

Abstract
The proportion of elderly people rises in the developed countries. The increased susceptibility of the elderly to infectious diseases is caused by immune dysfunction, especially T cell functional decline. Age-related hematopoietic stem cells deviate from lymphoid lineage to myeloid lineage. Thymus shrinks early in life, which is followed by the decline of naïve T cells. T-cell receptor repertoire diversity declines by aging, which is caused by cytomegalovirus-driven T cell clonal expansion. Functional decline of B cell induces antibody affinity declines by aging. Many effector functions including phagocytosis of myeloid cells are down regulated by aging. The studies of aging of myeloid cells have some controversial results. Although M1 macrophages have been shown to be replaced by anti-inflammatory (M2) macrophages by advanced age, many human studies showed that pro-inflammatory cytokines are elevated in older human. To solve this discrepancy here we divide age-related pathological changes into two categories. One is an aging of immune cell itself. Second is involvement of immune cells to age-related pathological changes. Cellular senescence and damaged cells in aged tissue recruit pro-inflammatory M1 macrophages, which produce pro-inflammatory cytokines and proceed to age-related diseases. Underlying biochemical and metabolic studies will open nutritional treatment.
Keywords: Elderly people, Damage associated molecular patterns, Immune dysfunction, Lymphoid lineage, Myeloid lineage, Shrinkage of thymus, Cytomegalovirus, Age-related tissue damage, Cellular senescence, Pro-inflammatory
Core tip: The authors divide immune cells, which are involved in age-related pathological changes, into two categories. First category is aging of immune cell-itself, which include age-related myeloid lineage deviation of hematopoietic stem cells, the shrinkage of thymus followed by the decline of naïve T cells, the cytomegalovirus infection-mediated decline of T-cell receptor repertoire diversity and functional decline of myeloid cells. Second category is the involvement of immune cells to age-related pathological changes. Age-related tissue damage and cellular senescence activate pro-inflammatory (M1) macrophages, which induce many age-related diseases. Biochemical works are needed to further elucidate age-related immune changes.
INTRODUCTION
The proportion of elderly people rise world wide, especial in the developed countries. Aging-related changes in the immune system contribute to the increased susceptibility of the elderly to infectious diseases, cardiovascular disease and stroke caused by atherosclerosis, autoimmune disease such as rheumatoid arthritis, cancer and late onset anemia and degenerative diseases including Alzheimer diseases[1,2]. Further metabolic syndrome, which is caused by obesity occurs from middle age and proceeds to tissue failure such as renal failure in advanced age, is tightly related to immune system. Chronic infections such as hepatitis virus and chronic tissue damages caused by tobacco inhalation induce tissue damage, which arouses immune responses and wound repair responses. Chronic inflammation follows tissue fibrosis in advanced age proceeding to tissue failure such as chronic obstructive pulmonary disease. Most prominent cause of age-related immune dysfunction is T cell immunosenescence. There are three causes of T cell immunosenescence. One is the age-related hematopoietic stem cells (HSCs) deviation from lymphoid lineage to myeloid lineage. Second is the shrinkage of thymus. Third is expansion of T cell clones to cytomegalovirus (CMV). Changes of HSCs also affect immunosenescence. HSCs deviate to myeloid lineage by aging. Both in mouse and human myeloid-lymphoid ratio elevates by aging[3,4], which induces the decline of lymphoid cells (T and B cells) and erythrocytes and contribute age-related anemia and decline of adaptive immunity[5,6]. The number of aged B cells decline and affinity and diversity of antibodies are low[7]. Ageing related myeloid deviation increases the number of myeloid cells. However, the oxidative burst and phagocytosis of both neutrophils and macrophages are decreased[8]. The antigen presentation of aged dendritic cells and the cytolysis of Natural killer cells are low[9]. Pro-inflammatory cytokines such as interleukin-6 (IL-6), tumor necrosis factor α (TNF-α) and IL-1β are elevated in elderly people, which are called inflammaging and induce metabolic syndrome such as obesity, type II diabetes, atherosclerosis, cardiac diseases[10]. However, other reports have shown that pro-inflammatory (M1) macrophages are replaced by anti-inflammatory (M2) macrophages by aging[11]. Here to solve this discrepancy we propose to classify age-related immune changes as follows. Age-related pathological changes are classified as immune cell intrinsic changes by aging and involvement of immune cells to age-related pathological changes. Increased susceptibility of the elderly to infectious diseases is mainly caused by age-related immune cell intrinsic changes usually called immunosenescence. Metabolic syndrome and other related diseases, which occur at aged people, are mainly caused by immune cell attack to age-related tissue changes. Age-related tissue failure is caused by repeated immune cell attack. Here we first describe the recent findings from the points of immune cell intrinsic changes by aging and then go to age-related tissue changes, which induce immune reaction (Figure 1).
Figure 1.
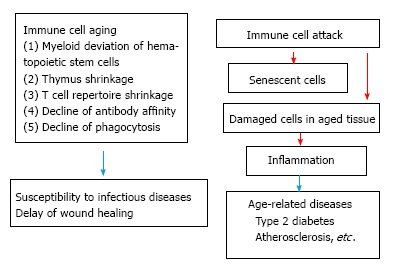
Two types of engagement of immune cells to aging.
IMMUNE CELL INTRINSIC CHANGES BY AGING (IMMUNOSENESCENCE)
HSCs aging
Adult immune cells develop from HSCs that originate from the bone marrow (BM) (Figure 2). From HSCs common lymphoid progenitor cells (CLPs) and myeloid precursor cells develop. T cells emigrate from BM to enter thymus, where they develop to naïve CD4 and CD8 T cells. B cells develop in bone marrow from pre-pro-B cells, pro-B cells and emigrate from bone marrow as pre-B cells to circulation and secondary lymph nodes and spleen. Finally B cells further differentiate to plasma cells to produce antibodies (Figures 2 and 3).
Figure 2.
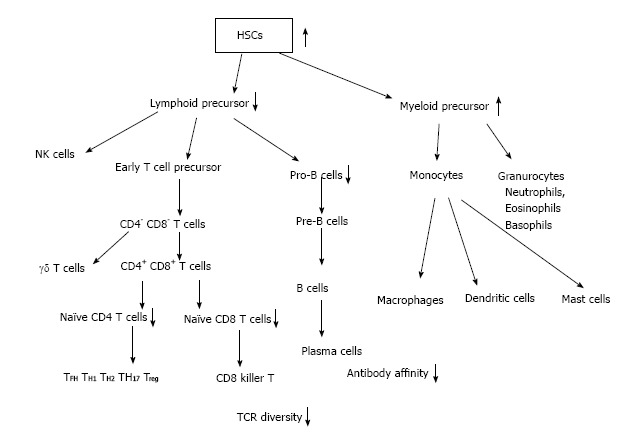
Hematopoietic stem cells to mature immune cells. HSCs: Hematopoietic stem cells; TCR: T-cell receptor.
Figure 3.
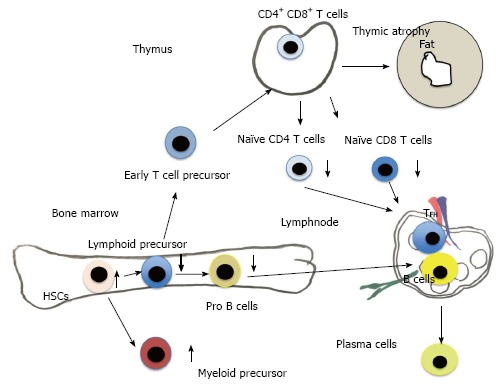
Differentiation of hematopoietic stem cells to mature immune cells.
Although the cellularity of BM decreases by aging[12], the number of HSCs in aged mice differ between mouse stains. The number of HSCs of aged-C57BL/6 mice increases, whereas other strain of mice decreases[13,14]. However, in HSCs of aged mice the self-renewal activity and homing activity to BM are two-fold less efficient compared with young HSCs[2,6,15-17]. It has been found by transplantation experiments that old HSCs tend to have a myeloid-skewed blood cell production caused by a decreased ability to produce lymphoid cells[3,18]. These age-related changes of HSCs induce age-related abnormalities such as myeloid leukemia[19], late onset anemia[20]. Biochemical mechanism underlining HSC aging has been elucidated recently. The small RhoGTPase Cdc42 in aged HSCs is elevated, which correlates with a loss of polarity in aged HSCs. It has been found that in young HSCs Cdc42 and tubulin are asymmetrically distributed at the same location, whereas in aged HSCs Cdc42 and tubulin were distributed throughout the cell body in an unpolarized fashion[21]. HSCs, which produce all mature blood cells, must counterbalance the deleterious effects of chronic stress by generating new cells to replace those that become damaged or destroyed. Cells adapt chronic stress by autophagy and apoptosis[22]. Autophagy adapts starvation by using degraded own cell organelles for nutrient. One report has shown that autophagy is suggested to be decreased by aging[23]. However, another report has shown that by electron microscopic analysis autophagy is increased by aged mice, and old HSCs retain an intact FoxO3a-driven pro-autophagy gene program[24]. FOXO genes are key regulators of longevity downstream of insulin and insulin like growth factor signaling[25]. MicroRNAs, a class of small-noncoding RNAs, regulate normal function of HSCs, including cell cycling and engraftment potential[26]. The microRNA-212/132 cluster (Mirc19) is enriched in HSCs and is up regulated during aging and plays a role in maintaining balanced hematopoietic output by buffering FOXO3 expression[27]. There exist many other molecules, which relate HSCs aging. For instance recent work done by Chang et al[28] showed that ABT263 (a specific inhibitor of the anti-apoptotic proteins BCL-2 and BCL-xL) rejuvenated aged HSCs.
T cell aging
One of the most early and dramatic changes in aging of immune system is the decrease of naïve T cells. Naïve T cells are derived from thymus and continuously recirculate through the peripheral lymphoid tissues. Naïve T cells differentiate into effector/memory T cells after primed with cognate antigen presented by dendritic cells. Naïve T cells originate from thymus. Naïve T cell production from thymus declines very early only after first year of life. These naïve T cell decline comes from the early thymic involution[29,30] (Figures 2 and 3). This dramatic decline of human naïve T cell output is compensated by homeostatic proliferation[31,32]. CD4+CD45RA+CCR7+ human naive T cells are recently found to be divided by CD31. CD31+ naïve T cells are recent thymic emigrants (RTEs). By aging CD31+ CD4+CD45RA+ decrease, while CD31- CD4+CD45RA+ increase during healthy aging by homeostatic proliferation[33-35]. Recent work using neonatal thymectomy has shown that total number of CD3+, CD4+ and CD8+ cell numbers decrease in the first year after neonatal thymectomy. CD4+CD45RA+CCR7+ and CD31+ CD4+CD45RA+ naive T cells decrease, while effector memory CD4+ T cells (CD45RA-CCR7-) and central memory CD4+ T cells (CD45RA-CCR7+) increase[36]. A human memory T cell subset with stem cell-like properties (CD4+CD45RA+CCR7+ CD28+CD27+FAS+), which has been shown recently[37] increase relatively. Wijk’s group has shown recently that after more than 10 years of thymectomy most children have thymic regeneration and show almost the same number of CD31+ naïve T cells compared to healthy control. Only small numbers of children have no thymic regeneration with lower T cell count and low percentage of naïve T cells compared to thymectonized CD31+ children[38]. Another report, which uses in vivo 2H2O labeling, has shown that compensatory increase of peripheral T- cell division rates are not required to compensate the age-related decrease of thymic output[34]. From these findings, Goronzy et al[39] summarized that even in early adulthood, thymic output contributes little to the maintenance of the size of the naïve T cell compartment. In order to build T cell repertoire, thymic output is necessary only in early life. To maintain T cell repertoire, thymic output is not necessary. Homeostatic proliferation of the existing T cell pool is enough to maintain CD4 T cell repertoire. Both IL-2 and IL-7 drive homeostatic proliferation and IL-2 receptor (CD25) expression is increased by aging. In contrast to CD4 naïve T cells, the CD8 naïve T cell compartment shrinks with age, which induces higher homeostatic proliferation in aged-CD8 naïve T cells than that in aged-CD4 naïve T cells[39]. Another cause of T cell dysfunction of aged people instead of decreased production of naïve T cells is the decline in TCR repertoire diversity, caused by cytomegalovirus-driven T cell clonal expansions[40]. The naïve T cell repertoire are in the range of approximately 2.5 × 107 in humans. These repertoires are hijacked by CMV infection[41]. Virus-specific clonally expanded CD8 T cells occupy more than 50% of memory pool and reached to 90% in aged mice[42]. Naïve T cells are thought to be the precursor of memory T cells. By infection memory T cells respond quickly by producing cytokines, granzyme and perforin. Recently, new type of CD8 T cells have been discovered called “memory T cells with a naive phenotype” (TMNP cells), increased with age. TMNP cells have Naïve T cell phenotypes, but quickly respond to infection by producing cytokines, granzyme and perforin. TMNP cells have unique V 14 T cell receptor. TMNP cells bound to multimers of CMV or EBV peptide and MHC class Ι[43]. These results mean that some population of CD8 T cells, which are expanded by CMV, work as effective protection of other virus infection. Further works are needed to elucidate the effects on age-related T cell dysfunction caused by CMV infection. Here we do not describe B cell dysfunction of aged people. B cell function decreases directly by B cell aging itself and by the result of age-related T cell functional decline, because helper T cell affect B cells by direct binding and secreting cytokines.
Myeloid cell aging
Innate immune cells include granulocytes (neutrophils, eosinophils, basophils), macrophages, dendritic cells, natural killer cells and innate lymphoid cells and others[44]. Among them “myeloid cell” here include granulocytes, macrophages and dendritic cells. We review recent findings, which relate to myeloid cell intrinsic aging. Phagocytosis is the most important effector function of myeloid cells especially neutrophils. Neutrophils play a crucial role in the immune defense against bacterial and fungal pathogens[45]. Neutrophils survive for only 8-12 h in the circulation and up to 1-2 d in tissues[46]. Neutrophils from aged human have impaired phagocytosis of bacteria[46,47] DHEA (Dehydroepiandrosterone sulfate), which increases the phosphorylation of p47phox via PKC-β and generate neutrophil superoxide to kill bacteria, decreases in old age[48]. Neutrophils also work as an important player in wound healing. Neutrophils normally begin arriving at the wound site within minutes of injury, continuing for several days. Molecules called damage associated molecular patterns (DAMPs) are produced from wounded tissue cells, which activate neutrophils. Neutrophils engulf damaged cells and produce inflammatory cytokines and chemokines, which facilitate wound repair. We have shown that in aged C57BL/6 mice, wound healing is relatively inefficient. Anti-Gri-1 treatment delayed wound healing in aged mice, whereas G-CSF injection increased wound repair[49]. In bacterial infection and wound healing, macrophages come to work after neutrophil and engulf dead cells including dead neutrophils.
Every tissue contains tissue macrophages, which are sometimes called tissue specific names such as microglia in brain, Langerhans cells in skin and kupper cells in liver. Important functions of tissue macrophages include innate defense against infection and wound healing. Origin and turnover of tissue macrophages depend on tissue types. Adult’s microglia derive from primitive myeloid progenitors of yolk sac that arise before embryonic day 8[50]. Further non-parenchymal macrophages such as perivascular, meningeal and choroid plexus macrophages, which mediate immune responses at brain boundaries, also arise from yolk sac precursors during embryonic development and remain a stable population[51]. Langerhans cells predominantly derived from embryonic fetal liver with a minor contribution of yolk sac[52,53]. Tissue-resident macrophages with Low turn over are self-maintain locally throughout adult life with minimal contribution from circulating monocytes[54] and self renew after birth[55]. In heart cardiac macrophages are primarily embryonic and are self-renewed, which reside throughout myocardium[56]. Recent data have shown that by advanced age these embryo-derived tissue macrophages are replaced by blood monocytes derived from HSCs. In the heart embryo-derived cardiac macrophages decline self-renewal by age and are progressively substituted by monocyte-derived macrophages[57]. Intestinal macrophages have a high turnover rate and are constantly replaced from bone marrow derived Ly6C+blood monocytes from early lifetime[58]. In peritoneal cavity, embryo-derived macrophages retain self-renewal at least until 4 mo in mice. By aging embryo-derived macrophages in peritoneal cavity are replaced by bone marrow-derived monocytes[59].
There exist two types of macrophages namely M1 macrophages and M2 macrophages. M1 (inflammatory) macrophages produce NO radical form arginine with IFNγ or LPS stimulation, whereas M2 macrophages produce TGFβ1 and inhibit NO synthase and stimulate arginase. Arginase converts arginine to ornithine, which induces cell division[60]. M2 macrophages are further subdivided into M2a and M2c. M2a is activated by IL-4/IL13, and relates to wound-healing process[61]. M2c is activated by IL10, which has anti-inflammatory activity[62]. It has been shown that macrophages of aged mice polarize to M2 macrophages[11,63]. M2 macrophages promote angiogenesis in age-related eye disease[11]. M2a macrophages have been shown to induce muscle aging by inducing fibrosis. Transplantation of young BM cells to aged mice shows fewer M2a macrophages and less accumulation of collagen[64]. These findings of age-related M2 predominance contradicts so called “inflammaging”, which predominates pro-inflammatory state.
AGE-RELATED TISSUE CHANGES, WHICH INDUCE IMMUNE CELL ATTACK
Although aged HSCs have reduced regenerative potential, aged HSCs are biased toward myeloid differentiation at the expense of lymphoid differentiation[65]. Chronic, low-grade, systemic inflammation is the primary risk factor for major human chronic diseases, including cardiovascular disorders, cancer, type 2 diabetes, and neurodegenerative disease. Increased production of inflammatory mediators that accompany this process, referred to as “inflammaging”[66]. This inflammatory tendency in aged human has wide variability. The mechanism of inflammaging has still not reach complete understanding.
Damaged macromolecules and self-debris released as a consequence of cell/organelle injury work as DAMPs, which may activate pro-inflammatory macrophages. These DAMPs include free radicals from oxidative stress and advanced glycation end products, ATP, fatty acids and amyloid. Another sources, which are up regulated by aging, are leaked microbial products from gut working as pathogen associated molecular patterns (PAMPs)[65]. These DAMPs and PAMPs may work as stimulator to M1 macrophages. These inflammaging and age-related M2 macrophage activation described previously seem to contradictive. How to consider this discrepancy? By acute infection or injury, neutrophils and M1 macrophages first predominate to the infected or injured site, then M2 macrophages resolve inflammation and induce tissue repair. In chronic inflammation, M1 macrophages continue to exist in inflamed tissue with late coming M2 macrophages. We speculate that both tissue fibrosis caused by M2 macrophages and pro-inflammatory status caused by M1 macrophages continue to exist in chronic inflammation. In acute infection or acute tissue damage, M2 macrophages compensate inflammation caused by M1 macrophages and induce tissue repair. However, in persistent infection such as hepatitis C or repeated tissue damage such as smoking, M1 macrophages, which work as inflammation and M2 macrophages, which resolve tissue damages by producing matrix materials, exist at the same time. Predominance of either M1 or M2 macrophages may dependent on disease process.
Metabolic syndrome and macrophages
Metabolic syndrome, which has the risk to proceed to Type 2 diabetes and atherosclerosis, is caused by obesity. Now it is established that visceral and subcutaneous white adipose tissues (WAT) are different concerning metabolic activity. Lipolytic effect of catecholamines is lower in visceral WAT than that in subcutaneous WAT[67]. Two main causes of insulin resistance (which causes type 2 diabetes) are saturated fatty acids and pro-inflammatory cytokines. Saturated fatty acids can bind to the Toll-like receptors (TLR) 2 and 4, especially in adipocytes and macrophages, which induce c-Jun N-terminal kinase (JNK), phosphorylate IRS1 and decrease insulin receptor signaling. Saturated fatty acids also produce pro-inflammatory cytokines[68,69]. The expansion of WAT leads to necrosis of hypertrophic adipocytes, which produce DAMPs. DAMPs recruit macrophages and produce pro-inflammatory cytokines, which aggravate insulin resistance[70]. Atherosclerosis is mediated by the recruitment of monocyte-derived macrophages into the subendothelial space, where they ingest the lipoproteins[71]. Type 2 diabetes proceeds to many other age-related diseases such as renal failure. Atherosclerosis also proceeds to stroke, coronary heart disease. Thus macrophages especially M1 macrophages are engaged in early stage of age-related diseases.
Clearance of senescent cells by immune cells
Not only M1 macrophages but also aged-other tissue cells produce pro-inflammatory cytokines. Vascular aging enhanced secretion of inflammatory mediators from aging endothelial cells and vascular smooth muscle cells from disease free animals[72]. One explanation of this age-related secretion of inflammatory mediators is senescence associated secretory phenotype (SASP). SASP is produced by senescent cells. From the studies using mice and aged human, aged innate immune system show elevation of basal inflammation[73]. Especially older human have elevated level of pro-inflammatory cytokines[74,75]. However, we should keep in mind that cellular senescence is an independent phenomenon to in vivo aging. Cellular senescence triggers tissue remodeling in embryonic tissue development and in tissue damage. Cellular senescence also engages in age associated loss of tissue functions[76,77]. Cellular senescence has been first described by Heyflick at 1961 as limited replicative potential of human fibroblasts in vitro[78]. Cellular senescence is cell cycle arrest with or without telomere shortening after cell division. There exist p53/ p21Waf1 pathways and p16INK4a/pRB pathways in cellular senescence[79]. Whether senescent cells contribute to age-related diseases remains unclear. Some reports have shown that senescent cells have a protective role in atherosclerosis. Mice lacking senescent markers such as p53, p21 or p19Arf show accelerated atherosclerosis[76]. Senescent cells produce SASPs, which attract immune cells and eliminate senescent cells. Senescent cells attract not only granulocytes and macrophages but also NK cells, CD8 T cells and CD4+ T helper 1 (Th1 CD4 T) cells[80,81]. However recent report showed that senescent foamy macrophages produced inflammatory cytokines (TNFα), monocyte recruiting chemokines (MCP1) and matrix proteases, which engaged in atherosclerosis[82]. In this case, macrophages themselves became senescent.
CONCLUSION
From the demand to solve worldwide increase of age-related diseases, the future biochemical studies are needed, which elucidate the phenomena of immunosenescence and immune cell attack to eliminate senescent cells or damaged cells by age-related stresses. In advanced age, diseases are caused by M1 or M2 macrophages as mentioned above. Which type of macrophages are involved depend on the situation. In order to prevent or treat age-related diseases we have to carefully evaluate the immune status. Because metabolic reprogramming of oxidative phosphorylation to glycolysis occurs in M1 pro-inflammatory macrophages and urea cycle intermediates such as arginine, ornithine, citrulline regulate M1/M2 polarization[60], the measurement of metabolic changes will elucidate the predominant type of macrophages. Studies of metabolic pathway of immune cells in aging will open the nutrient treatment of age-related diseases.
Footnotes
Conflict-of-interest statement: There are no conflict of interest.
Manuscript source: Invited manuscript
Specialty type: Biochemistry and molecular biology
Country of origin: Japan
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B, B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Peer-review started: August 24, 2016
First decision: November 19, 2016
Article in press: March 15, 2017
P- Reviewer: Freire-De-Lima CG, Goebel WS, Louboutin JP S- Editor: Kong JX L- Editor: A E- Editor: Lu YJ
References
- 1.Henry CJ, Marusyk A, DeGregori J. Aging-associated changes in hematopoiesis and leukemogenesis: what’s the connection? Aging (Albany NY) 2011;3:643–656. doi: 10.18632/aging.100351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Geiger H, de Haan G, Florian MC. The ageing haematopoietic stem cell compartment. Nat Rev Immunol. 2013;13:376–389. doi: 10.1038/nri3433. [DOI] [PubMed] [Google Scholar]
- 3.Sudo K, Ema H, Morita Y, Nakauchi H. Age-associated characteristics of murine hematopoietic stem cells. J Exp Med. 2000;192:1273–1280. doi: 10.1084/jem.192.9.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pang WW, Price EA, Sahoo D, Beerman I, Maloney WJ, Rossi DJ, Schrier SL, Weissman IL. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci USA. 2011;108:20012–20017. doi: 10.1073/pnas.1116110108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang J, Geiger H, Rudolph KL. Immunoaging induced by hematopoietic stem cell aging. Curr Opin Immunol. 2011;23:532–536. doi: 10.1016/j.coi.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 6.Morrison SJ, Wandycz AM, Akashi K, Globerson A, Weissman IL. The aging of hematopoietic stem cells. Nat Med. 1996;2:1011–1016. doi: 10.1038/nm0996-1011. [DOI] [PubMed] [Google Scholar]
- 7.Han S, Yang K, Ozen Z, Peng W, Marinova E, Kelsoe G, Zheng B. Enhanced differentiation of splenic plasma cells but diminished long-lived high-affinity bone marrow plasma cells in aged mice. J Immunol. 2003;170:1267–1273. doi: 10.4049/jimmunol.170.3.1267. [DOI] [PubMed] [Google Scholar]
- 8.Plowden J, Renshaw-Hoelscher M, Engleman C, Katz J, Sambhara S. Innate immunity in aging: impact on macrophage function. Aging Cell. 2004;3:161–167. doi: 10.1111/j.1474-9728.2004.00102.x. [DOI] [PubMed] [Google Scholar]
- 9.Uyemura K, Castle SC, Makinodan T. The frail elderly: role of dendritic cells in the susceptibility of infection. Mech Ageing Dev. 2002;123:955–962. doi: 10.1016/s0047-6374(02)00033-7. [DOI] [PubMed] [Google Scholar]
- 10.Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123:966–972. doi: 10.1172/JCI64098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kelly J, Ali Khan A, Yin J, Ferguson TA, Apte RS. Senescence regulates macrophage activation and angiogenic fate at sites of tissue injury in mice. J Clin Invest. 2007;117:3421–3426. doi: 10.1172/JCI32430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ogawa T, Kitagawa M, Hirokawa K. Age-related changes of human bone marrow: A histometric estimation of proliferative cells, apoptotic cells, T cells, B cells and macrophages. Mech Ageing Dev. 2000;117:57–68. doi: 10.1016/s0047-6374(00)00137-8. [DOI] [PubMed] [Google Scholar]
- 13.Geiger H, Rennebeck G, Van Zant G. Regulation of hematopoietic stem cell aging in vivo by a distinct genetic element. Proc Natl Acad Sci USA. 2005;102:5102–5107. doi: 10.1073/pnas.0408654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Haan G, Nijhof W, Van Zant G. Mouse strain-dependent changes in frequency and proliferation of hematopoietic stem cells during aging: correlation between lifespan and cycling activity. Blood. 1997;89:1543–1550. [PubMed] [Google Scholar]
- 15.Rossi DJ, Bryder D, Zahn JM, Ahlenius H, Sonu R, Wagers AJ, Weissman IL. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci USA. 2005;102:9194–9199. doi: 10.1073/pnas.0503280102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beerman I, Maloney WJ, Weissmann IL, Rossi DJ. Stem cells and the aging hematopoietic system. Curr Opin Immunol. 2010;22:500–506. doi: 10.1016/j.coi.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liang Y, Van Zant G, Szilvassy SJ. Effects of aging on the homing and engraftment of murine hematopoietic stem and progenitor cells. Blood. 2005;106:1479–1487. doi: 10.1182/blood-2004-11-4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takizawa H, Regoes RR, Boddupalli CS, Bonhoeffer S, Manz MG. Dynamic variation in cycling of hematopoietic stem cells in steady state and inflammation. J Exp Med. 2011;208:273–284. doi: 10.1084/jem.20101643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kiss TL, Sabry W, Lazarus HM, Lipton JH. Blood and marrow transplantation in elderly acute myeloid leukaemia patients - older certainly is not better. Bone Marrow Transplant. 2007;40:405–416. doi: 10.1038/sj.bmt.1705747. [DOI] [PubMed] [Google Scholar]
- 20.Beghé C, Wilson A, Ershler WB. Prevalence and outcomes of anemia in geriatrics: a systematic review of the literature. Am J Med. 2004;116 Suppl 7A:3S–10S. doi: 10.1016/j.amjmed.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 21.Florian MC, Dörr K, Niebel A, Daria D, Schrezenmeier H, Rojewski M, Filippi MD, Hasenberg A, Gunzer M, Scharffetter-Kochanek K, et al. Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell. 2012;10:520–530. doi: 10.1016/j.stem.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 24.Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R, Debnath J, Passegué E. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature. 2013;494:323–327. doi: 10.1038/nature11895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martins R, Lithgow GJ, Link W. Long live FOXO: unraveling the role of FOXO proteins in aging and longevity. Aging Cell. 2016;15:196–207. doi: 10.1111/acel.12427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao JL, Rao DS, O’Connell RM, Garcia-Flores Y, Baltimore D. MicroRNA-146a acts as a guardian of the quality and longevity of hematopoietic stem cells in mice. Elife. 2013;2:e00537. doi: 10.7554/eLife.00537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mehta A, Zhao JL, Sinha N, Marinov GK, Mann M, Kowalczyk MS, Galimidi RP, Du X, Erikci E, Regev A, et al. The microRNA-132 and microRNA-212 cluster regulates hematopoietic stem cell maintenance and survival with age by buffering FOXO3 expression. Immunity. 2015;42:1021–1032. doi: 10.1016/j.immuni.2015.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016;22:78–83. doi: 10.1038/nm.4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aspinall R, Andrew D. Thymic involution in aging. J Clin Immunol. 2000;20:250–256. doi: 10.1023/a:1006611518223. [DOI] [PubMed] [Google Scholar]
- 30.Bains I, Antia R, Callard R, Yates AJ. Quantifying the development of the peripheral naive CD4+ T-cell pool in humans. Blood. 2009;113:5480–5487. doi: 10.1182/blood-2008-10-184184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hazenberg MD, Otto SA, de Pauw ES, Roelofs H, Fibbe WE, Hamann D, Miedema F. T-cell receptor excision circle and T-cell dynamics after allogeneic stem cell transplantation are related to clinical events. Blood. 2002;99:3449–3453. doi: 10.1182/blood.v99.9.3449. [DOI] [PubMed] [Google Scholar]
- 32.Westera L, van Hoeven V, Drylewicz J, Spierenburg G, van Velzen JF, de Boer RJ, Tesselaar K, Borghans JA. Lymphocyte maintenance during healthy aging requires no substantial alterations in cellular turnover. Aging Cell. 2015;14:219–227. doi: 10.1111/acel.12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kilpatrick RD, Rickabaugh T, Hultin LE, Hultin P, Hausner MA, Detels R, Phair J, Jamieson BD. Homeostasis of the naive CD4+ T cell compartment during aging. J Immunol. 2008;180:1499–1507. doi: 10.4049/jimmunol.180.3.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kimmig S, Przybylski GK, Schmidt CA, Laurisch K, Möwes B, Radbruch A, Thiel A. Two subsets of naive T helper cells with distinct T cell receptor excision circle content in human adult peripheral blood. J Exp Med. 2002;195:789–794. doi: 10.1084/jem.20011756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kohler S, Wagner U, Pierer M, Kimmig S, Oppmann B, Möwes B, Jülke K, Romagnani C, Thiel A. Post-thymic in vivo proliferation of naive CD4+ T cells constrains the TCR repertoire in healthy human adults. Eur J Immunol. 2005;35:1987–1994. doi: 10.1002/eji.200526181. [DOI] [PubMed] [Google Scholar]
- 36.van Gent R, Schadenberg AW, Otto SA, Nievelstein RA, Sieswerda GT, Haas F, Miedema F, Tesselaar K, Jansen NJ, Borghans JA. Long-term restoration of the human T-cell compartment after thymectomy during infancy: a role for thymic regeneration? Blood. 2011;118:627–634. doi: 10.1182/blood-2011-03-341396. [DOI] [PubMed] [Google Scholar]
- 37.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C, et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17:1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van den Broek T, Delemarre EM, Janssen WJ, Nievelstein RA, Broen JC, Tesselaar K, Borghans JA, Nieuwenhuis EE, Prakken BJ, Mokry M, et al. Neonatal thymectomy reveals differentiation and plasticity within human naive T cells. J Clin Invest. 2016;126:1126–1136. doi: 10.1172/JCI84997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goronzy JJ, Fang F, Cavanagh MM, Qi Q, Weyand CM. Naive T cell maintenance and function in human aging. J Immunol. 2015;194:4073–4080. doi: 10.4049/jimmunol.1500046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blackman MA, Woodland DL. The narrowing of the CD8 T cell repertoire in old age. Curr Opin Immunol. 2011;23:537–542. doi: 10.1016/j.coi.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arstila TP, Casrouge A, Baron V, Even J, Kanellopoulos J, Kourilsky P. A direct estimate of the human alphabeta T cell receptor diversity. Science. 1999;286:958–961. doi: 10.1126/science.286.5441.958. [DOI] [PubMed] [Google Scholar]
- 42.Callahan JE, Kappler JW, Marrack P. Unexpected expansions of CD8-bearing cells in old mice. J Immunol. 1993;151:6657–6669. [PubMed] [Google Scholar]
- 43.Pulko V, Davies JS, Martinez C, Lanteri MC, Busch MP, Diamond MS, Knox K, Bush EC, Sims PA, Sinari S, et al. Human memory T cells with a naive phenotype accumulate with aging and respond to persistent viruses. Nat Immunol. 2016;17:966–975. doi: 10.1038/ni.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE, et al. Innate lymphoid cells--a proposal for uniform nomenclature. Nat Rev Immunol. 2013;13:145–149. doi: 10.1038/nri3365. [DOI] [PubMed] [Google Scholar]
- 45.Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6:173–182. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 46.Butcher SK, Chahal H, Nayak L, Sinclair A, Henriquez NV, Sapey E, O’Mahony D, Lord JM. Senescence in innate immune responses: reduced neutrophil phagocytic capacity and CD16 expression in elderly humans. J Leukoc Biol. 2001;70:881–886. [PubMed] [Google Scholar]
- 47.Simell B, Vuorela A, Ekström N, Palmu A, Reunanen A, Meri S, Käyhty H, Väkeväinen M. Aging reduces the functionality of anti-pneumococcal antibodies and the killing of Streptococcus pneumoniae by neutrophil phagocytosis. Vaccine. 2011;29:1929–1934. doi: 10.1016/j.vaccine.2010.12.121. [DOI] [PubMed] [Google Scholar]
- 48.Radford DJ, Wang K, McNelis JC, Taylor AE, Hechenberger G, Hofmann J, Chahal H, Arlt W, Lord JM. Dehydroepiandrosterone sulfate directly activates protein kinase C-beta to increase human neutrophil superoxide generation. Mol Endocrinol. 2010;24:813–821. doi: 10.1210/me.2009-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nishio N, Okawa Y, Sakurai H, Isobe K. Neutrophil depletion delays wound repair in aged mice. Age (Dordr) 2008;30:11–19. doi: 10.1007/s11357-007-9043-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goldmann T, Wieghofer P, Jordão MJ, Prutek F, Hagemeyer N, Frenzel K, Amann L, Staszewski O, Kierdorf K, Krueger M, et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol. 2016;17:797–805. doi: 10.1038/ni.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chorro L, Sarde A, Li M, Woollard KJ, Chambon P, Malissen B, Kissenpfennig A, Barbaroux JB, Groves R, Geissmann F. Langerhans cell (LC) proliferation mediates neonatal development, homeostasis, and inflammation-associated expansion of the epidermal LC network. J Exp Med. 2009;206:3089–3100. doi: 10.1084/jem.20091586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hoeffel G, Wang Y, Greter M, See P, Teo P, Malleret B, Leboeuf M, Low D, Oller G, Almeida F, et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J Exp Med. 2012;209:1167–1181. doi: 10.1084/jem.20120340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38:792–804. doi: 10.1016/j.immuni.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sieweke MH, Allen JE. Beyond stem cells: self-renewal of differentiated macrophages. Science. 2013;342:1242974. doi: 10.1126/science.1242974. [DOI] [PubMed] [Google Scholar]
- 56.Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, Brija T, Gautier EL, Ivanov S, Satpathy AT, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity. 2014;40:91–104. doi: 10.1016/j.immuni.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Molawi K, Wolf Y, Kandalla PK, Favret J, Hagemeyer N, Frenzel K, Pinto AR, Klapproth K, Henri S, Malissen B, et al. Progressive replacement of embryo-derived cardiac macrophages with age. J Exp Med. 2014;211:2151–2158. doi: 10.1084/jem.20140639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zigmond E, Varol C, Farache J, Elmaliah E, Satpathy AT, Friedlander G, Mack M, Shpigel N, Boneca IG, Murphy KM, et al. Ly6C hi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen-presenting cells. Immunity. 2012;37:1076–1090. doi: 10.1016/j.immuni.2012.08.026. [DOI] [PubMed] [Google Scholar]
- 59.Bain CC, Hawley CA, Garner H, Scott CL, Schridde A, Steers NJ, Mack M, Joshi A, Guilliams M, Mowat AM, et al. Long-lived self-renewing bone marrow-derived macrophages displace embryo-derived cells to inhabit adult serous cavities. Nat Commun. 2016;7:ncomms11852. doi: 10.1038/ncomms11852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AMM-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164:6166–6173. doi: 10.4049/jimmunol.1701141. [DOI] [PubMed] [Google Scholar]
- 61.He L, Marneros AG. Macrophages are essential for the early wound healing response and the formation of a fibrovascular scar. Am J Pathol. 2013;182:2407–2417. doi: 10.1016/j.ajpath.2013.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sene A, Khan AA, Cox D, Nakamura RE, Santeford A, Kim BM, Sidhu R, Onken MD, Harbour JW, Hagbi-Levi S, et al. Impaired cholesterol efflux in senescent macrophages promotes age-related macular degeneration. Cell Metab. 2013;17:549–561. doi: 10.1016/j.cmet.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Y, Wehling-Henricks M, Samengo G, Tidball JG. Increases of M2a macrophages and fibrosis in aging muscle are influenced by bone marrow aging and negatively regulated by muscle-derived nitric oxide. Aging Cell. 2015;14:678–688. doi: 10.1111/acel.12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Beerman I, Bhattacharya D, Zandi S, Sigvardsson M, Weissman IL, Bryder D, Rossi DJ. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc Natl Acad Sci USA. 2010;107:5465–5470. doi: 10.1073/pnas.1000834107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Franceschi C. Inflammaging as a major characteristic of old people: can it be prevented or cured? Nutr Rev. 2007;65:S173–S176. doi: 10.1111/j.1753-4887.2007.tb00358.x. [DOI] [PubMed] [Google Scholar]
- 67.Wajchenberg BL. Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocr Rev. 2000;21:697–738. doi: 10.1210/edrv.21.6.0415. [DOI] [PubMed] [Google Scholar]
- 68.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hirosumi J, Tuncman G, Chang L, Görgün CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 70.Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121:2111–2117. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13:709–721. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang M, Jiang L, Monticone RE, Lakatta EG. Proinflammation: the key to arterial aging. Trends Endocrinol Metab. 2014;25:72–79. doi: 10.1016/j.tem.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shaw AC, Goldstein DR, Montgomery RR. Age-dependent dysregulation of innate immunity. Nat Rev Immunol. 2013;13:875–887. doi: 10.1038/nri3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bruunsgaard H, Andersen-Ranberg K, Jeune B, Pedersen AN, Skinhøj P, Pedersen BK. A high plasma concentration of TNF-alpha is associated with dementia in centenarians. J Gerontol A Biol Sci Med Sci. 1999;54:M357–M364. doi: 10.1093/gerona/54.7.m357. [DOI] [PubMed] [Google Scholar]
- 75.Fagiolo U, Cossarizza A, Scala E, Fanales-Belasio E, Ortolani C, Cozzi E, Monti D, Franceschi C, Paganelli R. Increased cytokine production in mononuclear cells of healthy elderly people. Eur J Immunol. 1993;23:2375–2378. doi: 10.1002/eji.1830230950. [DOI] [PubMed] [Google Scholar]
- 76.Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15:482–496. doi: 10.1038/nrm3823. [DOI] [PubMed] [Google Scholar]
- 77.Vicente R, Mausset-Bonnefont AL, Jorgensen C, Louis-Plence P, Brondello JM. Cellular senescence impact on immune cell fate and function. Aging Cell. 2016;15:400–406. doi: 10.1111/acel.12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 79.Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev. 2014;28:99–114. doi: 10.1101/gad.235184.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sagiv A, Biran A, Yon M, Simon J, Lowe SW, Krizhanovsky V. Granule exocytosis mediates immune surveillance of senescent cells. Oncogene. 2013;32:1971–1977. doi: 10.1038/onc.2012.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, Zhao Z, Thapar V, Joyce JA, Krizhanovsky V, et al. Non-cell-autonomous tumor suppression by p53. Cell. 2013;153:449–460. doi: 10.1016/j.cell.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016;354:472–477. doi: 10.1126/science.aaf6659. [DOI] [PMC free article] [PubMed] [Google Scholar]