs2728 alleles were obtained only in AK55 using this plasmid-based method. Each time F1 rollers were screened, 9–29% were positive for the inserted BsiWI restriction site (except for fs2728, in which the restriction site was absent). fs2728 is an allele created as a result of non-homologous end joining in which 4 nt were deleted at the PAM site. Assuming this small deletion does not affect splicing, this 4 nt deletion (Δ79-82nuc) should result in a frameshift such that a truncated 44 amino acid protein is made, of which the last 18 amino acids are unique.
For the creation of the Glu29Ala, Glu29Val, Arg28Gly, Arg28Leu, Arg28Trp and the BsiWI control alleles, we injected N2 animals with purified Cas9 protein (PNA Bio, Thousand Oaks, CA, USA), tracrRNA and hlh-8 crRNA and dpy-10 crRNA (Dharmacon, Lafayette, CO, USA) as in (45) instead of using plasmid-derived Cas9 and guide RNA. However, the process of screening for successful lines was not altered. Using this method, F1 rollers were screened for the inserted BsiWI restriction site, and the desired mutations were confirmed by sequencing. In all but the single fs2728 case, the animals with a BsiWI site also had one of the desired mutations in the Arg28 or Glu29 codon and the silent mutation in the PAM site. All Arg28 and Glu29 mutants had a visible embryo retention (Ret) or Egl phenotype. The distinction between the two phenotypes is that both retain embryos as young adults, but Ret animals do eventually lay their embryos while Egl animals fail to lay any and the embryos hatch internally. The BsiWI control strain, which only contained the silent mutations, had no obvious phenotype and was only identified by PCR and restriction digestions with BsiWI.
Genetics/strains used
All strains were grown at 20 °C unless otherwise noted.
TY4236: him-8(e1489) IV; mIs10 [myo-2p::GFP + pes-10p::GFP + gut-promoter::GFP] V
AG278: hlh-8(av65) Glu29Ala
AG240: hlh-8(av43) Glu29Asp
AG237: hlh-8(av42) Glu29Gly
AG233: hlh-8(av39) Glu29Lys
AG245: hlh-8(av47) Glu29Gln
AG266: hlh-8(av58) Glu29Val
AG255: hlh-8(av53) fs2728
AG290: hlh-8(av70) Arg28Gly
AG285: hlh-8(av69) Arg28Leu
AG279: hlh-8(av66) Arg28Trp
AG315: hlh-8(av81) BsiWI only
AG316: arg-1::gfp (ccIs4444 II); mIs10; him-8(e1489)
AG317: hlh-8::gfp (ayIs7 IV); mIs10
AG318: egl-15::gfp (ayIs2 IV); mIs10
AK55: arg-1::gfp; hlh-8::gfp
AG250: arg-1::gfp; hlh-8::gfp; hlh-8(av49) Glu29Ala
AG253: arg-1::gfp; hlh-8::gfp; hlh-8(av52) Glu29Asp
AG241: arg-1::gfp; hlh-8::gfp; hlh-8(av44) Glu29Gly
AK176: arg-1::gfp; hlh-8::gfp; hlh-8(av39) Glu29Lys
AG249: arg-1::gfp; hlh-8::gfp; hlh-8(av48) Glu29Gln
AK177: arg-1::gfp; hlh-8::gfp; hlh-8(av58) Glu29Val
AG255: arg-1::gfp; hlh-8::gfp; hlh-8(av53) fs2728
AK167: egl-15::gfp
AK170: egl-15::gfp; hlh-8(av65) Glu29Ala
AK171: egl-15::gfp; hlh-8(av43) Glu29Asp
AK169: egl-15::gfp; hlh-8(av42) Glu29Gly
AK172: egl-15::gfp; hlh-8(av39) Glu29Lys
AK173: egl-15::gfp; hlh-8(av47) Glu29Gln
AK168: egl-15::gfp; hlh-8(av58) Glu29Val
AK174: egl-15::gfp; hlh-8(av53) fs2728
To generate Glu29†; egl-15::gfp homozygotes, Glu29† mutants were crossed with egl-15::gfp males. Glu29†/+; egl-15::gfp/+ heterozygotes were picked and egl-15::gfp homozygotes were identified. From these homozygotes, progeny were screened for the homozygous Glu29† mutation. Because many of the Glu29† mutants do not express their target genes well, it was first necessary to confirm that the gfp transgene was homozygous in WT siblings before homozygosing the Glu29† mutation. Strains that did not express the gfp reporter were also backcrossed to WT N2 males to ensure the transgene was present and homozygous.
To construct Glu29†; arg-1::gfp; hlh-8::gfp homozygotes, most Glu29† mutations were generated in the strain AK55 (arg-1::gfp; hlh-8::gfp) by the CRISPR/Cas9 method. Glu29Lys, fs2728 and Glu29Val were crossed into AK55. Because the Glu29Lys and Glu29V mutants do not express their target genes well, it was first necessary to confirm that the gfp transgenes were homozygous before homozygosing the Glu29† mutation.
To assay for semi-dominant embryo retention (Ret) phenotypes in a sensitized background, Glu29†/+ heterozygotes were generated by mating egl-15::gfp; mIs10 males or egl-15::gfp; mIs10; him-8 males with Glu29†; egl-15::gfp homozygotes. L4 hermaphrodite cross progeny were identified based on their green pharynx (mIs10 reporter). These animals were then scored 48 h later by observing the number of green (mIs10) embryos in the uterus of each animal. The pharyngeal gfp reporter is expressed in embryos at a late stage in development when they would have already been laid/expelled by their mother. As a control, egl-15::gfp; mIs10 or egl-15::gfp; mIs10; him-8 males were mated with egl-15::gfp animals and green (pharynx) cross progeny were selected for analysis as above.
To assay for semi-dominant phenotypes, Glu29†/+ heterozygotes were generated by mating mIs10; arg-1::gfp or mIs10; hlh-8::gfp males into Glu29†; arg-1::gfp; hlh-8::gfp animals or mIs10; egl-5::gfp males were crossed into Glu29†; egl-15::gfp hermaphrodites. The resulting F1 cross progeny were identified by their green pharynx. The Glu29†/+ animals were scored for the homozygous egl-15::gfp, arg-1::gfp or hlh-8::gfp signal to determine whether a single copy of the Glu29† mutation altered expression of these markers.
Assays to score phenotypes
The percentage of animals displaying the Egl and Con phenotypes was quantified by simply counting the number of animals on a plate displaying that phenotype. For those mutants that were not 100% Egl, animals were singled to separate plates to determine which ones were able to lay embryos the first day of adulthood. Development time (Fig. 3A) was determined by picking L4 larvae and recording when the F1 progeny of that animal became L4 larvae. Brood size was quantified by picking L4 animals and counting their brood after they bagged as adults and all of the progeny were outside of the parent or if the animals laid embryos, they were moved to new plates every 24 h until they stopped laying embryos to count the number of offspring.
M lineage assays: The M lineage was followed using Glu29†; hlh-8::gfp; arg-1::gfp animals and observing the hlh-8::gfp fluorescence pattern at various larval stages. The hlh-8::gfp reporter is an integrated multi-copy transgene containing 1.8 kb upstream of the hlh-8 start codon (3) This region of DNA does not depend on HLH-8 for expression (9). In L1 animals, the first division of the M mesoblast was scored as D/V, ventral–ventral (V) or dorsal–dorsal (D) based on the position of the two M descendants with respect to the anal opening on the ventral side of the animal. The SM cells that arise in the L2 stage are large, migratory blast cells that move from the posterior to the center of the animal. At the L3 stage, the SMs are the only cells from the M lineage that are expressing hlh-8::gfp, and the number of SMs were recorded at this stage. When the SMs reach the developing vulva, they each divide rapidly three times without intervening cell growth to make 16 small cells in two focal planes. Animals that had at least six small cells at the early L4 stage were scored as positive for SM descendants (SMd) and those with large cells the size/shape of the SMs were scored as having under or undivided SMs.
arg-1::gfp expression assays: Expression of the HLH-8 target gene, arg-1, was monitored using an integrated multi-copy transgene in which 2.8 kb of sequence upstream of the start codon was fused to gfp (46). Glu29†; arg-1::gfp; hlh-8::gfp homozygous animals were scored for the arg-1::gfp expression patterns in young adults (when the hlh-8::gfp transgene is no longer expressed). The percentage of the population expressing arg-1::gfp in the HMC, the vulval muscles and the enteric muscles was quantified. The intensity of the GFP was not scored; an animal was scored as expressing the transgene if any green signal was observed. For heterozygous Glu29†/+ animals, arg-1::gfp was scored in the vulval muscles 24 h past the L4 stage. The intensity of the GFP was scored for bright, medium, dim or lack of GFP expression (Fig. 6). For scoring GFP expression in the enteric muscles of heterozygotes, the percent of the population expressing in each muscle type was recorded.
egl-15::gfp expression assays: Expression of the HLH-8 target gene, egl-15, was monitored using an integrated multi-copy transgene in which 2.1 kb upstream of the egl-15 start codon was fused to gfp (3). Glu29†; egl-15::gfp homozygous animals were scored for percentage of the population expressing GFP in the vulval muscles of young adults. Animals were scored for bright, dim or lack of GFP expression. For heterozygous Glu29†/+ animals, egl-15::gfp was scored in the vulval muscles 24 h past the L4 stage. The intensity of the GFP was scored for bright, medium, dim or lack of GFP expression (Fig. 6).
Embryo retention assays in Glu29†/+ heterozygotes: To address whether heterozygous mutants were semi-dominant, assays were carried out in a sensitized egl-15::gfp background. Glu29†/+; egl-15::gfp; mIs10/+ animals were picked at the L4 stage and scored 48 h later. The mIs10 transgene is expressed in the developing pharynx during late embryogenesis. In WT animals, late stage embryos would be laid before the GFP could be observed. Animals that retain embryos, however, will have embryos with a green pharynx in their uterus. The number of green embryos was counted in each heterozygous mother.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
We are grateful to the families of the subjects for their cooperation and interest in this work. We thank members of the NIH Undiagnosed Diseases Program, including Shannon Marchegiani, Taylor Davis, Cornelius Boerkoel, Thomas Markello, and William Gahl for sharing their TWIST2 data prior to publication. We thank the staff at the high-throughput genomics facility of the Wellcome Trust Centre for Human Genetics (Oxford Genomics Centre) for exome sequencing, Hannah Larbalestier for analysis of the exome data, Amy Fabritius for helping with the CRISPR/Cas9 technology and Robyn Branicky (MRC, Cambridge, UK) who helped analyze the tail phenotypes of our mutants. For experimental assistance, we thank Maggie Nixon, and for advice regarding statistics, we thank Ekaterina Nestorovich. We appreciate the many comments from Shannon Marchegiani and Michael Krause on the manuscript. We thank the members of the Baltimore Worm Club who have given advice on the CRISPR/Cas9 technology and valuable feedback on this project. Some strains were provided by the Caenorhabditis Genetics Center, which is funded by National Institutes of Health Office of Research Infrastructure Programs (P40 OD010440).
Conflict of Interest statement. None declared.
Funding
This work was supported, in part, by the Intramural Research Program of the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases (S.K., A.C., T.J.H. and A.G.); The Catholic University of America (A.A., V.A.S. and A.K.C.); the MRC through the Weatherall Institute of Molecular Medicine Strategic Alliance (G0902418, MC_UU_12025); National Institute for Health Research Oxford Biomedical Research Centre Programme (A.O.M.W.); and Wellcome (Project Grant 093329 to A.O.M.W. and S.R.F.T.; Investigator Award 102731 to A.O.M.W.).
References
- 1. Ledent V., Paquet O., Vervoort M. (2002) Phylogenetic analysis of the human basic helix-loop-helix proteins. Genome Biol., 3, RESEARCH0030.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Simpson P. (1983) Maternal-zygotic gene interactions during formation of the dorsoventral pattern in Drosophila embryos. Genetics, 105, 615–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harfe B.D., Vaz Gomes A., Kenyon C., Liu J., Krause M., Fire A. (1998) Analysis of a Caenorhabditis elegans Twist homolog identifies conserved and divergent aspects of mesodermal patterning. Genes Dev., 12, 2623–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Franco H.L., Casasnovas J., Rodríguez-Medina J.R., Cadilla C.L. (2011) Redundant or separate entities? Roles of Twist1 and Twist2 as molecular switches during gene transcription. Nucleic Acids Res., 39, 1177–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tukel T., Šošić D., Al-Gazali L.I., Erazo M., Casasnovas J., Franco H.L., Richardson J.A., Olson E.N., Cadilla C.L., Desnick R.J. (2010) Homozygous nonsense mutations in TWIST2 cause Setleis syndrome. Am. J. Hum. Genet., 87, 289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marchegiani S., Davis T., Tessadori F., van Haaften G., Brancati F., Hoischen A., Huang H., Valkanas E., Pusey B., Schanze D.. et al. (2015) Recurrent mutations in the basic domain of TWIST2 cause Ablepharon Macrostomia and Barber-Say syndromes. Am. J. Hum. Genet., 97, 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. De Maria B., Mazzanti L., Roche N., Hennekam R.C. (2016) Barber-Say syndrome and Ablepharon-Macrostomia syndrome, an overview. Am. J. Med. Genet. A, 170, 1989–2001. [DOI] [PubMed] [Google Scholar]
- 8. Corsi A.K., Brodigan T.M., Jorgensen E.M., Krause M. (2002) Characterization of a dominant negative C. elegans Twist mutant protein with implications for human Saethre-Chotzen syndrome. Development, 129, 2761–2772. [DOI] [PubMed] [Google Scholar]
- 9. Corsi A.K., Kostas S.A., Fire A., Krause M. (2000) Caenorhabditis elegans twist plays an essential role in non-striated muscle development. Development, 127, 2041–2051. [DOI] [PubMed] [Google Scholar]
- 10. Jabs E.W. (2016) TWIST1 and the Saethre-Chotzen Syndrome In Erickson R.P., Wynshaw-Boris A. (eds), Epstein’s Inborn Errors of Development, The Molecular Basis of Clinical Disorders of Morphogenesis. Oxford University Press, Oxford, UK, pp. 479–484. [Google Scholar]
- 11. el Ghouzzi V., Le Merrer M., Perrin-Schmitt F., Lajeunie E., Benit P., Renier D., Bourgeois P., Bolcato-Bellemin A.L., Munnich A., Bonaventure J. (1997) Mutations of the TWIST gene in the Saethre-Chotzen syndrome. Nat. Genet., 15, 42–46. [DOI] [PubMed] [Google Scholar]
- 12. Howard T.D., Paznekas W.A., Green E.D., Chiang L.C., Ma N., Ortiz de Luna R.I., Garcia Delgado C., Gonzalez-Ramos M., Kline A.D., Jabs E.W. (1997) Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat. Genet., 15, 36–41. [DOI] [PubMed] [Google Scholar]
- 13. Johnson D., Horsley S.W., Moloney D.M., Oldridge M., Twigg S.R.F., Walsh S., Barrow M., Njølstad P.R., Kunz J., Ashworth G.J.. et al. (1998) A comprehensive screen for TWIST mutations in patients with craniosynostosis identifies a new microdeletion syndrome of chromosome band 7p21.1. Am. J. Hum. Genet., 63, 1282–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miller K.A., Twigg S.R.F., McGowan S.J., Phipps J.M., Fenwick A.L., Johnson D., Wall S.A., Noons P., Rees K.E., Tidey E.A.. et al. (2017) Diagnostic value of exome and whole genome sequencing in craniosynostosis. J. Med. Genet., 54, 260–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fu W., O'Connor T.D., Jun G., Kang H.M., Abecasis G., Leal S.M., Gabriel S., Rieder M.J., Altshuler D., Shendure J.. et al. (2013) Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature, 493, 216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rieder M.J., Green G.E., Park S.S., Stamper B.D., Gordon C.T., Johnson J.M., Cunniff C.M., Smith J.D., Emery S.B., Lyonnet S.. et al. (2012) A human homeotic transformation resulting from mutations in PLCB4 and GNAI3 causes auriculocondylar syndrome. Am. J. Hum. Genet., 90, 907–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gordon C.T., Vuillot A., Marlin S., Gerkes E., Henderson A., AlKindy A., Holder-Espinasse M., Park S.S., Omarjee A., Sanchis-Borja M.. et al. (2013) Heterogeneity of mutational mechanisms and modes of inheritance in auriculocondylar syndrome. J. Med. Genet., 50, 174–186. [DOI] [PubMed] [Google Scholar]
- 18. Dickinson D.J., Goldstein B. (2016) CRISPR-based methods for Caenorhabditis elegans genome engineering. Genetics, 202, 885–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Paznekas W.A., Cunningham M.L., Howard T.D., Korf B.R., Lipson M.H., Grix A.W., Feingold M., Goldberg R., Borochowitz Z., Aleck K.. et al. (1998) Genetic heterogeneity of Saethre-Chotzen syndrome, due to TWIST and FGFR mutations. Am. J. Hum. Genet., 62, 1370–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. de Heer I.M., de Klein A., van den Ouweland A.M., Vermeij-Keers C., Wouters C.H., Vaandrager J.M., Hovius S.E., Hoogeboom J.M. (2005) Clinical and genetic analysis of patients with Saethre-Chotzen syndrome. Plast. Reconstr. Surg., 115, 1894–1905. [DOI] [PubMed] [Google Scholar]
- 21. Stenirri S., Restagno G., Ferrero G.B., Alaimo G., Sbaiz L., Mari C., Genitori L., Maurizio F., Cremonesi L. (2007) Integrated strategy for fast and automated molecular characterization of genes involved in craniosynostosis. Clin. Chem., 53, 1767–1774. [DOI] [PubMed] [Google Scholar]
- 22. Thomas J.H. (1990) Genetic analysis of defecation in Caenorhabditis elegans. Genetics, 124, 855–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hodgkin J., Kuwabara P.E., Corneliussen B. (2000) A novel bacterial pathogen, Microbacterium nematophilum, induces morphological change in the nematode C. elegans. Curr. Biol., 10, 1615–1618. [DOI] [PubMed] [Google Scholar]
- 24. Sulston J.E., Horvitz H.R. (1977) Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev. Biol., 56, 110–156. [DOI] [PubMed] [Google Scholar]
- 25. Kostas S.A., Fire A. (2002) The T-box factor MLS-1 acts as a molecular switch during specification of nonstriated muscles in C. elegans. Genes Dev., 16, 257–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stern M.J., Horvitz H.R. (1991) A normally attractive cell interaction is repulsive in two C. elegans mesodermal cell migration mutants. Development, 113, 797–803. [DOI] [PubMed] [Google Scholar]
- 27. Wang P., Zhao J., Corsi A.K. (2006) Identification of novel target genes of CeTwist and CeE/DA. Dev. Biol., 293, 486–498. [DOI] [PubMed] [Google Scholar]
- 28. Jones N.C., Lynn M.L., Gaudenz K., Sakai D., Aoto K., Rey J.P., Glynn E.F., Ellington L., Du C., Dixon J.. et al. (2008) Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat. Med., 14, 125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Weaver K.N., Watt K.E., Hufnagel R.B., Navajas Acedo J., Linscott L.L., Sund K.L., Bender P.L., König R., Lourenco C.M., Hehr U.. et al. (2015) Acrofacial dysostosis, Cincinnati type, a mandibulofacial dysostosis syndrome with limb anomalies, is caused by POLR1A dysfunction. Am. J. Hum. Genet., 96, 765–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lehalle D., Wieczorek D., Zechi-Ceide R.M., Passos-Bueno M.R., Lyonnet S., Amiel J., Gordon C.T. (2015) A review of craniofacial disorders caused by spliceosomal defects. Clin. Genet., 88, 405–415. [DOI] [PubMed] [Google Scholar]
- 31. Twigg S.R.F., Wilkie A.O.M. (2015) New insights into craniofacial malformations. Hum. Mol. Genet., 24, R50–R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen Z.F., Behringer R.R. (1995) twist is required in head mesenchyme for cranial neural tube morphogenesis. Genes Dev., 9, 686–699. [DOI] [PubMed] [Google Scholar]
- 33. Rahbari R., Wuster A., Lindsay S.J., Hardwick R.J., Alexandrov L.B., Al Turki S., Dominiczak A., Morris A., Porteous D., Smith B.. et al. (2016) Timing, rates and spectra of human germline mutation. Nat. Genet., 48, 126–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Maerkl S.J., Quake S.R. (2009) Experimental determination of the evolvability of a transcription factor. Proc. Natl. Acad. Sci. U. S. A., 106, 18650–18655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Boisson B., Wang Y.D., Bosompem A., Ma C.S., Lim A., Kochetkov T., Tangye S.G., Casanova J.L., Conley M.E. (2013) A recurrent dominant negative E47 mutation causes agammaglobulinemia and BCR(-) B cells. J. Clin. Invest., 123, 4781–4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McGary K.L., Park T.J., Woods J.O., Cha H.J., Wallingford J.B., Marcotte E.M. (2010) Systematic discovery of nonobvious human disease models through orthologous phenotypes. Proc. Natl. Acad. Sci. U. S. A., 107, 6544–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Woods J.O., Singh-Blom U.M., Laurent J.M., McGary K.L., Marcotte E.M. (2013) Prediction of gene-phenotype associations in humans, mice, and plants using phenologs. BMC Bioinformatics, 14, 203.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Philogene M.C., Small S.G., Wang P., Corsi A.K. (2012) Distinct Caenorhabditis elegans HLH-8/twist-containing dimers function in the mesoderm. Dev. Dyn., 241, 481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Langmead B., Salzberg S.L. (2012) Fast gapped-read alignment with Bowtie 2. Nat. Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. (2009) The Sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang K., Li M., Hakonarson H. (2010) ANNOVAR, functional annotation of genetic variants from high-throughput sequencing data. Nucl. Acid Res., 38, e164.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim H., Ishidate T., Ghanta K.S., Seth M., Conte D. Jr, Shirayama M., Mello C.C. (2014) A co-CRISPR strategy for efficient genome editing in Caenorhabditis elegans. Genetics, 197, 1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dickinson D.J., Ward J.D., Reiner D.J., Goldstein B. (2013) Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat. Methods, 10, 1028–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Arribere J.A., Bell R.T., Fu B.X., Artiles K.L., Hartman P.S., Fire A.Z. (2014) Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics, 198, 837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Paix A., Folkmann A., Rasoloson D., Seydoux G. (2015) High efficiency, homology-directed genome editing in Caenorhabditis elegans using CRISPR-Cas9 ribonucleoprotein complexes. Genetics, 201, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhao J., Wang P., Corsi A.K. (2007) The C. elegans Twist target gene, arg-1, is regulated by distinct E box promoter elements. Mech. Dev., 124, 377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.