

Abstract
AIM
To identify neuron-selective androgen receptor (AR) signaling inhibitors, which could be useful in the treatment of spinal and bulbar muscular atrophy (SBMA), or Kennedy’s disease, a neuromuscular disorder in which deterioration of motor neurons leads to progressive muscle weakness.
METHODS
Cell lines representing prostate, kidney, neuron, adipose, and muscle tissue were developed that stably expressed the CFP-AR-YFP FRET reporter. We used these cells to screen a library of small molecules for cell type-selective AR inhibitors. Secondary screening in luciferase assays was used to identify the best cell-type specific AR inhibitors. The mechanism of action of a neuron-selective AR inhibitor was examined in vitro using luciferase reporter assays, immunofluorescence microscopy, and immunoprecipitations. Rats were treated with the most potent compound and tissue-selective AR inhibition was examined using RT-qPCR of AR-regulated genes and immunohistochemistry.
RESULTS
We identified the thiazole class of antibiotics as compounds able to inhibit AR signaling in a neuronal cell line but not a muscle cell line. One of these antibiotics, thiostrepton is able to inhibit the activity of both wild type and polyglutamine expanded AR in neuronal GT1-7 cells with nanomolar potency. The thiazole antibiotics are known to inhibit FOXM1 activity and accordingly, a novel FOXM1 inhibitor FDI-6 also inhibited AR activity in a neuron-selective fashion. The selective inhibition of AR is likely indirect as the varied structures of these compounds would not suggest that they are competitive antagonists. Indeed, we found that FOXM1 expression correlates with cell-type selectivity, FOXM1 co-localizes with AR in the nucleus, and that shRNA-mediated knock down of FOXM1 reduces AR activity and thiostrepton sensitivity in a neuronal cell line. Thiostrepton treatment reduces FOXM1 levels and the nuclear localization of beta-catenin, a known co-activator of both FOXM1 and AR, and reduces the association between beta-catenin and AR. Treatment of rats with thiostrepton demonstrated AR signaling inhibition in neurons, but not muscles.
CONCLUSION
Our results suggest that thiazole antibiotics, or other inhibitors of the AR-FOXM1 axis, can inhibit AR signaling selectively in motor neurons and may be useful in the treatment or prevention of SBMA symptoms.
Keywords: Androgen receptor, Selective androgen receptor modulator, Spinal and bulbar muscular atrophy, Kennedy’s disease
Core tip: Kennedy’s disease is caused by genetic expansion of the polyglutamine tract in the androgen receptor (AR). There is debate over whether the toxicity is due to expression of this mutant AR in motor neurons, muscle cells, or both. We have identified neuron-specific AR inhibitors, which could be used to help answer this question, and might be useful in the treatment or prevention of Kennedy’s disease. These inhibitors function via FOXM1 and beta-catenin, which are shown to have important roles in the regulation of AR in neurons.
INTRODUCTION
Spinal and bulbar muscular atrophy (SBMA), or Kennedy’s disease, is a neuromuscular disorder of males with a prevalence of approximately 1/50000[1]. The symptoms typically begin in the 4th or 5th decade and include progressive weakness due to degeneration of motor neurons in the brain stem and spinal cord[1]. Currently there is no means by which to prevent or treat the symptoms of SBMA. SBMA manifestations are dependent on androgen activation of a mutant androgen receptor (AR) with an expanded N-terminal polyglutamine tract[2]. Although the exact mechanism of toxicity is still under investigation, activation of AR in motor neurons causes them to die, leading to muscle atrophy. While studies in animal models demonstrate that inhibition of AR through androgen deprivation strategies (castration or suppression of testicular testosterone production) can ameliorate the disease manifestations associated with SBMA[3,4], similar studies in humans have not produced analogous results[5-7].
One reason for the lack of efficacy in humans may be that systemic AR inhibition can also lead to muscle mass decrease by inhibiting anabolic AR activity in muscle cells[8]. In essence, systemic AR inhibition might improve motor neuron disease but prevent rescue of muscle symptoms. Therefore, the ability to inhibit AR selectively in the motor neurons could ameliorate the symptoms of SBMA. However, there is an ongoing debate about the contribution of mutant AR in motor neurons vs muscle cells in the pathogenesis of SBMA[9-12]. Systemic treatment of BAC fxAR121 and AR113Q mice with antisense oligonucleotides (ASO) that silence mutant AR led to improvement of SBMA symptoms, suggesting a direct effect on mutant AR in muscle cells as ASO cannot cross the blood-brain barrier. Indeed, when administered directly into the CNS amelioration of the pathological phenotype was not achieved[9]. However, using a different transgenic mouse model of SBMA, AR97Q mice showed significant improvement after administration of ASO into the brain[13]. Thus, a neuron-selective AR inhibitor might not only have therapeutic application, but will also help to differentiate the tissue-specific role of AR in the etiology of the disease. Therefore, our goal was to screen for a drug that has cell specific antagonistic effects on AR. We applied our FRET-based AR conformation reporter assay[14] in several cell lines to screen for a selective AR modulator. We identified siomycin A and thiostrepton, thiazole antibiotics, as compounds with the ability to inhibit AR activity in a neuronal but not a muscle cell line.
Thiostrepton has been shown to down-regulate the transcription factor FOXM1, which is a member of the forkhead box (FOX) protein family. Since FOX proteins have been reported to interact with hormone receptors[15,16], we speculated that thiostrepton-induced AR inhibition may occur via FOXM1 regulation. In this study we demonstrate that FOXM1 expression correlates with the ability of thiostrepton to inhibit AR activity in cells lines and that the mechanism of AR inhibition involves disruption of a FOXM1/β-catenin/AR transcriptional complex. Additionally, we demonstrate that thiostrepton can cause decreased expression of FOXM1 and may inhibit AR activity selectively in motor neurons in vivo using our previously described panel of tissue-selective androgen-regulated genes[17].
MATERIALS AND METHODS
Cell lines
Most cells were obtained through the ATCC. The GT1-7 neuronal cell line was kindly provided by Dr. Mellon (UCSD). GT1-7 cells were maintained in DMEM (Hyclone) media enriched with 10% fetal bovine serum (FBS), 1% sodium pyruvate (CellGro, Mediatech, VA) and 1% non-essential amino acids (Irvine Scientific, CA). 293, 3T3L, L6, and C2C12 cells (all obtained from the ATCC) were cultured in DMEM supplemented with 10% FBS. PC12 (ARQ112) cells, which were a gift of Dr. Diane Merry, were cultured in DMEM supplemented with 10% FBS and 250 ng/mL doxycyclin to induce AR expression. LNCaP C4 cells were grown in RPMI-1640 (CellGro) containing 10% FBS. TM4 cells were grown in a 1:1 mixture of Ham's F12 medium and Dulbecco's modified Eagle’s medium with 1.2 g/L sodium bicarbonate and 15 mmol/L HEPES, 92.5%; horse serum, 5%; fetal bovine serum, 2.5%. ReNcell CX neural progenitor cells (Millipore) were grown on laminin coated plates in NSC maintenance media (Millipore) supplemented with EGF and FGF. All cultures were supplemented with 1% penicillin/streptomycin (Gibco, Life Technologies) and grown at 37 °C in humidified atmosphere containing 5% CO2.
Creation of stable C-AR-Y reporter cell lines
Cell lines listed above were either transfected with a plasmid or a virus containing the CFP-AR-YFP construct preceding an IRES and hygromycin resistance gene and clonal stable cell lines selected. Qualification criteria for the transfected cell lines were described previously[14].
Screen for AR-modulators
Screening was performed essentially as described previously[14]. Briefly, C-AR-Y reporter GTI-7 were transferred to 96 well plates and treated with library compounds for 24 h. Each compound was tested on four different plates at a concentration of 1 umol/L. To half of the plates DHT (5 nmol/L) was added in order to identify AR antagonists. The compounds to be tested were from the NCI/DTP Open Chemical Repository’s Approved Oncology Drugs Set II, Diversity Set II and Mechanistic Set. Control wells on each plate included non-transfected cells, no DHT (baseline signal), DHT (5 nmol/L) without library compound (maximal signal) plus DHT combined with hydroxyflutamide (1 umol/L) (AR antagonist). After 24 h, medium was aspirated and FRET was measured as previously described using a monochromator-based fluorescence plate reader (M1000, Tecan, Inc.)[14]. We used GT1-7 cells for primary screening and used other cell lines mentioned above for secondary evaluation of candidate hits.
Luciferase reporter assay
Cells were transfected using Lipofectamine Plus (Invitrogen) with a normalization plasmid (pRL-SV40, Promega) and an androgen responsive plasmid (MMTV-luciferase or PSA-luciferase) as previously described[14], and where indicated, with an shRNA plasmid targeting FOXM1 (a gift of Dr. May[18], adapted for mouse FOXM1 sequence targeting), a scrambled control shRNA plasmid, or ARQ24 or ARQ65 plasmids (gifts of Dr. Fischbeck). The following day, cells and drugs were distributed to 96-well plates. Twenty-four hours later, luciferase activity was assessed by a dual luciferase assay kit (Promega) on the Tecan plate reader. Samples were run in quadruplicates and a Student’s t test was performed to determine significance. P < 0.05 was considered significant.
Immunoprecipitation
The co-IP assay was performed as previously described[19]. In brief, cells were lysed on ice for 20 min in co-IP buffer containing 10 mmol/L Hepes (pH8), 300 mmol/L NaCL, 0.1 mmol/L EDTA, 20% glycerol, 0.2% NP-40 and protease inhibitor (Roche Diagnostics). Lysate was centrifuged at 10000 g for 10 min at 4 °C. Lysate was incubated with primary antibody (β-catenin: rabbit, 9562, Cell Signaling Technology) overnight at 4 °C followed by incubation with 30 uL of Protein A/G beads for 2 h at 4 °C. Collected protein complexes were washed and analyzed by Western blotting.
Western blot
Cell lysates was separated on a 6% SDS page gel and transferred to a nitrocellulose membrane. The membrane was blocked for one hour in 5% skim milk dissolved in TBS-T and probed with the following antibodies (1:1000): FoxM1 (rabbit, GeneTex), AR: 441 (mouse, Santa Cruz Biotechnology), β-catenin (rabbit 9562, Cell Signaling Technology), p84(mouse, GeneTex) and actin (mouse, Santa Cruz Biotechnology). Horse radish peroxidase linked secondary antibodies were used along with ECL reagents for visualization. Western blots were quantified using Quantity One analysis software (BioRad).
Animals and drugs
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All experiments were performed under the IUCAC approval of City of Hope. Animals were purchased from Jackson Laboratory (Bar Harbor, Main). For the pilot pharmacokinetic experiment, nine male Sprague Dawley rats (n = 3) were administered 1, 10, or 100 mg/kg intraperitoneal injections thrice per week for four weeks. One hour following the final dose, blood and spinal fluid were collected and analyzed by mass spectrometry. For the efficacy experiment, rats were divided into 3 groups (n = 7) as follows: Intact, castrated and thiostrepton. Thiostrepton was dissolved in 20% DMSO and 80% PEG400 (Sigma Aldrich) and administered at a concentration of 50 mg/kg per day via an IP osmotic pump implant (Alzet). Following a treatment period of 7 d, rats were euthanized and organs harvested, weighed and processed for quantitative RT-PCR. One-way analysis of variance was applied to determine statistical differences among groups using post-hoc Dunnett’s test. P < 0.05 was considered significant.
qRT-PCR
The quantitative RT-PCR method has been described previously[14]. In brief, dissected tissue was placed in RNA Later (Qiagen) before being homogenized using a bead homogenizer (TissueLyser, Qiagen). Following homogenization, RNA was isolated with an RNAEasy kit (Qiagen). RNA then was reverse transcribed (Promega, WI) and amplified (Qiagen Taq and reagents) on a StepOne PlusReal Time PCR System (Applied Biosystems), using SYBR green (Invitrogen) as the detecting dye and Rox (Invitrogen, CA) as the reference dye. Differences between experimental (x) and control (y) samples were normalized to RPL19 transcript levels (androgen unresponsive) and determined with the following calculation: [2^(Ctxgene1-Ctygene1))/(2^(CtxRPL19-CtyRPL19)). One-way analysis of variance was applied to determine statistical differences among groups using post-hoc Dunnett’s test. P < 0.05 was considered significant.
Immunofluorescence
Cells were fixed in 2% paraformaldehyde prior to incubating with primary antibody (FOXM1:mouse, Ab Cam,) overnight at 4 °C. Following washing, secondary antibody (Alexa 954 goat anti-mouse, Invitrogen) was applied for 45 min at 37 °C. After washing, slides were mounted and analyzed using a confocal microscope (Zeiss). The coloc-2 Image J plug-in (Fiji) was used to quantify co-localization of AR and FOXM1 from three independent samples using both Li’s Intensity Correlation Quotient (ICQ) and Manders split coefficients co-localization values. One-way analysis of variance was applied to determine statistical differences among groups. P < 0.05 was considered significant.
Immunohistochemistry
Spinal cords were fixed in 10% neutral buffered formalin, embedded in paraffin and sectioned. Following de-waxing, slides were blocked with 10% normal goat serum for 30 min. The primary antibody (1:500) (AR: PG21, Millipore; FoxM1: GeneTex) was applied and incubated overnight at 4 °C. Following three washes with TBS-T plus 0.1% Tween, slides were incubated with goat anti-rabbit antibody (1:200) for 30 min. Following three washes slides were incubated with Vectastain ABC reagent for 10 min at room temperature, then developed with DAB and counterstained with hematoxylin, dehydrated and mounted. A veterinary pathologist, blinded to the identity of the treatment groups, scored the spinal cord sections based on the intensity of FOXM1 expression, which was typically uniform across the entirety of a section, creating a scale ranging from 0-2 in both white matter and grey matter. Because few spinal cords were available for analysis, results from intact and castrated were grouped for analysis as “untreated”. The staining between castrate and intact animals was very similar and distinct from thiostrepton treated animals. As a secondary quantification method, Image J was used to determine pixel density of FOXM1 staining. Images of ten sections from four thiostrepton treated and four untreated rat slides were captured. Integrated density values were averaged (n = 40) and a Student’s t test was performed to determine significance. P < 0.05 was considered significant.
LC-MS/MS methods
Dried samples were resuspended in tetrahydrofuran spiked with 5 pg/μL reserpine, which was used as an instrument response control. Resuspended samples were separated with an Agilent 1290 UHPLC system (Agilent Technologies, Santa Clara, CA) and a Waters Acquity CSH C18 column (Waters, Millford, MA) using a linear gradient from 95% buffer A (0.1% formic acid in 60% water/40% acetonitrile) to 100% buffer B (0.1% formic acid in 90% isopropanol/10% acetonitrile) over 2.5 min at 380 μL/min. The UHPLC column eluent was sprayed into an Agilent 6490 triple quadrupole mass spectrometer equipped with a JetStream source (Agilent Technologies). Thiostrepton was detected by monitoring fragments from the doubly charged ion (m/z 832.9) at m/z 1247.4 and 1230.3. Data was analyzed using Agilent MassHunter quantitative analysis software.
RESULTS
Identification of GT1-7 specific AR inhibitors by high-throughput screening
In order to identify potential cell-type selective AR modulators, we built upon our FRET-based assay of AR conformation change. AR signaling is a complex process involving many steps in addition to ligand binding. One crucial step is the ligand-induced conformational change of AR, which brings the carboxy (C)- and amino (N)-termini of the receptor together. Full length AR with cyan fluorescent protein (CFP) or yellow fluorescent protein (YFP) fused to either end (CARY) produces a FRET signal upon AR conformation change (Figure 1A). This produces an accurate and quantifiable read-out of AR activity[14]. Because we were interested in the potential of neuron selective AR inhibitors for treatment of SBMA, we carried out our initial screen in CARY- transfected GT1-7 cells, which are a mouse model of hypothalamic neurons[20]. Cells were plated in 96 wells and treated with library compounds 24 h prior to accessing conformational changes by FRET. Non-CARY cells, CARY cells without DHT (baseline signal), with DHT (5 nmol/L) only (maximal signal) and DHT combined with 1 μmol/L hydroxyflutamide (OHF) (AR antagonist) served as controls (Figure 1A). Candidate hits were then further evaluated by dose response. Figure 1B shows a dose dependent inhibition of DHT-induced AR conformation change by the compound NSC285116 (siomycin A) in GT1-7 cells. Once a dose response was observed, candidate compounds were then cross examined in CARY-expressing cell lines representing other tissues of interest, including muscle (C2C12), testes (TM4), kidney (HEK293), prostate (LNCaP C4), and adipose (3T3L1) (Figure 1C). In addition to FRET, AR transcriptional activity was also assessed in several cell lines by transfecting cells with a luciferase reporter driven by the human PSA promoter[21]. After extensive testing in multiple cells lines (Figure 1D), we identified siomycin A as a potential neuron-selective AR inhibitor. It showed greatest potency in neuronal GT1-7 cells, with some activity in HEK293 kidney cells, but little to no activity in LNCaP C4 prostate cancer, C2C12 muscle or 3T3L1 adipose-derived cells.
Figure 1.
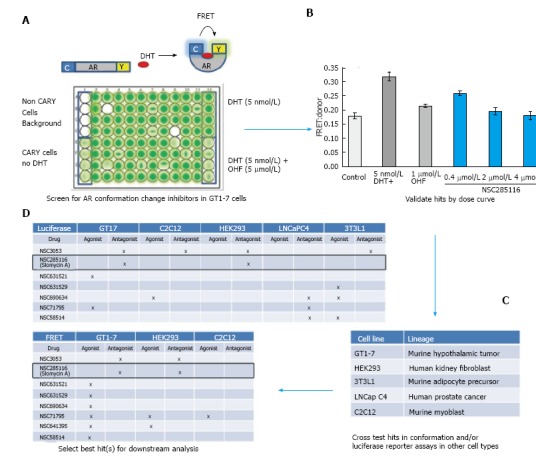
Screening for neuron-selective androgen receptor inhibitors. A: AR conformation change can be monitored by the CFP-AR-YFP FRET reporter (top). Screening for compounds that affect AR conformation change in GT1-7 cells was accomplished in 96 well plates; B: Hits were assessed for a dose response in the same assay. Shown is the dose response to NSC285116 (siomycin A); C, D: Hits were then assessed for agonist or antagonist activity in the indicated cell lines in a FRET or an androgen-responsive luciferase reporter assay. Siomycin A demonstrated antagonist activity in GT1-7 neuron cells but not C2C12 muscle cells and was therefore selected for further analysis. AR: Androgen receptor.
AR inhibition by thiazole antibiotics
Siomycin A is a thiazole class antibiotic. It has been shown to inhibit transcriptional activity and expression of the FOXM1 transcription factor[22-24]. Thiostrepton belongs to the same class of antibiotics and has been reported to function similarly to siomycin[25-27]. To test whether thiostrepton possesses similar AR antagonistic properties as siomycin, we assessed its ability to inhibit AR transcriptional activity via luciferase assay in GT1-7 and C2C12 cells (Figure 2A). Both thiostrepton and siomycin were found to selectively inhibit AR activity in GT1-7 cells, with thiostrepton having a greater potency. As thiazole antibiotics have also been found to have effects on proteosome function, we examined the activity of a recently developed, selective FOXM1 inhibitor, FDI-6[28]. We found this compound also inhibited AR activity in GT1-7 cells but not C2C12 cells, suggesting that anti-FOXM1 activity is the key to the ability to inhibit AR activity in these cells. Further experimentation was conducted primarily with thiostrepton as it was the most potent of the FOXM1 inhibitors. The ability of thiostrepton to inhibit AR activity in additional neuronal cells, including ReNcell CX cells[29] and PC12 cells[30], and in rat L6 skeletal muscle cells[31] was examined (Figure 2B). We again found that thiostrepton inhibited AR activity in neuronal but not muscle cells, strongly suggesting that AR inhibition by FOXM1 inhibitors is neuron-selective.
Figure 2.
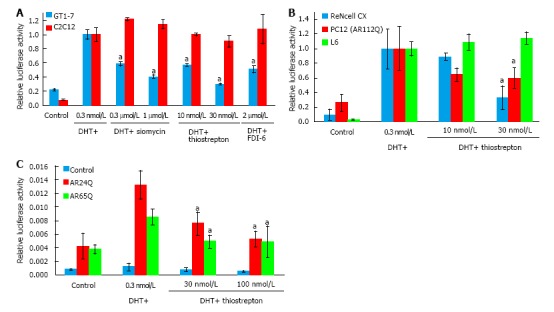
Thiazole antibiotics selectively inhibit androgen receptor in neuronal cells and have activity against polyQ-expanded androgen receptor. A: GT1-7 and C2C12 cells were transfected with androgen-responsive and control luciferase reporters and treated with the indicated drugs overnight. Both thiazole antibiotics siomycin and thiostrepton, as well as the selective FOXM1 inhibitor FDI-6, inhibited AR activity in GT1-7 but not C2C12 cells, with thiostrepton being the most potent of the drugs; B: Rat L6 skeletal muscle cells and ReNcell CX and PC12 (ARQ112) neuronal cells were transfected with androgen-responsive and control luciferase reporters and treated with thiostrepton overnight. Thiostrepton inhibited AR luciferase activity in neuronal, but not muscle cells; C: 293 cells were transfected with androgen-responsive and control luciferase reporters as well as plasmids containing AR with 24 (normal state) or 65 (disease state) glutamines and treated with drugs mentioned above overnight. Both normal and disease state AR activity was reduced by thiostrepton treatment. Error bars represent the mean ± SEM, aP < 0.05. AR: Androgen receptor.
SBMA is caused by the activity of polyglutamine-expanded AR. To investigate the effects of thiostrepton against polyglutamine-expanded AR we transfected AR-negative HEK293 cells with plasmids encoding AR with a 24 glutamine tract (normal) or with a 65 glutamine tract (disease associated). The polyglutamine-expanded AR was less transcriptionally active than the normal AR, as has been previously documented for such constructs[32]. Thiostrepton was able to inhibit the polyglutamine-expanded AR as well as the non-expanded AR (Figure 2C), suggesting that thiostrepton can inhibit the activity of a clinically relevant AR. In further support of this, the PC12 cells in Figure 2B express an AR with a 112 glutamine tract, and thiostrepton efficiently inhibited AR activity in those cells as well.
FOXM1 mediates AR inhibition by thiostrepton
FOX proteins can act as transcription factors and co-activators and have been shown to regulate AR transcriptional activity[33,34]. For instance, FOXA1 has been shown to interact and regulate transcription of both estrogen receptors and AR[35]. Because thiazole antibiotics are known to inhibit FOXM1, we speculated that the thiostrepton-induced AR antagonistic effects might be mediated via FOXM1. We first examined FOXM1 expression levels in different cell lines (Figure 3A) and found that FOXM1 expression levels were highest in GT1-7, followed by 293HEK and LNCaP C4 cells. Little or no expression was observed in the remaining cell lines, suggesting a correlation between FOXM1 expression and the inhibitory effect of thiostrepton on AR activity. Expression levels of the CARY construct did not correlate with sensitivity (Figure 3A).
Figure 3.
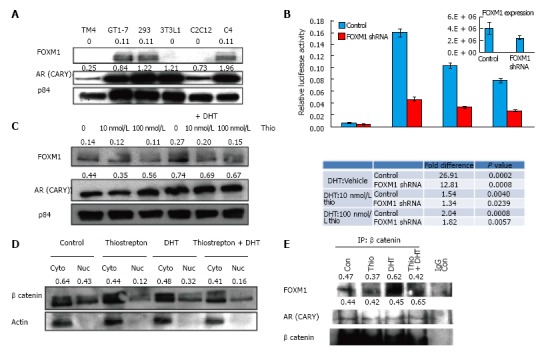
FOXM1 expression correlates with androgen receptor activity. A: Expression of FOXM1 and AR was assessed by Western blot in indicated cell lines; B: GT1-7 cells were transfected with a lentiviral plasmid shRNA targeting FOXM1 as well as luciferase plasmids, treated with drugs listed above, then assessed by luciferase assay. Decreased expression of FOXM1 (RT-qPCR in inset) reduced AR activity and diminished sensitivity to thiostrepton. Fold changes and P values are shown; C: Western blot analysis demonstrates that thiostrepton reduces FOXM1 levels in GT1-7 cells while DHT increases FOXM1 levels; D: GT1-7 cells were treated with the indicated drugs overnight, and nuclear and cytoplasmic fractions were probed by Western blot for the indicated proteins. β-catenin levels in nuclear fraction of thiostrepton treated cells are lower compared to untreated ones. Quantification of β-catenin levels relative to cytoplasmic actin expression is shown; E: β-catenin was immunoprecipitated from GT1-7 cell lysates and Western blot was performed to detect AR and FOXM1. Quantification of AR and FOXM1 levels relative to β-catenin levels in IP is shown. AR: Androgen receptor.
To further test the role of FOXM1 in association with AR activity, we decreased FOXM1 expression in GT1-7 cells using a lentiviral shRNA plasmid transfection. FOXM1 knock-down decreased the DHT-induced activity of AR, suggesting FOXM1 is an important AR coactivator in GT1-7 cells (Figure 3B). More importantly, decreasing FOXM1 expression diminished the potency of thiostrepton (compare DHT + thio/DHT alone ratio in control vs FOXM1-silenced cells). This strongly suggests that FOXM1 mediates the ability of thiostrepton to inhibit AR activity.
It has been previously reported that thiostrepton causes decreased levels of FOXM1[25,36]. Treatment of GT1-7 CARY cells with thiostrepton resulted in slightly decreased FOXM1 levels compared to p84 loading control protein levels, both in the presence and absence of DHT (Figure 3C), as shown from quantification of representative blots. In contrast to thiostrepton, DHT treatment increased FOXM1 levels, implying interplay between AR signaling pathway with FOXM1. AR levels themselves were not consistently decreased in response to thiostrepton treatment. FOXM1 has been shown to bind directly to β-catenin and facilitate its nuclear localization. The nuclear association between FOXM1 and β-catenin increases the transcriptional activity of both factors[19]. β-catenin is a key AR coactivator in a variety of cells[37,38]. We therefore reasoned that thiostrepton might inhibit AR activity by limiting access to nuclear β-catenin via FOXM1 depletion. Nuclear and cytoplasmic fractions were isolated from GT1-7 cells treated with or without thiostrepton and DHT. Thiostrepton treatment caused lower levels of nuclear β-catenin, both phosphorylated and unphosphorylated (active) fractions (see doublet in Figure 3D), suggesting that thiostrepton treatment leads to β-catenin inactivation via its effects on FOXM1. Control actin protein is observed primarily in cytoplasmic fraction, as expected.
Co-immunoprecipitation confirmed a direct interaction between β-catenin and FOXM1 as well as between β-catenin and AR (Figure 3E). The amount of FOXM1 pulled down was reduced in thiostrepton-treated cells. While this may be due in part to the effects of thiostrepton on total FOXM1 levels, which is difficult to control for in these experiments, the decreased interaction between FOXM1 and β-catenin could also have an effect on AR activity.
Using a co-IP assay in GT1-7 CARY cell lysates, we were unable to detect an association between AR and FOXM1 (data not shown). However, immunofluorescence staining demonstrated significant AR-FOXM1 co-localization in GT1-7 CARY cells (Figure 4). Untreated cells predominantly expressed AR in the cytoplasm, while DHT treatment caused a shift to the nucleus. FOXM1 expression was mainly confined to the nucleus, which was true for untreated and treated cells. Image analysis revealed little to no co-localization between FOXM1 and AR in untreated cells (Figure 4B). Addition of DHT increased association between FOXM1 and AR, which was not significantly affected by addition of thiostrepton. Although co-localization is not evidence of an interaction, our results suggest that FOXM1 and AR could plausibly function together in the presence of DHT. In conclusion, our data suggest a model in which thiostrepton decreases the levels of FOXM1, which leads to lower levels of nuclear β-catenin, an important AR coactivator, causing decreased AR transcriptional activity.
Figure 4.
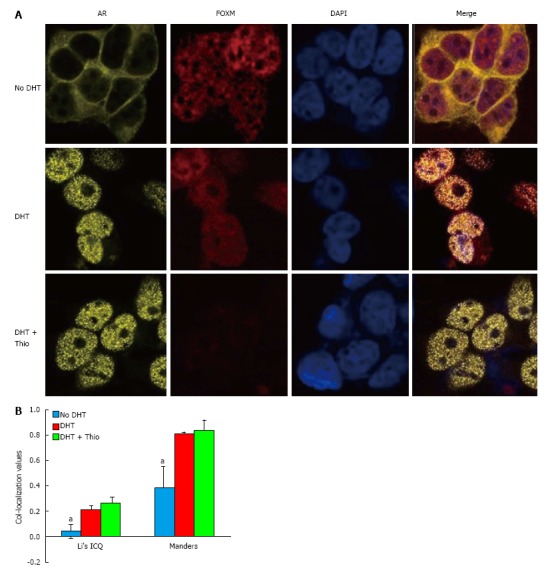
DHT promotes nuclear co-localization of androgen receptor and FOXM1. A: GT1-7 cells expressing a YFP-tagged AR were treated as indicated for 24 h prior to staining with anti-FOXM1 (red) and DAPI (blue); B: Quantification of co-localization using Image J demonstrates significant co-localization of FOXM1 and AR in the presence of DHT and DHT + thiostrepton by both Manders split coefficient and Li’s ICQ value. Error bars represent the mean ± SEM, aP < 0.05. AR: Androgen receptor; ICQ: Intensity Correlation Quotient.
Thiostrepton treatment inhibits in vivo AR activity preferentially in rat spinal cord compared to skeletal muscle
In order to achieve desired dosing and to determine if thiostrepton crosses the blood brain barrier, we first conducted a pilot pharmacokinetic study in rats. Rats (n = 3/group) received low (1 mg/kg), medium (10 mg/kg), or high (100 mg/kg) doses of thiostrepton by intraperitoneal (IP) injection, three times a week for four weeks. One hour after the final dose, animals were euthanized and blood as well as spinal fluid was collected and analyzed by LC-MS/MS (Figure 5A). We found a dose-dependent increase in plasma levels. Only the high dose of thiostrepton resulted in measurable levels of drug in the spinal fluid, suggesting that while the drug could cross the blood brain barrier, it exists in that compartment at much lower levels than the plasma. However, it did reach concentrations above its IC50 as determined by luciferase assay in cultured cells, suggesting that it could still be active in motor neurons of the spinal cord.
Figure 5.
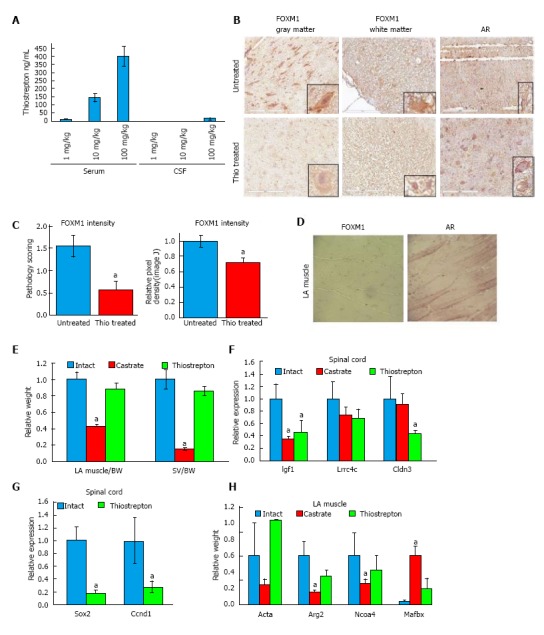
Thiostrepton treatment inhibits androgen receptor activity in rat spinal cord but not muscle. A: Rats (n = 3) were administered 1, 10, or 100 mg/kg intraperitoneal injections thrice per week for four weeks. One hour following the final dose, blood and spinal fluid were collected and analyzed by mass spectrometry. Thiostrepton displayed the expected dose response in the serum, but much less was found in the cerebral spinal fluid; B-F: Rats (n = 7) were treated with 100 mg/kg per day thiostrepton or vehicle for four weeks using intraperitoneally implanted osmotic pumps. Intact and castrate cohorts were included as controls; B, C: Immunohistochemistry revealed a reduction in the expression of FOXM1 in spinal cords compared to untreated (intact or castrate) animals. AR staining was also strong in the neurons; C: FOXM1 staining intensity was quantified by a veterinary pathologist (left) or by ImageJ (right) and found to be significantly reduced by thiostrepton treatment; D: IHC revealed weak to no FOXM1 staining in LA muscle tissue while AR staining was prevalent; E: Tissues were harvested and weighed. Castration significantly reduced the weight of the LA muscle (P < 0.01) and seminal vesicles (SV) (P < 0.01) compared to intact animals while thiostrepton treatment did not. (F-H) RT-qPCR was used to quantify the levels of androgen-regulated transcripts in spinal cord (F) or LA muscle tissue (H) or FOXM1-regulated transcripts in spinal cord (G). Thiostrepton was able to decrease the levels of androgen-regulated transcripts in spinal cords to a similar or greater extent than castration, suggesting inhibition of AR activity, while in LA muscle, castration had a much stronger effect on AR activity than thiostrepton treatment. Two FOXM1 target gene transcripts were decreased with thiostrepton treatment, demonstrating an on target effect of the drug. Error bars represent the mean ± SEM, aP < 0.05 (P < 0.08 in G). AR: Androgen receptor.
Based on our initial results, rats (n = 7) were treated with 100 mg/kg per day thiostrepton or vehicle for four weeks using IP implanted osmotic pumps. Intact and castrate cohorts were included as controls. To test the efficacy of the thiostrepton in the motor neurons, we examined the expression of FOXM1 in tissue sections of the spinal cord. FOXM1 was located in both gray and white matter of the spinal cord (Figure 5B), primarily in the nucleus, as had been shown previously[39]. In the grey matter, FOXM1 was mainly found in the neurons. In the white matter, FOXM1 was found in glial cells, at least some of which appeared to be astrocytes; however, further immunohistochemistry would be necessary to confirm the identity of the cells. As scored by a veterinary pathologist blinded to treatment groups, treatment with thiostrepton significantly reduced the expression of FOXM1 in the spinal cord (Figure 5C). As a secondary quantification method, Image J was used to determine pixel density of FOXM1 staining. Images of ten sections from four thiostrepton treated and four untreated slides were captured. Integrated density values were averaged and displayed relative to untreated control values (Figure 5C). Thiostrepton treatment caused lower intensity FOXM1 staining (P = 0.0036) by this method as well. This data suggest that we achieved functional levels of thiostrepton in the spinal cord and that it decreased FOXM1 levels in vivo, as it did in cultured cells. AR was clearly observed in the nucleus and cytoplasm of neurons in both white and gray matter, and levels were not affected by thiostrepton, which parallels our cell culture data. Immunohistochemistry also revealed low to no expression of FOXM1 in intact muscle tissue (Figure 5D), corroborating our cell culture data and in accordance with published literature where it has been reported that FOXM1 is highly expressed in embryonic muscle tissue, but is drastically reduced in adult skeletal muscle tissue[40]. AR expression is evident in the striated muscle tissue nuclei as expected and is not affected by thiostrepton treatment.
Androgens are known to control the growth and maintenance of the skeletal muscle and seminal vesicles[41]. In order to investigate the impact of thiostrepton on these tissues, we assessed their weights at the end of the study. Compared to castrated rats, which displayed significantly reduced levator ani (LA) muscle (P < 0.01) and seminal vesicles (SV) (P < 0.01) weights, thiostrepton did not change LA and SV weights when compared to controls (Figure 5B). This data suggest that thiostrepton does not significantly affect AR activity in muscle or SV, in line with our cell culture findings. Because an equivalent androgen-dependent change in weight cannot be tested in motor neurons, we assessed the levels of tissue-selective androgen regulate genes as a marker of AR activity. These genes were identified in our previous studies[17]. RNA was isolated from spinal cord and LA muscle tissue and transcript levels were measured by RT-qPCR (Figure 5F and H). Both castration and thiostrepton treatment caused decreases in three androgen-dependent genes, two of which were statistically significant with thiostrepton treatment, in the spinal cord compared to control treatment. In LA muscle, castration had a much greater effect than thiostrepton treatment at the four androgen-dependent genes analyzed, significantly altering the expression of 3 of 4 genes examined, while thiostrepton did not significantly affect the levels of any genes. Importantly, the transcript levels of FOXM1 target genes Ccnd1 and Sox2[42,43] were decreased in the spinal cords of thiostrepton-treated animals compared to intact controls, demonstrating on-target efficacy of the drug (Figure 5G). Our combined data suggest that thiostrepton inhibits AR activity in motor neurons but not LA muscle.
DISCUSSION
We originally created the FRET-based AR conformation change reporter assay to identify novel AR inhibitors for use in prostate cancer[14]. Our initial studied identified cell type selective AR inhibitors. Here, we demonstrate the utility of the assay on a broader scale to identify cell type selective AR modulators with potential for clinical use in realms other than prostate cancer. Our focus was to find a selective neuronal AR inhibitor which might have potential in treating SBMA. Our data demonstrate that thiostrepton selectively inhibits AR in different cell lines in vitro. Based on extensive literature demonstrating FOXM1 as a target of thiostrepton and other thiazole antibiotics[25,36,44], we speculated that the decreased AR activity is mediated by decreased levels of FOXM1, a transcription factor involved in many different molecular pathways. We showed that FOXM1 is differentially expressed in a panel of cell lines and that this expression mostly correlates with the ability of thiazole antibiotics to inhibit AR activity. LNCaP C4 cells, which expressed FOXM1, were not as sensitive to siomycin in our initial screen. This could be due perhaps to the expression of a mutant AR in these cells[45]; further investigation is warranted. We also showed that a chemically distinct FOXM1 inhibitor, FDI-6, inhibited AR activity in neuronal but not muscle cells. Furthermore, manipulation of FOXM1 levels affected AR activity and sensitivity to thiostrepton, strongly suggesting that thiostrepton acts through FOXM1 to inhibit AR.
We showed thiostrepton reduced FOXM1 levels, in accordance with published literature. While the effect was not as strong as some published data, FOXM1 levels were quantifiably lower. Likewise, we showed that thiostrepton quantifiably reduced the nuclear levels of β-catenin in GT1-7 cells. Although only one blot is shown in each figure, each of the IP and Western blot experiments was repeated several times, with high reproducibility. While we were unable to detect an interaction between AR and FOXM1 by co-IP, confocal imaging demonstrates the plausibility of these proteins functioning together in the nucleus in the presence of DHT. Our co-IP experiments also demonstrate interactions between β-catenin and both AR and FOXM1, both of which have been previously reported[19,37]. Our data suggest a model in which AR, FOXM1, and β-catenin form an active transcriptional complex in the nuclei of GT1-7 cells that is essential for full androgen-responsive transcription. Thiostrepton treatment reduces FOXM1 levels, which leads to lower levels of nuclear β-catenin, and thus reduced AR transcriptional activity.
Thiostrepton’s selective inhibition of AR appears to apply in vivo as well. Thiostrepton treatment did not inhibit AR activity in LA muscle or SV, as measured by changes in organ weights. It did cause decreased FOXM1 levels and inhibit androgen-regulated gene expression in spinal cord neurons. Although many of the RT-qPCR changes were not significant, this is likely due to the small number of animals in each group. While further testing is necessary to determine exactly how selective thiostrepton and other thiazole antibiotics are in vivo, our initial data suggest that we have identified a compound that inhibits AR in spinal cord neurons but not muscle, which was the goal of our study. Such a drug could be useful in the treatment of SBMA. In mouse models of SBMA, castration by physical or chemical means can prevent and/or ameliorate disease symptoms[39,40]. However, trials of androgen reducing agents in symptomatic humans have not had the same effect[42-44]. This could be in part because muscle atrophy has already occurred and androgens are needed to rebuild muscle tissue. Thus, a neuron selective AR inhibitor might inhibit degenerative AR activity while allowing the body’s natural androgens to rebuild atrophied muscle tissue. As mentioned above, there is evidence that polyglutamine expanded AR has direct toxic effects in muscle cells[10], in which case, a neuron-selective AR inhibitor would not benefit SBMA patients. However, other studies show that knocking down neuronal AR while keeping muscle levels intact rescues the disease phenotype[13]; genetic and RNA silencing approaches have not been able to settle the debate. Testing thiostrepton or similar compounds in mouse models of SBMA[46] could help answer this question.
There are several limitations to our study. The largest is the lack of testing of FOXM1 inhibitors in animal models of SBMA. We are working to establish collaborations to test these compounds in such models. The use of a phenotypic screen also presents challenges, especially in the identification of a specific mechanism of action. However, it is clear from our studies that FOXM1 is involved in the neuron-selective inhibition of AR that was originally observed in our screen. Finally, thiostrepton is a natural product, and its large, complex structure makes synthesis difficult. It is also highly hydrophobic, typically being used as a topical cream, and therefore not ideal for in vivo administration. Some work has been done to improve the synthesis and pharmacokinetics of thiostrepton, but the development of specific FOXM1/AR inhibitors with better drug like properties, including the ability to cross the blood brain barrier, would be beneficial. It is possible that FDI-6 and related compounds could be improved to become viable treatments for SBMA.
ACKNOWLEDGMENTS
We would like to thank Kurt Fischbeck, Naemeh Pourshafie, and Diane Merry for guidance and critical reading of the manuscript. We also thank Dan Campbell, Andrew Yeudall, Vu Ngo, Alvaro Aytes Meneses, and William May for providing reagents and advice.
COMMENTS
Background
Kennedy’s disease is a neuromuscular disorder of males with a prevalence of approximately 1/50000 that is caused by genetic expansion of the polyglutamine tract in the androgen receptor (AR). The authors have identified neuron-specific AR inhibitors, which could be used to help answer this question, and might be useful in the treatment or prevention of Kennedy’s disease. These inhibitors function via FOXM1 and beta-catenin, which are shown to have important roles in the regulation of AR in neurons.
Research frontiers
There is debate over whether the toxicity of polyglutamine expanded AR in Kennedy’s disease is due to expression of this mutant AR in motor neurons, muscle cells, or both. There are also no effective treatments for this disease.
Innovations and breakthroughs
This is the first report of an in vitro screen for selective AR modulators and the first report of a neuron-selective AR inhibitor. It also finds FOXM1 as an important co-factor for AR activity in neurons, and suggests that FOXM1 and beta-catenin may play a role in Kennedy’s disease.
Applications
The neuron-specific AR inhibitors the authors identified could be used to help determine the contribution of AR activity in neurons vs muscle to Kennedy’s disease and they might be useful in the treatment or prevention of this disease.
Terminology
Polyglutamine expansion refers to the genetic process in which the CAG repeat in genes, which corresponds to the amino acid glutamine, is inappropriately copied, resulting in an increasing number of glutamine residues in the corresponding protein. At a certain length, which varies by gene, this expansion at the protein level causes disease.
Peer-review
This is an interesting study, the paper is well written and results could be potentially relevant.
Footnotes
Institutional review board statement: No human studies.
Institutional animal care and use committee statement: All procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of the City of Hope (IACUC protocol number: 10009).
Conflict-of-interest statement: No authors have conflicts of interest to declare.
Data sharing statement: Additional data available from the corresponding author at jjones@coh.org.
Manuscript source: Invited manuscript
Specialty type: Biochemistry and molecular biology
Country of origin: United States
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B, B, B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Peer-review started: February 12, 2017
First decision: March 10, 2017
Article in press: May 4, 2017
P- Reviewer: Baldi E, Bruzzone S, Gupta VK, Rey R, Sukocheva OA S- Editor: Song XX L- Editor: A E- Editor: Lu YJ
References
- 1.Brooks BP, Fischbeck KH. Spinal and bulbar muscular atrophy: a trinucleotide-repeat expansion neurodegenerative disease. Trends Neurosci. 1995;18:459–461. doi: 10.1016/0166-2236(95)94497-s. [DOI] [PubMed] [Google Scholar]
- 2.Fischbeck KH, Lieberman A, Bailey CK, Abel A, Merry DE. Androgen receptor mutation in Kennedy’s disease. Philos Trans R Soc Lond B Biol Sci. 1999;354:1075–1078. doi: 10.1098/rstb.1999.0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Katsuno M, Adachi H, Kume A, Li M, Nakagomi Y, Niwa H, Sang C, Kobayashi Y, Doyu M, Sobue G. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron. 2002;35:843–854. doi: 10.1016/s0896-6273(02)00834-6. [DOI] [PubMed] [Google Scholar]
- 4.Katsuno M, Adachi H, Doyu M, Minamiyama M, Sang C, Kobayashi Y, Inukai A, Sobue G. Leuprorelin rescues polyglutamine-dependent phenotypes in a transgenic mouse model of spinal and bulbar muscular atrophy. Nat Med. 2003;9:768–773. doi: 10.1038/nm878. [DOI] [PubMed] [Google Scholar]
- 5.Fischbeck KH, Bryan WW. Anti-androgen treatment for spinal and bulbar muscular atrophy. Ann Neurol. 2009;65:119–120. doi: 10.1002/ana.21633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fernández-Rhodes LE, Kokkinis AD, White MJ, Watts CA, Auh S, Jeffries NO, Shrader JA, Lehky TJ, Li L, Ryder JE, et al. Efficacy and safety of dutasteride in patients with spinal and bulbar muscular atrophy: a randomised placebo-controlled trial. Lancet Neurol. 2011;10:140–147. doi: 10.1016/S1474-4422(10)70321-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weydt P, Sagnelli A, Rosenbohm A, Fratta P, Pradat PF, Ludolph AC, Pareyson D. Clinical Trials in Spinal and Bulbar Muscular Atrophy-Past, Present, and Future. J Mol Neurosci. 2016;58:379–387. doi: 10.1007/s12031-015-0682-7. [DOI] [PubMed] [Google Scholar]
- 8.Storer TW, Miciek R, Travison TG. Muscle function, physical performance and body composition changes in men with prostate cancer undergoing androgen deprivation therapy. Asian J Androl. 2012;14:204–221. doi: 10.1038/aja.2011.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sobue G, Hashizume Y, Mukai E, Hirayama M, Mitsuma T, Takahashi A. X-linked recessive bulbospinal neuronopathy. A clinicopathological study. Brain. 1989;112(Pt 1):209–232. doi: 10.1093/brain/112.1.209. [DOI] [PubMed] [Google Scholar]
- 10.Cortes CJ, Ling SC, Guo LT, Hung G, Tsunemi T, Ly L, Tokunaga S, Lopez E, Sopher BL, Bennett CF, et al. Muscle expression of mutant androgen receptor accounts for systemic and motor neuron disease phenotypes in spinal and bulbar muscular atrophy. Neuron. 2014;82:295–307. doi: 10.1016/j.neuron.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lieberman AP, Yu Z, Murray S, Peralta R, Low A, Guo S, Yu XX, Cortes CJ, Bennett CF, Monia BP, et al. Peripheral androgen receptor gene suppression rescues disease in mouse models of spinal and bulbar muscular atrophy. Cell Rep. 2014;7:774–784. doi: 10.1016/j.celrep.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rinaldi C, Malik B, Greensmith L. Targeted Molecular Therapies for SBMA. J Mol Neurosci. 2016;58:335–342. doi: 10.1007/s12031-015-0676-5. [DOI] [PubMed] [Google Scholar]
- 13.Sahashi K, Katsuno M, Hung G, Adachi H, Kondo N, Nakatsuji H, Tohnai G, Iida M, Bennett CF, Sobue G. Silencing neuronal mutant androgen receptor in a mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet. 2015;24:5985–5994. doi: 10.1093/hmg/ddv300. [DOI] [PubMed] [Google Scholar]
- 14.Jones JO, Diamond MI. A cellular conformation-based screen for androgen receptor inhibitors. ACS Chem Biol. 2008;3:412–418. doi: 10.1021/cb800054w. [DOI] [PubMed] [Google Scholar]
- 15.Madureira PA, Varshochi R, Constantinidou D, Francis RE, Coombes RC, Yao KM, Lam EW. The Forkhead box M1 protein regulates the transcription of the estrogen receptor alpha in breast cancer cells. J Biol Chem. 2006;281:25167–25176. doi: 10.1074/jbc.M603906200. [DOI] [PubMed] [Google Scholar]
- 16.Lam EW, Brosens JJ, Gomes AR, Koo CY. Forkhead box proteins: tuning forks for transcriptional harmony. Nat Rev Cancer. 2013;13:482–495. doi: 10.1038/nrc3539. [DOI] [PubMed] [Google Scholar]
- 17.Otto-Duessel M, He M, Adamson TW, Jones JO. Enhanced evaluation of selective androgen receptor modulators in vivo. Andrology. 2013;1:29–36. doi: 10.1111/j.2047-2927.2012.00006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christensen L, Joo J, Lee S, Wai D, Triche TJ, May WA. FOXM1 is an oncogenic mediator in Ewing Sarcoma. PLoS One. 2013;8:e54556. doi: 10.1371/journal.pone.0054556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang N, Wei P, Gong A, Chiu WT, Lee HT, Colman H, Huang H, Xue J, Liu M, Wang Y, et al. FoxM1 promotes β-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell. 2011;20:427–442. doi: 10.1016/j.ccr.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liposits Z, Merchenthaler I, Wetsel WC, Reid JJ, Mellon PL, Weiner RI, Negro-Vilar A. Morphological characterization of immortalized hypothalamic neurons synthesizing luteinizing hormone-releasing hormone. Endocrinology. 1991;129:1575–1583. doi: 10.1210/endo-129-3-1575. [DOI] [PubMed] [Google Scholar]
- 21.Bolton EC, So AY, Chaivorapol C, Haqq CM, Li H, Yamamoto KR. Cell- and gene-specific regulation of primary target genes by the androgen receptor. Genes Dev. 2007;21:2005–2017. doi: 10.1101/gad.1564207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gartel AL. The oncogenic transcription factor FOXM1 and anticancer therapy. Cell Cycle. 2012;11:3341–3342. doi: 10.4161/cc.21841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Radhakrishnan SK, Bhat UG, Hughes DE, Wang IC, Costa RH, Gartel AL. Identification of a chemical inhibitor of the oncogenic transcription factor forkhead box M1. Cancer Res. 2006;66:9731–9735. doi: 10.1158/0008-5472.CAN-06-1576. [DOI] [PubMed] [Google Scholar]
- 24.Gartel AL. A new target for proteasome inhibitors: FoxM1. Expert Opin Investig Drugs. 2010;19:235–242. doi: 10.1517/13543780903563364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hegde NS, Sanders DA, Rodriguez R, Balasubramanian S. The transcription factor FOXM1 is a cellular target of the natural product thiostrepton. Nat Chem. 2011;3:725–731. doi: 10.1038/nchem.1114. [DOI] [PubMed] [Google Scholar]
- 26.Jiang L, Wang P, Chen L, Chen H. Down-regulation of FoxM1 by thiostrepton or small interfering RNA inhibits proliferation, transformation ability and angiogenesis, and induces apoptosis of nasopharyngeal carcinoma cells. Int J Clin Exp Pathol. 2014;7:5450–5460. [PMC free article] [PubMed] [Google Scholar]
- 27.Kwok JM, Myatt SS, Marson CM, Coombes RC, Constantinidou D, Lam EW. Thiostrepton selectively targets breast cancer cells through inhibition of forkhead box M1 expression. Mol Cancer Ther. 2008;7:2022–2032. doi: 10.1158/1535-7163.MCT-08-0188. [DOI] [PubMed] [Google Scholar]
- 28.Gormally MV, Dexheimer TS, Marsico G, Sanders DA, Lowe C, Matak-Vinković D, Michael S, Jadhav A, Rai G, Maloney DJ, et al. Suppression of the FOXM1 transcriptional programme via novel small molecule inhibition. Nat Commun. 2014;5:5165. doi: 10.1038/ncomms6165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Donato R, Miljan EA, Hines SJ, Aouabdi S, Pollock K, Patel S, Edwards FA, Sinden JD. Differential development of neuronal physiological responsiveness in two human neural stem cell lines. BMC Neurosci. 2007;8:36. doi: 10.1186/1471-2202-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Batistatou A, Merry DE, Korsmeyer SJ, Greene LA. Bcl-2 affects survival but not neuronal differentiation of PC12 cells. J Neurosci. 1993;13:4422–4428. doi: 10.1523/JNEUROSCI.13-10-04422.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yaffe D. Retention of differentiation potentialities during prolonged cultivation of myogenic cells. Proc Natl Acad Sci USA. 1968;61:477–483. doi: 10.1073/pnas.61.2.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kazemi-Esfarjani P, Trifiro MA, Pinsky L. Evidence for a repressive function of the long polyglutamine tract in the human androgen receptor: possible pathogenetic relevance for the (CAG)n-expanded neuronopathies. Hum Mol Genet. 1995;4:523–527. doi: 10.1093/hmg/4.4.523. [DOI] [PubMed] [Google Scholar]
- 33.Sahu B, Laakso M, Ovaska K, Mirtti T, Lundin J, Rannikko A, Sankila A, Turunen JP, Lundin M, Konsti J, et al. Dual role of FoxA1 in androgen receptor binding to chromatin, androgen signalling and prostate cancer. EMBO J. 2011;30:3962–3976. doi: 10.1038/emboj.2011.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu X, Gupta A, Wang Y, Suzuki K, Mirosevich J, Orgebin-Crist MC, Matusik RJ. Foxa1 and Foxa2 interact with the androgen receptor to regulate prostate and epididymal genes differentially. Ann N Y Acad Sci. 2005;1061:77–93. doi: 10.1196/annals.1336.009. [DOI] [PubMed] [Google Scholar]
- 35.Robinson JL, Carroll JS. FoxA1 is a key mediator of hormonal response in breast and prostate cancer. Front Endocrinol (Lausanne) 2012;3:68. doi: 10.3389/fendo.2012.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gartel AL. Thiazole Antibiotics Siomycin a and Thiostrepton Inhibit the Transcriptional Activity of FOXM1. Front Oncol. 2013;3:150. doi: 10.3389/fonc.2013.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang F, Li X, Sharma M, Sasaki CY, Longo DL, Lim B, Sun Z. Linking beta-catenin to androgen-signaling pathway. J Biol Chem. 2002;277:11336–11344. doi: 10.1074/jbc.M111962200. [DOI] [PubMed] [Google Scholar]
- 38.Truica CI, Byers S, Gelmann EP. Beta-catenin affects androgen receptor transcriptional activity and ligand specificity. Cancer Res. 2000;60:4709–4713. [PubMed] [Google Scholar]
- 39.Zhang S, Teng H, Ding Q, Fan J, Shi W, Zhou Y, Zhang C. FoxM1 involvement in astrocyte proliferation after spinal cord injury in rats. J Mol Neurosci. 2013;51:170–179. doi: 10.1007/s12031-013-9972-0. [DOI] [PubMed] [Google Scholar]
- 40.Bolte C, Zhang Y, Wang IC, Kalin TV, Molkentin JD, Kalinichenko VV. Expression of Foxm1 transcription factor in cardiomyocytes is required for myocardial development. PLoS One. 2011;6:e22217. doi: 10.1371/journal.pone.0022217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hershberger LG, Shipley EG, Meyer RK. Myotrophic activity of 19-nortestosterone and other steroids determined by modified levator ani muscle method. Proc Soc Exp Biol Med. 1953;83:175–180. doi: 10.3181/00379727-83-20301. [DOI] [PubMed] [Google Scholar]
- 42.Lee Y, Kim KH, Kim DG, Cho HJ, Kim Y, Rheey J, Shin K, Seo YJ, Choi YS, Lee JI, et al. FoxM1 Promotes Stemness and Radio-Resistance of Glioblastoma by Regulating the Master Stem Cell Regulator Sox2. PLoS One. 2015;10:e0137703. doi: 10.1371/journal.pone.0137703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Z, Banerjee S, Kong D, Li Y, Sarkar FH. Down-regulation of Forkhead Box M1 transcription factor leads to the inhibition of invasion and angiogenesis of pancreatic cancer cells. Cancer Res. 2007;67:8293–8300. doi: 10.1158/0008-5472.CAN-07-1265. [DOI] [PubMed] [Google Scholar]
- 44.Bhat UG, Halasi M, Gartel AL. Thiazole antibiotics target FoxM1 and induce apoptosis in human cancer cells. PLoS One. 2009;4:e5592. doi: 10.1371/journal.pone.0005592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tan J, Sharief Y, Hamil KG, Gregory CW, Zang DY, Sar M, Gumerlock PH, deVere White RW, Pretlow TG, Harris SE, et al. Dehydroepiandrosterone activates mutant androgen receptors expressed in the androgen-dependent human prostate cancer xenograft CWR22 and LNCaP cells. Mol Endocrinol. 1997;11:450–459. doi: 10.1210/mend.11.4.9906. [DOI] [PubMed] [Google Scholar]
- 46.Katsuno M, Adachi H, Inukai A, Sobue G. Transgenic mouse models of spinal and bulbar muscular atrophy (SBMA) Cytogenet Genome Res. 2003;100:243–251. doi: 10.1159/000072860. [DOI] [PubMed] [Google Scholar]