

Abstract
Epithelial-Mesenchymal Transition (EMT) is a dynamic process through which epithelial cells transdifferentiate from an epithelial phenotype into a mesenchymal phenotype. Previous studies have also demonstrated that both mechanical signaling and soluble growth factor signaling facilitate this process. One possible point of integration for mechanical and growth factor signaling is the extracellular matrix. Here we investigate the role of the extracellular matrix (ECM) protein fibronectin (FN) in this process. We demonstrate that inhibition of FN fibrillogenesis blocks activation of the Transforming Growth Factor-Beta (TGF-β) signaling pathway via Smad2 signaling, decreases cell migration and ultimately leads to inhibition of EMT. Results show that soluble FN, FN fibrils, or increased contractile forces are insufficient to independently induce EMT. We further demonstrate that inhibition of latent TGF-β1 binding to FN fibrils via either a monoclonal blocking antibody against the growth factor binding domain of FN or through use of a FN deletion mutant that lacks the growth factor binding domains of FN blocks EMT progression, indicating a novel role for FN in EMT in which the assembly of FN fibrils serves to localize TGF-β1 signaling to drive EMT.
Keywords: Epithelial-Mesenchymal Transition, Extracellular Matrix, Fibronectin, TGF-β1
1. Introduction
EMT is a morphogenic process characterized by a phenotypic shift in epithelial cell monolayers to motile and oftentimes invasive mesenchymal cells [1]. Epithelial cell sheets typically serve as a protective barrier, lining cavities and organs throughout the body. However, upon transdifferentiation, mesenchymal cells begin to fill interstitial spaces, driving growth and assembly of new tissues. This tightly regulated process is fundamental in the generation of new tissues and organs during embryogenesis and is a key factor in tissue remodeling and wound healing [2-4]. Although the process is fundamental in development, EMT can also be misregulated, leading to its implication in several disease states, including cancer and organ fibrosis [5, 6]. Epithelial cells exhibit strong cell-cell adhesions, an apicobasal polarized distribution of organelles and cytoskeletal components, and cortical actin surrounding the cell periphery. Upon induction of EMT, a continuous monolayer of cuboidal epithelial cells disassemble their adherens junctions and transform into spindle-like, elongated mesenchymal cells [6]. This transition is accompanied by a switch to a front/back polarity that allows the cells to migrate [7]. There is also a reorganization of actin into stress fibers, which may lead to increased cell motility and invasive properties [8]. EMT has also been shown to enhance angiogenesis close to the tumor microenvironment [9]. This misregulated transdifferentiation has been attributed to the process by which cancer stem cells detach from their primary tumor, penetrate the ECM and migrate through the blood stream to form metastatic colonies in distant tissues or organs [10].
EMT can be induced via either soluble growth factors, including TGF-β1 [11-13], epidermal growth factor (EGF) [14, 15], fibroblast growth factor (FGF), hepatocyte growth factor (HGF), platelet-derived growth factor (PDGF), Jagged, Delta-like and Wnt ligands [16, 17] &/or by mechanical signaling, including increased substrate stiffness [18-25] and increased contractile force [17, 26-28].
While many recent studies have highlighted the importance of mechanical signaling in cellular events [17, 29], it is not entirely clear how the combination of mechanical and soluble signals trigger this complex transdifferentiation. One possible point of integration is the ECM. Many ECM fibrils are assembled in response to cell-applied contractile force and many ECM proteins contain binding sites for growth factors that allow the matrix to serve as either a sink or reservoir for soluble growth factors [30-32].
One particularly attractive target for mechanical-chemical signal integration is the ECM protein fibronectin (FN). FN is a soluble protein found in the blood plasma that is assembled by cells into insoluble elastic fibrils [33, 34]. FN expression is increased in wound healing [35, 36], embryonic development [37, 38] and malignant tumors [39-42], all of which have been associated with EMT. FN fibrils require cellular contractile forces to assemble. These forces stretch FN dimers from a compact conformation to an extended conformation, which exposes a cryptic binding site that facilitates fibril growth [43, 44]. This open conformation also exposes the 12-14th Type III domains of FN, which is a heparin-binding region of FN that binds upwards of 40 soluble growth factors, including TGF-β1, HGF, CTGF, many FGFs and several PDGFs, with nM affinity [45]. These studies have demonstrated that bound growth factors are still capable of binding their receptors, suggesting that FN serves as a growth factor delivery system as opposed to a growth factor sequestering mechanism. It is known that the inactive form of TGF-β1 bound to its Latent TGF-β Binding Protein 1 (LTBP-1) integrates into the FN matrix [46, 47], and that this binding is heparin mediated [48].
Given the prominent role of TGF-β1 in inducing EMT [11-13, 45] and the evidence of latent TGF-β1 localization to FN fibrils [46, 47, 49], we investigated whether FN fibrils may serve as an integration point for mechanical and chemical signals by probing the role of FN fibrils in facilitating TGF-β1-induced EMT.
2. Results
2.1. FN fibril assembly is increased during Epithelial-Mesenchymal Transition
As an initial investigation, we explored whether FN fibril assembly was altered in response to EMT that was induced by either exogenous active TGF-β1 or conditioned media from MDA-MB-231 malignant breast cancer cells (Fig. S1), which have been shown to express TGF-β1 [50-52]. While previous studies have demonstrated that FN mRNA levels and protein expression are both upregulated in response to TGF-β1 [53-56], it is not clear whether this translates into increased assembly of FN fibrils, which require application of cellular contractile forces to soluble FN. MCF10A human breast epithelial cells were EGF and serum starved for 2 hours, then either exposed to 2 ng/ml TGF-β1 or left untreated in EGF and serum-free conditions for 48 hours. Cells that were cultured in EGF-free/serum-free media maintained an epithelial phenotype, as evidenced by the cobblestone morphology visible in actin immunofluorescence images; these cells assembled no detectable FN fibrils. When exposed to TGF-β1, cells transformed to a mesenchymal phenotype, as depicted through the loss of cobblestone morphology and the acquisition of prominent F-actin stress fibers. Under these conditions, FN fibril assembly was significantly increased. While these results show that FN fibril assembly is induced during EMT, they do not indicate a causal relationship. We next probed for a role for FN assembly in EMT through inhibition of FN fibrillogenesis.
2.2. Inhibition of FN fibril assembly inhibits aspects of EMT, TGF-β1 signaling, changes in cell area and cell density, and cell migration
To test whether FN fibrils play a functional role in facilitating EMT, we inhibited FN assembly with a 49 amino acid peptide from the functional upstream domain of the S. pyogenes protein Adhesin F1, which has previously been used to inhibit FN fibril formation [57, 58]. To examine the efficacy of this peptide (referred to subsequently as ‘FUD’), we incubated TGF-β1-treated MCF10As with increasing concentrations of the FUD inhibitor (Fig. S2). The total fibril area was summed for each image and normalized to the control case in which no FUD was administered, as described in materials and methods. Data indicate that FUD treatment inhibited roughly 70% of FN fibril formation at concentrations of 125 nM or greater (Fig. S2).
MCF10As were then cultured with either FUD, TGF-β1, or the combination of the two to determine the effects of FN fibril inhibition on EMT. Cells were EGF/serum starved for 2 hours, at which point FUD was added. TGF-β1 was then added 1 hour after FUD inhibition to ensure that FN fibril assembly was inhibited prior to addition of TGF-β1. Fig. 1A shows representative images from MCF10As cultured +/- 125 nM FUD and +/- 2 ng/ml TGF-β1. In the absence of TGF-β1 or FUD, cells exhibited a classic epithelial phenotype. Addition of TGF-β1 disrupted adherens junctions, induced F-actin stress fibers and induced assembly of FN fibrils. Cells cultured in both TGF-β1 and FUD had no significant FN fibril formation and also maintained the key characteristics of the epithelial phenotype, including E-cadherin containing junctions and peripheral actin. Note that under these conditions, there were some changes in actin organization; however, these cells lacked the prominent stress fibers observed when treated with TGF-β1 alone. Treatment with FUD alone had no detectable effects on the epithelial phenotype. Quantification of the total FN fibril area per image was accomplished for each condition as described in materials and methods (Fig. 1B). In comparison with the positive control (treatment with TGF-β1), the average fibril area for the negative control and the co-treated condition decreased significantly by approximately 90% and 50% respectively. EMT marker presence was also quantified via an ordinate scale for actin and E-cadherin images, described in the materials and methods section below (Fig. 1C). These showed a statistically significant change in response to TGF-β1 that was inhibited by co-treatment with FUD.
Fig 1.
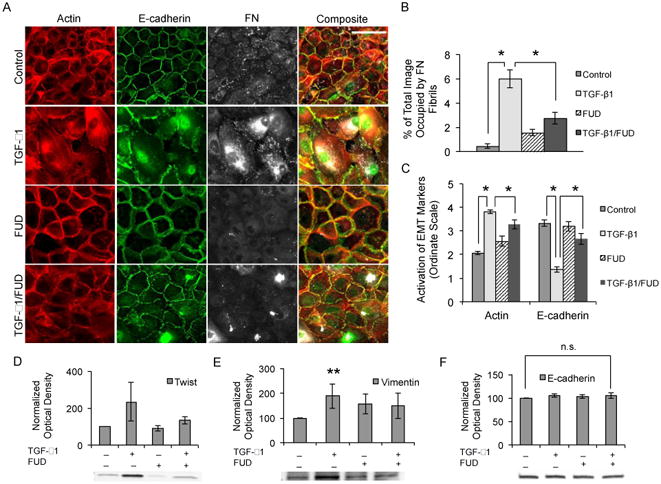
Inhibition of FN assembly blocks TGF-β1-induced EMT in MCF10As. (A) Immunofluorescence images of MCF10As after 48 hours of treatment with TGF-β1 and/or FUD. Ab staining for F-actin (red), E-cadherin (green), and FN (white). (B) Quantification of average fibril area per image. N ≥ 9 for each condition. (C) Quantification of EMT markers actin and E-cadherin via ordinate scale. N ≥ 4 for each condition. (D-F) MCF10As were treated with TGF-β1 and/or FUD for 48 hours and analyzed for EMT markers. Whole-cell lysates (15-30 μg total protein) were probed by Western blotting for (D) Twist, (E) Vimentin, and (F) E-cadherin markers. Band optical density was quantified and normalized to total lane protein. Data is presented as percentage of negative control optical density. n.s., not significant; N ≥ 4 for each condition. *p ≤ 0.01, and **p ≤ 0.1 significantly different from control or TGF-β1, Student's t-test. Scale bar is 50 μm.
To further quantify the effects of FN fibrils on EMT, we measured protein expression levels for several key mesenchymal markers (Fig. 1D-F). Expression of the intermediate filament protein vimentin, a classic markers of EMT [45, 59], significantly increased in response to TGF-β1; this increase was inhibited when co-treated with FUD and TGF-β1. We observed a strong but non-significant increase in the transcription factor Twist (p = 0.17) in the presence of TGF-β1. This increase in Twist was downregulated upon co-treatment with FUD and TGF-β1. Twist is known to downregulate the expression of E-cadherin [60, 61]; as such, while we saw no significant change in E-cadherin expression, downregulation of E-cadherin may occur at later stages of EMT. These data suggest that inhibition of FN fibrils inhibits mesenchymal marker expression in mammary epithelial cells.
To ensure that FN fibril inhibition blocks TGF-β1-induced EMT via the TGF-β pathway, we immunofluorescently labeled cells with an anti-Smad2 antibody. Smad2 is part of the Smad2/3 complex, which is phosphorylated by the TGF-βRII receptor following ligation of TGF-β1 to the receptor [62, 63]. Upon phosphorylation, Smad2 translocates to the nucleus, where it acts to repress E-cadherin expression and trigger EMT. MCF10As were cultured under four conditions; EGF/serum-free media, treatment with FUD alone, TGF-β1 alone and a co-treatment of the two, for 48 hours (Fig. 2A). In the control condition, Smad2 is distributed throughout the cytoplasm; upon addition of TGF-β1, it translocates to the nucleus. Co-treatment with both TGF-β1 and FUD inhibited the nuclear localization of Smad2, indicating that blocking FN fibrillogenesis specifically inhibits the TGF-β signaling pathway. FUD treatment alone had no effect on Smad2 (Fig. 2A). Quantification of nuclear colocalization of Smad2 was accomplished for each condition as described in materials and methods (Fig. 2C). After 48 hours of treatment with TGF-β1, the nuclear intensity of the Smad2 protein was much greater than the cytoplasmic intensity. These effects were blocked with co-treatment of FUD and TGF-β1.
Fig 2.
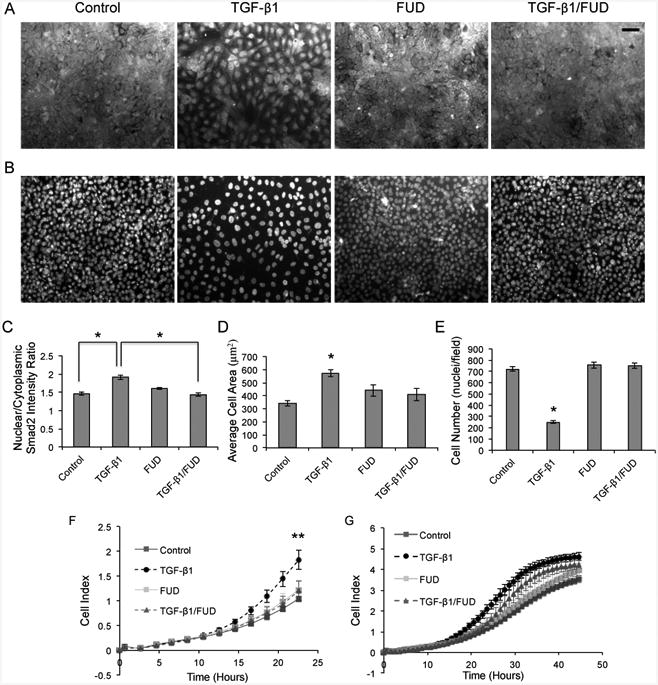
FN fibril inhibition blocks the TGF-β pathway, inhibits cell size, cell number, and decreases migration in MCF10As. (A) Representative immunofluorescence images of Smad2 in MCF10As after 48 hours of treatment with TGF-β1 and FUD. (B) Immunofluorescence images of nuclei from part A (C) Quantification of nuclear colocalization of Smad2. N ≥ 16 for each condition. *p < 0.01 significantly different from TGF-β1, Student's t-test. (D) Quantification of the average cell size per condition. N ≥ 16 for each condition. *p < 0.01 significantly different from TGF-β1, Student's t-test. (E) Quantification of cell density. N ≥ 16 for each condition. *p < 0.01 significantly different from TGF-β1, Student's t-test. (F-G) Migration kinetics of MCF10A cells by continuous monitoring of live cell migration for approximately 48 hours. Direct comparison of FN fibril inhibitor &/or TGF-β1 effects on cell migration (F) after 24 hours, and (G) after 48 hours. Control samples served as the baseline cell index levels for comparison with wells containing a combination of FUD &/or TGF-β1. Cell index values from 4 wells per condition ± SD correspond to impedance from microelectrode sensor. **p < 0.05 significantly different from control or TGF-β1, Student's t-test. Scale bar is 50 μm.
TGF-β1 has been shown to induce increases in mammary epithelial cell size through activation of mammalian target of rapamycin (mTOR) [64]. We show that after 48 hours of treatment with TGF-β1, MCF10As exhibit larger cell area, and that co-treatment with FUD and TGF-β1 blocks this increase in size. Quantification of the average cell area was accomplished for each condition as described in materials and methods (Fig. 2D). In addition, treatment with TGF-β1 decreases the cell density. This effect is blocked through co-treatment with FUD and TGF-β1 (Fig. 2B). Quantification of cell density was accomplished for each condition as described in materials and methods (Fig. 2E).
Previous studies have shown that TGF-β1 inhibits cell proliferation [65, 66]. Therefore, we investigated the effect of FUD treatment on cell proliferation via Ki67 imaging. Treatment with TGF-β1 alone showed a significant decrease in proliferation as compared to the control (Fig. S3A). These effects were blocked upon co-treatment with FUD and TGF-β1. Quantification of nuclear colocalization of Ki67 was accomplished for each condition as described in materials and methods (Fig. S3B).
An important implication of EMT in disease states is its association with increased migration [67]. To assess whether MCF10A migration is increased during TGF-β1-induced EMT, we measured migration using an xCELLigence RTCA real-time migration system. Cells were plated into wells within the system and migration was tracked in real time for approximately 48 hours after TGF-β1 and/or FUD treatment. Cell migration was determined by changes in impedance measurements of the microelectrode sensor as cells travelled from the upper chamber, through the microporous membrane, to the lower chamber and reported as cell index. Increases in cell index represent enhanced migratory kinetics. Each condition was repeated in quadruplicate wells. After 24 hours, there was very little migration exhibited by the control, whereas migration increased 57% when MCF10As were exposed to TGF-β1; this increase was inhibited when cells were co-treated with FUD and TGF-β1, indicating that blocking FN fibril growth inhibits EMT-induced migration (Fig. 2F). In the time frame between 24 and 48 hours, the FUD inhibitory effect wanes, as exhibited by the increase in cell index of the TGF-β1/FUD condition (Fig. 4G).
Fig 4.
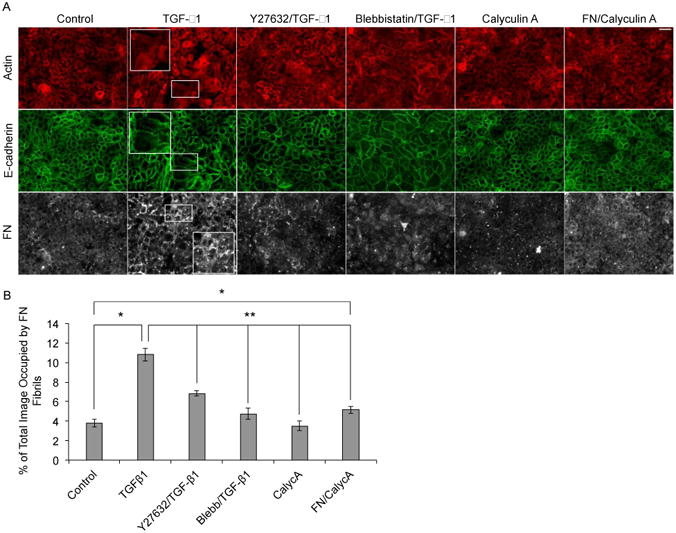
Manipulation of contractile forces affects both assembly of FN fibrils and EMT. (A) Representative immunofluorescence images of MCF10As cultured for 48 hours in EGF/serum-free medium with (column 1) no additions, (column 2) 2 ng/ml TGF-β1, (column 3) 20 μM Y27632 with 2 ng/ml TGF-β1, (column 4) 20 μM blebbistatin with 2 ng/ml TGF-β1, (column 5) 0.3 nM Calyculin A and (column 6) 10 nM FN with 0.3 nM Calyculin A. Ab staining for F-actin, (red) E-cadherin (green) and FN (white). (B) Quantification of average fibril area per image. N ≥ 13 for each condition. *p < 0.01 significantly different from control, **p < 0.05 significantly different from TGF-β1, Student's t-test. Scale bar is 50 μm.
2.3. EMT is not dependent solely on the presence of soluble FN, contractile forces, or FN fibrils
Having established a prominent role for FN fibrils in TGF-β1 signaling, aspects of EMT, and epithelial cell migration, we next investigated the mechanism that underlies this effect. We envisioned four possible mechanisms: 1) elevated levels of soluble FN alone drive EMT; 2) FN fibrils alter the contractile state of the cell, thus driving EMT; 3) the presence of FN fibrils drive EMT, independently of TGF-β1; or 4) FN fibrils cluster newly synthesized, endogenous latent TGF-β1 at the cell surface to upregulate TGF-β signaling.
Previous studies have shown that TGF-β1 induces increased expression of soluble FN [53-56] and that soluble FN expression is increased in many malignant tumor cells [39, 41]. Assembled FN fibrils are comprised of both FN secreted from the cells (cellular FN) and FN within the serum (plasma FN) [68]. Initially, we investigated the role of FUD on the expression of cellular FN. MCF10As treated with TGF-β1 exhibited elevated expression of FN compared to controls. Cells that were co-treated with FUD and TGF-β1 also exhibited elevated expression of cellular FN (Fig. 3A), suggesting that TGF-β1-induced changes in FN expression are upstream of FN fibril-dependent signaling.
Fig 3.
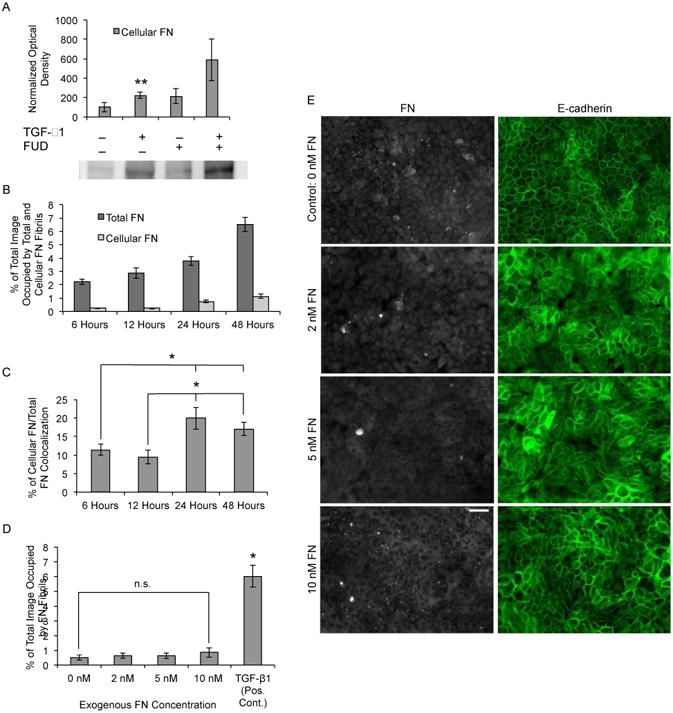
Increased exposure to soluble FN is insufficient to drive EMT. (A) MCF10As were treated with TGF-β1 and/or FUD for 48 hours and whole-cell lysates (15-30 μg total protein) were probed by Western blotting for expression of cellular FN. Band optical density was quantified and normalized to total lane protein. Data is presented as percentage of negative control optical density. Two way ANOVA (+/- FUD, +/- TGF-β1), N ≥ 12 for each condition. **p ≤ 0.05 significantly different for effect of TGF-β1. (B) Quantification of cellular FN fibril area and total FN fibril area per image over time. N ≥ 12 for each condition. (C) Quantification of percentage of overlap between cellular FN fibrils and total FN fibrils. N ≥ 12 for each condition. *p ≤ 0.01 significantly different from 6 hours or 12 hours, Student's t-test. (D) Quantification of percent of total image area occupied by FN fibrils for MCF10As cultured with increasing concentrations of exogenous FN. n.s., not significant; one way ANOVA, N ≥ 5 for each condition. *p ≤ 0.01 significantly different from TGF-β1, Student's t-test. (E) Immunofluorescence images of MCF10As cultured with increasing concentrations of soluble FN for 48 hours. Ab staining for FN (white), and E-cadherin (green). Scale bar is 50 μm.
Next, we sought to determine the source of FN in fibrils assembled by MCF10As (Fig. 3B-D). Cells were incubated with plasma FN and TGF-β1 and the source of FN was probed through the use of a monoclonal Ab (3E2) specific to cellular FN and a polyclonal Ab that reacts with all FN. The concentration of exogenous soluble FN was selected based on previously measured concentrations in the plasma of patients with malignant tumors [69-71]. MCF10As were EGF and serum starved and then exposed to 10 nM soluble FN and 2 ng/ml TGF-β1 for 6, 12, 24 and 48 hours (images not shown). Quantification of total and cellular FN fibril area per image was accomplished as described in materials and methods (Fig. 3B). Data indicate that after 6 hours, TGF-β1 began to stimulate FN fibril assembly, which continued to increase upon prolonged exposure to TGF-β1. Cellular FN fibril assembly area however, remained low at early time points, increasing slightly after 24 and 48 hours. Colocalization of cellular and total FN fibrils also remained low at early time points, with a significant increase at 24 and 48 hours (Fig. 5C). These results indicate that at early time points, FN fibrils are primarily comprised of exogenous plasma FN present in serum. However, cellular FN content increases at later time points (24 and 48 hours). This is consistent with the expectation that de novo cellular FN is not synthesized and secreted until 24 hours after TGF-β1 exposure.
Fig 5.
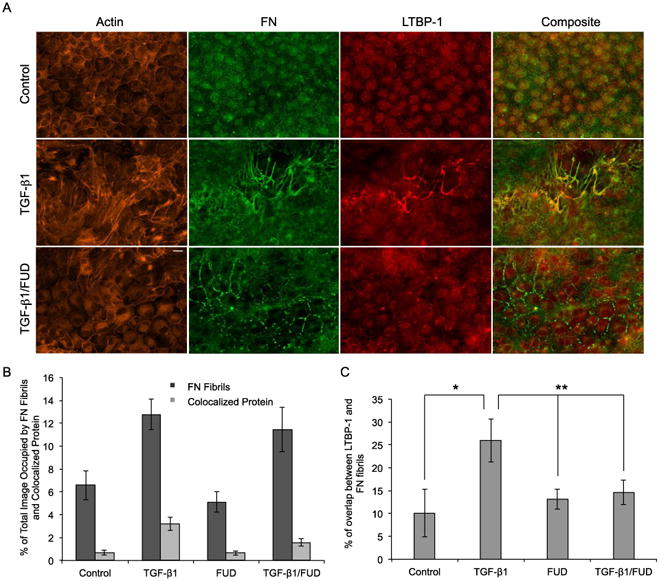
Inhibition of FN fibrillogenesis blocks TGF-β1-induced colocalization of LTBP-1 on FN fibrils in MDCKII cells. (A) Immunofluorescence images of MDCKII cells after 48 hours of treatment with TGF-β1 and FUD. Ab staining for F-actin (orange), FN (green) and LTBP-1 (red). Composite displays colocalization of FN and LTBP-1 in yellow. (B) Quantification of percentage of image occupied by FN fibrils and colocalized LTBP-1. N ≥ 9 for each condition. (C) Quantification of percentage of overlap between LTBP-1 and FN fibrils. N ≥ 9 for each condition. *p < 0.01 significantly different from control, **p < 0.05 significantly different from TGF-β1, Student's t-test. Scale bar is 20 μm.
We next tested whether soluble FN could drive EMT independently of TGF-β signaling. Exogenous plasma FN was added to MCF10A culture medium in increasing concentrations from 0 – 10 nM in the absence of EGF, serum, or TGF-β1. As compared to cells treated with TGF-β1, those treated with exogenous, soluble FN neither induced FN fibril assembly (Fig. 3D), nor induced EMT, as observed through E-cadherin staining in immunofluorescent images (Fig. 3E). This indicates that soluble FN alone is insufficient to induce EMT in these cells.
We next examined a role for contractile forces in the induction of FN-fibril induced EMT, since it is well established that FN fibrils require cell-generated contractile forces to be assembled. MCF10As were treated with one of three force-altering compounds: 1) Rho Kinase inhibitor Y27632, which decreases myosin phosphorylation and thus cell contractility; 2) blebbistatin, a myosin II inhibitor also shown to decrease cell contractility [72-74]; or 3) Calyculin A, a phosphatase inhibitor which has previously been shown to activate acto-myosin contractility [75]. To examine the effect of inhibiting cell contractility on the induction of EMT, MCF10As were co-cultured with either 20 μM Y27632 and 2 ng/mL TGF-β1 or 20 μM blebbistatin and 2 ng/mL TGF-β1 for 48 hours. Results showed that inhibition of contractile forces blocked the phenotypic changes seen with TGF-β1 treatment alone (Fig. 4A) and significantly lowered FN fibril assembly (Fig. 4B). These results indicate that inhibition of contractile forces blocks TGF-β1-induced EMT, which is consistent with previous data that demonstrates that TGF-β1 acts through the Rho/ROCK pathway [76, 77]. Since FN polymerization and matrix assembly have been shown to be mediated through the RhoA pathway [78], we investigated whether stimulating cell contractility in a soluble FN-rich environment would be sufficient to induce FN fibril assembly and subsequent EMT. Therefore, we cultured MCF10As with either 0.3 nM Calyculin A or co-treated with both 0.3 nM Calyculin A and 10 nM soluble FN. Increasing contractile forces had no effect on actin organization nor did it cause disruption of cell-cell junctions, indicating that these cells did not undergo EMT (Fig. 4A). Cells treated with both FN and Calyculin A showed a significant increase in FN fibril assembly as compared to cells cultured in the absence of EGF/serum. However, FN fibril area was significantly lower in cells treated with Calyculin A and co-treated with Calyculin A and FN as compared to cells treated with TGF-β1 alone (Fig. 4B). This indicates that increasing contractile force alone does not induce FN assembly or EMT independently.
Finally, we sought to determine whether the presence of FN fibrils alone was capable of inducing EMT. This investigation was aided by a fortuitous discovery while exploring EMT in other epithelial cell lines. MDCK II cells exhibit moderate FN fibrillogenesis even in the absence of growth factor induction. Interestingly, this naturally occurring FN fibrillogenesis did not induce EMT in these cells as exhibited through the presence of cortical actin around the periphery of the cell (Fig. 5A). MDCK II cells cultured in medium containing exogenous active TGF-β1 increase deposition of LTBP-1, which is a protein in the latent TGF-β1 complex. Given that LTBP-1 is incorporated into the ECM upon secretion from the cell, specifically binding to FN fibrils [46, 47, 49], we investigated whether MDCK II cells would increase secretion of LTBP-1 and integrate LTBP-1 into the FN matrix upon stimulation from active TGF-β1. In control conditions, there was no notable LTBP-1 expression and colocalization with FN fibrils. However, when exogenous active TGF-β1 was added to the media, LTBP-1 deposition and colocalization with FN fibrils significantly increased (Fig. 5B-C). Actin was also reorganized into parallel stress fibers (Fig. 5A). Co-treatment with FUD and TGF-β1 blocked actin reorganization; LTBP-1 colocalization with FN fibrils significantly decreased in comparison to treatment with TGF-β1 alone. These data suggest that FN fibrils alone are incapable of facilitating EMT. We also show that manipulation of contractile forces, as described in earlier experiments, had little effect on LTBP-1 deposition as compared to samples treated with TGF-β1 alone (Fig. S4A-B). Inhibition of contractile forces decreased LTBP-1 deposition and colocalization with FN fibrils, while increasing contractile forces did not independently alter LTBP-1 deposition or colocalization with FN fibrils (Fig. S4C). Therefore, we have shown that in the absence of external growth factors, increased FN fibrillogenesis has a minimal effect on the induction of EMT.
2.4. TGF-β1-induced EMT requires localization of latent TGF-β1 to FN fibrils
Given that neither elevated levels of soluble FN alone, contractile forces alone, or FN fibrils alone induced EMT, we focused on the requirement of growth factor localization to FN fibrils as the critical event in TGF-β1-induced EMT. To determine whether TGF-β1 localization is essential to the induction of EMT, we incubated MCF10As with 11 μg/mL of Fibronectin Antibody A32, a monoclonal antibody that binds to FNIII12-14 [79], which is the region of FN that contains the growth factor binding site [45, 80]. This region is also responsible for mediating electrostatic interactions in the compact conformation of soluble FN [81]. As such, this site is not exposed in soluble FN but is exposed in assembled fibrils [79]. Quantification of colocalization (data not shown) reveals that the total image area containing FN fibrils occupied by colocalized monoclonal ab is approximately 40 percent. Cells under these conditions exhibit cortical actin as compared to those treated with TGF-β1 alone, which exhibit stress fibers, and also show reduced transcription of the mesenchymal markers αSMA and vimentin (Fig. 6). We hypothesize that exogenous active TGF-β1 initiates EMT by upregulating expression and secretion of FN and the TGF-β1 complex, and that localization of de novo endogenous latent TGF-β1 complex to assembled FN fibrils is necessary for complete EMT. To confirm this, mRNA transcription of FN and LTBP-1 were quantified in response to TGF-β1 and/or the monoclonal FN blocking antibody. Results show that both FN and LTBP-1 transcription are increased in response to TGF-β1 (Fig. 6B, C). Neither of these responses is inhibited by treatment with the monoclonal antibody, suggesting that these are upstream of FN fibril signaling. We hypothesize that downstream signaling requires both assembly of FN fibrils and localization of newly synthesized latent TGF-β1 complex to the fibrils. We show that blocking the growth factor binding site on FN fibrils inhibits TGF-β1-induced EMT, indicating that TGF-β1 localization to FN fibrils is necessary for EMT.
Fig 6.
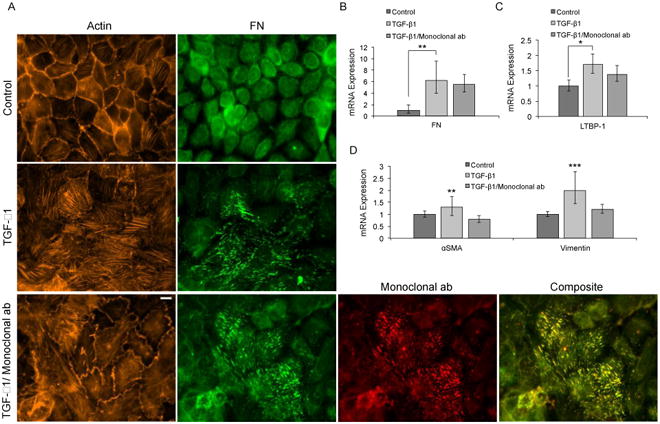
Blocking the LTBP-1/FN binding site inhibits TGF-β1–induced EMT in MCF10A cells. (A) Representative immunofluorescence images of MCF10A cells cultured for 48 hours with TGF-β1 and/or monoclonal ab. Ab staining for F-actin (orange) FN (green), and monoclonal ab (red). Composite displays colocalization of FN and monoclonal ab in yellow. mRNA levels for (B) FN (C) LTBP-1, and (D) EMT markers αSMA and vimentin, were determined by means of RT-qPCR. N = 3 for each condition. *p ≤ 0.01, **p ≤ 0.05, and ***p ≤ 0.1 significantly different from TGF-β1, Student's t-test. Scale bar is 10 μm.
To further confirm these results, we cultured cells with either wild type recombinant FN, with a FN deletion mutant in which the 11th through 14th Type III domains have been deleted (FN/A11-14), or with no exogenously added FN, in the presence of TGF-β1. Since the 11th through 14th Type III domains encompass the growth factor binding domains, we hypothesized that deletion of these domains should inhibit TGF-β1 localization and EMT. Given that TGF-β1 increases expression of FN, experiments with the deletion mutant FN/Δ11-14 will not eliminate all growth factor binding sites in fibrils, but should result in a significantly reduced population of binding sites. Results indicated that cells cultured in the presence of FN/Δ11-14 had less LTBP-1 localization to fibrils relative to either no exogenous FN or exogenous wild type FN (Fig. 7A). Cells cultured in FN/Δ11-14 also exhibited less stress fiber formation and more cortical actin, compared to samples with either no exogenous FN or with wild type recombinant FN. Transcription of mesenchymal markers Twist and vimentin were also quantified in response to co-culture with TGF-β1 and FN/Δ11-14. Results show that FN/Δ11-14 cultured cells exhibited decreased transcription levels compared to cells treated with TGF-β1 alone or with wild type recombinant FN and TGF-β1 (Fig. 7B). Therefore, we show that exposing cells to FN fibrils lacking the growth factor binding domain inhibits both colocalization with LTBP-1 and TGF-β1-induced EMT, further indicating that the LTBP-1/latent TGF-β1 complex localization to FN fibrils is necessary for EMT.
Fig 7.
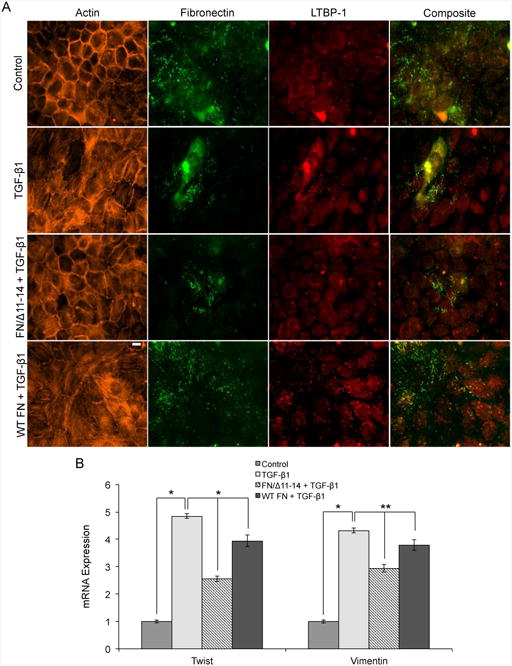
FN lacking the growth factor binding domains III 11-14 inhibits TGF-β1–induced EMT in MCF10A cells. (A) Representative immunofluorescence images of MCF10A cells cultured for 24 hours with TGF-β1 and/or FN/Δ11-14. Ab staining for F-actin (orange), FN (green), and LTBP-1 (red). Composite displays colocalization of FN and LTBP-1 in yellow. mRNA levels for (B) EMT markers twist and vimentin, were determined by means of RT-qPCR. N = 3 for each condition. *p < 0.005, and **p < 0.05 significantly different from TGF-β1, Student's t-test. Scale bar is 10 μm.
As additional support for this hypothesis, we cultured cells in increasing concentration of TGF-β1 in the presence of the FN assembly inhibitor. If FN fibrils indeed act as a mechanism to concentrate latent TGF-β1, then increasing exogenous active TGF-β1 should begin to overcome the FN fibril dependent responses. Indeed, increasing the soluble concentration of active TGF-β1 10-fold from the baseline value showed some evidence of EMT, even in the presence of FUD (Fig. S5).
2.5. FN fibrils that are pre-assembled in the presence of TGF-pi are capable of inducing EMT in the absence of exogenous active TGF-pi
To further investigate our hypothesis that EMT requires both assembly of FN fibrils and localization of growth factors to these fibrils, we conducted a bank of experiments using pre-assembled ECM. MCF10As cultured on pre-assembled ECM in the presence of TGF-β1could be induced to undergo EMT in the absence of additional TGF-β1. MCF10As were cultured with or without TGF-β1 for 48 hours, at which point cells were extracted from the ECM (Fig. 8A). New MCF10As were then grown on the pre-assembled ECM and cultured for an additional 24 hours, in the absence of active TGF-β1. Cells cultured on ECM that was assembled by TGF-β1-treated cells appeared to be transformed, as indicated by disrupted adherens junctions and reorganized F-actin stress fibers, compared to those cultured on pre-formed ECM cultured in the absence of TGF-β1 (Fig. 8B). These results indicate that pre-assembled ECM from cells cultured with TGF-β1 have the ability to transform cells without additional growth factor treatment.
Fig 8.
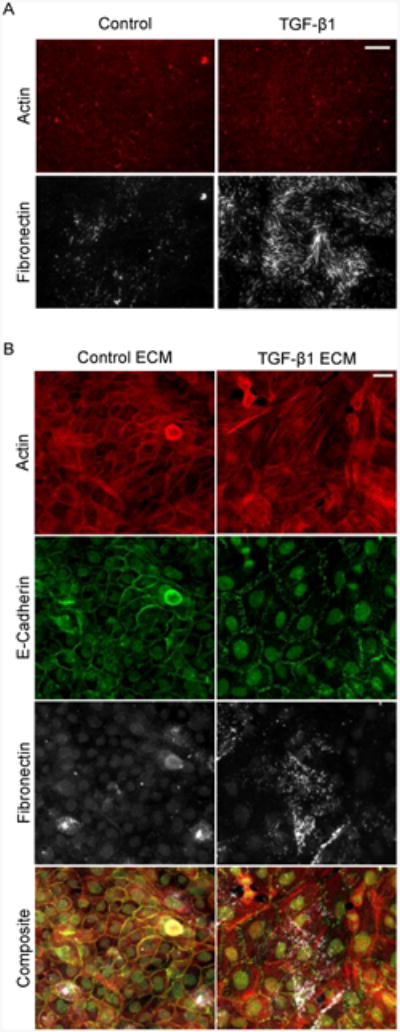
MCF10As on cell-extracted ECM. (A) Representative immunofluorescence images of the resulting MCF10A matrix after treatment with or without TGF-β1 for 48 hours. (B) Representative immunofluorescence images of MCF10As cultured on pre-assembled ECM from cells treated with or without TGF-β1 for 48 hours and cultured for an additional 24 hours. Ab staining for F-actin (red), E-cadherin (green), and FN (white). Scale bar is 20 μm.
3. Discussion
TGF-β1 has been implicated as a driver of EMT and inducer of soluble FN expression; here we demonstrate that TGF-β1-induced EMT is associated with an increase in FN fibril assembly and that blocking FN assembly with a bacterial protein fragment can significantly inhibit aspects of EMT, including adherens junction disassembly and increased expression of mesenchymal markers. Our studies further demonstrate that EMT cannot be induced in epithelial cells simply by increasing exogenous FN, increasing contractile forces, or by the presence of FN fibrils. It is worth noting that isolating FN fibril signaling and contractile force signaling is difficult; the two are symbiotic and previous studies from our group and others have shown that the two have reciprocal effects [22, 82] as such, inhibiting FN will alter forces and vice versa. However, the current study suggests that force changes alone are not sufficient to induce EMT. Furthermore, we demonstrate that latent TGF-β1 complex expression is upregulated and localized to FN fibrils upon treatment with exogenous active TGF-β1, and that inhibiting this interaction with either a FN blocking antibody or a FN deletion mutant that lacks the growth factor binding domains of FN inhibits aspects of EMT, including mesenchymal marker expression and actin reorganization.
Taken together, these data indicate that TGF-β1 induces FN fibril formation and increases the expression of endogenous latent TGF-β1 complex, then localizes new latent TGF-β1 to FN fibrils. While this provides a novel insight into a mechanism by which an ECM protein facilitates soluble growth factor signaling, there are still many questions as to how the signaling events occur. First, the results suggest a two-staged signaling event: that is, the initial addition of exogenous active TGF-β1 must induce the assembly of FN, but only activates EMT pathways once FN assembly has proceeded. We hypothesize that the first stage of signaling is actin rearrangement and that this requires low concentrations of TGF-β1 to be initiated; indeed, FUD inhibition blocks most aspects of EMT, but appears to have the least effect on actin rearrangement, suggesting that this may be upstream of FN fibril assembly. Once assembled, the TGF-β1 signal is amplified, which is presumably necessary for EMT signaling. Of interest is the source of TGF-β1 in the FN localization data (Fig, 5); by labeling LTBP-1, we only label TGF-β1 complex that has been secreted by the cells and not TGF-β1 that is exogenously added (this TGF-β1 is the active conformation, which is cleaved from LTBP-1). This indicates that the addition of exogenous TGF-β1 is inducing an increased expression and/or secretion of endogenous TGF-β1. Future studies will delve into the temporal dynamics and positive feedback mechanisms of this system.
Also of interest is the potential mechanism of how localized growth factors upregulate signaling pathways: we have hypothesized three potential mechanisms and will investigate these in future studies: 1) clustered growth factors have an increased local concentration, which increases the frequency of receptor-ligand binding; 2) clustered growth factors lead to increased clustering of receptors, thus upregulating the frequency of receptor autophosphorylation; and/or 3) ECM-bound growth factors are internalized and endocytosed at a lower rate, resulting in an elevated time of signal transduction. This third mechanism is the most intriguing to us; published models have suggested that this may be an important signaling mechanism in VEGF [83] and we plan to extensively investigate this possibility by probing signal transduction from tethered TGF-β1 substrates.
Several papers have suggested that FN alone can drive EMT [84, 85]. However, we have shown that in breast epithelial cells, this does not occur. In previous studies, cells were plated onto FN as opposed to our studies in which we plate on laminin. We have repeated these experiments on FN-coated coverslips and observe similar effects to what is observed on laminin coated coverslips (data not shown). There are several possible explanations for these conflicting results, but the most plausible is that there is a difference in the composition of growth factors secreted from the cells in other studies; that is, if the cells in question secrete elevated levels of TGF-β1 (or other FN-binding, EMT-inducing growth factors), then the mere presence of FN fibrils may be sufficient to induce EMT. Future studies will also investigate the broad applicability of these findings to both cells from other tissues as well as other growth factor signaling pathways.
The effectiveness of FUD in disrupting EMT indicates a potential therapeutic intervention for growing solid tumors and/or fibrotic diseases. By blocking FN assembly and/or growth factor localization, the growth factor expression of resident cells is not disrupted; instead, the mechanism of clustering and localization at the cell surface is disrupted. This may be a strategy that allows for global blockage of disease progression in diseases associated with EMT (such as cancer [86, 87], cirrhosis [88], or kidney fibrosis [89], regardless of which particular growth factor signaling pathway is misregulated.
4. Materials and Methods
4.1. Cell Culture and Reagents
All cells were cultured in a humidified atmosphere at 37°C with 5% CO2. Human MCF10A mammary epithelial cells were obtained from the National Cancer Institute Physical Sciences in Oncology Bioresource Core Facility, in conjunction with American Type Culture Collection (Manassas, VA). MDCK II cells were a gift from Dr. Daniel Conway (VCU Biomedical Engineering, Richmond, VA). MCF10As were maintained under standard culture conditions in DMEM/F-12 HEPES (Life Technologies, Carlsbad, CA), supplemented with 5% horse serum, 0.05% hydrocortisone, 0.01% cholera toxin, 0.1% insulin, 0.02% EGF and 1% antibiotics. MDCK II cells were maintained under standard culture conditions in DMEM (Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum and 1% antibiotics. Purified recombinant active TGF-β1 was purchased from Sigma Aldrich (St. Louis, MO). Immunofluorescence imaging was conducted using primary antibodies: Ms anti-Hu E-cadherin (HECD-1, Abcam, Cambridge, United Kingdom), Rb anti-Hu FN (Abcam, Cambridge, United Kingdom), Ms anti-Hu FN (Sigma Aldrich, St. Louis, MO), Ms anti-Hu III12-14 FN (Thermo Scientific, Rockford, IL), Ms anti-Hu LTBP-1 (R&D Systems, Minneapolis, MN), Rb anti-Hu Smad2 (86F7, Cell Signaling Technology, Danvers, MA), and Rb anti-Ki-67 (Santa Cruz Biotechnology, Dallas Texas). F-actin images were acquired by labeling cells with AlexaFluor555 Phalloidin (Life Technologies, Carlsbad, CA). Western blot analysis was conducted using primary antibodies against Twist (Abcam, Cambridge, United Kingdom), Vimentin (Cell Signaling Technology, Danvers, MA) and E-cadherin (HECD-1, Abcam, Cambridge, United Kingdom).
4.2. Immunofluorescence imaging
For immunofluorescence experiments, glass coverslips were ethanol washed and coated with 50 μg/ml laminin (Sigma Aldrich, St. Louis MO) for 2 hours at 37 °C. MCF10A and MDCKII cells were plated on laminin-coated 12-mm glass coverslips at densities resulting in near confluent monolayers. Cells were grown overnight then EGF/serum starved for 2 hours. Incubated with appropriate inhibitors/stimulators for 1 hour was then followed by treatment with or without 2 ng/mL of TGF-β1 for an additional 48 hours. Cells were permeabilized with 0.5% Triton in 4% paraformaldehyde for 2 minutes, then incubated in 4% paraformaldehyde for 20 minutes. Several PBS-rinses were performed, followed by blocking in 0.1% BSA and labeling with primary antibody for 30 minutes at 37 °C. Cells were then blocked again in 0.1% BSA and incubated with the appropriate secondary antibody for 30 minutes. Images were acquired on a Zeiss AxioObserver Z1 fluorescence microscope using ZEN2011 software.
4.3. Protein quantification
MCF10As were plated on laminin-coated 25-mm glass coverslips at densities resulting in near confluent monolayers. Cells were treated as described above. After 48 hours, cells were lysed with l mL of RIP A Buffer (Thermo Scientific, Rockford, IL) containing 1% Halt Protease Inhibitor (Thermo Scientific, Rockford, IL) for one minute. The protein solution was quantified with a BCA protein assay (Thermo Scientific, Rockford, IL) and a NanoDrop 2000 uv-vis spectrophotometer (ThermoFisher, Rockford, IL). Standard polyacrylamide gel electrophoresis and Western blot procedures were employed using the BIO-RAD Mini Trans-Blot Electrophoretic Transfer Cell apparatus, Any-kD, 4-15%, and 7.5% bis-acrylamide crosslinked TGX Stain-Free gels and polyvinylidene fluoride (PVDF) microporous membranes. Chemiluminescent images of the PVDF membranes were captured via the BIO-RAD ChemiDoc Touch Imaging System. Total protein normalization was performed on post-transfer membranes and quantification of western blotting signals was conducted with ImageLab software.
4.4. Expression and purification of FUD
cDNA for FUD was inserted into a bacterial expression vector that contains a C-terminal polyhistidine tag and maltose binding protein (MBP), both of which facilitate protein purification. cDNA was obtained from Dr. Harold Erickson, Duke University Medical Center, with permission from Dr. Deane Mosher at the University of Wisconsin. FUD expression, purification and determination of recombinant protein concentration has been previously described [9]. FN fibril formation was inhibited by addition of 125 nM FUD in MCF10As and 300 nM FUD in MDCK II cells.
4.5 Expression and purification of FN lacking growth factor binding domains III 11-14
FN/Δ11-14 cDNA was a gift from Dr. Harold Erickson (Duke University) and was expressed in Human Embryonic Kidney 293 cells (HEK293) as previously described [90]. Briefly, FN/Δ11-14 DNA was incubated with poly-ethylimine (PEI) at a 1:2 ratio for 10 min, and transfected into Human Embryonic Kidney 293 cells in serum-free Hybridoma media. Cells were incubated for 10 days, and conditioned media was collected from the cell cultures. FN/Δ11-14 was isolated from conditioned media by flowing over a gelatin agarose column, which binds FN via its gelatin binding domain. The column was then washed with PBS, and FN/Δ11-14 was eluted with 1 M L-arginine. L-arginine was subsequently removed by buffer exchange via PD-10 desalting columns. Expression was confirmed by polyacrylamide gel electrophoresis on a 7.5% bis-acrylamide crosslinked TGX Stain-Free gel.
4.6. Migration assay
A two-day migration assay was conducted according to xCELLigence RTCA DP System's general cell migration protocol. MCF10As were EGF starved in low serum (0.5%) overnight and then harvested in 0.05% trypsin and trypsin neutralizing solution. The CIM-Plate 16 is a modified Boyden chamber that contains three essential components, a lid, an upper chamber, equipped with a microporous polyethylene terephthalate (PET) membrane and a gold microelectrode sensor and a lower chamber, which easily and securely fit together. The upper chamber (containing low serum/EGF free media) and lower chamber (containing media with normal serum/EGF levels to serve as a chemoattractant) of the CIM-Plate 16 were equilibrated with the appropriate combination of 2 ng/mL TGF-β1 and 125 nM FUD to avoid creating a gradient of either additive. Approximately 0.4×105 cells were seeded in each well. The four conditions (with or without FUD &/or TGF-β1) were carried out in quadruplicate. Cells were either stimulated or inhibited to travel through the membrane (average pore size 8 um) on the bottom of the upper chamber into the well of the lower chamber by the chemoattractant and combination of FUD/TGF-β1. Migration was monitored for 48 hours and the RTCA DP Instrument recorded cell index. Once the cells translocated from the upper chamber through the microporous membrane, they adhered to the microelectrode sensor surface before migrating to the lower chamber. The microelectrode sensor generated higher impedance values as more cells came into contact, which corresponded to higher cell index levels. These values were sensed by the RTCA DP analyzer, which was placed inside a tissue culture incubator along with the CIM-Plate 16.
4.7. Ordinal scale scoring of images
To quantify the effect of fibrillogenesis on the initiation of EMT by TGF-β1, we analyzed immunofluorescence image morphology via an ordinate scale. Three independent scientists were asked to blindly rank immunofluorescence images for each experimental condition. Scores ranged from 1 to 4 and were based on separate criteria for actin images and E-cadherin images. For actin images a score of 1 corresponded to cortical actin surrounding the periphery of the cell, a score of 2 corresponded to mostly cortical actin, some stress fiber assembly throughout the middle of the cell, a score of 3 corresponded to 50 percent cortical actin and 50 percent randomly oriented stress fibers and a score of 4 corresponded to distinct stress fibers oriented parallel to one another, across multiple cells, with no cortical actin. For the E-cadherin images a score of 1 corresponded to E-cadherin within a thin line around each cell with the formation of a uniform sheets of cells, a score of 2 corresponded to some E-cadherin drifting towards the nucleus with junctions partially intact, a score of 3 corresponded to a zigzag pattern around the junctions and a score of 4 corresponded to complete disassembly with no E-cadherin surrounding the cell.
4.8. FN fibril quantification
To quantify FN fibrillogenesis, immunofluorescence images of FN were analyzed via an author-written image processing algorithm in Matlab that creates a binary mask of each FN image and determines the total area occupied by FN fibrils per image. These values were normalized by the total image size.
4.9. Average cell area quantification
To quantify the average cell area, immunofluorescence images of F-actin and nuclei were analyzed via an author-written image processing algorithm in Matlab. Total cell area was determined through creation of a binary mask of each F-actin image followed by calculation of the total area occupied by F-actin per image. Cell number was determined by the total number of nuclei per image. The algorithm then divided the total cell area by the cell number to generate the average cell area.
4.10. Nuclear signaling quantification
Smad2 and Ki67 were measured based on localization within the cell nucleus. Immunofluorescence images of Smad2 or Ki67 and nuclei were analyzed via an author-written image processing algorithm in Matlab. The intensity of the protein inside the nuclei was quantified using mean intensity within the nuclear region, as defined by the complementary nuclei image. Cytoplasmic intensity was determined by the mean intensity of the protein outside of the nuclear boundaries. Nuclear colocalization is represented by the ratio of nuclear intensity to cytoplasmic intensity.
4.11. Cell density quantification
Values for proliferation were based on correlation with the nuclei count. Immunofluorescent images of nuclei were analyzed via an automated counting function in ImageJ.
4.12 Cell extraction from extracellular matrix
MCF10As were plated on laminin-coated 12-mm glass coverslips at densities resulting in near-confluent monolayers. Cells were treated as described in Section 4.2 with or without 2 ng/mL TGF-β1 and cultured for 48 hours. Cell extraction was performed as described in [91]. Briefly, cells were treated with extraction buffer (0.5% (v/v) Triton-X-100, and 20 nM NH4OH in PBS) for 5 minutes, followed by gentile PBS rinsing. Samples were either 1) fixed with 1% gluteraldehyde and 1M ethanolamine for 30 minutes each, rinsed with PBS, treated with 1mg/mL sodium borohydride to reduce autofluorescence due to glutaraldehyde and stained for F-actin and fibronectin as described in 4.2, or 2) cultured with new MCF10As [92]. Cells cultured on pre-assembled ECM were EGF and serum starved 3-6 hours after plating and grown for an additional 24 hours, at which point they were fixed and stained as described in 4.2.
4.13 Real time quantitative PCR analysis
Total RNA was collected using an RNeasy kit (Qiagen). cDNA was converted using reverse transcriptase from an iScript cDNA Synthesis Kit (Bio-rad). The cDNA was selectively amplified by predesigned primer sets to the gene of interest (IDT). An 18s specific primer set (IDT) was used as a normalization control. A Power SYBR reporter (Fisher) was used for the qRT-PCR assays which were measured using a CFX Connect Real-time System (Bio-rad). Experiments were completed using the comparative Ct model in triplicate and analyzed by normalizing the values to the -/- control using Microsoft Excel and Bio-rad CFX Manager.
Supplementary Material
Highlights.
Inhibition of FN fibril formation blocks Epithelial-Mesenchymal Transition (EMT)
Increasing soluble FN or assembling FN fibrils without TGF--b1 doesn't induce EMT
Inhibiting the localization of TGF--b1 to assembled FN fibrils blocks EMT
These suggest that FN fibrils facilitate EMT by clustering TGF--b1 at the cell surface
Acknowledgments
The authors wish to thank Dr. Daniel Conway, Jiten Narang and Lewis Scott for thoughtful discussions and suggestions on this work. We would like to thank Dr. Harold Erickson for the generous gift of the FUD cDNA and FN/D11-14 cDNA, and we would also like to thank Dr. Daniel Conway for the gift of the MDCK II cells. This work was supported in part by the National Science Foundation under award number CMMI-1537168, the National Institutes of Health under NRSA Award Number F31GM121035 (LAG), the VCU Dean's Undergraduate Research Initiative (RSM), the Initiative for Maximizing Student Diversity (BAM) and the Alice T. and William H. Goodwin Endowment for Undergraduate Education (NTH).
Abbreviations
- EMT
Epithelial-Mesenchymal Transition
- FN
Fibronectin
- LTBP-1
Latent Transforming Growth Factor-Binding Protein 1
- TGF-β1
Transforming Growth Factor-Beta 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nature reviews Molecular cell biology. 2006;7(2):131–42. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 2.Hay ED. The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it. Developmental dynamics : an official publication of the American Association of Anatomists. 2005;233(3):706–720. doi: 10.1002/dvdy.20345. [DOI] [PubMed] [Google Scholar]
- 3.Yan C, Grimm WA, Garner WL, Qin L, Travis T, Tan N, Han YP. Epithelial to mesenchymal transition in human skin wound healing is induced by tumor necrosis factor-alpha through bone morphogenic protein-2. Am J Pathol. 2010;176(5):2247–58. doi: 10.2353/ajpath.2010.090048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shook D, Keller R. Mechanisms, mechanics and function of epithelial-mesenchymal transitions in early development. Mechanisms of development. 2003;120(11):1351–83. doi: 10.1016/j.mod.2003.06.005. [DOI] [PubMed] [Google Scholar]
- 5.Nieto MA. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annual review of cell and developmental biology. 2011;27:347–76. doi: 10.1146/annurev-cellbio-092910-154036. [DOI] [PubMed] [Google Scholar]
- 6.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 7.Hay ED. An overview of epithelio-mesenchymal transformation. Acta anatomica. 1995;154(1):8–20. doi: 10.1159/000147748. [DOI] [PubMed] [Google Scholar]
- 8.Bhowmick NA, Ghiassi M, Bakin A, Aakre M, Lundquist CA, Engel ME, Arteaga CL, Moses HL. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell. 2001;12(1):27–36. doi: 10.1091/mbc.12.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell research. 2009;19(2):156–72. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nature medicine. 2009;15(9):1010–2. doi: 10.1038/nm0909-1010. [DOI] [PubMed] [Google Scholar]
- 11.Massagué J. TGFbeta in Cancer. Cell. 2008;134(2):215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thiery JP. Epithelial–mesenchymal transitions in development and pathologies. Current Opinion in Cell Biology. 2003;15(6):740–746. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 13.Derynck R, Akhurst RJ. Differentiation plasticity regulated by TGF-beta family proteins in development and disease. Nature cell biology. 2007;9(9):1000–1004. doi: 10.1038/ncb434. [DOI] [PubMed] [Google Scholar]
- 14.Mats G, Åsa F, Jan-Olof K, Lars EE, Nils-Erik H, Mikael N. Transforming growth factor-β and epidermal growth factor synergistically stimulate epithelial to mesenchymal transition (EMT) through a MEK-dependent mechanism in primary cultured pig thyrocytes. Journal of cell science. 2002;115(22):4227–4236. doi: 10.1242/jcs.00091. [DOI] [PubMed] [Google Scholar]
- 15.Hui-Wen L, Sheng-Chieh H, Weiya X, Xinyu C, Jin-Yuan S, Yongkun W, James LA, Gabriel NH, Mien-Chie H. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer research. 2007;67(19):9066–9076. doi: 10.1158/0008-5472.CAN-07-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aristidis M, Carl-Henrik H. Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Science. 2007;98(10) doi: 10.1111/j.1349-7006.2007.00550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Connor JW, Gomez EW. Biomechanics of TGFβ-induced epithelial-mesenchymal transition: implications for fibrosis and cancer. Clinical and translational medicine. 2013;3:23. doi: 10.1186/2001-1326-3-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O'Connor JW, Riley PN, Nalluri SM, Ashar PK, Gomez EW. Matrix Rigidity Mediates TGFbeta1-Induced Epithelial-Myofibroblast Transition by Controlling Cytoskeletal Organization and MRTF-A Localization. Journal of cellular physiology. 2015;230(8):1829–39. doi: 10.1002/jcp.24895. [DOI] [PubMed] [Google Scholar]
- 19.McGrail DJ, Kieu QM, Dawson MR. The malignancy of metastatic ovarian cancer cells is increased on soft matrices through a mechanosensitive Rho-ROCK pathway. Journal of cell science. 2014;127(Pt 12):2621–6. doi: 10.1242/jcs.144378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee K, Chen QK, Lui C, Cichon MA, Radisky DC, Nelson CM. Matrix compliance regulates Rac1b localization, NADPH oxidase assembly, and epithelial-mesenchymal transition. Mol Biol Cell. 2012;23(20):4097–108. doi: 10.1091/mbc.E12-02-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Markowski MC, Brown AC, Barker TH. Directing epithelial to mesenchymal transition through engineered microenvironments displaying orthogonal adhesive and mechanical cues. Journal of biomedical materials research Part A. 2012;100(8):2119–27. doi: 10.1002/jbm.a.34068. [DOI] [PubMed] [Google Scholar]
- 22.Brown AC, Fiore VF, Sulchek TA, Barker TH. Physical and chemical microenvironmental cues orthogonally control the degree and duration of fibrosis-associated epithelial-to-mesenchymal transitions. The Journal of pathology. 2013;229(1):25–35. doi: 10.1002/path.4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leight JL, Wozniak MA, Chen S, Lynch ML, Chen CS. Matrix rigidity regulates a switch between TGF-beta1-induced apoptosis and epithelial-mesenchymal transition. Mol Biol Cell. 2012;23(5):781–91. doi: 10.1091/mbc.E11-06-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spencer CW, Laurent F, Jeff HT, Yurong G, Vincent HP, Hannah EM, Albert CC, Robert LS, Susan ST, Adam JE, Jing Y. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nature Cell Biology. 2015;17(5) doi: 10.1038/ncb3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hinz B. Tissue stiffness, latent TGF-beta1 activation, and mechanical signal transduction: implications for the pathogenesis and treatment of fibrosis. Current rheumatology reports. 2009;11(2):120–126. doi: 10.1007/s11926-009-0017-1. [DOI] [PubMed] [Google Scholar]
- 26.Gomez EW, Chen QK, Gjorevski N, Nelson CM. Tissue geometry patterns epithelial-mesenchymal transition via intercellular mechanotransduction. Journal of cellular biochemistry. 2010;110(1):44–51. doi: 10.1002/jcb.22545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giacomini M, Travis M, Kudo M, Sheppard D. Epithelial cells utilize cortical actin/myosin to activate latent TGF-β through integrin α(v)β(6)-dependent physical force. Experimental cell research. 2012;318(6):716–722. doi: 10.1016/j.yexcr.2012.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masszi A, Di Ciano C, Sirokmány G, Arthur WT, Rotstein OD, Wang J, McCulloch CA, Rosivall L, Mucsi I, Kapus A. Central role for Rho in TGF-beta1-induced alpha-smooth muscle actin expression during epithelial-mesenchymal transition. American journal of physiology Renal physiology. 2003;284(5):24. doi: 10.1152/ajprenal.00183.2002. [DOI] [PubMed] [Google Scholar]
- 29.Gjorevski N, Boghaert E, Nelson CM. Regulation of Epithelial-Mesenchymal Transition by Transmission of Mechanical Stress through Epithelial Tissues. Cancer microenvironment : official journal of the International Cancer Microenvironment Society. 2012;5(1):29–38. doi: 10.1007/s12307-011-0076-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326(5957):1216–9. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen CS, Tan J, Tien J. Mechanotransduction at cell-matrix and cell-cell contacts. Annual review of biomedical engineering. 2004;6:275–302. doi: 10.1146/annurev.bioeng.6.040803.140040. [DOI] [PubMed] [Google Scholar]
- 32.Butcher DT, Alliston T, Weaver VM. A tense situation: forcing tumour progression. Nature reviews Cancer. 2009;9(2):108–122. doi: 10.1038/nrc2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Geiger B, Bershadsky A, Pankov R, Yamada KM. Transmembrane crosstalk between the extracellular matrix--cytoskeleton crosstalk. Nature reviews Molecular cell biology. 2001;2(11):793–805. doi: 10.1038/35099066. [DOI] [PubMed] [Google Scholar]
- 34.Pankov R. Fibronectin at a glance. Journal of cell science. 2002;115(20):3861–3863. doi: 10.1242/jcs.00059. [DOI] [PubMed] [Google Scholar]
- 35.Jill CS, Elof E, Steven PS, Noelynn O. Fibronectin gene expression differs in normal and abnormal human wound healing. Wound Repair and Regeneration. 1994;2(1) doi: 10.1046/j.1524-475X.1994.20104.x. [DOI] [PubMed] [Google Scholar]
- 36.Clark RA. Fibronectin matrix deposition and fibronectin receptor expression in healing and normal skin. The Journal of investigative dermatology. 1990;94(6 Suppl) doi: 10.1111/1523-1747.ep12876104. [DOI] [PubMed] [Google Scholar]
- 37.George EL, Georges-Labouesse EN, Patel-King RS, Rayburn H, Hynes RO. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development. 1993;119(4):1079–91. doi: 10.1242/dev.119.4.1079. [DOI] [PubMed] [Google Scholar]
- 38.Takayoshi S, Melinda L, Kenneth MY. Fibronectin requirement in branching morphogenesis. Nature. 2003;423(6942):876–881. doi: 10.1038/nature01712. [DOI] [PubMed] [Google Scholar]
- 39.Bae YK, Kim A, Kim MK, Choi JE, Kang SH, Lee SJ. Fibronectin expression in carcinoma cells correlates with tumor aggressiveness and poor clinical outcome in patients with invasive breast cancer. Hum Pathol. 2013 doi: 10.1016/j.humpath.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 40.Castellani P, Siri A, Rosellini C, Infusini E, Borsi L, Zardi L. Transformed human cells release different fibronectin variants than do normal cells. The Journal of cell biology. 1986;103(5):1671–7. doi: 10.1083/jcb.103.5.1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Svante S, Antti V. Fibronectin in human solid tumors. International Journal of Cancer. 1981;27(4):427–435. doi: 10.1002/ijc.2910270403. [DOI] [PubMed] [Google Scholar]
- 42.Kadar A, Tõkés AMM, Kulka J, Robert L. Extracellular matrix components in breast carcinomas. Seminars in cancer biology. 2002;12(3):243–257. doi: 10.1016/s1044-579x(02)00027-5. [DOI] [PubMed] [Google Scholar]
- 43.Zhong C, Chrzanowska-Wodnicka M, Brown J, Shaub A, Belkin AM, Burridge K. Rho-mediated contractility exposes a cryptic site in fibronectin and induces fibronectin matrix assembly. The Journal of cell biology. 1998;141(2):539–51. doi: 10.1083/jcb.141.2.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baneyx G, Baugh L, Vogel V. Fibronectin extension and unfolding within cell matrix fibrils controlled by cytoskeletal tension. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(8):5139–5143. doi: 10.1073/pnas.072650799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martino MM, Hubbell JA. The 12th-14th type III repeats of fibronectin function as a highly promiscuous growth factor-binding domain. FASEB J. 2010;24(12):4711–21. doi: 10.1096/fj.09-151282. [DOI] [PubMed] [Google Scholar]
- 46.Dallas SL, Keene DR, Bruder SP, Saharinen J, Sakai LY, Mundy GR, Bonewald LF. Role of the latent transforming growth factor beta binding protein 1 in fibrillin-containing microfibrils in bone cells in vitro and in vivo. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2000;15(1):68–81. doi: 10.1359/jbmr.2000.15.1.68. [DOI] [PubMed] [Google Scholar]
- 47.Dallas SL, Sivakumar P, Jones CJ, Chen Q, Peters DM, Mosher DF, Humphries MJ, Kielty CM. Fibronectin regulates latent transforming growth factor-beta (TGF beta) by controlling matrix assembly of latent TGF beta-binding protein-1. The Journal of biological chemistry. 2005;280(19):18871–80. doi: 10.1074/jbc.M410762200. [DOI] [PubMed] [Google Scholar]
- 48.Chen Q, Sivakumar P, Barley C, Peters DM, Gomes RR, Farach-Carson MC, Dallas SL. Potential role for heparan sulfate proteoglycans in regulation of transforming growth factor-beta (TGF-beta) by modulating assembly of latent TGF-beta-binding protein-1. The Journal of biological chemistry. 2007;282(36):26418–30. doi: 10.1074/jbc.M703341200. [DOI] [PubMed] [Google Scholar]
- 49.Taipale J, Miyazono K, Heldin CH, Keski-Oja J. Latent transforming growth factor-beta 1 associates to fibroblast extracellular matrix via latent TGF-beta binding protein. The Journal of cell biology. 1994;124(1-2):171–81. doi: 10.1083/jcb.124.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jeng MH, ten Dijke P, Iwata KK, Jordan VC. Regulation of the levels of three transforming growth factor beta mRNAs by estrogen and their effects on the proliferation of human breast cancer cells. Molecular and cellular endocrinology. 1993;97(1-2):115–23. doi: 10.1016/0303-7207(93)90217-8. [DOI] [PubMed] [Google Scholar]
- 51.Arrick BA, Korc M, Derynck R. Differential regulation of expression of three transforming growth factor beta species in human breast cancer cell lines by estradiol. Cancer research. 1990;50(2):299–303. [PubMed] [Google Scholar]
- 52.Martinez-Carpio PA, Mur C, Fernandez-Montoli ME, Ramon JM, Rosel P, Navarro MA. Secretion and dual regulation between epidermal growth factor and transforming growth factor-beta1 in MDA-MB-231 cell line in 42-hour-long cultures. Cancer letters. 1999;147(61-2):25–9. doi: 10.1016/s0304-3835(99)00261-x. [DOI] [PubMed] [Google Scholar]
- 53.Wikner NE, Persichitte KA, Baskin JB, Nielsen LD, Clark RA. Transforming growth factor-beta stimulates the expression of fibronectin by human keratinocytes. The Journal of investigative dermatology. 1988;91(3):207–12. doi: 10.1111/1523-1747.ep12464997. [DOI] [PubMed] [Google Scholar]
- 54.Ignotz RA, Endo T, Massague J. Regulation of fibronectin and type I collagen mRNA levels by transforming growth factor-beta. The Journal of biological chemistry. 1987;262(14):6443–6. [PubMed] [Google Scholar]
- 55.Roberts CJ, Birkenmeier TM, McQuillan JJ, Akiyama SK, Yamada SS, Chen WT, Yamada KM, McDonald JA. Transforming growth factor beta stimulates the expression of fibronectin and of both subunits of the human fibronectin receptor by cultured human lung fibroblasts. The Journal of biological chemistry. 1988;263(10):4586–92. [PubMed] [Google Scholar]
- 56.Ignotz RA, Massague J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. The Journal of biological chemistry. 1986;261(9):4337–45. [PubMed] [Google Scholar]
- 57.Tomasini-Johansson BR, Kaufman NR, Ensenberger MG, Ozeri V, Hanski E, Mosher DF. A 49-residue peptide from adhesin F1 of Streptococcus pyogenes inhibits fibronectin matrix assembly. The Journal of biological chemistry. 2001;276(26):23430–9. doi: 10.1074/jbc.M103467200. [DOI] [PubMed] [Google Scholar]
- 58.Altrock E, Sens C, Wuerfel C, Vasel M, Kawelke N, Dooley S, Sottile J, Nakchbandi IA. Inhibition of fibronectin deposition improves experimental liver fibrosis. Journal of hepatology. 2015;62(3):625–33. doi: 10.1016/j.jhep.2014.06.010. [DOI] [PubMed] [Google Scholar]
- 59.Christine G, Erik WT. The Epithelial to Mesenchymal Transition and Metastatic Progression in Carcinoma. The Breast Journal. 1996;2(1) [Google Scholar]
- 60.Kang Y, Massague J. Epithelial-mesenchymal transitions: twist in development and metastasis. Cell. 2004;118(3):277–9. doi: 10.1016/j.cell.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 61.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117(7):927–39. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 62.Massague J. How cells read TGF-beta signals. Nature reviews Molecular cell biology. 2000;1(3):169–78. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 63.Derynck R, Feng XH. TGF-beta receptor signaling. Biochimica et biophysica acta. 1997;1333(2):F105–50. doi: 10.1016/s0304-419x(97)00017-6. [DOI] [PubMed] [Google Scholar]
- 64.Lamouille S, Derynck R. Cell size and invasion in TGF-beta-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. The Journal of cell biology. 2007;178(3):437–51. doi: 10.1083/jcb.200611146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barnard JA, Bascom CC, Lyons RM, Sipes NJ, Moses HL. Transforming growth factor beta in the control of epidermal proliferation. The American journal of the medical sciences. 1988;296(3):159–63. doi: 10.1097/00000441-198809000-00003. [DOI] [PubMed] [Google Scholar]
- 66.Moses H, Barcellos-Hoff MH. TGF-beta biology in mammary development and breast cancer. Cold Spring Harb Perspect Biol. 2011;3(1):a003277. doi: 10.1101/cshperspect.a003277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. The Journal of clinical investigation. 2009;119(6):1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Peters DM, Portz LM, Fullenwider J, Mosher DF. Co-assembly of plasma and cellular fibronectins into fibrils in human fibroblast cultures. The Journal of cell biology. 1990;111(1):249–56. doi: 10.1083/jcb.111.1.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Haglund C, Ylatupa S, Mertaniemi P, Partanen P. Cellular fibronectin concentration in the plasma of patients with malignant and benign diseases: a comparison with CA 19-9 and CEA. Br J Cancer. 1997;76(6):777–83. doi: 10.1038/bjc.1997.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ylatupa S, Haglund C, Mertaniemi P, Vahtera E, Partanen P. Cellular fibronectin in serum and plasma: a potential new tumour marker? Br J Cancer. 1995;71(3):578–82. doi: 10.1038/bjc.1995.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Choate JJ, Mosher DF. Fibronectin concentration in plasma of patients with breast cancer, colon cancer, and acute leukemia. Cancer. 1983;51(6):1142–7. doi: 10.1002/1097-0142(19830315)51:6<1142::aid-cncr2820510628>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 72.Das T, Safferling K, Rausch S, Grabe N, Boehm H, Spatz JP. A molecular mechanotransduction pathway regulates collective migration of epithelial cells. Nat Cell Biol. 2015;17(3):276–87. doi: 10.1038/ncb3115. [DOI] [PubMed] [Google Scholar]
- 73.Wozniak MA, Desai R, Solski PA, Der CJ, Keely PJ. ROCK-generated contractility regulates breast epithelial cell differentiation in response to the physical properties of a three-dimensional collagen matrix. The Journal of cell biology. 2003;163(3):583–95. doi: 10.1083/jcb.200305010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Poincloux R, Collin O, Lizarraga F, Romao M, Debray M, Piel M, Chavrier P. Contractility of the cell rear drives invasion of breast tumor cells in 3D Matrigel. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(5):1943–8. doi: 10.1073/pnas.1010396108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ishihara H, Martin BL, Brautigan DL, Karaki H, Ozaki H, Kato Y, Fusetani N, Watabe S, Hashimoto K, Uemura D, et al. Calyculin A and okadaic acid: inhibitors of protein phosphatase activity. Biochemical and biophysical research communications. 1989;159(3):871–7. doi: 10.1016/0006-291x(89)92189-x. [DOI] [PubMed] [Google Scholar]
- 76.Kamaraju AK, Roberts AB. Role of Rho/ROCK and p38 MAP kinase pathways in transforming growth factor-beta-mediated Smad-dependent growth inhibition of human breast carcinoma cells in vivo. The Journal of biological chemistry. 2005;280(2):1024–36. doi: 10.1074/jbc.M403960200. [DOI] [PubMed] [Google Scholar]
- 77.Fleming YM, Ferguson GJ, Spender LC, Larsson J, Karlsson S, Ozanne BW, Grosse R, Inman GJ. TGF-beta-mediated activation of RhoA signalling is required for efficient (V12)HaRas and (V600E)BRAF transformation. Oncogene. 2009;28(7):983–93. doi: 10.1038/onc.2008.449. [DOI] [PubMed] [Google Scholar]
- 78.Zhong CL, Chrzanowska-Wodnicka M, Brown J, Shaub A, Belkin AM, Burridge K. Rho-mediated contractility exposes a cryptic site in fibronectin and induces fibronectin matrix assembly. Journal of Cell Biology. 1998;141(2):539–551. doi: 10.1083/jcb.141.2.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hubbard B, Buczek-Thomas JA, Nugent MA, Smith ML. Heparin-dependent regulation of fibronectin matrix conformation. Matrix biology : journal of the International Society for Matrix Biology. 2014;34:124–31. doi: 10.1016/j.matbio.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhu J, Clark RA. Fibronectin at select sites binds multiple growth factors and enhances their activity: expansion of the collaborative ECM-GF paradigm. The Journal of investigative dermatology. 2014;134(4):895–901. doi: 10.1038/jid.2013.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Johnson KJ, Sage H, Briscoe G, Erickson HP. The compact conformation of fibronectin is determined by intramolecular ionic interactions. The Journal of biological chemistry. 1999;274(22):15473–9. doi: 10.1074/jbc.274.22.15473. [DOI] [PubMed] [Google Scholar]
- 82.Scott LE, Mair DB, Narang JD, Feleke K, Lemmon CA. Fibronectin fibrillogenesis facilitates mechano-dependent cell spreading, force generation, and nuclear size in human embryonic fibroblasts. Integrative biology : quantitative biosciences from nano to macro. 2015;7(11):1454–65. doi: 10.1039/c5ib00217f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tan WH, Popel AS, Mac Gabhann F. Computational model of VEGFR2 pathway to ERK activation and modulation through receptor trafficking. Cell Signal. 2013;25(12):2496–510. doi: 10.1016/j.cellsig.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, Sheppard D, Chapman HA. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(35):13180–13185. doi: 10.1073/pnas.0605669103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Park J, Schwarzbauer JE. Mammary epithelial cell interactions with fibronectin stimulate epithelial-mesenchymal transition. Oncogene. 2013 doi: 10.1038/onc.2013.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Marcello G, Barbara R, Gianmario B. The role of epithelial-mesenchymal transition in cancer pathology. Pathology. 2007;39(3):305–318. doi: 10.1080/00313020701329914. [DOI] [PubMed] [Google Scholar]
- 87.Thompson EW, Newgreen DF, Tarin D. Carcinoma invasion and metastasis: a role for epithelial-mesenchymal transition? Cancer research. 2005;65(14):5991–5. doi: 10.1158/0008-5472.CAN-05-0616. discussion 5995. [DOI] [PubMed] [Google Scholar]
- 88.Wells RG. The epithelial-to-mesenchymal transition in liver fibrosis: here today, gone tomorrow? Hepatology. 2010;51(3):737–40. doi: 10.1002/hep.23529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Youhua L. Epithelial to mesenchymal transition in renal fibrogenesis: pathologic significance, molecular mechanism, and therapeutic intervention. Journal of the American Society of Nephrology : JASN. 2004;15(1):1–12. doi: 10.1097/01.asn.0000106015.29070.e7. [DOI] [PubMed] [Google Scholar]
- 90.Lemmon CA, Ohashi T, Erickson HP. Probing the folded state of fibronectin type III domains in stretched fibrils by measuring buried cysteine accessibility. The Journal of biological chemistry. 2011;286(30):26375–82. doi: 10.1074/jbc.M111.240028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Beacham DA, Amatangelo MD, Cukierman E. Preparation of extracellular matrices produced by cultured and primary fibroblasts. Current protocols in cell biology / editorial board, Juan S Bonifacino … et al. 2007;Chapter 10 doi: 10.1002/0471143030.cb1009s33. Unit 10 9. [DOI] [PubMed] [Google Scholar]
- 92.Beisker W, Dolbeare F, Gray JW. An improved immunocytochemical procedure for high-sensitivity detection of incorporated bromodeoxyuridine. Cytometry. 1987;8(2):235–9. doi: 10.1002/cyto.990080218. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.