

Summary
We have developed a general progressive procedure, Active Interaction Mapping, to guide assembly of the hierarchy of functions encoding any biological system. Using this process, we assemble an ontology of functions comprising autophagy, a central recycling process implicated in numerous diseases. A first-generation model, built from existing gene networks in Saccharomyces, captures most known autophagy components in broad relation to vesicle transport, cell cycle and stress response. Systematic analysis identifies synthetic-lethal interactions as most informative for further experiments; consequently, we saturate the model with 156,364 such measurements across autophagy-activating conditions. These targeted interactions provide more information about autophagy than all previous datasets, producing a second-generation ontology of 220 functions. Approximately half are previously unknown; we confirm roles for Gyp1 at the phagophore-assembly site, Atg24 in cargo engulfment, Atg26 in cytoplasm-to-vacuole targeting, and Ssd1, Did4 and others in selective and non-selective autophagy. The procedure and autophagy hierarchy are at http://atgo.ucsd.edu/.
Graphical abstract
Introduction
A key promise of Systems Biology is to advance our understanding of biological systems by combining genome-scale data with model building in a virtuous cycle (Ideker et al., 2001a; Kitano, 2002). New genomics data inform the biological model and, in turn, analysis of the model informs collection of new datasets. Systems Biology typically surveys the literature to assemble knowledge of specific molecules, interactions and reactions involved in a biological system, then uses this knowledge to initialize a computational model (Covert et al., 2004; Davidson et al., 2002; Ideker et al., 2001b; Karr et al., 2012; King et al., 2004; Malleshaiah et al., 2010). Model predictions are systematically compared to laboratory measurements, with discrepancies used to improve the model and design further experiments.
Formulating a model can be laborious, requires that much is already known about the system, and may be biased towards well-studied components. Alternatively, many studies have sought to construct models of biological systems directly from systematic datasets (Ergun et al., 2007; Ho et al., 2002). Genome-scale data, including profiles of mRNAs, proteins or metabolites and gene and protein interactions, are analyzed to infer a network of genes describing the system (Behrends et al., 2010; Havugimana et al., 2012; Kim et al., 2011; Lefebvre et al., 2012; Picotti et al., 2013). Such network models have been increasingly used to describe key biological systems, including tissue specificity (Greene et al., 2015), cell differentiation (Cahan et al., 2014), and diseases such as cancer (Carro et al., 2010; Creixell et al., 2015; Leiserson et al., 2015).
At this juncture, we see two challenges. First, biological systems do not merely consist of flat networks of genes– they are exquisitely hierarchical in nature. Models should seek to capture this hierarchy, which extends over multiple scales from nucleotides (1nm scale) to proteins (1–10nm), protein complexes (10–100nm), cellular processes (100nm), organelles (1μm), cells (1–10μm), tissues (100μm–100mm) and complex organisms (>1m). Second, while the field has taken a foundational step by enabling the construction of models directly from data rather than only literature, a next step will be to determine how to iteratively improve these models with the most informative new data. The ability to rationally motivate and design new -omics data sets is sorely needed in genomics; far too many large-scale experiments are performed simply because one can, without justification or guidance for why those experiments might lead to an improved biological understanding.
Towards the first challenge, a hierarchical biological model already in widespread use is the Gene Ontology (GO) (Ashburner et al., 2000). Through extensive literature curation efforts, GO aims to factor the cell into a complete hierarchy of cellular components and processes, each represented by a GO “term”. Recently, we showed that an ontology of the cell very much like GO could be automatically constructed from diverse genome-scale datasets (Dutkowski et al., 2013; Kramer et al., 2014). Using this method we built a data-driven ontology of yeast, the Network eXtracted Ontology (NeXO), which recapitulated approximately 60% of known cellular components in GO. However, it was also clear that our hierarchical understanding of cell biology is far from complete, leaving open the second challenge: how to most efficiently improve cell function hierarchies with high value experiments. Thus far, methods have been devised for selecting the optimal next experiment considering a particular kind of model (e.g. a gene network), data type (e.g. growth phenotyping), and a constrained repertoire of treatments (e.g. single or double gene knockouts) (Atias et al., 2014; Barrett and Palsson, 2006; Ideker et al., 2000; King et al., 2009; King et al., 2004). However, such questions of experimental design have not been approached for building general hierarchical models, and given the plethora of -omics data types that can now be generated, guidance is needed on which type of data one should collect next to improve the models.
Here, we describe a general program for elucidating the hierarchy of functions underlying a cellular process, based on progressive cycles of mapping and modeling which we call Active Interaction Mapping or AI-MAP (Figure 1A). We apply this approach to develop a hierarchical model of autophagy, the conserved process of “self-eating” by which cells respond to starvation and other stress by degrading macromolecules and recycling their constituent building blocks (Levine and Yuan, 2005). During autophagy, cellular contents are enclosed in a double-walled vesicle, the autophagosome, and delivered to the yeast vacuole or mammalian lysosome for degradation and recycling; such contents can include non-selective cytoplasmic contents (macroautophagy) or selective organelles, such as peroxisomes (pexophagy) or mitochondria (mitophagy). All non-selective and selective autophagy pathways share a core set of molecular machinery (Jin and Klionsky, 2013), which has wide-ranging effects on pathways such as tumor suppression (Liang et al., 1999), neurodegeneration (Rubinsztein et al., 2005), and aging (Rubinsztein et al., 2011).
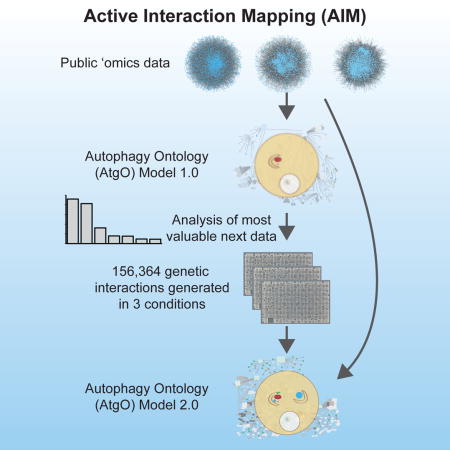
Figure 1. Program for Active Interaction Mapping.

(A) After initialization with public data sets (i), the approach proceeds by progressive iterations of data integration (ii), hierarchical model assembly (iii), and adaptive generation of new data (iv). (B) Pairwise gene similarity scores from curated knowledge (GO) versus the integrated data. Curves track percentiles in GO similarity scores at a given level of data similarity. (C) Selection of 492 autophagy-related genes. Twenty genes (red) are assigned to “core autophagy” by (Jin and Klionsky, 2013); another 102 genes (yellow) are annotated to GO:autophagy (GO:0006914). For all other genes (green) the average similarity score to core autophagy genes is calculated from the network data; a similarity threshold (vertical line) is set to select 370 genes at least as similar to core genes as those in GO:autophagy. (D) Inference of the hierarchical model from (idealized) pairwise similarity scores using CliXO.
In what follows, we [1] construct a first-generation hierarchical model of autophagy from publicly available interaction networks for the budding yeast, Saccharomyces cerevisiae, [2)] identify synthetic-lethal genetic interactions as the most useful type of data for systematically improving the model, [3] generate static and differential genetic networks involving growth measurements of over 156,000 yeast genotypes across autophagy-relevant conditions, and [4] integrate these new with previous datasets to arrive at a substantially improved understanding of autophagy. The Active Interaction Mapping process creates a ‘living’ ontology of cell function, which actively suggests new data types for self-improvement and highlights new functions discovered as data are added. The process has general utility for any biological process.
Results
Integrating diverse ‘omics data constructs a first-generation model of autophagy
To initialize the AI-MAP process, we sought to construct a ‘first-generation’ hierarchical model of autophagy, called the Autophagy Ontology (AtgO), based on publicly available molecular interaction networks in S. cerevisiae. Although not all datasets have been specifically designed to study autophagy, non-selective or selective autophagy pathways have some activity in nearly all conditions (Reggiori et al., 2012). In total, we obtained a compendium of 78 networks of 9 different data types, including protein interactions, genetic interactions, gene co-expression and gene-gene similarity based on shared protein sequence and structural information, as previously curated for YeastNet (Kim et al., 2014) (Figure 1A i).
To build an ontology from these heterogeneous datasets, we first integrated all data into a single pairwise gene similarity network (Figure 1A ii) and then analyzed the hierarchical structure of this network to infer a gene ontology (Figure 1A iii). Pairwise gene-gene similarities in the similarity network were determined using a statistical regression procedure that combines available interaction datasets into a single quantitative similarity score. To learn how the different data types should be combined, the regression was trained to model a ‘standard reference’ gene similarity network derived from GO (gene-gene similarity scores based on relatedness in GO of the two genes, see Experimental Procedures). In this way, the data-derived similarity network captured known biological processes and components in GO when supported by data (Pearson r = 0.4 versus GO, p < 10−300, out-of-bag prediction); similar data combinations then identify new biological processes, components, and relationships not previously documented (Figure 1B).
For assembly of the ontology, we selected 492 candidate genes with potential relation to autophagy based on literature or data (Figure 1C). A broad net was intentionally cast to allow later steps of the AI-MAP process to dictate which of these 492 genes are most related to core autophagy functions. The CliXO algorithm (Clique Extracted Ontologies, Kramer et al., 2014) was then applied to analyze the hierarchical structure of the data-derived gene similarity network among these 492 genes (Figure 1A iii). By this method, nested communities of genes apparent in the pairwise similarity data were identified, resulting in a hierarchy of 218 terms and 310 term relations which we call the Autophagy Ontology (AtgO 1.0, Figure 1D). To determine which terms represent known biological functions and which represent potential new biology, AtgO 1.0 was aligned with GO according to a previously described alignment procedure (Dutkowski et al., 2013). This process seeks a one-to-one mapping between terms in AtgO and terms in GO; for aligned terms, term names and descriptions are transferred from the GO reference. Term AtgO:9, which aligned to GO:0006914 (autophagy), encapsulated 234 genes organized into 82 sub-terms (Figure 2A–C), including 19 of the 20 core autophagy genes and 67 other genes annotated to GO: “autophagy” (Figure 2D). Also included were 148 genes newly associated with autophagy; within these we observed a significant enrichment for functions in cell cycle (51 genes, p < 10−12), cellular response to stress (42 genes, p < 1012) and vesicle transport (47 genes, p < 10−24), placing these functions in broad relation to autophagy.
Figure 2. First-generation autophagy ontology.

(A–C) AtgO 1.0 model of terms (rectangles), genes (ovals), and hierarchical interrelations among these (links). Rectangle size shows number of genes annotated to that term; its color shows term alignment to GO: darker blue indicates higher similarity to GO, white terms/red outlines do not align. Oval color indicates gene status. For compactness, gene annotations are displayed only for terms AtgO:15 (B, no GO alignment) and AtgO:18 (C, aligned to GO: “macroautophagy”). (D) Comparison to the Gene Ontology as curated from literature, showing GO terms and annotations for GO:0006914:“autophagy” and its descendants. Links of the same color are relations with the same parent term.
This AI-MAP protocol is available in a Jupyter Notebook at http://atgo.ucsd.edu; notebook execution constructs the AtgO model described herein; however, general ontology models can be constructed by running the notebook with new data supplied by the user.
Analysis of model reveals genetic interactions as most effective new data
Next, we determined which new network data would be most capable of improving the AtgO model in further experiments (Figure 1A iv). For this purpose, we removed all data of a given type and evaluated the resulting decrease in model performance, measured as the ability to capture the GO reference (Figure 3A). The largest contribution was from protein-protein interactions, closely followed by genetic interactions; that is, recovery of GO depends most strongly on data of these two types, making these more desirable. Beyond this absolute importance measure, we were also interested in the importance of data relative to the present time, i.e., the extent to which adding another dataset of a given type is expected to improve the model. Therefore, we measured the decrease in model performance as individual studies of a particular type were sequentially removed (Figure 3B). The unit of one ‘study’ was used to normalize for different temporal and financial costs of generating each type of data – it roughly estimates the amount of data one ‘omics lab can produce in one study on average (i.e. data per lab-study). Removing single studies of genetic interactions caused the steepest instantaneous decrease in performance, suggesting that such studies may continue to provide the most information. A similar result was obtained when individual interactions rather than whole studies were progressively removed (Figure S1). Interestingly, gene expression data provided very little information not captured by other data types, either when removed as a group (Figure 3A) or one study at a time (Figure 3B).
Figure 3. Active analysis of data types leads to conditional genetic interaction maps.

(A) Performance decrease in AtgO 1.0 when all data of one type are excluded from the model. Performance is measured as the Pearson correlation between pairwise similarity scores derived from integrated data versus GO, focusing on gene pairs within the 492 autophagy-related genes (see main text). (B) Performance degradation as single studies of the indicated type (color) are cumulatively excluded. The mean (points) and standard deviation (error bars) are calculated over 50 random sets of removed studies. See also Figure S1. (C–D) New genetic interaction maps between 52 autophagy query genes and 3007 non-essential array genes in untreated conditions (rich media, C) as well as rapamycin and starvation conditions (D). Differential maps between each pair of conditions are also displayed. See also Table S1. (E) Differential genetic interactions (rapamycin – untreated) between core autophagy genes and implicated array genes. (F) Pho8Δ60 enzymatic activity measurement of gene deletions in (E). Samples were collected from growing cells (0h; mid-log phase in YPD) and after nitrogen starvation (4h SD–N). All activity measurements were normalized to Pho8Δ60 activity in the WT 4h sample (100%). Error bars indicate SD of three replicates. (G–H) Performance of reconstructing GO, using new data only (G) or integrating new with prior data (H). Performance is measured over gene pairs within the set of 52 autophagy query genes, in comparison to randomized data or GO (mean of 100 trials).
Conditional genetic interaction networks incorporate new autophagy genes
Guided by the above analysis, we designed a systematic screen for genetic interactions targeted at genes and conditions relevant to autophagy (Figure 3C, D). Synthetic Genetic Array technology (SGA) (Tong and Boone, 2006) was used to query 52 autophagy-related genes for genetic interactions against an array of 3,007 genes, covering approximately two-thirds of the non-essential yeast genome. SGA uses high-throughput robotic colony pinning on agar to create and score growth of many double gene deletion strains in parallel, here yielding 52 × 3,007 = 156,364 tests for gene-gene interaction (Table S1). These SGA networks were created in three conditions: rapamycin, which pharmacologically induces autophagy; amino-acid starvation, which metabolically induces autophagy; and an untreated control. An established computational workflow (Bean et al., 2014) was used to assign quantitative S-scores to all gene pairs, with positive S-scores indicating faster than expected growth (epistatic or suppressive interaction) and negative S-scores indicating slower than expected growth (synthetic-sick or lethal interaction). SGA networks were also computed in ‘differential’ configurations (Bandyopadhyay et al., 2010), based on the differences in S-scores between 1) rapamycin and untreated, 2) nitrogen starvation and untreated, and 3) nitrogen starvation and rapamycin.
Several array genes displayed strong differential interactions with core autophagy genes, most specifically upon rapamycin treatment (Figure 3E). These included SSD1, encoding an mRNA-binding protein which represses translation (Jansen et al., 2009); DID4 and STP22 encoding subunits of the ESCRT complexes, which are required for autophagy in humans but had not yet been examined in yeast (Rusten and Stenmark, 2009); GYP1, a GTPase-activating protein (GAP) (Du and Novick, 2001); IRA2, an inhibitor of RAS-cAMP (Rodkaer and Faergeman, 2014); PIB2, a phosphatidylinositol(3)-phosphate binding protein of unknown function (Shin et al., 2001); and YPL247C, an uncharacterized open reading frame. To investigate whether these genes play a direct role in macroautophagy, we scored their null mutants by the Pho8Δ60 assay, which provides a quantitative marker of autophagic flow into the vacuole (Noda et al., 1995). With the exception of YPL247C, autophagy showed a clear dependence on all of these genes, as predicted (Figure 3F).
New targeted interactions markedly improve the hierarchical model
Next, we evaluated the extent to which the new interactions improved the AtgO ontology. When used as the sole source of data, networks from each of the static conditions showed some ability to reconstruct GO (r = 0.13 – 0.18, Figure 3G). The differential networks showed better performance (r = 0.23 – 0.31), consistent with previous findings on the utility of differential interaction mapping (Bandyopadhyay et al., 2010; Ideker and Krogan, 2012). Integrating all static and differential conditions into a single network yielded the best correspondence with GO (r = 0.42), suggesting that multiple conditions reveal different aspects of cell biology. Remarkably, this performance was better than all previous data combined (r = 0.30; Figure 3H). Integrating previous with new data performed best of all (r = 0.48). We designate the ‘second-generation’ ontology of 220 terms resulting from these integrated data as AtgO 2.0 (Figure 4).
Figure 4. Second-generation ontology.

(A) AtgO 2.0 hierarchical model. AtgO:220:“autophagy and related processes” and its descendants are displayed. Gene annotations are shown for branches beneath AtgO:11:“membrane trafficking, cell growth and stress responses II” (B) and AtgO:14:“vesicle (especially golgi) and organelle traffic/cell cycle and stress response” (C). Term names have been manually curated by our team, thus may differ from previous versions of AtgO or GO. See also Table S2, Table S3, Figure S2.
Beyond its improved ability to reconstruct GO, we found that the hierarchical structure of this model captured many potential new biological functions and relationships. In particular, analysis of AtgO 2.0 revealed that the majority of terms (56%) involved previously unknown biological findings, which we categorized into four broad types: [1] terms representing previously unknown subfunctions of autophagy (previously unknown groupings of primarily known autophagy genes), [2] terms representing previously unknown subfunctions related to autophagy (groupings of genes previously attributed to diverse functions), [3] terms representing superprocesses which integrate known processes, and [4] terms representing a known process but expanded by adding genes (Figure 5A, Table S2). In what follows, we survey brief, but suggestive, experimental findings from several of these types. We have constructed a web portal to AtgO 2.0 (http://atgo.ucsd.edu/) which permits exploration of the autophagy hierarchy with visualization of the network data supporting each term.
Figure 5. Characterization of AtgO 2.0.

(A) Summary of AtgO terms by types of biological findings. (B) Pairwise gene similarity matrix and resulting AtgO 2.0 hierarchy for a subset of core autophagy genes.
A key regulator of vesicular trafficking, Gyp1, is required for autophagy-related pathways
AtgO 2.0 suggested a broader involvement of vesicle trafficking, docking and fusion pathways in autophagy (Figure 4C): for example, we noted AtgO:18 (“vesicle transport machinery of ER-EE-MVB-vacuole and autophagy”, 37 genes) which integrated core autophagy or pexophagy genes with many Rab GTPases and GAPs known primarily for their function in Golgi transport (GYP1, YPT1, YPT31-32 and SEC4) (Jean and Kiger, 2012) (Figure 4C i). GYP1, the GTPase-activating protein (GAP) of this pathway, displayed significant genetic interactions with core autophagy genes under starvation and rapamycin treatment, including positive interactions between Gyp1 and the Atg9-recycling system (Atg9, Atg18), as well as with the two core Ubiquitin-like conjugation systems (Atg3, Atg5, Atg7) (Figure 3E); these interactions were not observed in untreated conditions (Table S1). Gyp1 had been previously localized to the Golgi (Du and Novick, 2001); however, we observed that GFP-Gyp1 is also localized to the phagophore assembly site (PAS), the location of the autophagosome-generating machinery (Figure 6A). Functional experiments with a gyp1Δ strain revealed a requirement for Gyp1 in PAS formation, as deletion of GYP1 caused mislocalization of GFP-Atg8 (Figure 6B) and an Atg8-processing defect (Figure 6C); reduced maturation of prApe1 into mApe1 via selective autophagy (Figure 6D); and impaired macroautophagy under nitrogen-starvation as measured by the Pho8Δ60 assay (Figure 3F). These assays confirm a general requirement of Gyp1 in autophagy-related pathways.
Figure 6. Analysis of Gyp1 and Atg8/Atg24.

(A) Co-localization (yellow arrows) of Gyp1 and the PAS marker Ape1 in wild-type cells under nominal conditions (SD) or after rapamycin (SD+rapamycin) treatment, using fluorescence microscopy. (B) Atg8 localization in wild-type and gyp1Δ cells. (C) Atg8 cleavage and (D) prApe1 maturation assay in wild-type, atg6Δ (positive control), gyp1Δ and vps1Δ strains. (E) Atg8 localization in context of wild-type, Ypt1 overexpression, and gyp1Δ with Ypt1 overexpression after rapamycin treatment for 1 hour. (F) Model proposing the conversion of GDP-bound Ypt1 into the GTP-bound, active form, catalyzed by guanidine nucleotide exchange factor TRAPPIII during autophagy. Ypt1-GTP is recognized by at least two autophagy effectors, Atg11 and the COG complex, and is converted back to the GDP-bound, inactive form by the GTPase-activating protein Gyp1. (G) Localization of Atg24 in wild-type, atg7Δ and atg8Δ strains of P. pastoris under pexophagy conditions. Bar scale is 5 μm.
The Rab GTPase Ypt1 has also been implicated in autophagy (Wang et al., 2015); Gyp1 may act as the heretofore unknown GAP on Ypt1 at the PAS. To test this possibility, we used a Ypt1 overexpression system which leads to increased PAS formation and rescues the autophagy defect of a Ypt1 GEF mutant (Trs85; upstream of Ypt1), but not of a Ypt1 effector mutant (Atg11; downstream of Ypt1) (Lipatova et al., 2012). When Ypt1 was overexpressed in a gyp1Δ strain, the gyp1Δ defect in PAS formation was not rescued (Figure 6E). This indicates that Gyp1 acts downstream of Ypt1 during autophagy, similar to its function as the GAP for Ypt1 in ER-to-Golgi protein trafficking (Figure 6F).
Finally, some of the same autophagy-deficient phenotypes were observed for a vps1Δ strain (Figures 6B, C); VPS1 is also annotated to AtgO:18 and had been formerly implicated in pexophagy (Mao et al., 2014); these results indicate it may have a more general role in autophagy-related pathways.
Engulfment of selective autophagy cargos involves Atg8 and Atg24
AtgO:247 was a new term in AtgO 2.0 that grouped ATG8, encoding a ubiquitin-like protein that localizes to isolation membranes and mature autophagosomes (Noda et al., 2010), with ATG24, a regulator of membrane fusion (Ano et al., 2005; Kanki and Klionsky, 2008), based on a combination of strong genetic interaction profile similarity and co-expression (Figure 4B i, Figure 5B). Working with the yeast Pichia pastoris, where the very large peroxisomes serve as a model cargo for imaging of autophagy (Oku and Sakai, 2008), we followed Atg24-GFP in both wild-type and atg8Δ cells. We noted a change in Atg24 localization specific to the atg8Δ genotype, in which Atg24 occupies a large perimeter of the peroxisome cargo, as opposed to only a few punctate structures in wild-type cells (Figure 6G). Atg8 function has been thought to depend on Atg7, an E1-like enzyme that conjugates Atg8 to phosphatidylethanolamine (PE), a lipid. Therefore, we also examined the localization of Atg24 in a atg7Δ mutant, predicting that it would have the same phenotype as that in atg8Δ cells. Surprisingly, Atg24 localization was not affected in atg7Δ cells; thus, the new term appears to represent a separate activity of Atg8, independent of Atg8 conjugation to PE. Although further investigations will be needed to fully understand the Atg8-Atg24 relationship, we have provisionally named AtgO:247 “engulfment of selective autophagy cargo”.
Atg26 is required for processing of large aggregates in the Cvt pathway
Term AtgO:185 expanded from two genes, Atg11 and Atg19 (Figure 2C i, Figure 7A), to include Atg26 and Atg27 (Figure 4A i, Figure 7B), based on strong genetic interaction profile similarity across the new conditions. Three of these genes (Atg11, Atg19, Atg27; Yen et al., 2007; Yorimitsu and Klionsky, 2005) have known roles in the Cytoplasm-to-vacuole targeting (Cvt) pathway, by which aggregates of the aminopeptidase precursor prApe1 are transported to, and processed in the vacuole to mature Ape1. AtgO:185 implied involvement of the remaining gene, Atg26, in transport of prApe1 aggregates to the vacuole. In the yeast Pichia pastoris, the Atg26 ortholog participates in pexophagy, where it is required for the degradation of large peroxisomes; hence, its designation as an ‘Atg’ gene (Nazarko et al., 2009; Oku et al., 2003).
Figure 7. Analysis of Atg26 involvement in the transport of Atg19-receptor cargos.

(A–B) The refinement of AtgO 1.0 (A) to AtgO 2.0 (B), resulting in expansion of AtgO:185 which we name “transport of Atg19-receptor cargos”. (C) Co-localization of dTomato-Atg26 and GFP-Atg19 in wild-type cells before and after 1 hour of rapamycin treatment. (D) Western blot of prApe1 and Ape1 across genetic backgrounds (wild-type, atg1Δ, atg26Δ) with prApe1 expressed from a copper-inducible promoter and incubated 16 hours in SD–Cu media plus copper (CuSO4) followed by removal from CuSO4 and treatment with rapamycin for indicated times. (E) Fluorescence micrographs of cells expressing endogenous prApe1, prApe1-BFP from the endogenous promoter, and prApe1 from the copper-inducible promoter. Cells incubated 16 hours in SD–Cu +50μm CuSO4 followed by 14 hours in SD–Cu media +rapamycin. (F) Analysis of cells with constructs as in (E). Fraction of cells with observed prApe1 aggregates (light blue) and large prApe1 aggregates >1μM (red) after 16 hours incubation in SD–Cu media + indicated CuSO4, followed by 14 hours incubation in SD–Cu + rapamycin. >250 cells were analyzed. Asterisk indicates significant difference from wild-type under the same condition as judged by Fisher’s exact test. (G) Same as (F) but pre-treatment in CuSO4 followed by 14 hours incubation in SD–Cu +rapamycin. (H, I) Quantitative analysis of processed GFP for GFP-Ape4 and GFP-GAG Ty processing assay. Error bars represent SD of three replicates. Asterisks indicate significant difference compared to wild-type as judged by the one sided t-test (p < 0.05).
Although previous work in S. cerevisiae had found no involvement of Atg26 in standard autophagy assays (Cao and Klionsky, 2007), we observed that Atg26 co-localizes in distinct, overlapping puncta with Atg19, the known receptor for prApe1 aggregates (Figure 7C). We then monitored processing of prApe1 in wild-type and atg26Δ strains, as well as in atg1Δ cells, as a positive control for disruption of prApe1 processing (Cheong et al., 2008). Normal maturation of prApe1 was observed in wild-type, but not atg1Δ, cells (Figure 7D). The atg26Δ mutant also showed processing defects, especially in cells overexpressing prApe1, which produce larger aggregates (50 μM CuSO4, Figure 7D). Furthermore, we found that atg26Δ cells contained significantly more large prApe1 aggregates than did wild-type cells (p = 0.03, Figure 7E, F); overexpression exacerbated this defect (5 μM CuSO4, p = 0.02; 50 μM CuSO4, p = 0.0003). Using rapamycin to induce macroautophagy, wild-type cells successfully processed all large prApe1 aggregates even when they were greatly overexpressed (50 μM CuSO4, Figure 7F). In contrast, many aggregates remained in the atg26Δ mutant after rapamycin treatment (Figure 7G). Finally, in addition to prApe1, the Cvt pathway transports the vacuolar hydrolases aminopeptidase 4 (Ape4) and the Ty1 transposon to the vacuole (Suzuki et al., 2011; Yuga et al., 2011). We found that atg26Δ strains had reduced processing for each of these cargos (Figure 7H, I). These results support a role for Atg26 in the transport of several cargos which use Atg19 as their receptor, including large prApe1-containing aggregates, Ty1, and Ape4; hence we gave AtgO:185 the name “transport of Atg19-receptor cargos”.
AtgO as an automated literature-curation device
By incorporating evidence from all available ‘omics studies, AtgO 2.0 recovered many functions already reported in literature but which had not yet been curated by GO (Figure 5A; representing 23% of all terms). For example, AtgO:138 recovered the Ubp3p-Bre5p ubiquitin protease complex implicated in ribophagy (Figure 4B ii) (Kraft et al., 2008); AtgO:160 recovered the Pho85-Pho80 CDK-cyclin complex which regulates autophagy (Figure 4B iii) (Yang et al., 2010); AtgO:106 recovered Skm1, Ste20 and Cla4, part of a complex that downregulates sterol uptake (Figure 4B iv) (Lin et al., 2009); and AtgO:140 recovered the Vma12-Vma22 assembly complex (Figure 4B v) (Graham et al., 1998). We submitted each of these terms to GO curators, and given the ample prior evidence, all were accepted for inclusion in the Gene Ontology (GO:1990861, GO:1990860, GO:1990872 and GO:1990871 respectively). Other terms missing from GO, but supported by literature, include AtgO:246, which captured an E1/E2 enzyme complex that activates Atg8 in a ubiquitin-like cascade (Figure 4B vi, Figure 5B), and AtgO:240, a second ubiquitin-like cascade activating Atg12 (Figure 4B vii, Figure 5B) (Kaiser et al., 2012). Numerous new AtgO gene annotations were also supported by prior studies: for instance, term AtgO:96 was assigned Vac8, Nvj1, and Swh1, which form the nucleus-vacuole junction (Figure 4B viii) during piecemeal microautophagy of the nucleus (Dawaliby and Mayer, 2010; Kvam and Goldfarb, 2004). Two of these genes (NVJ1, SWH1) were not annotated as such in GO and thus at our request were annotated to this term (GO:0034727). As another example, we found that eight proteins annotated to autophagy by AtgO but not GO already had literature support for autophagy phenotypes: Hog1 (Prick et al., 2006), Pep12 (Kanki et al., 2009), Snf1 (Wang et al., 2001), Stt4 (Wang et al., 2012), Tpk1 (Budovskaya et al., 2004), Vps4 (Nebauer et al., 2007), Vps51 (Reggiori et al., 2003), and Vps52 (Reggiori et al., 2003). Here too, these new gene annotations were accepted for inclusion in GO. Thus, AI-MAP can automatically mine, structure, and unify knowledge of a cellular process to create a central research resource that is complementary to literature curated models.
Application to human autophagy
To test the feasibility of the Active Interaction Mapping approach in the study of human biology, we applied the same procedure described in yeast to a compendium of human data including gene expression profiling, protein-protein interactions, genetic interactions, co-localization and sequence similarity. As in yeast, we began with a seed set of (33) genes designated as having core functions common to non-selective and selective autophagy (Jin and Klionsky, 2013). This process resulted in a human Autophagy Ontology (hAtgO 1.0) of 1,452 genes and 1,664 terms, 173 of which align significantly to existing terms in the human GO, and the rest of which represent potential new autophagy subprocesses and components (Figure S2A, Table S3). Compared to yeast, a smaller percentage of the ontology (10% vs 35%) aligned to existing GO terms, which may be due to better GO annotation of yeast than human, more complete data in yeast than human, or both. As in yeast, much of the core machinery of autophagy was grouped together (hAtgO:2962), which contains 14 core and 8 other genes (Figure S2A). Also as before, our confidence in the model is increased by the presence of recently discovered autophagy biology which is not yet captured by GO; for example, the Huntingtin protein, Htt, is placed amongst core autophagy proteins, consistent with the recent discovery that this important disease gene acts physiologically as a scaffold for selective autophagy (Rui et al., 2015). Turning to the data most valuable for constructing hAtgO, we see a striking difference as compared to yeast, as the Active Interaction Mapping process relies primarily on gene expression profiling to construct the model of the human autophagy system (Figure S2B). This result suggests the generality of Active Interaction Mapping, as it can quickly be adapted to a new species (human) and can rely on the data most available and useful for that species and system.
Discussion
This study provides a roadmap for how to progressively elucidate the ontology of cellular functions underlying any biological process. Studying a new process is straightforward, requiring starting knowledge of some of the genes involved and access to public ‘omics data sets. At all points of the study, virtually any genomic data can be analyzed since almost any data linked to genes or proteins can inform gene-gene similarity networks.
The current AtgO 2.0 model, assembled entirely from network data, is larger and has differences in content and structure from the literature-curated GO. Many of these differences imply new biological findings, as reported above. Other differences between the models may arise due to differences in policy between the AI-MAP process and human curation. For example, AtgO groups core autophagy genes under the single term “core machinery of autophagy”, whereas GO repetitively annotates each core gene to all non-selective and selective autophagy pathways including “macroautophagy,” “microautophagy,” and “nucleophagy.” Furthermore, GO generally does not group paralogous genes of similar function, whereas AtgO creates a term for functionally similar paralogs, the same as for any functional gene set (e.g., “PDK homolog kinases,” “ADP ribosylation factors”).
An important aspect of Active Interaction Mapping is that heterogeneous new and existing data are interwoven in the same modeling framework. This enables a comprehensive assessment of the current support for a biological model (Figure 3A), as well as what type of information is most needed next (Figure 3B). After data generation, the value of the new dataset is systematically evaluated in the context of all existing data (Figures 3G, H). Here, such assessments pointed us to genetic interactions, leading us to generate a new targeted dataset, which was more powerful at recovering knowledge about autophagy in GO than all previous public datasets combined (Figure 3H). In this regard, the significant information gained by including multiple conditions (Figure 3G) argues that condition-specific and differential interaction maps have provided a particularly worthwhile bolus of data.
In future many research communities, each focused on a cellular process of interest, may find it useful to organize around shared hierarchical models as a means for representing cell biological knowledge, communicating new findings, and designing targeted datasets. Once a baseline model of a process is created, the value of any new dataset can be evaluated by its ability to improve this baseline. The value of new interaction mapping efforts can thus be rigorously evaluated rather than assumed, and the design of these experiments can be guided rationally, based on current knowledge and data.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
As Lead Contact, Trey Ideker is responsible for all reagent and resource requests. Please contact Trey Ideker at tideker@ucsd.edu with requests and inquiries.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast strains
Saccharomyces cerevisiae and Pichia pastoris laboratory strains as described in Key Resources table with growth conditions as appropriate for experiments as described in Method Details.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-vacuolar aminopeptidase 1 (Ape1) | Klionsky et al., 1992 | N/A |
| Anti-β-actin (C4) | Santa Cruz Biotechnology | Cat# sc-47778 |
| Anti-GFP (JL8) | Clontech | Cat# 632381 |
| Anti-Mouse IgG-HRP | BioRad | Cat#170-6516 |
| Anti-Rabbit IgG-HRP | BioRad | Cat#170-1019 |
| Biological Samples | ||
| Saccharomyces cerevisiae (BY4741) knockout library | ThermoFisher | Cat# 95400.BY4741 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Rapamycin | Sigma | Cat# R0395 |
| Nourseothricin (clonNAT) | Jena Bioscience | Cat# AB-102L |
| Geneticin selective antibiotic (G418 sulfate) | ThermoFisher | Cat# 11811031 |
| Phenylmethanesulfonyl fluoride (PMSF) | Sigma | Cat# P7626 |
| ρ-nitrophenyl phosphate (pNPP) | Sigma | Cat# N9389 |
| ρ-nitrophenol | Sigma | Cat# N7660 |
| Pierce BCA Protein Assay Kit | ThermoFisher | Cat# 23225 |
| Critical Commercial Assays | ||
| Deposited Data | ||
| YeastNet v3 | Kim et al., 2014 | http://www.inetbio.org/yeastnet/ |
| GeneMania | Warde-Farley et al., 2010 | http://pages.genemania.org/data/ |
| Gene Expression Omnibus (GEO) | Barrett et al., 2013 | https://www.ncbi.nlm.nih.gov/geo/ |
| Gene Network Analysis Tool (GNAT) | Pierson et al., 2015 | http://mostafavilab.stat.ubc.ca/gnat/ |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| MATa leu2–3,112, trp1, ura3–52, pho8::pho8Δ60, pho13::URA3 | Bicknell et al., 2010 | MNY1121; WT Pho8Δ60 |
| MATa leu2–3,112, trp1, ura3–52, pho8::pho8Δ60, pho13::URA3 atg8Δ::kanMX | Bicknell et al., 2010 | MNY1123; atg8Δ Pho8Δ60 |
| MATa leu2–3,112, trp1, ura3–52, pho8::pho8Δ60, pho13::URA3 ypl247cΔ::kanMX | This study | sAtg0-186; ypl247cΔ Pho8Δ60 |
| MATa, leu2–3,112, trp1, ura3–52, pho8::pho8Δ60, pho13::URA3 gyp1Δ::kanMX | This study | sAtg0-187; gyp1Δ Pho8Δ60 |
| MATa leu2–3,112, trp1, ura3–52, pho8::pho8Δ60, pho13::URA3 pib2Δ::kanMX | This study | sAtg0-188; pib2Δ Pho8Δ60 |
| MATa leu2–3,112, trp1, ura3–52, pho8::pho8Δ60, pho13::URA3 ira2Δ::kanMX | This study | sAtg0-189; ira2Δ Pho8Δ60 |
| MATa leu2–3,112, trp1, ura3–52, pho8::pho8Δ60, pho13::URA3 ssd1Δ::kanMX | This study | sAtg0-190; ssd1Δ Pho8Δ60 |
| MATa leu2–3,112, trp1, ura3–52, pho8::pho8Δ60, pho13::URA3 did4Δ::kanMX | This study | sAtg0-191; did4Δ Pho8Δ60 |
| MATa leu2–3,112, trp1, ura3–52, pho8::pho8Δ60, pho13::URA3 stp22Δ::kanMX | This study | sAtg0-192; stp22Δ Pho8Δ60 |
| MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0, pDP103 (BFP-Ape1), NRB1322 (GFP-Gyp1) | This study | sAtg0-52′; WT+BFP-Ape1+ GFP-Gyp1 |
| MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0, pRS315-GFP-Atg8 | This study | sAtg0-1; WT+GFP-Atg8 |
| MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 atg6Δ::kanMX, pRS315-GFP-Atg8 | This study | sAtg0-12; atg6Δ+GFP-Atg8 |
| MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 gyp1Δ::kanMX, pRS315-GFP-Atg8 | This study | sAtg0-39; gyp1Δ+GFP-Atg8 |
| MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 vps1Δ::kanMX, pRS315-GFP-Atg8 | This study | sAtg0-48; vps1Δ+GFP-Atg8 |
| MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 gyp1Δ::kanMX, pRS315-GFP-Atg8, NRB167 (Ypt1 Overexpression) | This study | sAtg0-150; gyp1Δ+GFP-Atg8+OverYpt1 |
| MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0, pRS315-GFP-Atg8, NRB167 (Ypt1 Overexpression) | This study | sAtg0-152; WT+GFP-Atg8+OverYpt1 |
| MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0, pTS551 (GFP-Ape4) | This study | sAtg0-79; WT+GFP-Ape4 |
| MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 atg11Δ::kanMX, pTS551 (GFP-Ape4) | This study | sAtg0-80; atg11Δ+GFP-Ape4 |
| MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 atg19Δ::kanMX, pTS551 (GFP-Ape4) | This study | sAtg0-81; atg19Δ+GFP-Ape4 |
| MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 atg26Δ::kanMX, pTS551 (GFP-Ape4) | This study | sAtg0-82; atg26Δ+GFP-Ape4 |
| MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 atg27Δ::kanMX, pTS551 (GFP-Ape4) | This study | sAtg0-83; atg27Δ+GFP-Ape4 |
| MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0, pYEX-BX (GFP–Ty1 Gag) | This study | sAtg0-111; WT+GFP-Ty1 Gag |
| MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 atg11Δ::kanMX, pYEX-BX (GFP–Ty1 Gag) | This study | sAtg0-112; atg11Δ+GFP-Ty1 Gag |
| MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 atg19Δ::kanMX, pYEX-BX (GFP–Ty1 Gag) | This study | sAtg0-113; atg19Δ+GFP-Ty1 Gag |
| MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 atg26Δ::kanMX, pYEX-BX (GFP–Ty1 Gag) | This study | sAtg0-114; atg26Δ+GFP-Ty1 Gag |
| MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 atg27Δ::kanMX, pYEX-BX (GFP–Ty1 Gag) | This study | sAtg0-115; atg27Δ+GFP-Ty1 Gag |
| MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0, pCK770 (pRS415-GFP-Atg19), pCU416-tdTomato-Atg26 | This study | sAtg0-194; WT+GFP-Atg19+tdTomato-Atg26 |
| MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0, pCK782 (Ape1 overexpression) | This study | sAtg0-3; WT+OverApe1 |
| MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0, pCK782 (Ape1 overexpression) | This study | sAtg0-4; WT+OverApe1+BFP-Ape1 |
| MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 atg26Δ::kanMX, pCK782 (Ape1 overexpression) | This study | sAtg0-28; atg26Δ+OverApe1 |
| MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 atg26Δ::kanMX, pDP105 (Ape1 overexpression+BFP-Ape1) | This study | sAtg0-29; atg26Δ+OverApe1+BFP-Ape1 |
| MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 atg1Δ::kanMX, pCK782 (Ape1 overexpression) | This study | sAtg0-195; atg1Δ+OverApe1 |
| MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 atg1Δ::kanMX, pDP105 (Ape1 overexpression+BFP-Ape1) | This study | sAtg0-196; atg1Δ+OverApe1+BFP-Ape1 |
| PPY12 his4 arg4 pPIC6a-BFP-SKL::Blasticidinr, pJCF210(Atg24-GFP)::HIS4 | This study | sJCF322; WT+BFP-SKL+Atg24-GFP |
| atg7Δ::ScARG4 his4 arg4 pPICz-BFP-SKL::Zeocinr, pJCF210(Atg24-GFP)::HIS4 | This study | sJCF351; atg7Δ+BFP-SKL+Atg24-GFP |
| atg8Δ::kanMX his4 arg4 pPICz-BFP-SKL::Zeocinr, pJCF210(Atg24-GFP)::HIS4 | This study | sJCF353; atg8Δ+BFP-SKL+Atg24-GFP |
| MATα can1Δ::MFA1pr-HIS3::MFα1pr-LEU2 ura3Δ0 leu2Δ0 his3Δ1 lys2Δ0) | Tong et al., 2004 | Y3656, query strain |
| Recombinant DNA | ||
| BFP-Ape1 | Pfaffenwimmer et al., 2014 | pDP103 |
| pRS306 pADH1-GFP-HA-GYP1-CYC1term | Rivera-Molina and Novick, 2009 | NRB1322 |
| pRS315-GFP-Atg8 | This study | pRS315-GFP-Atg8 |
| Ypt1 Overexpression | Bacon et al., 1989 | pNB167 |
| GFP-Ape4 | Yuga et al., 2011 | pTS551 |
| pRS415-GFP-Atg19 | Pfaffenwimmer et al., 2014 | pCK770 |
| GFP–Ty1 Gag | Suzuki et al., 2011 | pYEX-BX |
| pCU416-tdTomato-Atg26 | This study | pCU416-tdTomato-Atg26 |
| Ape1 overexpression | Pfaffenwimmer et al., 2014 | pCK782 |
| Ape1 overexpression+BFP-Ape1 | Pfaffenwimmer et al., 2014 | pDP105 |
| Atg24-GFP | This study | pJCF210 |
| pPIC6a-BFP-SKL | This study | pPIC6a-BFP-SKL |
| pPICz-BFP-SKL | This study | pPICz-BFP-SKL |
| Sequence-Based Reagents | ||
| Software and Algorithms | ||
| Active Interaction Mapping | This Study | http://atgo.ucsd.edu/download.html |
| CliXO v0.3 | Kramer et al., 2014 | https://mhk7.github.io/clixo_0.3/ |
| Ontology Alignment Algorithm | Dutkowski et al., 2013 | https://mhk7.github.io/alignOntology/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Bean Colony Analyzer Toolkit | Bean et al., 2014 | https://github.com/brazilbean/Matlab-Colony-Analyzer-Toolkit |
| R | R Project | http://www.R-project.org. |
| Jupyter | Jupyter Project | http://jupyter.org/ |
| Python | https://www.python.org/ | |
| Scikitlearn | http://scikit-learn.org/stable/ | |
| Other | ||
METHOD DETAILS
Public data for S. cerevisiae
Data from YeastNet v3 (Kim et al., 2014) were obtained from http://www.inetbio.org/yeastnet/, consisting of the following numbers of datasets classified by type: 50 co-expression, 1 domain co-occurrence, 1 genomic neighbor, 10 genetic interaction, 12 high-throughput protein-protein interaction, 1 phylogenetic profile, 1 protein network tertiary structure, 1 low-throughput protein-protein interaction, and 1 co-citation.
Data integration
Input datasets were provided as features to a random forest regression system for prediction of gene-gene pairwise similarity, trained using pairwise gene similarities in GO as a bronze standard. The network of predicted similarities we call the ‘data-derived gene similarity network’ We aimed to use the Gene Ontology (GO) as a standard reference for learning to integrate data in a supervised fashion. Previously, we showed that a weighted network of gene-gene similarities derived from a gene ontology can be analyzed to reconstruct the full hierarchy of terms and term relations with near perfect precision and recall (Kramer et al., 2014). Because the network and the ontology can be interconverted, both contain the same information. Based on this equivalence, we guided our integration of data using a gene similarity network derived from GO instead of using GO directly. We derived this network by calculating the Resnik semantic similarity for each pair of genes (Resnik, 1995). Semantic similarities are calculated across the Biological Process and Cellular Component branches of GO, downloaded on 6/2/2015 from www.geneontology.org.
The 78 input datasets were integrated into a single network by using them as features in a supervised learning of the GO gene similarity network. Learning was performed using random forest regression (Breiman, 2001) from the Python scikit-learn package (Pedregosa et al., 2011). Predictions were made “out of bag”, i.e., the similarity of a gene pair was predicted based on information learned from other gene pairs. In effect, the random forest learns patterns in the networks which recapitulate information in GO. Hence, only relations in GO that can be systematically explained from data are included, any relations not justified by the data are excluded, and new relations not in GO are added when the network data support them (Figure 1B).
Construction of Autophagy Ontology (AtgO) 1.0
We analyzed the data-derived gene similarity network using the Clique Extracted Ontology (CliXO) algorithm, version 0.3 (http://mhk7.github.io/clixo_0.3/; Kramer et al., 2014), with parameters α=0.1 and β=0.5. We identified significantly aligned terms between AtgO and GO using a previously described ontology alignment procedure with FDR threshold of 10% and minimum alignment score of 0.24 (http://mhk7.github.io/alignOntology/; Dutkowski et al., 2013). Alignment was performed first against the sub-branch of GO rooted at GO: “autophagy” and then the entire GO, prepared as in Dutkowski et al., 2013; significant alignments to the autophagy sub-branch were given first priority.
Genetic interaction mapping
Strain construction, plating of mutants, mutant selection, and scoring of genetic interactions in each condition were performed using a previously defined protocol (Collins et al., 2006; Schuldiner et al., 2006). Using a replica pinning robot (Singer Instruments; UK), haploid double mutants representing crosses of 52 autophagy-related query genes and 3007 array mutants were grown on agar plates that were either untreated, rapamycin treated or nitrogen deficient. Plates were photographed and colony sizes normalized, spatially corrected and quantified using the Colony Analyzer Toolkit (Bean et al., 2014). For the three replicates per double mutant, the resulting experimental data were used to assign a quantitative S-score based on a modified T-test that compares the observed double mutant growth rate to that expected assuming no interaction exists, again using the Colony Analyzer Toolkit. Differential interactions were calculated by subtracting the S-scores for the same double mutant pair across conditions. Resulting S-scores available in Table S1.
Construction of Autophagy Ontology (AtgO) 2.0
The data-derived gene similarity network was recalculated incorporating both prior data and the new genetic interaction screen. The CliXO algorithm was applied with previous parameters. Term names in AtgO 2.0 were manually curated by our team, considering the names of aligned terms in GO, when available, as well as an extensive literature review. Reasoning and citations for term names are included in Table S2.
Biochemical studies
The prApe1 processing assay
The prApe1 processing assays for Figure 6D: cells were grown for 16 h in YPD medium (1% yeast extract, 2% peptone and 2% glucose) without exceeding exponential phase (OD600 above 1), 1 OD600 equivalents of cells were collected and Trichloroacetic acid (TCA) precipitated using a final concentration of 12.5% TCA and incubated at least for 30 min at −80°C, next TCA-treated cells were pelleted by centrifugation (10 min at 21,000 g, room temperature), washed twice with ice-cold 80% acetone, and air-dried. After dissolving the pellets in 100 μl of 1% SDS/0.1 N sodium hydroxide, 20 μl of 6× SDS sample buffer was added. Samples were boiled for 5 min. For SDS-polyacrylamide gel electrophoresis, 10 μl of each sample was used per lane and blotted onto nitrocellulose membranes. Western blots were blocked with 5% dry skim milk in Tris-buffered saline with 0.1% tween-20 (TTBS) and, probed with anti-Ape1 (1:5000; rabbit; a gift from Dr. Daniel Klionsky) and anti-β-actin antibodies (1:5000; mouse; C4, Santa Cruz Biotechnology) diluted in TTBS with 5% dry skim milk and incubated overnight at 4°C. Membranes were probed with anti-rabbit or anti-mouse IgG HRP (1:5000; BioRad). All blots were detected with HyGLO Quick Spray (Denville Scientific Inc.) on a Medical film processor SRX-101A (Konica).
The prApe1 processing assays for Figure 7D: cells harboring the plasmid overexpressing prApe1 (pCK782, a gift from Dr. Claudine Kraft; Papinski et al., 2014) were grown for 16 h in SD-Cu medium (0.67% yeast nitrogen base [YNB] without amino acids and copper, 2% glucose plus any required amino acid, nucleotide, or vitamin supplement) without exceeding exponential phase (OD600 above 1) and transferred to SD-Cu medium with 200 nM Rapamycin and the indicated CuSO4 concentration at an OD600 of 1. One ml of cells was collected at different times, TCA precipitated and analyzed as described above.
Pho8Δ60 assay
The Pho8Δ60 assay (WT and atg8Δ strains were a gift from Dr. Maho Niwa) was performed as described previously (Manjithaya et al., 2010; Noda and Klionsky, 2008). For the alkaline phosphatase assay, five OD600 equivalents of yeast cells were harvested, washed once with cold water and once with wash buffer (0.85% NaCl and 1mM PMSF) and resuspended in 500 μl lysis buffer (20 mM Pipes, pH 7.0, 0.5% Triton X-100, 50 mM KCl, 100 mM potassium acetate, 10 mM MgSO4, 10 μM ZnSO4, and 1 mM PMSF). The cells were lysed by vortexing at full speed 10 times with 250 μl equivalents of glass beads for 1 min and incubated for 1 min on ice in between. The lysate was centrifuged at 14,000 g for 5 min at 4°C and the supernatant was recovered without disturbing the pellet. 100 μl of this supernatant was added to 400 μl reaction buffer (250 mM Tris-HCl, pH 8.5, 0.4% Triton X-100, 10 mM MgSO4, and 1.25 mM p-nitrophenyl phosphate [pNNP]), and samples were incubated for 10–15 min at 30°C before terminating the reaction by adding 500 μl of stop buffer (2 M glycine, pH 11). Production of p–nitrophenol was monitored by measuring the absorbance at 400 nm (A400) using a spectrophotometer (DU-730; Beckman Coulter), and the concentration in nmol of p–nitrophenol in the samples was calculated by graphing the adjusted A400 values relative to a standard curve of commercial p–nitrophenol (0 to 100 nmol). Protein concentration in the extracts was measured with the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific), and the specific activity was calculated as nmol p–nitrophenol/min/mg protein.
The GFP-Atg8, GFP-Ape4 and GFP-Ty1 Gag processing assays
For the GFP-Atg8, GFP-Ape4 and GFP-Ty1 Gag processing assays, yeast strains harboring the plasmids GFP-Atg8 (pRS315-GFP-Atg8, in house plasmid), GFP-Ape4 (pTS551, a gift from Dr. Takahiro Shintani; Yuga et al., 2011) and GFP-Ty1 Gag (pYEX-BX[GFP–Ty1 Gag], a gift from Dr. Yoshinori Ohsumi; Suzuki et al., 2011) were grown to mid log phase in SD medium (0.67% YNB without amino acids, 2% glucose plus any required amino acid, nucleotide, or vitamin supplement) lacking auxotrophic amino acids and then shifted to SD-N medium (0.17% YNB without amino acids and ammonium sulfate, and 2% glucose) at an OD600 of 1 for the indicated time. At each indicated time point, 1 ml of cell culture was removed and TCA precipitated as described above. The protein extracts were resolved by SDS-PAGE and proteins were revealed by Western blotting with anti-GFP (1:2500; mouse; JL8, Clontech) and anti-β-actin (1:5000; mouse; C4, Santa Cruz Biotechnology) antibodies. Densitometry was performed using ImageJ software.
Fluorescence microscopy studies
Cell culture conditions for microscopy were performed as described for biochemical studies. Fluorescence microscopy images were acquired at indicated times using a motorized fluorescence microscope (Axioskop 2 MOT, Carl Zeiss MicroImaging, Thornwood, NY) coupled to a monochrome digital camera (AxioCam MRm, Carl Zeiss MicroImaging) and processed using the AxioVision 4.8.2 software. To quantify prApe1 aggregates images, the fluorescence microscopy parameters such as exposition time, gain and binning were kept constant. Pexophagy condition used for Pichia pastoris in Figure 6G: cells were grown in methanol medium (0.67% YNB without amino acids and 1% methanol plus any required amino acid) starting an OD600 of 0.2 for 15–16 hours and shifted to SD for 1 hour, then images were acquired.
Other methods
Cloning, gene deletion and yeast transformation were performed using standard methods.
Human Autophagy Ontology (hAtgO)
Datasets utilized are listed in Table S3. All quantitative interaction/co-expression data were linearly transformed into 8 bit integers prior to data integration. Ontology constructed using available Active Interaction Mapping Jupyter notebook downloadable at atgo.ucsd.edu/download.html
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical tests performed using R and python. Significance of pearson correlation calculated using pearsonr function in python’s scipy package. Statistical GO gene set enrichments in the main text calculated using the hypergeometric test in R. For Figure 7H and I, statistical comparison of processed GFP for GFP-Ape4 and GFP-GAG Ty processing assay were calculated by comparing 3 replicates of each strain to 3 replicates of wild-type using the one sided t-test in R.
DATA AND SOFTWARE AVAILABILITY
Genetic interaction S-scores as measured for 52 autophagy-related query genes and 3007 array genes under 3 static and 3 differential conditions: Table S1.
Active Interaction Mapping (AIM) software and data necessary to perform AIMin S. cerevisiae and H. Sapiens are available for download as a Jupyter notebook at atgo.ucsd.edu/download.html. The AtgO 2.0 model is also available for browsing and exploration at atgo.ucsd.edu.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health to TI (R01-ES014811, R01-GM084279, P50-GM085764), to SS (R01-DK41737 and a Chancellor’s Associates Chair) and to JMC (U41-HG001315, U41-HG002273). Additional funding in support of trainees was provided to MHK (F30-HG007618, T32-GM007198) and MY (T32-GM008806). We wish to thank Dr. Claudine Kraft, Dr. Maho Niwa, Dr. Yoshinori Ohsumi, Dr. Scott Emr and Dr. Takahiro Shintani for providing reagents. We acknowledge Zlatan Hodzic for technical work. The authors declare no competing financial interests related to this work.
Footnotes
Author Contributions
MHK, MKY and TI designed the study and developed conceptual ideas. MHK and MF constructed AtgO. KM, JCF and KL performed the genetic interaction screens. MHK and JCF curated AtgO 2.0 term names. RB and JMC curated new Gene Ontology terms and annotations. KO and BD created the atgo.ucsd.edu web portal. JCF, MK and SS designed and performed biochemical experiments. MHK, JCF, SS and TI wrote the manuscript with input from other authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ano Y, Hattori T, Oku M, Mukaiyama H, Baba M, Ohsumi Y, Kato N, Sakai Y. A sorting nexin PpAtg24 regulates vacuolar membrane dynamics during pexophagy via binding to phosphatidylinositol-3-phosphate. Molecular biology of the cell. 2005;16:446–457. doi: 10.1091/mbc.E04-09-0842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atias N, Gershenzon M, Labazin K, Sharan R. Experimental design schemes for learning Boolean network models. Bioinformatics. 2014;30:i445–452. doi: 10.1093/bioinformatics/btu451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacon RA, Salminen A, Ruohola H, Novick P, Ferro-Novick S. The GTP-binding protein Ypt1 is required for transport in vitro: the Golgi apparatus is defective in ypt1 mutants. The Journal of cell biology. 1989;109:1015–1022. doi: 10.1083/jcb.109.3.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay S, Mehta M, Kuo D, Sung MK, Chuang R, Jaehnig EJ, Bodenmiller B, Licon K, Copeland W, Shales M, et al. Rewiring of genetic networks in response to DNA damage. Science. 2010;330:1385–1389. doi: 10.1126/science.1195618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett CL, Palsson BO. Iterative reconstruction of transcriptional regulatory networks: an algorithmic approach. PLoS computational biology. 2006;2:e52. doi: 10.1371/journal.pcbi.0020052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, et al. NCBI GEO: archive for functional genomics data sets--update. Nucleic acids research. 2013;41:D991–995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean GJ, Jaeger PA, Bahr S, Ideker T. Development of ultra-high-density screening tools for microbial “omics”. PloS one. 2014;9:e85177. doi: 10.1371/journal.pone.0085177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bicknell AA, Tourtellotte J, Niwa M. Late phase of the endoplasmic reticulum stress response pathway is regulated by Hog1 MAP kinase. The Journal of biological chemistry. 2010;285:17545–17555. doi: 10.1074/jbc.M109.084681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L. Random forests. Mach Learn. 2001;45:5–32. [Google Scholar]
- Budovskaya YV, Stephan JS, Reggiori F, Klionsky DJ, Herman PK. The Ras/cAMP-dependent protein kinase signaling pathway regulates an early step of the autophagy process in Saccharomyces cerevisiae. The Journal of biological chemistry. 2004;279:20663–20671. doi: 10.1074/jbc.M400272200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahan P, Li H, Morris SA, Lummertz da Rocha E, Daley GQ, Collins JJ. CellNet: network biology applied to stem cell engineering. Cell. 2014;158:903–915. doi: 10.1016/j.cell.2014.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Klionsky DJ. Atg26 is not involved in autophagy-related pathways in Saccharomyces cerevisiae. Autophagy. 2007;3:17–20. doi: 10.4161/auto.3371. [DOI] [PubMed] [Google Scholar]
- Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, Sulman EP, Anne SL, Doetsch F, Colman H, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463:318–325. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong H, Nair U, Geng J, Klionsky DJ. The Atg1 kinase complex is involved in the regulation of protein recruitment to initiate sequestering vesicle formation for nonspecific autophagy in Saccharomyces cerevisiae. Molecular biology of the cell. 2008;19:668–681. doi: 10.1091/mbc.E07-08-0826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SR, Schuldiner M, Krogan NJ, Weissman JS. A strategy for extracting and analyzing large-scale quantitative epistatic interaction data. Genome biology. 2006;7:R63. doi: 10.1186/gb-2006-7-7-r63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covert MW, Knight EM, Reed JL, Herrgard MJ, Palsson BO. Integrating high-throughput and computational data elucidates bacterial networks. Nature. 2004;429:92–96. doi: 10.1038/nature02456. [DOI] [PubMed] [Google Scholar]
- Creixell P, Schoof EM, Simpson CD, Longden J, Miller CJ, Lou HJ, Perryman L, Cox TR, Zivanovic N, Palmeri A, et al. Kinome-wide decoding of network-attacking mutations rewiring cancer signaling. Cell. 2015;163:202–217. doi: 10.1016/j.cell.2015.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson EH, Rast JP, Oliveri P, Ransick A, Calestani C, Yuh CH, Minokawa T, Amore G, Hinman V, Arenas-Mena C, et al. A genomic regulatory network for development. Science. 2002;295:1669–1678. doi: 10.1126/science.1069883. [DOI] [PubMed] [Google Scholar]
- Dawaliby R, Mayer A. Microautophagy of the nucleus coincides with a vacuolar diffusion barrier at nuclear-vacuolar junctions. Molecular biology of the cell. 2010;21:4173–4183. doi: 10.1091/mbc.E09-09-0782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du LL, Novick P. Yeast rab GTPase-activating protein Gyp1p localizes to the Golgi apparatus and is a negative regulator of Ypt1p. Molecular biology of the cell. 2001;12:1215–1226. doi: 10.1091/mbc.12.5.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutkowski J, Kramer M, Surma MA, Balakrishnan R, Cherry JM, Krogan NJ, Ideker T. A gene ontology inferred from molecular networks. Nature biotechnology. 2013;31:38–45. doi: 10.1038/nbt.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ergun A, Lawrence CA, Kohanski MA, Brennan TA, Collins JJ. A network biology approach to prostate cancer. Molecular systems biology. 2007;3:82. doi: 10.1038/msb4100125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham LA, Hill KJ, Stevens TH. Assembly of the yeast vacuolar H+-ATPase occurs in the endoplasmic reticulum and requires a Vma12p/Vma22p assembly complex. The Journal of cell biology. 1998;142:39–49. doi: 10.1083/jcb.142.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene CS, Krishnan A, Wong AK, Ricciotti E, Zelaya RA, Himmelstein DS, Zhang R, Hartmann BM, Zaslavsky E, Sealfon SC, et al. Understanding multicellular function and disease with human tissue-specific networks. Nature genetics. 2015;47:569–576. doi: 10.1038/ng.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havugimana PC, Hart GT, Nepusz T, Yang H, Turinsky AL, Li Z, Wang PI, Boutz DR, Fong V, Phanse S, et al. A census of human soluble protein complexes. Cell. 2012;150:1068–1081. doi: 10.1016/j.cell.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams SL, Millar A, Taylor P, Bennett K, Boutilier K, et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- Ideker T, Galitski T, Hood L. A new approach to decoding life: systems biology. Annual review of genomics and human genetics. 2001a;2:343–372. doi: 10.1146/annurev.genom.2.1.343. [DOI] [PubMed] [Google Scholar]
- Ideker T, Krogan NJ. Differential network biology. Molecular systems biology. 2012;8:565. doi: 10.1038/msb.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideker T, Thorsson V, Ranish JA, Christmas R, Buhler J, Eng JK, Bumgarner R, Goodlett DR, Aebersold R, Hood L. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science. 2001b;292:929–934. doi: 10.1126/science.292.5518.929. [DOI] [PubMed] [Google Scholar]
- Ideker TE, Thorsson V, Karp RM. Discovery of regulatory interactions through perturbation: inference and experimental design. Pacific Symposium on Biocomputing Pacific Symposium on Biocomputing. 2000:305–316. doi: 10.1142/9789814447331_0029. [DOI] [PubMed] [Google Scholar]
- Jean S, Kiger AA. Coordination between RAB GTPase and phosphoinositide regulation and functions. Nature reviews Molecular cell biology. 2012;13:463–470. doi: 10.1038/nrm3379. [DOI] [PubMed] [Google Scholar]
- Jin M, Klionsky DJ. The Core Molecular Machinery of Autophagosome Formation. In: Wang HG, editor. Autophagy and cancer. New York: Springer; 2013. pp. 25–45. [Google Scholar]
- Kaiser SE, Mao K, Taherbhoy AM, Yu S, Olszewski JL, Duda DM, Kurinov I, Deng A, Fenn TD, Klionsky DJ, et al. Noncanonical E2 recruitment by the autophagy E1 revealed by Atg7-Atg3 and Atg7-Atg10 structures. Nature structural & molecular biology. 2012;19:1242–1249. doi: 10.1038/nsmb.2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki T, Klionsky DJ. Mitophagy in yeast occurs through a selective mechanism. The Journal of biological chemistry. 2008;283:32386–32393. doi: 10.1074/jbc.M802403200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki T, Wang K, Baba M, Bartholomew CR, Lynch-Day MA, Du Z, Geng J, Mao K, Yang Z, Yen WL, et al. A genomic screen for yeast mutants defective in selective mitochondria autophagy. Molecular biology of the cell. 2009;20:4730–4738. doi: 10.1091/mbc.E09-03-0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karr JR, Sanghvi JC, Macklin DN, Gutschow MV, Jacobs JM, Bolival B, Jr, Assad-Garcia N, Glass JI, Covert MW. A whole-cell computational model predicts phenotype from genotype. Cell. 2012;150:389–401. doi: 10.1016/j.cell.2012.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Shin J, Kim E, Kim H, Hwang S, Shim JE, Lee I. YeastNet v3: a public database of data-specific and integrated functional gene networks for Saccharomyces cerevisiae. Nucleic acids research. 2014;42:D731–736. doi: 10.1093/nar/gkt981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YA, Wuchty S, Przytycka TM. Identifying causal genes and dysregulated pathways in complex diseases. PLoS computational biology. 2011;7:e1001095. doi: 10.1371/journal.pcbi.1001095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RD, Rowland J, Oliver SG, Young M, Aubrey W, Byrne E, Liakata M, Markham M, Pir P, Soldatova LN, et al. The automation of science. Science. 2009;324:85–89. doi: 10.1126/science.1165620. [DOI] [PubMed] [Google Scholar]
- King RD, Whelan KE, Jones FM, Reiser PG, Bryant CH, Muggleton SH, Kell DB, Oliver SG. Functional genomic hypothesis generation and experimentation by a robot scientist. Nature. 2004;427:247–252. doi: 10.1038/nature02236. [DOI] [PubMed] [Google Scholar]
- Kitano H. Systems biology: a brief overview. Science. 2002;295:1662–1664. doi: 10.1126/science.1069492. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Cueva R, Yaver DS. Aminopeptidase I of Saccharomyces cerevisiae is localized to the vacuole independent of the secretory pathway. The Journal of cell biology. 1992;119:287–299. doi: 10.1083/jcb.119.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft C, Deplazes A, Sohrmann M, Peter M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nature cell biology. 2008;10:602–610. doi: 10.1038/ncb1723. [DOI] [PubMed] [Google Scholar]
- Kramer M, Dutkowski J, Yu M, Bafna V, Ideker T. Inferring gene ontologies from pairwise similarity data. Bioinformatics. 2014;30:i34–42. doi: 10.1093/bioinformatics/btu282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvam E, Goldfarb DS. Nvj1p is the outer-nuclear-membrane receptor for oxysterol-binding protein homolog Osh1p in Saccharomyces cerevisiae. Journal of cell science. 2004;117:4959–4968. doi: 10.1242/jcs.01372. [DOI] [PubMed] [Google Scholar]
- Lefebvre C, Rieckhof G, Califano A. Reverse-engineering human regulatory networks. Wiley interdisciplinary reviews Systems biology and medicine. 2012;4:311–325. doi: 10.1002/wsbm.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiserson MD, Vandin F, Wu HT, Dobson JR, Eldridge JV, Thomas JL, Papoutsaki A, Kim Y, Niu B, McLellan M, et al. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nature genetics. 2015;47:106–114. doi: 10.1038/ng.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Yuan J. Autophagy in cell death: an innocent convict? The Journal of clinical investigation. 2005;115:2679–2688. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- Lin M, Unden H, Jacquier N, Schneiter R, Just U, Hofken T. The Cdc42 effectors Ste20, Cla4, and Skm1 down-regulate the expression of genes involved in sterol uptake by a mitogen-activated protein kinase-independent pathway. Molecular biology of the cell. 2009;20:4826–4837. doi: 10.1091/mbc.E09-01-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipatova Z, Belogortseva N, Zhang XQ, Kim J, Taussig D, Segev N. Regulation of selective autophagy onset by a Ypt/Rab GTPase module. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:6981–6986. doi: 10.1073/pnas.1121299109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malleshaiah MK, Shahrezaei V, Swain PS, Michnick SW. The scaffold protein Ste5 directly controls a switch-like mating decision in yeast. Nature. 2010;465:101–105. doi: 10.1038/nature08946. [DOI] [PubMed] [Google Scholar]
- Manjithaya R, Jain S, Farre JC, Subramani S. A yeast MAPK cascade regulates pexophagy but not other autophagy pathways. The Journal of cell biology. 2010;189:303–310. doi: 10.1083/jcb.200909154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao K, Liu X, Feng Y, Klionsky DJ. The progression of peroxisomal degradation through autophagy requires peroxisomal division. Autophagy. 2014;10:652–661. doi: 10.4161/auto.27852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarko TY, Farre JC, Subramani S. Peroxisome size provides insights into the function of autophagy-related proteins. Molecular biology of the cell. 2009;20:3828–3839. doi: 10.1091/mbc.E09-03-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebauer R, Rosenberger S, Daum G. Phosphatidylethanolamine, a limiting factor of autophagy in yeast strains bearing a defect in the carboxypeptidase Y pathway of vacuolar targeting. The Journal of biological chemistry. 2007;282:16736–16743. doi: 10.1074/jbc.M611345200. [DOI] [PubMed] [Google Scholar]
- Noda NN, Ohsumi Y, Inagaki F. Atg8-family interacting motif crucial for selective autophagy. FEBS letters. 2010;584:1379–1385. doi: 10.1016/j.febslet.2010.01.018. [DOI] [PubMed] [Google Scholar]
- Noda T, Klionsky DJ. The quantitative Pho8Delta60 assay of nonspecific autophagy. Methods in enzymology. 2008;451:33–42. doi: 10.1016/S0076-6879(08)03203-5. [DOI] [PubMed] [Google Scholar]
- Noda T, Matsuura A, Wada Y, Ohsumi Y. Novel system for monitoring autophagy in the yeast Saccharomyces cerevisiae. Biochemical and biophysical research communications. 1995;210:126–132. doi: 10.1006/bbrc.1995.1636. [DOI] [PubMed] [Google Scholar]
- Oku M, Sakai Y. Pexophagy in Pichia pastoris. Methods in enzymology. 2008;451:217–228. doi: 10.1016/S0076-6879(08)03215-1. [DOI] [PubMed] [Google Scholar]
- Oku M, Warnecke D, Noda T, Muller F, Heinz E, Mukaiyama H, Kato N, Sakai Y. Peroxisome degradation requires catalytically active sterol glucosyltransferase with a GRAM domain. The EMBO journal. 2003;22:3231–3241. doi: 10.1093/emboj/cdg331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papinski D, Schuschnig M, Reiter W, Wilhelm L, Barnes CA, Maiolica A, Hansmann I, Pfaffenwimmer T, Kijanska M, Stoffel I, et al. Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Molecular cell. 2014;53:471–483. doi: 10.1016/j.molcel.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. Scikit-learn: Machine Learning in Python. J Mach Learn Res. 2011;12:2825–2830. [Google Scholar]
- Pfaffenwimmer T, Reiter W, Brach T, Nogellova V, Papinski D, Schuschnig M, Abert C, Ammerer G, Martens S, Kraft C. Hrr25 kinase promotes selective autophagy by phosphorylating the cargo receptor Atg19. EMBO reports. 2014;15:862–870. doi: 10.15252/embr.201438932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picotti P, Clement-Ziza M, Lam H, Campbell DS, Schmidt A, Deutsch EW, Rost H, Sun Z, Rinner O, Reiter L, et al. A complete mass-spectrometric map of the yeast proteome applied to quantitative trait analysis. Nature. 2013;494:266–270. doi: 10.1038/nature11835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson E, Consortium GT, Koller D, Battle A, Mostafavi S, Ardlie KG, Getz G, Wright FA, Kellis M, Volpi S, et al. Sharing and Specificity of Co-expression Networks across 35 Human Tissues. PLoS computational biology. 2015;11:e1004220. doi: 10.1371/journal.pcbi.1004220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prick T, Thumm M, Haussinger D, Vom Dahl S. Deletion of HOG1 leads to Osmosensitivity in starvation-induced, but not rapamycin-dependent Atg8 degradation and proteolysis: further evidence for different regulatory mechanisms in yeast autophagy. Autophagy. 2006;2:241–243. doi: 10.4161/auto.2743. [DOI] [PubMed] [Google Scholar]
- Reggiori F, Komatsu M, Finley K, Simonsen A. Autophagy: more than a nonselective pathway. International journal of cell biology. 2012;2012:219625. doi: 10.1155/2012/219625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reggiori F, Wang CW, Stromhaug PE, Shintani T, Klionsky DJ. Vps51 is part of the yeast Vps fifty-three tethering complex essential for retrograde traffic from the early endosome and Cvt vesicle completion. The Journal of biological chemistry. 2003;278:5009–5020. doi: 10.1074/jbc.M210436200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnik P. Using information content to evaluate semantic similarity in a taxonomy. Int Joint Conf Artif. 1995:448–453. [Google Scholar]
- Rivera-Molina FE, Novick PJ. A Rab GAP cascade defines the boundary between two Rab GTPases on the secretory pathway. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:14408–14413. doi: 10.1073/pnas.0906536106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodkaer SV, Faergeman NJ. Glucose- and nitrogen sensing and regulatory mechanisms in Saccharomyces cerevisiae. FEMS yeast research. 2014;14:683–696. doi: 10.1111/1567-1364.12157. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, DiFiglia M, Heintz N, Nixon RA, Qin ZH, Ravikumar B, Stefanis L, Tolkovsky A. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy. 2005;1:11–22. doi: 10.4161/auto.1.1.1513. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- Rui YN, Xu Z, Patel B, Chen Z, Chen D, Tito A, David G, Sun Y, Stimming EF, Bellen HJ, et al. Huntingtin functions as a scaffold for selective macroautophagy. Nature cell biology. 2015;17:262–275. doi: 10.1038/ncb3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusten TE, Stenmark H. How do ESCRT proteins control autophagy? Journal of cell science. 2009;122:2179–2183. doi: 10.1242/jcs.050021. [DOI] [PubMed] [Google Scholar]
- Schuldiner M, Collins SR, Weissman JS, Krogan NJ. Quantitative genetic analysis in Saccharomyces cerevisiae using epistatic miniarray profiles (E-MAPs) and its application to chromatin functions. Methods. 2006;40:344–352. doi: 10.1016/j.ymeth.2006.07.034. [DOI] [PubMed] [Google Scholar]
- Shin ME, Ogburn KD, Varban OA, Gilbert PM, Burd CG. FYVE domain targets Pib1p ubiquitin ligase to endosome and vacuolar membranes. The Journal of biological chemistry. 2001;276:41388–41393. doi: 10.1074/jbc.M105665200. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Morimoto M, Kondo C, Ohsumi Y. Selective autophagy regulates insertional mutagenesis by the Ty1 retrotransposon in Saccharomyces cerevisiae. Developmental cell. 2011;21:358–365. doi: 10.1016/j.devcel.2011.06.023. [DOI] [PubMed] [Google Scholar]
- Tong AH, Boone C. Synthetic genetic array analysis in Saccharomyces cerevisiae. Methods in molecular biology. 2006;313:171–192. doi: 10.1385/1-59259-958-3:171. [DOI] [PubMed] [Google Scholar]
- Tong AH, Lesage G, Bader GD, Ding H, Xu H, Xin X, Young J, Berriz GF, Brost RL, Chang M, et al. Global mapping of the yeast genetic interaction network. Science. 2004;303:808–813. doi: 10.1126/science.1091317. [DOI] [PubMed] [Google Scholar]
- Wang J, Davis S, Menon S, Zhang J, Ding J, Cervantes S, Miller E, Jiang Y, Ferro-Novick S. Ypt1/Rab1 regulates Hrr25/CK1delta kinase activity in ER-Golgi traffic and macroautophagy. The Journal of cell biology. 2015;210:273–285. doi: 10.1083/jcb.201408075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Yang Z, Liu X, Mao K, Nair U, Klionsky DJ. Phosphatidylinositol 4-kinases are required for autophagic membrane trafficking. The Journal of biological chemistry. 2012;287:37964–37972. doi: 10.1074/jbc.M112.371591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Wilson WA, Fujino MA, Roach PJ. Antagonistic controls of autophagy and glycogen accumulation by Snf1p, the yeast homolog of AMP-activated protein kinase, and the cyclin-dependent kinase Pho85p. Molecular and cellular biology. 2001;21:5742–5752. doi: 10.1128/MCB.21.17.5742-5752.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic acids research. 2010;38:W214–220. doi: 10.1093/nar/gkq537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Geng J, Yen WL, Wang K, Klionsky DJ. Positive or negative roles of different cyclin-dependent kinase Pho85-cyclin complexes orchestrate induction of autophagy in Saccharomyces cerevisiae. Molecular cell. 2010;38:250–264. doi: 10.1016/j.molcel.2010.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen WL, Legakis JE, Nair U, Klionsky DJ. Atg27 is required for autophagy-dependent cycling of Atg9. Molecular biology of the cell. 2007;18:581–593. doi: 10.1091/mbc.E06-07-0612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorimitsu T, Klionsky DJ. Atg11 links cargo to the vesicle-forming machinery in the cytoplasm to vacuole targeting pathway. Molecular biology of the cell. 2005;16:1593–1605. doi: 10.1091/mbc.E04-11-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuga M, Gomi K, Klionsky DJ, Shintani T. Aspartyl aminopeptidase is imported from the cytoplasm to the vacuole by selective autophagy in Saccharomyces cerevisiae. The Journal of biological chemistry. 2011;286:13704–13713. doi: 10.1074/jbc.M110.173906. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.