

Abstract
Loss of TP53 and RB1 function have both been linked to poor response to DNA damaging drugs in breast cancer patients. We inactivated TP53 and/or RB1 by siRNA mediated knockdown in breast cancer cell lines varying with respect to ER/PgR and Her-2 status as well as TP53 and RB1 mutation status (MCF-7, T47D, HTB-122 and CRL2324) and determined effects on cell cycle arrest, apoptosis and senescence with or without concomitant treatment with doxorubicin. In T47D cells, we found the cell cycle phase distribution to be altered when inactivating TP53 (P=0.0003) or TP53 and RB1 concomitantly (P≤0.001). No similar changes were observed in MCF-7, HTB-122 or CRL2324 cells. While no significant change was observed for the CRL2324 cells upon doxorubicin treatment, MCF-7, T47D as well as HTB-122 cells responded to knockdown of TP53 and RB1 in concert, with a decrease in the fraction of cells in G1/G0-phase (P=0.042, 0.021 and 0.027, respectively). Inactivation of TP53 and/or RB1 caused no change in induction of apoptosis. Upon doxorubicin treatment, inactivation of TP53 or RB1 separately caused no induction of apoptosis in MCF-7 and HTB-122 cells; however, concomitant inactivation leads to a slightly reduced activation of apoptosis. Interestingly, upon doxorubicin treatment, concomitant inactivation of TP53 and RB1 caused a decrease in senescence in MCF-7 cells (P=0.027). Comparing the effects of concomitant knockdown on apoptosis and senescence, we observed a strong interaction (P=0.001). We found concomitant inactivation of TP53 and RB1 to affect various routes of response to doxorubicin treatment in breast cancer cells.
Introduction
Chemotherapy resistance is the main cause of therapy failure and death among cancer patients; yet the mechanisms behind resistance remains poorly understood.1
Doxorubicin causes DNA double helical breaks leading to activation of cell cycle arrest, apoptosis or entry of a permanent state of cell cycle arrest, senescence.2–4
In regulation of these processes, the tumor suppressor’s p53 (encoded by the TP53 gene) and the retinoblastoma protein, pRb (encoded by the RB1 gene) are both known to have key roles.5–7
TP53 mutations have been associated with resistance to DNA damaging chemotherapy treatment across different malignancies.8–12 However, the finding that anthracyclines may work on some tumors harboring TP53 mutations while other tumors respond to therapy despite lack of TP53 function has lead us to postulate that redundant gene pathways may act in concert.1,13
pRb, regulating the transition between G1/G0- and S-phase in the cell cycle, has a key role executing cell cycle arrest in response to DNA damage.14,15 While conflicting evidence has linked RB1 mutations to lack of, as well as an improved response to chemotherapy;16,17 previously we reported mutations in RB1 to be associated with a poor response to anthracyclines and mitomycin in breast cancer.18,19
Experimental evidence has linked concomitant inactivation of the p53- and the Rb-pathway to lymphoma development and resistance toward cyclophosphamide in mice through prevention of cellular senescence,20 as well as immortalization of keratinocytes.21 While we found concomitant inactivation of the p53- and Rb-functional pathways to be associated with resistance to DNA damaging drugs in breast cancer patients, the mechanism behind this effect has not been elucidated.22
In order to further characterize the mechanisms behind chemotherapy resistance, we here performed functional analyzes of the cellular responses, cell cycle arrest, apoptosis and senescence upon siRNA mediated inactivation of TP53 and RB1, alone or combined, in unstressed cells and after doxorubicin treatment in different breast cancer cell lines.
Results
Establishment of a concomitant knockdown model
In order to establish a model appropriate for studying the effects of lack of p53 and/or pRb on response to doxorubicin treatment, we applied siRNA-mediated knockdown. The siRNAs used effectively and selectively suppressed both TP53 and RB1 in untreated MCF-7 cells as well as in doxorubicin treated cells (Figure 1). Notably, TP53 inactivation lead to a slight upregulation of pRb in chemotherapy treated cells, supporting the hypothesis that the Rb-pathway may act as compensatory pathway in TP53 inactivated tumor cells in response to cellular stress. Efficient knockdown was also confirmed by western blot analysis in the additional cell lines T47D, HTB-122 and CRL2324 (with the expected exception that pRB could not be detected in HTB-122 cells, since these cells are known to harbor a homozygous frameshift deletion within the RB1 gene).
Figure 1.

Protein expression after siRNA transfection of 4 cell lines. Left panel shows results of western blotting from untreated cells (DMSO), while the right panel show the corresponding results from cells treated with 1 μM doxorubicin for 24 h. The lanes show results from cells subjected to siRNA-control and siRNA-mediated knockdown of TP53, RB1 and these two genes concomitantly. The top rows presents pRb (110 kDa) and the middle lanes p53 (53 kDa). Actin is used at loading control (42 kDa).
Subsequently, we assessed the potential impact of knockdown of TP53 or RB1, as well as both genes concomitantly, on three different cellular endpoints potentially induced by doxorubicin treatment: cell cycle arrest, apoptosis and senescence.
Effect of TP53 and/or RB1 inactivation on cell cycle progression
First we assessed the impact of TP53 and RB1 inactivation in MCF-7 cells not treated with any chemotherapy. Inactivation of TP53, RB1 or both genes in concert, did not alter the cell cycle phase distribution significantly (Figure 2a). However, in a second ER-positive cell line, T47D, TP53 knockdown lead to a significant difference in the cell cycle distribution compared to the siRNA-control (P≤0.001; Figure 2b). Knockdown of RB1 caused no changes in cell cycle distribution in T47D cells, while concomitant inactivation of TP53 and RB1 lead to a more pronounced shift in the cell cycle compared to knockdown of TP53 alone (analysis of variance with all phases included; P≤0.001). Performing the same experiments on triple negative cell lines (HTB-122 and CRL2324), we observed no direct effect with respect to cell cycle distribution upon knockdown of either TP53 or RB1 or by the two genes in concert (Figures 2c and d).
Figure 2.

Effect on cell cycle progression. Cell cycle analysis after siRNA knockdown of untreated (DMSO) or 1 μM doxorubicin treated (a) MCF-7, (b) T47D, (c) HTB-122 and (d) CRL2324 breast cancer cells. Cells were treated with siRNA-Control, siRNA against TP53, siRNA for RB1 or concomitantly against TP53 and RB1. Analysis was performed by NucleoCounter 3000. Bars present the percent of cell debris (purple), cells in G1/G0-phase (green), in S phase (pink) and in G2/M phase (blue), respectively. The experiment was repeated in triplicate, including three independent siRNA transfections and treatment with three different batches of DMSO or chemotherapy. Error bars indicate standard deviations. *P≤0.05, ***P≤0.001.
Doxorubicin treatment of the cell lines lead to a significant shift in the cell cycle phase distribution in MCF-7 cells (P=0.001; comparison of untreated and doxorubicin treated siRNA-control cells), while the T47D cells seemed not to respond to the dose applied and revealed no shift in the cell cycle. Regarding the triple negative cell lines, we found HTB-122 to have a significant shift in the cell cycle phase distribution upon doxorubicin treatment (variance analysis with all phases included; P≤0.001), while CRL2324 appeared to not respond to the treatment.
Assessing the impact of TP53 inactivation on the response to doxorubicin treatment in MCF-7, we found the cell cycle phase distribution to alter significantly when compared with the siRNA-control cells (28% decrease in cells in the G1/G0-phase with a 20% increase of cells in the G2/M-phase: P=0.024; Figure 2a). A similar albeit non-significant trend was observed in HTB-122 cells (P=0.073); in contrast, no effects were recorded in T47D or CRL2324 cells (Figures 2b–d). RB1 inactivation alone, on the other hand, had no effects on cell cycle distribution across the different cell lines.
Importantly, in doxorubicin treated MCF-7 cells, concomitant inactivation of TP53 and RB1 lead to decrease of cells arrested in G1/G0-phase and increase of cells in G2/M-phase compared to the siRNA-control cells (P=0.042), however, not significantly when compared to cells with TP53 inactivated alone. Similar to MCF-7, in T47D cells, in which doxorubicin had revealed no effect on cell cycle in the siRNA-control cells, concomitant inactivation of TP53 and RB1 caused the same effect (P=0.021), indicating the cell cycle to be strongly affected by TP53 and RB1 inactivation under doxorubicin treatment. Among the triple negative cell lines, similar results were observed for HTB-122 (P=0.027), while no significant change was observed for the CRL2324 cells.
Effect TP53 and/or RB1 inactivation on induction of apoptosis
Based on the changes observed in the cell cycle profiles, we further assessed the degree of initiated apoptosis after separate or concomitant inactivation of TP53 and RB1.
In untreated cells, knockdown of TP53, RB1 or both genes simultaneously did not reveal any major changes in the induction of apoptosis in either of the cell lines analyzed (Figures 3a–d).
Figure 3.

Induction of apoptosis by Annexin V analysis. (a) MCF-7, (b) T47D, (c) HTB-122 and (d) CRL2324 breast cancer cells were treated with siRNA-Control, siRNA for TP53, siRNA for RB1 or siRNA against both TP53 and RB1 together. Graphs show the percentage of apoptotic cells; untreated (DMSO; purple bars) or treated with 1 μM doxorubicin (green bars) for 24 h, analyzed by Annexin V assay 48 h post knockdown. The pillars represent both the cells that are early apoptotic and apoptotic. The experiment was repeated in triplicate, with three independent siRNA transfections and three different DMSO or doxorubicin batches. Error bars indicate standard deviations. *P≤0.05, **P≤0.01. The percentage change in untreated vs doxorubicin treated cells is presented for (a) MCF-7 and (c) HTB-122 (lower panel, blue bars).
Upon treatment with doxorubicin, the fraction of apoptotic MCF-7 cells increased significantly (P=0.002, comparison of siRNA-control treated cells; Figure 3a). Similar to our observations when assessing cell cycle progression, treatment with doxorubicin did not influence apoptosis in T47D and CRL2324 cells, while HTB-122 revealed a higher general level of apoptosis than MCF-7, but a more modest increase in apoptosis in response to doxorubicin (Figures 3b–d).
In cells treated with doxorubicin, inactivation of TP53 or RB1 alone or combined, caused an unexpected, slight increase in the fraction of apoptotic MCF-7 cells as compared to the siRNA-control. In contrast, no change was observed in HTB-122 cells (Figures 3a and c).
Interestingly, in MCF-7 cells, concomitant inactivation of TP53 and RB1 lead to a slightly reduced induction of apoptosis upon doxorubicin treatment as compared with when the two genes were inactivated separately (Figure 3a). A similar observation was made in the HTB-122 cells (Figure 3c). Taking the lack of reduced apoptosis (when comparing concomitant inactivation with siRNA-control) into account, these data indicate apoptosis not to be a cellular endpoint strongly affected by TP53 and/or RB1 inactivation.
Effect of TP53 and/or RB1 inactivation on induction of senescence
In order to test a more long term effect of TP53 and RB1 inactivation and a possible alternative mechanism of response to doxorubicin, we assessed the induction of cellular senescence in MCF-7 cells with and without doxorubicin treatment.
In cells not exposed to doxorubicin, TP53 and/or RB1 inactivation (alone or combined) did not influence the fraction of cells undergoing senescence (Figure 4).
Figure 4.
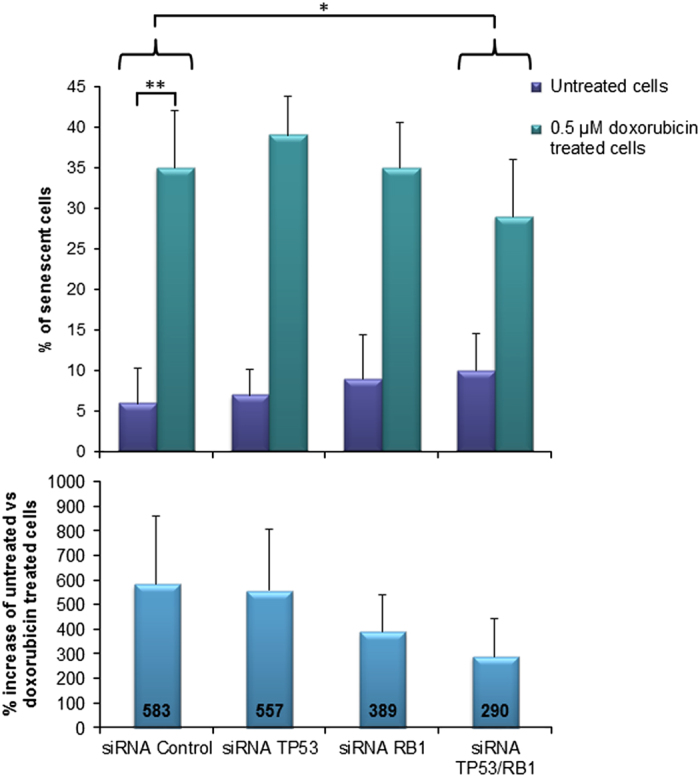
Induction of senescence measured by β-galactosidase activity staining. Bars indicate the percentage of senescent MCF-7 cells: untreated (0.5 μM DMSO; purple bars) or cells treated with 0.5 μM doxorubicin (green bars) for 7 days. Cells were treated with siRNA-Control, siRNA against TP53, RB1 or both against TP53 and RB1 simultaneously. The percentage change in induced senescence of untreated vs doxorubicin treated cells is presented (lower panel, blue bars). Error bars indicate standard deviations. *P≤0.05, **P≤0.01.
Upon treatment of siRNA-control cells with doxorubicin, we observed a large increase in the fraction of senescent cells (583% increase when compared to the untreated siRNA-control cells, P=0.004; Figure 4).
Most importantly, upon concomitant inactivation of TP53 and RB1, doxorubicin treatment lead to a much smaller increase of senescent cells as compared to siRNA-control (290%; P=0.027 for interaction between concomitant knockdown and reduced induction of senescence).
Notably, we found a significant interaction when comparing the increase in apoptosis and the decrease in senescence in doxorubicin treated MCF-7 cells with concomitant inactivation of TP53 and RB1 (P=0.001).
Discussion
The mechanism behind chemotherapy resistance is still poorly understood, despite extensive experimental research. While TP53 and/or RB1 mutational status have been linked to lack of sensitivity to anthracyclines,8,9,12,18,19,22 the mechanisms remains poorly understood. Here, we aimed at systematically exploring the effect of TP53 and RB1 inactivation, separately and in concert, on the cellular response to doxorubicin treatment using a panel of cell lines representing various breast cancer subtypes (luminal, tipple negative and Her-2-classes), respectively.23 We used MCF-7 (ER+, PgR+ and Her-2 negative) being WT for TP53 as well as for RB1, T47D (ER+, PgR+ and Her-2 negative) harboring mutated TP53 (L194F) and WT RB1, HTB-122 (triple negative) harboring mutated TP53 (R249S) and additionally a partly deleted RB1, likely causing a non-functional pRb protein and finally, CRL2324 (triple negative) also with mutated TP53 (R175H) and WT RB1. For all three TP53 mutated cell lines, the mutations are previously characterized as GOF mutations, and all three cell lines have lost their wild-type allele.
In our study, we applied siRNA-mediated knockdown of TP53 and RB1 in all the four cell lines, regardless of their mutational status. This was done partly in order to obtain a uniform comparison of the cell lines, however most importantly because it is unclear which (if any) normal p53 functions that are retained, and which additional functions that are gained by the different GOF mutations. siRNA mediated knockdown would therefore assure complete loss of p53 function, even in the cell lines harboring TP53 mutations.
The effect and biological outcome of doxorubicin treatment in vitro is context dependent and the concentration of drug and time of exposure is crucial.24,25 The dosage of the doxorubicin treatment and the time of exposure used in the present study were chosen based on in vivo concentrations under treatment of breast cancer patients. Although this dose is somewhat lower than what is often used for in vitro studies, the finding of substantial effects of doxorubicin at this concentration on cell cycle progression in MCF-7 and HTB-122 cells revealed the drug concentration to be of biological importance.
In the present study we found inactivation of TP53 and RB1 in concert in general to have a larger effect on cell cycle distribution than inactivation of either of the two genes individually in MCF-7 cells. When addressing activation of apoptosis, however, upon concomitant inactivation of the two genes, we observed a somewhat unexpected increase in apoptotic cells; this may indicate that apoptosis might not be the most important cellular endpoint affected by TP53 and/or RB1 inactivation in MCF-7 cells. As an alternative endpoint of response to chemotherapy treatment, we also examined the induction of senescence in these cells. Interestingly, we observed a strong effect of concomitant inactivation of TP53 and RB1 on senescence. Notably, our finding of reduced senescence upon concomitant inactivation of TP53 and RB1 is in agreement with results from experimental studies revealing concomitant inactivation of the TP53 and RB1 pathways to be associated with lymphogenesis as well as resistance toward chemotherapy due to loss of senescence.20 More recently, Laine and colleagues have shown that in breast cancer cells with inactivated p53-p21 pathway, the feedback mechanisms between E2F1 and CIP2A are essential for activation of senescence and response to chemotherapy, providing further evidence for the combined roles of the p53- and the pRb functional pathways in this respect.26
As a response to DNA damage and stress, both apoptosis and cellular senescence can be activated in parallel, but also separately. Why some cells respond with apoptosis, while other respond with senescence upon the same treatment is still not fully understood. It has previously been reported that treatment of a colorectal cancer cell line with low doses of doxorubicin induces senescence rather than apoptosis,27 but it has also been suggested that in cells that are unable to induce apoptosis, senescence may act as a ‘backup’ response, strongly contributing to the treatment outcome.28 Furthermore, our results are supported by several in vivo studies, indicating that upon chemotherapy treatment, the role of p53 as a tumor suppressor may be mediated through induction of senescence rather than apoptosis.29,30
The MCF-7 cells may be regarded as the most suitable model in our experiments since they are wild type for both TP53 and RB1. One may assume that cell lines with mutations in TP53 and or RB1 would adapt to the situation and activate compensatory signaling pathways, making them independent of TP53/RB1 function. However, in our experiments, we found the results in HTB-122 cells to resemble the results form MCF-7, even if HTB-122 harbors a mutated TP53 (R249S): Upon chemotherapy treatment, similar changes were observed in cell cycle distribution and, further, when assessing activation of apoptosis in HTB-122 cells, the effects of TP53 and/or RB1 inactivation were limited in this cell line as well. This may indicate that some of the functions of TP53 R249S could influence the cells response to cytotoxic stress.
Taken together, the results from these two cell lines support our previous data from breast cancer patients in vivo, strengthening the proposed model which states that resistance to chemotherapy might depend on a ‘two-pathway-hit’, involving inactivation on both the p53- and the Rb-pathway.22 Notably, this may also influence resistance to chemotherapy in other tumor types: Zhu et al recently suggested, based on studies of primary and metastatic prostate cancer, that genetic co-inactivation of both pRb and p53 could explain why more advanced prostate cancers become chemotherapy resistant as the tumors evolve.25,31,32
Although p53 and pRb are considered to be two of the most important proteins responsible for induction of apoptosis and cell cycle arrest, it is important to note that the involvement and functionality of other alternative pathways could also be involved in the response to chemotherapy in a setting where both p53 and pRb are inactivated. Such alternative pathways could be the cause for why our results from MCF-7 and HTB-122 were not properly mirrored in T47D and CRL2324. Regarding T47D and CRL2324 cells, the overall percentage of apoptotic cells were very low in our experiments. However, it should be noted that, despite the apparent resistance to the applied doxorubicin dose, T47D cells did actually reveal some changes in cell cycle distribution, resembling the results in MCF-7 cells. Alternative pathways, independent of p53, such as ATM-Chk2-cdc25 signaling, has been found to influence the cellular responses to genotoxic stress,33 and it may be that such a signaling route is active in this cell line.
To conclude, the present study shows that concomitant inactivation of the p53- and the Rb-pathway after treatment with a DNA damaging chemotherapeutic drug, leads to altered cell cycle, minor changes in apoptosis and a decrease of cells that are entering a senescent state, compared to inactivation of the two pathways individually. Additionally, our findings indicate these effects to be independent of the cells ER- and PgR-status.
Materials and methods
Cell lines and cultures
The breast cancer cell lines used in the present study are listed in Table 1. The cell lines were preferred based on their ER, PgR, Her-2 characteristic, in addition to their TP53 and RB1 mutational status. Except for MCF-7, being WT for TP53 as well as RB1, the other cell lines (T47D, HTB-122 and CRL2324) each harboring a TP53 mutation previously characterized as a ‘gain of function’ (GOF) mutation and with loss of the normal allele. HTB-122 in addition harbor partly deleted RB1, possibly causing a non-functional pRb protein.34–37
Table 1. Breast cancer cell lines characterizations.
| Cell line | ATCC no. | Medium | Subtype | Tumor type | ER | PgR | Her-2 | TP53 | RB1 |
|---|---|---|---|---|---|---|---|---|---|
| MCF-7 | HTB-22 | EMEM | L | Met AC | + | + | − | WT | WT |
| T47D | HTB-133 | McCoys | L | IDC | + (low) | + (high) | − | GOF Mutation (L194F) Homozygous | WT |
| HTB-122 | BT-549 | RPMI | B | IDC | − | − | − | GOF Mutation (R249S) Homozygous | Frameshift (c.265_607-del343) Homozygous |
| CRL2324 | HCC1395 | RPMI | B | DC | − | − | − | GOF Mutation (R175H) Homozygous | WT |
Abbreviations: B, Basal B subtype; L, luminal subtype; DC, ductal carcinoma; IDC, invasive ductal carcinoma; Met AC, metastatic adenocarcinoma.
Before use, the cell lines identities were confirmed by STR based DNA fingerprinting using the AmpFlSTR Profiler Plus and Cofiler kits (Applied biosystems by Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions. All cell lines and medium are purchased from ATCC, and all of the different medium were supplemented with 10% FBS (Lonza, Basel, Switzerland), 1% l-Glatamine (Lonza) and 1% Penicillin Streptavidin (Lonza), and in addition McCoys were supplemented with 1 μl/ml insulin (Sigma, St Louis, MO, USA) for the T47D cells. In all the assays the cells were transfected with siRNA for 24 h, followed by treatment with dimethyl sulfoxide (DMSO, referred to as untreated cells, as negative control, Sigma) or 1 μM doxorubicin (Nycomed, Zurich, Switzerland) for 24 h, with the exception of the senescence assay (see below).
siRNA treatment
Cells were treated with ON Target siRNA smart pool (Darmacon, Lafayette, CO, USA) against TP53 and/or RB1 or control siRNA. Transfection was performed using Lipofectamin RNAiMAX (Invitrogen, Carlsbad, CA, USA) according to manufacturer’s instructions and monitored by Western blot analysis. We applied siRNA-mediated knockdown of TP53 and RB1 in all the cell lines, for more uniform comparison.
Doxorubicin treatment
Cells were treated with 1 μM doxorubicin for assessment of cell cycle phase distribution and induction of apoptosis. This fixed dose was applied across all cell lines in order to mimic the effective doses of doxubicin used in patients. For the assessment of senescence, the long incubation time made this dose non-feasible and a reduced dose of 0.5 μM doxorubicin was applied for this assay (see details for the assays below).
Western blot analysis
The cells were harvested with Trypsin EDTA (Lonza) and lysed with IPH buffer containing protease and phosphatase inhibitor (Roche, Basel, Switzerland). Further, the cells were washed with 1xPBS and loaded on to 12% polyacrylamide gels (BioRad, Hercules, CA, USA) with PS11 protein ladder (GeneOn, Ludwigshafen, Germany) and blotted for 7 min with Trans-blot Turbo system (BioRad) on to 0.2 μM nitrocellulose membranes (BioRad). Membranes were blocked with 5% non-fat milk in TBS-Tween0.05%, and visualized by anti-p53 (Sigma), anti-pRb (Cell Signaling, Danvers, MA, USA) and HRP-tagged secondary antibody (Sigma) in 1% dry milk in TBS-Tween0.05% on Fuji-LAS 4000 (GE Healthcare Life Science, Little Chalfont, UK). As loading control, anti-Actin (Thermo Scientific, Carlsbad, CA, USA) was used as primary antibody.
Cell cycle progression analysis
The siRNA transfected cells, treated with DMSO or 1 μM doxorubicin were incubated 5 min at 37 °C in lysis buffer containing Hoechst (Chemometec, Allerød, Denmark). Cells were further added stabilizing buffer (Chemometec) before analyzing with the NucleoCounter 3000 for cell cycle status by DNA quantification. The analysis was repeated in three independent experiments with three independent transfections.
Apoptosis analysis by Annexin V staining
siRNA transfected cells were treated with either DMSO or 1 μM doxorubicin before harvesting with Trypsin EDTA (Lonza). The cells were then washed in 1xPBS, incubated for 15 min at 37 °C in Annexin V buffer with Annexin V (Biotium) and Hoechst (Chemometec). Next, the cells were washed once in Annexin V buffer (Biotium, Fremont, CA, USA), before they were re-suspended in Annexin V buffer with 4% PI (Chemometec). Apoptosis assay was performed on a NucleoCounter 3000 instrument (Chemometec) with identification of apoptotic cells. The assay was repeated in triplicate with three independent transfections. In order to avoid major overlap with signals from PI (535/617 nm), or from potential remains of doxorubicin molecules (480/~570 nm) we applied the CF488A Annexin V conjugate (490/515 nm; Chemometec) and a custom filter setting on the NucleoCounter instrument.
Senescence assay: β-galactosidase activity staining
Transfected MCF-7 cells were treated with DMSO or 0.5 μM doxorubicin for 7 days before staining for β-galactosidase activity with the Senescence β-galactosidase Staining Kit (Cell Signaling), according to the manufacturer’s recommendations. Staining of the cells was performed by incubation of the cells in staining solution for 16 h, in a cultivation incubator with humidified atmosphere, without CO2 at 37 °C. The senescent cells were observed as blue β-galactosidase positive cells upon microscopy (Nikon eclipse TS100) and quantitated by counting performed by two independent investigators, blinded to sample identity and each other’s results. The experiment was repeated in triplicates and in each experiment a minimum of 164 cells was counted for each sample treated with doxorubicin and 383 cells for the untreated sample.
Statistics
Potential differences in cell cycle distribution and apoptosis, as well as for the interaction between the induction of senescence and activation of apoptosis, were assessed by univariate analysis of variance (assessing the cell cycle distribution, all phases were included in the univariate analysis of variance). All P-values are reported as two-sided.
Acknowledgments
We thank Gjertrud Titlestad Iversen for excellent technical assistance. The work conducted in this project was performed at the Mohn Cancer Research Laboratory with support by grants from the Norwegian Cancer Society, Bergen Research Foundation and the Norwegian Health Region West.
Footnotes
Edited by N Barlev
The authors declare no conflict of interest.
References
- Lonning PE, Knappskog S. Mapping genetic alterations causing chemoresistance in cancer: identifying the roads by tracking the drivers. Oncogene 2013; 32: 5315–5330. [DOI] [PubMed] [Google Scholar]
- Gewirtz DA. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol 1999; 57: 727–741. [DOI] [PubMed] [Google Scholar]
- Di Leonardo A, Linke SP, Clarkin K, Wahl GM. DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev 1994; 8: 2540–2551. [DOI] [PubMed] [Google Scholar]
- Kim HS, Lee YS, Kim DK. Doxorubicin exerts cytotoxic effects through cell cycle arrest and Fas-mediated cell death. Pharmacology 2009; 84: 300–309. [DOI] [PubMed] [Google Scholar]
- Enoch T, Norbury C. Cellular responses to DNA damage: cell-cycle checkpoints, apoptosis and the roles of p53 and ATM. Trends Biochem Sci 1995; 20: 426–430. [DOI] [PubMed] [Google Scholar]
- Knudsen ES, Knudsen KE. Tailoring to RB: tumour suppressor status and therapeutic response. Nat Rev Cancer 2008; 8: 714–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer 2002; 2: 594–604. [DOI] [PubMed] [Google Scholar]
- Chrisanthar R, Knappskog S, Lokkevik E, Anker G, Ostenstad B, Lundgren S et al. CHEK2 mutations affecting kinase activity together with mutations in TP53 indicate a functional pathway associated with resistance to epirubicin in primary breast cancer. PLoS One 2008; 3: e3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Borresen-Dale AL, Johnsen H, Aas T, Geisler J, Akslen LA et al. TP53 gene mutations predict the response to neoadjuvant treatment with 5-fluorouracil and mitomycin in locally advanced breast cancer. Clin Cancer Res 2003; 9: 5582–5588. [PubMed] [Google Scholar]
- Geisler S, Lonning PE, Aas T, Johnsen H, Fluge O, Haugen DF et al. Influence of TP53 gene alterations and c-erbB-2 expression on the response to treatment with doxorubicin in locally advanced breast cancer. Cancer Res 2001; 61: 2505–2512. [PubMed] [Google Scholar]
- Preudhomme C, Dervite I, Wattel E, Vanrumbeke M, Flactif M, Lai JL et al. Clinical significance of p53 mutations in newly diagnosed Burkitt's lymphoma and acute lymphoblastic leukemia: a report of 48 cases. J Clin Oncol 1995; 13: 812–820. [DOI] [PubMed] [Google Scholar]
- Zenz T, Eichhorst B, Busch R, Denzel T, Habe S, Winkler D et al. TP53 mutation and survival in chronic lymphocytic leukemia. J Clin Oncol 2010; 28: 4473–4479. [DOI] [PubMed] [Google Scholar]
- Lonning PE. Genes causing inherited cancer as beacons to identify the mechanisms of chemoresistance. Trends Mol Med 2004; 10: 113–118. [DOI] [PubMed] [Google Scholar]
- Wang JY, Knudsen ES, Welch PJ. The retinoblastoma tumor suppressor protein. Adv Cancer Res 1994; 64: 25–85. [DOI] [PubMed] [Google Scholar]
- Harrington EA, Bruce JL, Harlow E, Dyson N. pRB plays an essential role in cell cycle arrest induced by DNA damage. Proc Natl Acad Sci USA 1998; 95: 11945–11950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer 2008; 8: 671–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkiewicz AK, Ertel A, McFalls J, Valsecchi ME, Schwartz G, Knudsen ES. RB-pathway disruption is associated with improved response to neoadjuvant chemotherapy in breast cancer. Clin Cancer Res 2012; 18: 5110–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berge EO, Knappskog S, Geisler S, Staalesen V, Pacal M, Borresen-Dale AL et al. Identification and characterization of retinoblastoma gene mutations disturbing apoptosis in human breast cancers. Mol Cancer 2010; 9: 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berge EO, Knappskog S, Lillehaug JR, Lonning PE. Alterations of the retinoblastoma gene in metastatic breast cancer. Clin Exp Metastasis 2011; 28: 319–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 2002; 109: 335–346. [DOI] [PubMed] [Google Scholar]
- Smeets SJ, van der Plas M, Schaaij-Visser TB, van Veen EA, van Meerloo J, Braakhuis BJ et al. Immortalization of oral keratinocytes by functional inactivation of the p53 and pRb pathways. Int J Cancer 2011; 128: 1596–1605. [DOI] [PubMed] [Google Scholar]
- Knappskog S, Berge EO, Chrisanthar R, Geisler S, Staalesen V, Leirvaag B et al. Concomitant inactivation of the p53- and pRB- functional pathways predicts resistance to DNA damaging drugs in breast cancer in vivo. Mol Oncol 2015; 9: 1553–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perou CM. Molecular stratification of triple-negative breast cancers. Oncologist 2010; 15: 39–48. [DOI] [PubMed] [Google Scholar]
- Lupertz R, Watjen W, Kahl R, Chovolou Y. Dose- and time-dependent effects of doxorubicin on cytotoxicity, cell cycle and apoptotic cell death in human colon cancer cells. Toxicology 2010; 271: 115–121. [DOI] [PubMed] [Google Scholar]
- Zhu L, Lu Z, Zhao H. Antitumor mechanisms when pRb and p53 are genetically inactivated. Oncogene 2015; 34: 4547–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine A, Sihto H, Come C, Rosenfeldt MT, Zwolinska A, Niemela M et al. Senescence sensitivity of breast cancer cells is defined by positive feedback loop between CIP2A and E2F1. Cancer Discov 2013; 3: 182–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliwinska MA, Mosieniak G, Wolanin K, Babik A, Piwocka K, Magalska A et al. Induction of senescence with doxorubicin leads to increased genomic instability of HCT116 cells. Mech Ageing Dev 2009; 130: 24–32. [DOI] [PubMed] [Google Scholar]
- Brown JM, Wouters BG. Apoptosis, p53, and tumor cell sensitivity to anticancer agents. Cancer Res 1999; 59: 1391–1399. [PubMed] [Google Scholar]
- Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 2011; 145: 571–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L et al. Restoration of p53 function leads to tumour regression in vivo. Nature 2007; 445: 661–665. [DOI] [PubMed] [Google Scholar]
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010; 18: 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012; 487: 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartek J, Falck J, Lukas J. CHK2 kinase--a busy messenger. Nat Rev Mol Cell Biol 2001; 2: 877–886. [DOI] [PubMed] [Google Scholar]
- Kao J, Salari K, Bocanegra M, Choi YL, Girard L, Gandhi J et al. Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PLoS One 2009; 4: e6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006; 10: 515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- T'Ang A, Varley JM, Chakraborty S, Murphree AL, Fung YK. Structural rearrangement of the retinoblastoma gene in human breast carcinoma. Science 1988; 242: 263–266. [DOI] [PubMed] [Google Scholar]
- Wasielewski M, Elstrodt F, Klijn JG, Berns EM, Schutte M. Thirteen new p53 gene mutants identified among 41 human breast cancer cell lines. Breast Cancer Res Treat 2006; 99: 97–101. [DOI] [PubMed] [Google Scholar]