

Abstract
The Runx1 transcription factor plays an important role in tissue homeostasis through its effects on stem/progenitor cell populations and differentiation. The effect of Runx1 on epithelial differentiation of the secretory cell lineage of the colon was recently demonstrated. This study aimed to examine the role of Runx1 in tumor development in epithelial cells of the gastrointestinal tract. Conditional knockout mice that lacked Runx1 expression in epithelial cells of the GI tract were generated. These mice were crossed onto the Apc Min background, killed and their intestinal tumor phenotypes were compared with Apc Min Runx1 wild‐type control mice. Apc‐wild‐type Runx1‐mutant mice were also examined for tumor development. Colons from Runx1 knockout and wild‐type mice were used for genome‐wide mRNA expression analyses followed by gene‐specific quantitative RT‐PCR of whole colon and colon epithelium to identify Runx1 target genes. Runx1 deficiency in intestinal epithelial cells significantly enhanced tumorigenesis in Apc Min mice. Notably, epithelial Runx1 deficiency in Apc‐wild‐type mice was sufficient to cause tumor development. Absence of Runx1 was associated with global changes in the expression of genes involved in inflammation and intestinal metabolism, and with gene sets indicative of a metastatic phenotype and poor prognosis. Gene‐specific analysis of Runx1‐deficient colon epithelium revealed increased expression of genes linked to an expansion of the stem/progenitor cell population. These results identify Runx1 as a novel tumor suppressor gene for gastrointestinal tumors and support a role for Runx1 in maintaining the balance between the intestinal stem/progenitor cell population and epithelial differentiation of the GI tract. (Cancer Sci 2012; 103: 593–599)
The human and mouse family of RUNX transcription factors are composed of three members, RUNX1, RUNX2 and RUNX3. They function as either transcriptional activators or repressors depending on the target gene, cell type and the presence of co‐factors. RUNX1 has been implicated in homeostasis of the hematopoietic stem cell number, as well as in developmental lineage specification, thereby being a critical factor for hematopoiesis and hematopoietic function.( 1 , 2 ) In mice, Runx1 is essential for mammalian development as germline knockouts (KO) are embryonic lethal due to a complete failure of hematopoiesis.( 3 ) In humans, polymorphisms that reduce the binding of RUNX1 to specific target genes have been associated with susceptibility to the autoimmune diseases psoriasis, systemic lupus erythematosus (SLE) and rheumatoid arthritis.( 4 ) Moreover, RUNX1 is frequently mutated in a common subtype of human acute myeloid leukemia (AML), where often the rate‐limiting step is formation of mutant fusion genes/proteins that combine the N‐terminal DNA‐binding domain of RUNX1 (RHD) with the full‐length activation domains of oncogenes.( 5 ) An example is the eight twenty‐one (ETO) t(8;21) translocation that is present in approximately 40% of human AML. Notably, these fusion proteins can function as dominant negative mutants that silence RUNX1 target genes leading to a loss of function of RUNX1 in AML.( 6 , 7 ) An example is derepression of vascular endothelial growth factor alpha (VEGFA) (an independent prognostic factor for relapse‐free survival in AML) expression in human AML caused by t(8;21) translocations.( 8 ) Knockin of a Runx1‐ETO fusion transgene causes embryonic lethality in mice similar to the germline Runx1 KO, further consistent with the fusion protein having a dominant negative effect on Runx1. ( 9 ) Small intragenic mutations in RUNX1 have also been reported to exert a dominant negative effect on RUNX1 function in AML,( 10 ) in myelodysplasia and familial platelet disorders that can lead to AML( 11 , 12 , 13 , 14 ) in chronic myelomonocytic leukemia that predisposes to AML( 15 ) and in T‐lymphoblastic lymphomas.( 10 )
Less is known about the role of RUNX1 in epithelial cancers. RUNX1 has been reported to be overexpressed in human endometrial cancer and in a mouse skin cancer model.( 16 ) In contrast, RUNX1 expression has been reported to be downregulated in some human pituitary cancers,( 17 ) recurrent deletions of RUNX1 were found in human pancreatic adenocarcinomas( 18 ) and homozygous deletions in RUNX1 have been observed in esophageal adenocarcinoma cell lines and xenografts.( 19 ) RUNX1 downregulation is associated with an increase in FOXO1 expression and hyperproliferation of breast cancer cells, and has been reported in hormone‐negative breast cancers lacking HER2 amplification.( 20 , 21 ) In the gastrointestinal tract, RUNX1 has been reported to be downregulated in human gastric cancer cells,( 22 ) and very recently, Runx1 was identified as an intestinal cancer susceptibility gene in a Sleeping Beauty transposon‐mediated mutagenesis screen in mice.( 23 )
The role of another family member, RUNX3, is better characterized in epithelial cancers as it has been reported to act as either an oncogene or tumor suppressor, with a predominance of reports describing its function as a tumor suppressor gene in several human epithelial cancers, such as colorectal, gastric, esophageal, lung, liver and melanomas.( 24 , 25 , 26 , 27 , 28 , 29 , 30 ) In the intestinal tract, dysregulation of RUNX3 is linked to both inflammation and cancer. Targeted Runx3 deficiency in mouse leukocytes leads to inflammatory bowel disease (IBD),( 31 , 32 ) while hypermethylation of the RUNX3 promoter in human intestinal epithelial cells has been observed in ulcerative colitis.( 33 , 34 ) Silencing of RUNX3 by hypermethylation has also been reported in human microsatellite instable/CpG island methylator phenotype (MSI/CIMP) colorectal cancer (CRC).( 35 , 36 , 37 ) Further, in the gastrointestinal (GI) tract, both RUNX1 and RUNX3 are directly involved in the transforming growth factor beta (TGFβ) signaling pathway and act to promote expression of TGFβ and its pathway genes such as BMPs and SMADs. ( 38 ) Thus, there is evidence that RUNX1 and RUNX3 expression, function and role in cancer overlap in some tissues, an observation that has been further supported by the recent report of single nucleotide polymorphisms (SNP) in both RUNX1 and RUNX3 that confer susceptibility to CRC.( 39 ) Finally, in contrast, a third RUNX family member, RUNX2, is proposed to largely function as an oncogene in bone and other cancers.( 5 , 40 )
Recently, inducible Mx‐Cre‐mediated conditional deletion of Runx1 in mouse colon resulted in decreased expression of Klf4 and goblet cell depletion, demonstrating a role for Runx1 in regulation of intestinal epithelial differentiation of the secretory cell lineage.( 41 ) Several lines of evidence indicate that RUNX1 can both positively and negatively interact with Notch signaling,( 42 , 43 , 44 ) which is important for the proper differentiation and organization of cells in the GI tract via crosstalk with Wnt/β‐catenin and other canonical signaling pathways.( 45 , 46 ) Upregulation of Notch signaling is implicated in both gastric and colorectal cancers, where it represses goblet cell differentiation.( 45 , 47 , 48 ) Inhibition of Notch activity converts proliferative enterocytes into goblet cells( 45 ) possibly via activation of KLF4 gene expression.( 49 )
Because of the reported role of RUNX1 in KLF4 signaling and goblet cell differentiation and its interaction with Notch signaling activity, here we investigated the role of epithelial expression of Runx1 in intestinal tumorigenesis. We found that deficiency of Runx1 significantly enhanced tumorigenesis in Apc Min mice, thus providing the first direct evidence that Runx1 is a tumor suppressor gene in the mammalian GI tract. Notably, Apc‐wild‐type Runx1 knockout mice also developed intestinal tumors when aged >10 months. Loss of epithelial expression of Runx1 was associated with increased expression in the whole colon of genes involved in inflammation and intestinal metabolism, and with gene sets indicative of a metastatic phenotype and poor prognosis. Gene‐specific analysis in Runx1‐deficient colonic epithelial cells revealed increased expression of several genes implicated in stem cells and Notch signaling.
Materials and Methods
Mice. C57BL/6J (later referred to as wildtype [WT]) and C57BL/6J‐Apc Min mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). C57BL/6‐Runx1 floxed mice( 3 ) were obtained from Dr Daniel Littman (New York University, New York, NY, USA). Villin‐Cre transgenic mice that are fully congenic on the C57BL/6 genetic background were obtained from a colony maintained in our laboratory. The Runx1 floxed allele was introgressed into an Apc Min strain of mice that carried a hemizygous copy of the Villin‐Cre transgene. For details of mouse husbandry see the Data S1.
Genotyping. DNA was isolated from tail snips using a Qiagen DNA Easy kit (Qiagen, Valencia, CA, USA). The genotype of the Apc, Villin‐Cre and Runx1 loci was determined by PCR assays as previously described.( 3 , 50 , 51 ) The PCR was performed on an Applied Biosystems GeneAmp 9700 Thermal Cycler (Applied Biosystems, Foster City, CA, USA).
Oligonucleotide microarray and data analysis. Two‐color microarray‐based gene expression analysis was carried out using Agilent 4x44K whole mouse genome microarrays (G4122F; Agilent Technologies, Wilmington, DE, USA). This array includes more than 41 000 mouse genes and transcripts. Per sample, 500 ng of RNA was spiked, labeled, amplified and hybridized using Agilent reagents, according to Agilent Gene Expression oligo microarrays protocol (version 5.0.1). For each group (Villin‐Cre transgenic and non‐transgenic), half of the samples were labeled with Cy3 and half of the samples with Cy5 to prevent bias of the data due to labeling with these fluorescent dyes. Arrays were scanned using an Agilent Microarray Scanner, and the intensity of fluorescent images was quantified using Agilent Feature Extraction software (version 9.5.1). Pre‐processing of data, quality checks and significance analysis were all performed as previously described.( 52 ) Differential expression of genes was quantified by the moderated t‐statistic. Gene Set Enrichment Analysis (GSEA) was used as a computational method to identify sets of genes with coordinate differences in gene expression (GSEA v2.0, http://www.broad.mit.edu/gsea/).( 53 , 54 ) Gene sets analyzed were obtained from the Molecular Signatures Database (MSigDB_v2.5, http://www.broad.mit.edu/gsea/msigdb/index.jsp) using settings as previously described.( 55 ) Gene expression data has been submitted to the Gene Expression Omnibus. Accession number GSE34292.
Statistical analysis. Two‐sided P‐values for tumor counts were determined using the Wilcoxon rank‐sum test comparing gender and age‐matched classes produced in the same genetic crosses.
Histopathology, immunohistochemistry, RNA extraction, isolation of intestinal epithelium, qRT‐PCR and tumor analysis (See Data S1, Supplemental Methods).
Results
Phenotype of intestinal‐specific Runx1‐deficient mice. Homozygous Runx1 floxed mice expressing the intestinal‐specific Villin‐Cre transgene were compared with Villin‐Cre‐negative littermates. We observed no differences in survival, gender ratio, growth, adult size or weight. Formalin‐fixed intestinal tissues from all regions were examined using histopathological analysis and no differences were discernable. Tissues were further tested via immunohistochemistry for Ki‐67 (cell proliferation), lysozyme (Paneth cells), Muc2 and Alcian blue (goblet cells) and no differences were observed (Fig. S1). Knockout of Runx1 expression was confirmed using immunohistochemistry (Fig. S2) and qRT‐PCR (Fig. S3).
Runx1 deficiency results in enhanced tumorigenesis in ApcMin mice. We tested the role of Runx1 in intestinal tumorigenesis by creating Apc Min mice that were homozygous for a Runx1 floxed allele and were either positive or negative for the intestinal‐specific Villin‐Cre transgene. Direct comparison of littermate test (Villin‐Cre positive, Runx1 KO) and control mice (Villin‐Cre negative, Runx1 wild type) provided strong evidence that Runx1 is a tumor suppressor in the mouse intestine. Intestinal tumor multiplicity in the Runx1 KO mice was threefold higher in the colon (P < 0.05), greater than fourfold higher in the cecum (P < 0.05) and almost twofold higher in the small intestine (P < 0.05). More than one‐third of the Runx1 KO mice developed more than 200 tumors, with the highest tumor count at 321 tumors. In contrast, the majority of Runx1 wild‐type mice developed <100 tumors and <5% developed more than 200 tumors (Table 1). We also measured tumor sizes and found that tumors in the small intestine of Runx1 KO mice were larger than Runx1 wild‐type mice, with the difference strongest in the ileum (Fig. S4). All of these phenotypic differences were observed in both male and female mice. All tumors examined using histopathology were adenomas, and thus resemble the same types of tumors normally observed in Apc Min mice. Moreover, in contrast to Apc Min mice that do not normally develop gastric tumors, approximately 10% of Apc Min Runx1 KO mice developed 1, 2, 3 or 4 small tumors in the pyloric canal (Fig. S5A). Histological analysis revealed the tumors to be small, polypoid adenomas in the terminal pylorus or at the junction of the pylorus with the duodenum (Fig. S5B).
Table 1.
Deficiency for Runx1 enhances tumorigenesis in ApcMin mice
| Runx1 genotype | Gender | n | Colon tumors | Colon tumor incidence (%) | Cecal tumors | Small intestine tumors |
|---|---|---|---|---|---|---|
| Wild type | F | 30 | 1.3 ± 1.9 | 78 | 0.2 ± 0.6 | 90 ± 59 |
| Knockout | F | 31 | 4.0 ± 2.5* | 100 | 1.0 ± 1.6* | 155 ± 63* |
| Wild type | M | 36 | 1.5 ± 1.7 | 78 | 0.3 ± 0.6 | 66 ± 37 |
| Knockout | M | 38 | 4.5 ± 3.6* | 100 | 1.2 ± 1.2* | 135 ± 60* |
Apc Min Runx1 KO mice and littermate Apc Min Runx1 wild‐type mice were killed at 100 days of age. Tumors were isolated and scored as described in the Materials and Methods section. Two‐sided P‐values for tumor counts were determined using the Wilcoxon rank‐sum test comparing gender and age‐matched classes produced in the same genetic crosses. *P‐value: <0.05.
Runx1 deficiency results in tumorigenesis in Apc+/+ mice. We aged a group of approximately 20 Apc +/+ Runx1 KO and Runx1 wild‐type mice to approximately 12 months to evaluate the intestinal phenotype of Runx1 deficiency alone. We found that approximately 33% of Runx1 KO mice developed 1, 2 or 3 tumors in the duodenum, while no tumors were observed in Runx1 wild‐type control mice (Table 2). All tumors examined using histopathology were adenomas. Four tumors were examined using light microscopy; all were relatively small, approximately 3–4 mm diameter. All tumors were classified as sessile or flat adenomas, with one tumor showing a tendency towards pedunculation (fibrovascular expansion of the lamina propria) at one margin. Three tumors overlay, wholly or in part, the underlying Brunner’s glands, and in places the epithelial component appeared to be contiguous with the Brunner’s glands (Fig. S6A). No evidence of local invasion or metastasis was observed. Immunohistochemical analysis indicated that more than half of these duodenal tumors demonstrated upregulation of β‐catenin protein (Fig. S6B).
Table 2.
Deficiency for Runx1 causes duodenal tumors in Apc+/+ mice
| Genotype | n | Mean age | Incidence (%) | Tumor multiplicity |
|---|---|---|---|---|
| Runx1 wild type | 10 | 12 months | 0 | 0 |
| Runx1 knockout | 9 | 12 months | 33 | 0.7 |
Runx1 knockout mice and littermate wild‐type mice were aged to approximately 12 months, killed and the tissues were formalin fixed. The entire intestinal tract was examined for tumors as described in the Materials and Methods section.
Gene expression analysis of epithelial Runx1 KO and Runx1 WT colon. To identify epithelial Runx1‐mediated alterations in gene expression in the mouse GI tract, RNA was isolated from the medial colon of Runx1 KO mice and from gender and age‐matched Runx1 wild‐type littermate control mice. Gene expression analysis was then carried out using Agilent 4x44K whole mouse genome microarrays, followed by quantitative RT‐PCR of select target genes. Examination of the 300 most significant genes in the array study identified several functional clusters of genes, with the largest group consisting of genes involved in inflammation, immune responses and the hematopoietic system. This group included Duoxa2, C4bp, Hemt1, Slc7a11, Cxcl14, Crisp3, Cd55, Clca3, Tnip3, Indo, Pou2af1, Runx2, Mst1, Immunoglobulins (many), Cd300lf, Rorc, Cd40, Ehf, Mmp9, Cd2, Cebpd, Retnlg, Il1rl1, Ccl8, Icos, Nfkbiz, Ccr10, Il1rl2, Gpr17, Cxcr4 and Tnfrsf13b. The second largest group of Runx1 target genes was comprised of genes implicated in intestinal metabolism, including several lipases and genes associated with goblet cell function. This group included Mptx, Pnliprp2, Retnlb, Pnliprp1, Clps, Es1, Clca3, Tgn, Slc1a1, Itlna, Spink4, Scin, Ero1l, Pax6, Mmp9, Hsp110 and Retnlg. Figure S7 lists the most significant differentially expressed genes. Differential expression was confirmed by qRT‐PCR analysis (Fig. 1). Notably, for all six genes out of the top seven genes (ranked by P‐value) for which reliable primers could be generated, differential expression was confirmed, thereby validating the array data.
Figure 1.
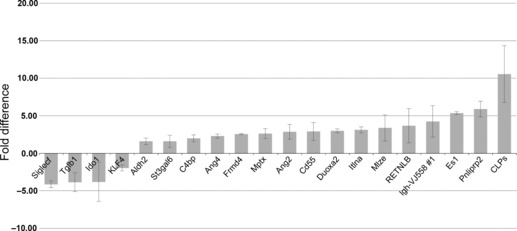
Change in gene expression in Runx1 knockout (KO) whole colon. List of 20 Runx1 target genes examined using qRT‐PCR. Details of the qRT‐PCR assay are described in the Materials and Methods section. Each bar represents the mean and standard error of multiple experiments that measured fold differences in the mRNA expression of specific target genes in whole colon tissue from gender and aged matched pairs of Apc +/+ Runx1 KO and Runx1 wild‐type mice. All tissues were from adult mice generally approximately 100 days of age, and the mRNA were isolated from 1‐cm sections of colon from the same region of the distal colon. All mRNA values were normalized to 18S values. Four replicates of each assay were performed for each matched pair of mRNA (KO versus wild type) and these sets of assays were repeated at least twice for each pair of mRNA). At least two matched pairs of mRNA were tested for each gene, with most genes tested in at least three matched pairs of mRNA. To be included in this figure, genes met the flowing criteria: (i) the mean fold difference was at least 1.5 (and almost all of the 20 genes showed at least a twofold difference); and (ii) each gene showed a change in gene expression in the same direction in each matched pair of mRNA (i.e. all upregulated or all downregulated in each matched pair of mRNA).
Gene set enrichment analysis. Changes in biological processes are accompanied by coordinate variation in expression of sets of genes. Gene Set Enrichment Analysis (GSEA) was applied to identify associations with gene sets differentially expressed between Runx1 KO and Runx1 WT colon (Table 3). Interestingly, a significant association was found with the gene set that distinguishes pediatric acute myeloid leukemia (AML) subtype inv16[CBFβ‐MYH11] (gene set #2)( 56 ) thereby illustrating that genes expressed on mutation of CBFβ in human AML overlap significantly with the genes expressed on epithelial Runx1 deficiency in mouse colon. Because Runx1 binds CBFβ to form a functional heterodimeric transcription factor,( 9 ) these data support the validity of our approach. The significant association with the ‘tryptophan metabolism’ gene set (gene set #9) is in accordance with alteration of expression of genes involved in inflammation and immune responses that might lead to IBD and increased CRC risk.( 57 ) The association with the ‘peroxisome proliferator‐activated receptor (PPAR) signaling pathway’ (gene set #11) is in accordance with the alteration of expression of genes involved in intestinal metabolism,( 58 ) and also predisposes to increased CRC risk( 50 ) Moreover, two gene sets indicate that Runx1 deficiency is associated with expansion of a pluripotent (stem) cell population at the expense of more differentiated cells (gene set #1( 59 ) and #7( 60 )) while other gene sets indicate that Runx1 deficiency is associated with more metastases (gene set #4( 61 )) and poor prognosis in breast cancer (gene set #3( 62 )) and hepatocellular cancer (gene set #10). Taken together, these data imply that tumor‐free colons from Runx1‐deficient mice express gene profiles related to expanded stem/pluripotent progenitor cell populations compared with colon from Runx1 wild‐type mice.
Table 3.
Gene Set Enrichment Analysis (GSEA) gene sets ‘UP or DOWN’ in Runx1 knockout compared with Runx1 wild type (FWER < 0.05)
| # | Collection† | Name of Gene Set† | Size (number of genes) | ES | NES | FDR | FWER |
|---|---|---|---|---|---|---|---|
| q‐value | P‐value | ||||||
| Gene sets ‘UP’ in Runx1 knockout compared with Runx1 wild type (FWER < 0.05) | |||||||
| 1 | Curated (c2) | BOQUEST_CD31PLUS_VS_CD31MINUS_DN | 155 | 0.55 | 2.40 | 0 | 0 |
| 2 | Curated (c2) | ROSS_CBF_MYH | 32 | 0.68 | 2.18 | 0.009 | 0.012 |
| 3 | Curated (c2) | BRCA_PROGNOSIS_NEG | 68 | 0.58 | 2.17 | 0.009 | 0.017 |
| 4 | Curated (c2) | EMT_UP | 43 | 0.62 | 2.16 | 0.007 | 0.018 |
| 5 | Curated (c2) | AGEING_KIDNEY_UP | 203 | 0.48 | 2.13 | 0.009 | 0.028 |
| 6 | Curated (c2) | AGEING_KIDNEY_SPECIFIC_UP | 103 | 0.53 | 2.13 | 0.007 | 0.029 |
| 7 | Curated (c2) | BRUNO_IL3_DN | 44 | 0.61 | 2.11 | 0.009 | 0.039 |
| 8 | Curated (c2) | PRMT5_KD_UP | 126 | 0.51 | 2.10 | 0.008 | 0.041 |
| Gene sets ‘DOWN’ in Runx1 knockout compared with Runx1 wild type (FWER < 0.05) | |||||||
| 9 | Curated (c2) | TRYPTOPHAN_METABOLISM | 24 | −0.72 | −2.16 | 0.004 | 0.010 |
| 10 | Curated (c2) | HCC_SURVIVAL_GOOD_VS_POOR_UP | 89 | −0.52 | −2.08 | 0.006 | 0.029 |
| 11 | Curated (c2) | HSA03320_PPAR_SIGNALING_PATHWAY | 51 | −0.57 | −2.05 | 0.006 | 0.044 |
†Obtained from the Molecular Signatures Database (MSigDB_v2.5, http://www.broad.mit.edu/gsea/msigdb/index.jsp). ES, GSEA enrichment score; FDR, false discovery rate; FWER, family wise error rate; NES, GSEA normalized enrichment score.
Gene‐specific expression analysis of Runx1 KO and Runx1 WT colon epithelium. Considering the reported interactions between Runx1 and Notch signaling activity combined with the whole colon GSEA results indicating putative effects of Runx1 on regulation of the size of the stem/progenitor cell compartment, we decided to examine expression of individual genes associated with intestinal stem/progenitor cells and intestinal differentiation. Colon epithelium was isolated from Runx1 KO and Runx1 WT colon and gene expression was analyzed using qRT‐PCR for genes analyzed in the whole colon plus the putative stem cell marker Aldh1a1, and the Notch pathway genes Notch1, Jag1 and Hes1 (Fig. 2). Our data from the analysis of Runx1‐deficient colon epithelium confirmed the expression patterns for many of the whole colon genes, but also revealed increased expression of Aldh1a1, Notch1, Jag1 and Hes1, thereby supporting a role for Runx1 in direct regulation of the expression of Notch signaling pathway genes.
Figure 2.
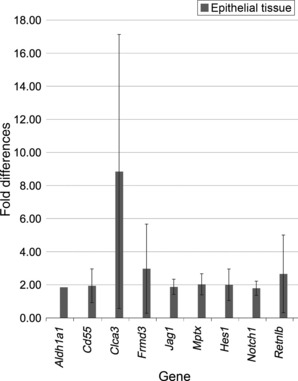
Changes in gene expression in Runx1 knockout (KO) colon epithelial cells. List of nine Runx1 target genes examined using qRT‐PCR. Details of the qRT‐PCR assay are described in the Materials and Methods section. Each bar represents the mean and standard error of multiple experiments that measured fold differences in the mRNA expression of specific target genes in colon epithelial tissue from gender and age‐matched pairs of Apc +/+ Runx1 KO and Runx1 wild‐type mice. All tissues were from adult mice generally approximately 100 days of age, and the mRNA were isolated from 1‐cm sections of colon from the same region of the distal colon. All mRNA values were normalized to 18S values. Four replicates of each assay were performed for each matched pair of mRNA (KO versus wild type) and these sets of assays were repeated at least twice for each pair of mRNA). At least two matched pairs of mRNA were tested for each gene with most genes tested in at least three matched pairs of mRNA. To be included in this figure, genes met the flowing criteria: (i) the mean fold difference was at least 1.5 (and almost all of the nine genes showed at least a twofold difference); and (ii) each gene showed a change in gene expression in the same direction in each matched pair of mRNA (i.e. all upregulated or all downregulated in each matched pair of mRNA).
Discussion
Here we showed that Runx1 is a novel tumor suppressor gene in the GI tract. Conditional ablation of Runx1 expression in Apc Min mice caused a significant increase in the number of tumors, in all regions of the intestine, and in both male and female mice. At a low frequency we also observed small tumors in the pyloric canal leading from the stomach to the duodenum, a type of tumor that is not normally seen in Apc Min mice at 100 days.( 63 ) It is very possible that these gastric tumors arose from the combination of Apc deficiency and excision of Runx1 in a rare population of progenitor cells that express villin in the antral glands of the pylorus.( 64 , 65 ) Moreover, Runx1 deficiency also caused the development of duodenal tumors in Apc wild‐type mice that were aged to approximately 12 months. To examine the effects of Runx1 epithelial deficiency on gut homeostasis preceding colon tumor development, genome‐wide mRNA expression profiles of colon from Runx1 KO mice were compared with Runx1 WT mice. The largest clusters of differentially expressed genes were implicated in: (i) inflammation, immune responses and hematopoeitic processes; and (ii) intestinal metabolism, such as lipases, goblet cell proteins and transporters. Virtually all of the most significantly regulated genes identified (ranked by P‐values) were confirmed using qRT‐PCR, lending confidence to the array data (Fig. 1). Analysis of the differential expression of gene sets using GSEA revealed a significant association with genes expressed on mutation of CBFβ, a key partner of RUNX1 in transcriptional activation, further supporting the validity of our approach. Other gene sets confirmed the effects of Runx1 deficiency on inflammation and immune responses and on intestinal metabolism (Table 3).
Two of the top genes implicated in inflammation and immune cell responses are involved in complement activity: C4bp and Cd55, ( 66 ) which inhibit complement by differing mechanisms.( 67 , 68 ) As both C4bp and Cd55 are upregulated in Runx1 KO colon, these changes suggest that Runx1 promotes complement recruitment and activity. C4bp has been reported to be increased in the plasma of colon cancer patients,( 55 ) while Cd55 expression has been reported to be increased in the intestine of Apc Min mice( 69 ) and in human colorectal cancer( 70 ) where its expression is associated with tumor recurrence, metastasis and a poor prognosis.( 71 ) Thus, Runx1 inhibition of C4bp and Cd55 expression in the colon is consistent with its tumor‐suppressive role in the GI tract.
A second large cluster of Runx1 target genes in mouse colon are involved in intestinal cell metabolism, in particular genes expressed in goblet and Paneth cells. An example is Retnlb, resistin‐like beta, which is expressed in intestinal goblet cells where it is involved in lipid metabolism, host defense and immune responses.( 72 ) Retnlb expression is increased in inflammatory bowel disease, gastric cancer and colorectal cancer, where it is associated with upregulation of interferon (IFN) gamma activity.( 73 , 74 ) Retnlb was also reported to be highly upregulated in tumors isolated from Apc Min mice.( 75 ) Our gene expression studies indicated that Retnlb expression is inhibited by Runx1 in mouse colon (1, 2). Two other genes involved in intestinal metabolism that are upregulated in Runx1 KO colon are Ang4 (angiogenin 4) and Spink4 (serine peptidase inhibitor, Kazal type 4). Both of these genes have been described as transcriptional targets of β‐catenin signaling in Paneth cells whose expression is upregulated in mouse Apc −/− cells,( 76 , 77 ) thereby connecting Runx1 to Wnt/β‐catenin signaling.
In addition to the putative link between Runx1 and Wnt/β‐catenin signaling, Runx1 has also been connected to Notch signaling. Recently, Runx1 was reported to be implicated in regulation and differentiation of the intestinal secretory cell lineage, identifying the Kruppel like factor 4 (Klf4) gene as a target of Runx1. ( 40 ) Klf4 is a zinc‐finger transcription factor that is strongly expressed in terminally differentiated intestinal epithelial cells. Klf4 has also been associated with differentiation of the secretory cell lineage and with inhibition of intestinal cell proliferation via blocking progression of the cell cycle.( 78 , 79 ) Haploinsufficiency for Klf4 enhanced tumorigenesis in the Apc Min mouse( 80 ) and KLF4 has been reported to be downregulated in human colorectal cancer.( 81 ) In the GI tract, expression of KLF4 is inhibited by Notch signaling and inhibition of Notch signaling results in a shift of intestinal crypt cells in favor of a goblet cell fate and away from an absorptive cell fate.( 48 , 80 ) Moreover, it is known that Hes1, a key Notch pathway gene, represses expression of Math1, a gene required for goblet cell differentiation.( 82 ) In the present study we confirmed using qRT‐PCR that Runx1 positively regulated expression of Klf4 in mouse colon (Fig. 1) and found that Runx1 appeared to regulate expression of Notch pathway genes (Notch1, Jag1 and Hes1) in colon epithelial cells (Fig. 2). Taken together, these data suggest that positive regulation of Klf4 by Runx1 might result from inhibition of Notch signaling and thus de‐repression of Klf4 by Notch. Of interest, Runx3 has been reported to directly interact with Notch1 and suppress Notch signaling in hepatocellullar cancer cells,( 83 , 84 ) hence it is possible that Runx3 and Runx1 exert similar suppressive effects on Notch signaling in the intestinal tract.
Both Wnt and Notch signaling pathways are involved in regulation of the stem‐/pluripotent progenitor cell compartment.( 45 ) Moreover, Runx1 itself has been described as a regulator of stem cell proliferation, with isoform‐specific positive and negative effects on the cell cycle.( 85 ) Therefore, one mechanism through which Runx1 might exert its suppressive effects on tumor development is by limiting expansion of the stem‐/pluripotent progenitor cell compartment. Aldh1a1 (Aldh1) was recently implicated as a putative biomarker for colon cancer stem cells.( 86 ) In the present study, Runx1 negatively regulated expression of Aldh1a1 in colonic epithelial cells (Fig. 2). This finding is consistent with a putative role for Runx1 in regulation of the stem/progenitor population, further supported by Runx1 negative regulation of Notch1, Jag1 and Hes1 in colonic epithelial cells (Fig. 2).
Moreover, GSEA revealed significant associations of Runx1 deficiency with gene sets representing an increased metastatic phenotype (Table 3, gene set #4) and poor prognosis in breast and hepatocellular cancer (Table 3, gene sets #3 and #10), phenomena that might be related to (cancer) stem cell characteristics. Considered together, these data imply that colon from Runx1 KO mice expresses genes involved in expanded (cancer) stem/pluripotent progenitor cell populations compared with Runx1 wild‐type mice.
In summary, we have provided the first evidence that Runx1 is a tumor‐suppressor gene in the mammalian GI tract. Deficiency for Runx1 in intestinal epithelial cells significantly enhanced tumorigenesis in Apc Min mice, while Runx1 deficiency alone was sufficient to cause tumorigenesis in Apc‐wild‐type mice. We have identified a set of Runx1 target genes in mouse colon indicative of changes in gut homeostasis, that is, genes involved in inflammation and intestinal metabolism, which are known to increase the risk of tumor development. Moreover, the results of the present study support a role for Runx1 in suppressing Notch pathway signaling, a finding that is consistent with Runx1 regulating the stem cell/progenitor cell compartment in intestinal crypts and consequent Notch‐mediated effects on intestinal cell differentiation and proliferation. Further studies are required to explore the genetic and epigenetic status and role of Runx1 in human colorectal cancers.
Disclosure Statement
All authors declare that they do not have any competing financial interest in relation to the work described.
Supporting information
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Acknowledgments
The authors thank Dr Daniel Littman, New York University, for generously providing the Runx1 floxed mice. This research was funded by a grant (and American Recovery and Reinvestment Act [ARRA] supplement) from the National Cancer Institute, CA137756‐01 (PI, R.T.C.).
References
- 1. Coffman JA. Is Runx a linchpin for developmental signaling in metazoans? J Cell Biochem 2009; 107: 194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cohen MM Jr. Perspectives on RUNX genes: an update. Am J Med Genet 2009; 149A: 2629–46. [DOI] [PubMed] [Google Scholar]
- 3. Taniuchi I, Osato M, Egawa T et al. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell 2002; 111: 621–33. [DOI] [PubMed] [Google Scholar]
- 4. Yamada R, Ymamoto K. Recent findings on genes associated with inflammatory disease. Mutat Res 2005; 573: 136–51. [DOI] [PubMed] [Google Scholar]
- 5. Blyth K, Cameron ER, Neil JC. The RUNX genes: gain or loss of function in cancer. Nat Rev Cancer 2005; 5: 376–87. [DOI] [PubMed] [Google Scholar]
- 6. Niebuhr B, Fischer M, Tager M, Cammenga J, Stocking C. Gatekeeper function of the RUNX1 transcription factor in acute leukemia. Blood Cells Mol Dis 2008; 40: 211–8. [DOI] [PubMed] [Google Scholar]
- 7. Cheng CK, Li L, Cheng SH et al. Secreted‐frizzled related protein 1 is a transcriptional repression target of the t(8;21) fusion protein in acute myeloid leukemia. Blood 2011; 118: 6638–48. [DOI] [PubMed] [Google Scholar]
- 8. ter Elst A, Ma B, Scherpen FJG et al. Repression of vascular endothelial growth factor expression by the runt‐related transcription factor 1 in acute myeloid leukemia. Cancer Res 2011; 71(7): 2761–71. [DOI] [PubMed] [Google Scholar]
- 9. Speck NA, Gilliland DG. Core‐binding factors in haematopoiesis and leukaemia. Nat Rev Cancer 2002; 2: 502–13. [DOI] [PubMed] [Google Scholar]
- 10. Kundu M, Compton S, Garrett‐Beal L et al. Runx1 deficiency predisposes mice to T‐lymphoblastic lymphoma. Blood 2005; 106: 3621–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beri‐Dexheimer M, Latger‐Cannard V, Philippe C et al. Clinical phenotype of germline RUNX1 haploinsufficiency: from point mutations to large genomic deletions. Eur J Hum Genet 2008; 16: 1014–8. [DOI] [PubMed] [Google Scholar]
- 12. Owen CJ, Toze CL, Koochin A et al. Five new pedigrees with inherited RUNX1 mutations causing familial platelet disorder with propensity to myeloid malignancy. Blood 2008; 112: 4639–45. [DOI] [PubMed] [Google Scholar]
- 13. Preudhomme C, Renneville A, Bourdon V et al. High frequency of RUNX1 biallelic alteration in acute myeloid leukemia secondary to familial platelet disorder. Blood 2009; 113: 5583–7. [DOI] [PubMed] [Google Scholar]
- 14. Kuo MC, Liang DC, Huang CF et al. RUNX1 mutations are frequent in chronic myelomonocytic leukemia and mutations at the C‐terminal region might predict acute myeloid leukemia transformation. Leukemia 2009; 23: 1426–31. [DOI] [PubMed] [Google Scholar]
- 15. Schegelberger B, Gohring G, Thol F, Heuser M. Update on cytogenetic and molecular changes in myelodysplastic syndromes. Leuk Lymphoma 2011; DOI: 10.3109/10428194.2011.618235 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 16. Hoi CS, Lee SE, Lu SY et al. Runx1 directly promotes proliferation of hair follicle stem cells and epithelial tumor formation in mouse skin. Mol Cell Biol 2010; 30: 2518–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dudley KJ, Revill K, Whitby P, Clayton RN, Farrell WE. Genome‐wide analysis in a murine Dnmt1 knockdown model identifies epigenetically silenced genes in primary human pituitary tumors. Mol Cancer Res 2008; 6: 1567–74. [DOI] [PubMed] [Google Scholar]
- 18. Birnbaum DJ, Adelaide J, Mamessier E et al. Genome profiling of pancreatic adenocarcinoma. Genes Chromosom Cancer 2011; 50: 456–65. [DOI] [PubMed] [Google Scholar]
- 19. Boonstra JJ, van Marion R, Douben HJCW et al. Mapping of homozygous deletions in verified esophageal adenocarcinoma cell lines and xenografts. Genes Chromosom Cancer 2011; DOI: 10.1002/gcc.20952 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 20. Janes KA. RUNX1 and its understudied role in breast cancer. Cell Cycle 2011; 10(20): 3461–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang L, Brugge JS, Janes KA. Intersection of FOXO‐ and RUNX1‐mediated gene expression programs in single breast epithelial cells during morphogenesis and tumor progression. Proc Natl Acad Sci USA 2011; 108: E803–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sakakura C, Hagiwara A, Miyagawa K et al. Frequent downregulation of the runt domain transcription factors RUNX1, RUNX3 and their cofactor CBFB in gastric cancer. Int J Cancer 2005; 113: 221–8. [DOI] [PubMed] [Google Scholar]
- 23. March HN, Rust AG, Wright NA et al. Insertional mutagenesis identifies multiple networks of cooperating genes driving intestinal tumorigenesis. Nat Genet 2011; 43: 1202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen W, Gao N, Shen Y, Cen JN. Hypermethylation downregulates Runx3 gene expression and its restoration suppresses gastric epithelial cell growth by inducing p27 and caspase3 in human gastric cancer. J Gastroenterol Hepatol 2010; 25: 823–31. [DOI] [PubMed] [Google Scholar]
- 25. Chuang LS, Ito Y. RUNX3 is multifunctional in carcinogenesis of multiple solid tumors. Oncogene 2010; 29: 2605–15. [DOI] [PubMed] [Google Scholar]
- 26. Ito K, Inoue KI, Bae SC, Ito Y. Runx3 expression in gastrointestinal tract epithelium: resolving the controversy. Oncogene 2009; 28: 1379–84. [DOI] [PubMed] [Google Scholar]
- 27. Lee CW, Ito K, Ito Y. Role of RUNX3 in bone morphogenetic protein signaling in colorectal cancer. Cancer Res 2010; 70: 4243–52. [DOI] [PubMed] [Google Scholar]
- 28. Shiraha H, Nishina S, Yamamoto K. Loss of Runt‐related transcription factor 3 causes development and progression of hepatocellular carcinoma. J Cell Biochem 2011; 112: 745–9. [DOI] [PubMed] [Google Scholar]
- 29. Lim B, Ju H, Kim M, Kang C. Increased genetic susceptibility to intestinal‐type gastric cancer is associated with increased activity of the RUNX3 distal promoter. Cancer 2011; 117: 5161–71. [DOI] [PubMed] [Google Scholar]
- 30. Hu SL, Kong XY, Cheng ZD et al. Promoter methylation of p16, Runx3, DAPK and CHFR genes is frequent in gastric carcinoma. Tumori 2010; 96: 726–33. [DOI] [PubMed] [Google Scholar]
- 31. Brenner O, Levanon D, Negreanu V et al. Loss of Runx3 function in leukocytes is associated with spontaneously developed colitis and gastric mucosal hyperplasia. Proc Natl Acad Sci USA 2004; 101: 16016–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sugai M, Aoki K, Osato M et al. Runx3 is required for full activation of regulatory T cells to prevent colitis‐associated tumor formation. J Immunol 2011; 186: 6515–20. [DOI] [PubMed] [Google Scholar]
- 33. Garrity‐Park M, Loftus EV, Sandborn WJ, Smyrk T. Myeloperoxidase immunohistochemistry as a measure of disease activity in ulcerative colitis: association with ulcerative colitis‐colorectal cancer, tumor necrosis factor polymorphism and RUNX3 methylation. Inflamm Bowel Dis 2011; DOI: 10.1002/ibd.21681 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 34. Watanabe T, Kobunai T, Ikeuchi H et al. RUNX3 copy number predicts the development of UC‐associated colorectal cancer. Int J Oncol 2011; 38: 201–7. [PubMed] [Google Scholar]
- 35. Ibrahim AE, Arends MJ, Silva AL et al. Sequential DNA methylation changes are associated with DNMT3B overexpression in colorectal neoplastic progression. Gut 2011; 60: 499–508. [DOI] [PubMed] [Google Scholar]
- 36. Nishio M, Sakakura C, Nagata T et al. RUNX3 promoter methylation in colorectal cancer: its relationship with microsatellite instability as a novel serum marker. Anticancer Res 2010; 30: 2673–82. [PubMed] [Google Scholar]
- 37. Kodach LL, Jacobs RJ, Heijmans J et al. The role of EZH2 and DNA methylation in the silencing of the tumour suppressor RUNX3 in colorectal cancer. Carcinogenesis 2010; 31: 1567–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Slattery ML, Lundgreen A, Herrick JS et al. Associations between genetic variation in RUNX1, RUNX2, RUNX3, MAPK1 and eIF4E and risk of colon and rectal cancer: additional support for a TGF‐β‐signaling pathway. Carcinogenesis 2011; 32: 318–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Slattery ML, Lundgreen A, Herrick JS, Wolff RK, Caan BJ. Genetic variation in the transforming growth factor‐beta signaling pathway and survival after diagnosis with colon and rectal cancer. Cancer 2011; 117: 4175–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Blyth K, Vaillant F, Jenkins A et al. Runx2 in normal tissues and cancer cells: a developing story. Blood Cells Mol Dis 2010; 45: 117–23. [DOI] [PubMed] [Google Scholar]
- 41. Buchert M, Darido C, Lagerqvist E et al. The symplekin/ZONAB complex inhibits intestinal cell differentiation by the repression of AML1/Runx1. Gastroenterology 2009; 137: 156–64. [DOI] [PubMed] [Google Scholar]
- 42. Burns CE, Traver D, Mayhall E, Shepard JL, Zon LI. Hematopoietic stem cell fate is established by the Notch‐Runx pathway. Genes Dev 2005; 19: 2331–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Salat D, Liefke R, Wiedenmann J, Borggrefe T, Oswald F. ETO, but not leukemogenic fusion protein AML1/ETO, augments RBP‐Jkappa/SHARP‐mediated repression of notch target genes. Mol Cell Biol 2008; 28: 3502–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nakagawa M, Ichikawa M, Kumano K et al. AML1/Runx1 rescues Notch1‐null mutation‐induced deficiency of para‐aortic splanchnopleural hematopoiesis. Blood 2006; 108: 3329–34. [DOI] [PubMed] [Google Scholar]
- 45. van Es JH, van Gijn ME, Riccio O et al. Notch/gamma‐secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 2005; 435: 959–63. [DOI] [PubMed] [Google Scholar]
- 46. Crosnier C, Stamataki D, Lewis J. Organizing cell renewal in the intestine: stem cells, signals and combinatorial control. Nat Rev Genet 2006; 7: 349–59. [DOI] [PubMed] [Google Scholar]
- 47. Sikandar SS, Pate KT, Anderson S et al. NOTCH signaling is required for formation and self‐renewal of tumor‐initiating cells and for repression of secretory cell differentiation in colon cancer. Cancer Res 2010; 70: 1469–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chu D, Li Y, Wang W et al. High level of Notch1 protein is associated with poor overall survival in colorectal cancer. Ann Surg Oncol 2010; 17: 1337–42. [DOI] [PubMed] [Google Scholar]
- 49. Zheng H, Pritchard DM, Yang X et al. KLF4 gene expression is inhibited by the notch signaling pathway that controls goblet cell differentiation in mouse gastrointestinal tract. Am J Physiol 2009; 296: G490–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McAlpine CA, Barak Y, Matise I, Cormier RT. Intestinal‐specific PPARgamma deficiency enhances tumorigenesis in ApcMin/+ mice. Int J Cancer 2006; 119: 2339–46. [DOI] [PubMed] [Google Scholar]
- 51. Cormier RT, Hong KH, Halberg RB et al. Secretory phospholipase Pla2g2a confers resistance to intestinal tumorigenesis. Nat Genet 1997; 17: 88–91. [DOI] [PubMed] [Google Scholar]
- 52. Fijneman RJ, Bade LK, Peham JR et al. Pla2g2a attenuates colon tumorigenesis in azoxymethane‐treated C57BL/6 mice; expression studies reveal Pla2g2a target genes and pathways. Cell Oncol 2009; 31: 345–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mootha VK, Lindgren CM, Eriksson KF et al. PGC‐1alpha‐responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003; 34: 267–73. [DOI] [PubMed] [Google Scholar]
- 54. Subramanian A, Tamayo P, Mootha VK et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 2005; 102: 15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Battistelli S, Stefanoni M, Lorenzi B et al. Coagulation factor levels in non‐metastatic colorectal cancer patients. Int J Biol Markers 2008; 23: 36–41. [PubMed] [Google Scholar]
- 56. Ross ME, Mahfouz R, Onciu M et al. Gene expression profiling of pediatric acute myelogenous leukemia. Blood 2004; 104: 3679–87. [DOI] [PubMed] [Google Scholar]
- 57. Cherayil BJ. Indoleamine 2,3‐dioxygenase in intestinal immunity and inflammation. Inflamm Bowel Dis 2009; 15: 1391–6. [DOI] [PubMed] [Google Scholar]
- 58. Kanehisa M, Araki M, Goto S et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res 2008; 36: D480–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Boquest AC, Shahdadfar A, Fronsdal K et al. Isolation and transcription profiling of purified uncultured human stromal stem cells: alteration of gene expression after in vitro cell culture. Mol Biol Cell 2005; 16: 1131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bruno L, Hoffmann R, McBlane F et al. Molecular signatures of self‐renewal, differentiation, and lineage choice in multipotential hemopoietic progenitor cells in vitro . Mol Cell Biol 2004; 24: 741–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jechlinger M, Grunert S, Tamir IH et al. Expression profiling of epithelial plasticity in tumor progression. Oncogene 2003; 22: 7155–69. [DOI] [PubMed] [Google Scholar]
- 62. van‘t Veer LJ, Dai H, van de Vijver MJ et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002; 415: 530–6. [DOI] [PubMed] [Google Scholar]
- 63. Tomita H, Yamada Y, Oyama T et al. Development of gastric tumors in ApcMin/+ mice by the activation of the β‐Catenin/Tcf signaling pathway. Cancer Res 2007; 67: 4079–87. [DOI] [PubMed] [Google Scholar]
- 64. Qiao X, Ziel JW, McKimpson W et al. Prospective identification of a multi‐lineage progenitor in murine stomach epithelium. Gastroenterology 2007; 133: 1989–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Li Q, Wang L, Jia Z, Wei D, Le X, Xie K. Conditional inactivation of Klf4 gene in distinct cell population of stomach mucosa renders mice susceptible to gastric carcinogenesis. Published Abstract # 0954, International Society of Gastrointestinal Oncology, 2009 Gastrointestinal Oncology Conference, 1–3 October 2009.
- 66. Hoek RM, de Launay D, Kop EN et al. Deletion of either CD55 or CD97 ameliorates arthritis in mouse models. Arthritis Rheum 2010; 62: 1036–42. [DOI] [PubMed] [Google Scholar]
- 67. Blom AM, Villoutreix BO, Dahlback B. Functions of human complement inhibitor C4b‐binding protein in relation to its structure. Arch Immunol Ther Exp 2004; 52: 83–95. [PubMed] [Google Scholar]
- 68. Kim DD, Song WC. Membrane complement regulatory proteins. J Clin Immunol 2006; 118: 127–36. [DOI] [PubMed] [Google Scholar]
- 69. Holla VR, Wang D, Brown JR, Mann JR, Katkuri S, DuBois RN. Prostaglandin E2 regulates the complement inhibitor CD55/decay‐accelerating factor in colorectal cancer. J Biol Chem 2005; 280: 476–83. [DOI] [PubMed] [Google Scholar]
- 70. Mikesch JH, Buerger H, Simon R, Brandt B. Decay‐accelerating factor (CD55): a versatile acting molecule in human malignancies. Biochim Biophys Acta 2006; 1766: 42–52. [DOI] [PubMed] [Google Scholar]
- 71. Han SL, Xu C, Wu XL, Li JL, Liu Z, Zeng QQ. The impact of expressions of CD97 and its ligand CD55 at the invasion front on prognosis of rectal adenocarcinoma. Int J Colorectal Dis 2010; 25: 695–702. [DOI] [PubMed] [Google Scholar]
- 72. Herbert DR, Yang JQ, Hogan SP et al. Intestinal epithelial cell secretion of RELM‐beta protects against gastrointestinal worm infection. J Exp Med 2009; 206: 2947–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Nair MG, Guild KJ, Du Y et al. Goblet cell‐derived resistin‐like molecule beta augments CD4 + T cell production of IFN‐gamma and infection‐induced intestinal inflammation. J Immunol 2008; 181: 4709–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zheng LD, Tong QS, Weng MX et al. Enhanced expression of resistin‐like molecule beta in human colon cancer and its clinical significance. Dig Dis Sci 2009; 54: 274–81. [DOI] [PubMed] [Google Scholar]
- 75. Leclerc D, Deng L, Trasler J, Rozen R. ApcMin/+ mouse model of colon cancer: gene expression profiling in tumors. J Cell Biochem 2004; 93: 1242–54. [DOI] [PubMed] [Google Scholar]
- 76. Andreu P, Peignon G, Slomianny C et al. A genetic study of the role of the Wnt/beta‐catenin signalling in Paneth cell differentiation. Dev Biol 2008; 324: 288–96. [DOI] [PubMed] [Google Scholar]
- 77. Gregorieff A, Stange DE, Kujala P et al. The ets‐domain transcription factor Spdef promotes maturation of goblet and paneth cells in the intestinal epithelium. Gastroenterology 2009; 137: 1333–45. [DOI] [PubMed] [Google Scholar]
- 78. Flandez M, Guilmeau S, Blache P, Augenlicht LH. KLF4 regulation in intestinal epithelial cell maturation. Exp Cell Res 2008; 314: 3712–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ghaleb AM, McConnell BB, Kaestner KH, Yang VW. Altered intestinal epithelial homeostasis in mice with intestine‐specific deletion of the Krüppel‐like factor 4 gene. Dev Biol 2011; 349: 310–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ghaleb AM, McConnell BB, Nandan MO, Katz JP, Kaestner KH, Yang VW. Haploinsufficiency of Kruppel‐like factor 4 promotes adenomatous polyposis coli dependent intestinal tumorigenesis. Cancer Res 2007; 67: 7147–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wei D, Kanai M, Huang S, Xie K. Emerging role of KLF4 in human gastrointestinal cancer. Carcinogenesis 2006; 27: 23–31. [DOI] [PubMed] [Google Scholar]
- 82. Yang Q, Bermingham NA, Finegold MJ, Zoghbi HY. Requirement of Math1 for secretory cell lineage commitment in the mouse intestine. Science 2001; 294: 2155–8. [DOI] [PubMed] [Google Scholar]
- 83. Gao J, Chen Y, Wu KC et al. RUNX3 directly interacts with intracellular domain of Notch1 and suppresses Notch signaling in hepatocellular carcinoma cells. Exp Cell Res 2010; 316: 149–57. [DOI] [PubMed] [Google Scholar]
- 84. Nishina SI, Shiraha H, Nakanishi Y et al. Restored expression of the tumor suppressor gene RUNX3 reduces cancer stem cells in hepatocellular carcinoma by suppressing Jagged1‐Notch signaling. Oncol Rep 2011; 26: 523–31. [DOI] [PubMed] [Google Scholar]
- 85. Wang CQ, Jacob B, Nah GS, Osato M. Runx family genes, niche, and stem cell quiescence. Blood Cells Mol Dis 2010; 44: 275–86. [DOI] [PubMed] [Google Scholar]
- 86. Huang EH, Hynes MJ, Zhang T et al. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res 2009; 69: 3382–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item