

Abstract
Aims
CD1d-restricted natural killer T (NKT) cells function by regulating numerous immune responses during innate and adaptive immunity. Depletion of all populations of CD1d-dependent NKT cells has been shown by several groups to reduce atherosclerosis in two different mouse models of the disease. In this study, we determined if removal of a single (Vα14) NKT cell population protects mice from the disease.
Methods and results
Targeted deletion of the Jα18 gene results in selective depletion of CD1d-dependent Vα14 NKT cells in C57BL/6 mice without affecting the population of other NKT, NK, and conventional T cells. Therefore, to study the effect of Vα14 NKT cell depletion on the progression of atherosclerosis, we examined the extent of lesion formation using paired littermate LDL receptor null mice that were either +/+ or −/− for the Jα18 gene following the feeding of these mice a cholesterol- and fat-enriched diet for 8 weeks. At the end of the study, we found no difference in either serum total- or lipoprotein-cholesterol distributions between groups. However, quantification of atherosclerosis revealed that Vα14 NKT cell deficiency significantly decreased lesion size in the aortic root (20–28%) and arch (28–38%) in both genders of mice. By coupling the techniques of laser capture microdissection with quantitative real-time RT–PCR, we found that expression of the proatherogenic cytokine interferon (IFN)-γ was significantly reduced in lesions from Jα18−/− mice.
Conclusion
This study is the first to identify a specific subpopulation of NKT cells that promotes atherosclerosis via a mechanism appearing to involve IFN-γ expression.
Keywords: Atherosclerosis, Cytokines, Histo(patho)logy, Immunology, Leukocytes
1. Introduction
Atherosclerosis is an inflammatory vascular disease that is now known to involve components of both the innate and acquired immune systems.1–4 Studies aimed at defining the role of distinct populations of lymphocytes in the development of atherosclerosis have indicated that natural killer (NK) cells and T helper 1 (Th1) cells are proatherogenic,5–9 whereas Th2 cells and B cells are antiatherogenic.7,8,10,11 These findings are further supported by research which has found that cytokines released from NK cells and Th1 cells [e.g. interferon (IFN)-γ and interleukin (IL)-6] are proatherogenic,9,12–16 and that among cytokines released by Th2 cell, IL-10 is antiatherogenic.17–21
Lipid antigens are presented to unique groups of T cells as part of a complex with the non-classical, non-polymorphic MHC class Ib molecule CD1d displayed on certain antigen presenting cells such as dendritic cells and monocyte-derived macrophages.22 The ability of one unique subset of T cells to interact with CD1d results from the expression on these cells of a highly biased, evolutionarily conserved, T cell receptor (TCR) consisting of an invariant α-chain (a Vα14 segment joined to a Jα18 segment) that pairs preferentially with one of three β-chains (Vβ8.2, Vβ-7, and Vβ-2).23,24 This unique subset of T cells also expresses the classical NK cell marker NK1.1 (CD161) and, as such, these T cells are known as invariant NKT cells25 or Vα14 NKT cells.26
Upon stimulation, all NKT cell populations have the capacity to exert immunoregulatory functions by releasing large amounts of inflammatory cytokines, including the proatherogenic cytokine IFN-γ.9,12–14 This release of cytokines will in turn cause the activation of adjacent NK cells, B cells, conventional CD4+ and CD8+ T cells,24,27 as well as adjacent antigen presenting cells.28 Although the natural ligand(s) for CD1d-restricted NKT cells (including Vα14 NKT cells) still remain to be characterized, NKT cells have a strong response to and are selectively activated by the exogenous synthetic glycolipid α-galactosylceramide (α-GalCer), which binds specifically to CD1d.29 A number of recent studies have shown that α-GalCer specifically enhances atherosclerosis in apolipoprotein E null (Apoe−/−) mice30–32 and has a significant impact on the cytokine profile (including, but not limited to, IFN-γ) of these mice. Interestingly, not only have NKT cells been shown to be present in the atherosclerotic lesions of Apoe−/−31 and LDL receptor null (Ldlr−/−)33 mice but CD1d has also been detected in human atherosclerotic lesions,34 underscoring the probable involvement of NKT cells in the disease process. These findings, and the capacity of NKT cells to produce IFN-γ, an established proatherogenic cytokine,9,14 suggest that CD1d-restricted NKT cells might play a participatory role in the atherogenic process. In this current study, we use Ldlr−/− mice to further examine the potential proatherogenic role of one unique population of NKT cells that recognizes CD1d; these cells are the Vα14 NKT cells and are reported to represent the largest population of CD1d-restricted NKT cells.35
2. Methods
2.1. Animals and diet
Male Jα18−/− (formally called Jα281−/−) mice were a generous gift from Drs Malcolm S. Duthie and Stuart J. Kahn, Infectious Disease Research Institute, Seattle, WA, USA. Jα18−/− mice were found to reproduce normally and otherwise are healthy in appearance. As cited by Cui et al.,36 the development of the lymphoid organs in Jα18−/− mice is macroscopically normal, and the numbers of total lymphocytes are not significantly different from Jα18+/+ mice with the exception of a complete loss of the Vα14 NKT subpopulation of NKT cells. Male Jα18−/− mice were bred with female Ldlr−/− mice originally obtained from The Jackson Laboratory, USA, and maintained in the Animal Care Facility at the University of Ottawa Heart Institute. Both strains of mice have been backcrossed for more than 10 generations to the C57BL/6 background. F1 heterozygotes were mated with Ldlr−/− mice to obtain an F2 generation of breeding mice that are Jα18+/− × Ldlr−/−. Genetic screening for the LDL receptor and Jα18 genes was carried out by PCR on DNA from mouse-tail samples (see Supplementary material online).
Ldlr −/− mice that were either Jα18+/+ or −/− were fed a standard laboratory mouse diet supplemented with 21% (wt/wt) butterfat and 0.15% (wt/wt) cholesterol, 19.5% (wt/wt) casein and no sodium cholate (#112286, Dyets Inc., Bethlehem, PA, USA) for 8 weeks. Ldlr−/− mice fed this particular diet for 8 weeks have previously been shown to develop early-stage atherosclerotic lesions.37 Both genders of mice were used in this study, and all mice started the atherogenic diet just after being weaned at 4 weeks of age. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
2.2. Blood collection
Terminal blood samples were collected by puncture of the right ventricle. Blood was allowed to clot at room temperature for 30 min and then centrifuged at 1000 g for 25 min at 4°C.
2.3. Plasma cholesterol and lipoprotein profiles
See legend to Figure 1, and Supplementary material online.
Figure 1.
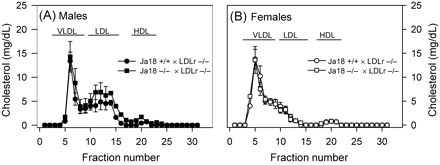
Serum (60 µL) from male (A) and female (B) Ldlr−/− mice that were either Jα18+/+ (circular symbol) or Jα18−/− (square symbol) was resolved by size exclusion chromatography using a Superose 6 column. Total cholesterol concentrations were determined in fraction numbers 11–40, with each fraction having a total volume of 500 µL. Symbols represent the means and bars the SEM of values obtained from the serum of 5 mice/curve.
2.4. Tissue collection
Mice were perfused with PBS via a cannula placed in the left ventricle, with perfusate drained from a severed right atrium. Hearts were separated from the aorta at the base, embedded in optimum cutting temperature medium, and snap-frozen on a metal plate that was cooled with liquid nitrogen.
2.5. Quantification of atherosclerotic lesions in tissue sections
The size of atherosclerotic lesions in the ascending aorta was determined from four Sudan IV stained serial sections, cut 10 µm thick and separated by 100 µm. Lesion analysis began with the first section of tissue that contained the ostia for the coronary arteries; region defining the boundary between the aortic sinus and ascending aorta. Using the Sudan IV staining as a guide, lesion area defined as intimal tissue within the internal elastic lamina was determined using Image-Pro Plus software (V6.2, Media Cybernetics, Silver Springs, MD, USA) on images that were created using a digital CoolSNAP cf camera (Roper Scientific Inc., Duluth, GA, USA). The mean lesion area derived from the four serial sections was taken as the average lesion size for each animal as described previously.5,13,14,38,39
2.6. Quantification of atherosclerotic lesions in the aortic arch
The percentage of atherosclerotic lesion area covering the aortic arch in an en face preparation of the vessel was quantified as described previously.14,40,41 In this process, no lipophylic dye was used to aid in the visualization of discernable lesions.
2.7. Histological staining
It was performed as described previously,5,14 using sequential sections of the ascending aorta to detect neutral lipid, macrophages, CD3+ T cells, MHC class II, IFN-γ, and extracellular collagen (for more detail, see Supplementary material online). Since immunostaining for CD3 and MHC class II leads to discrete staining of definable cells, lesion-associated cells expressing either antigen were counted and the mean number of cells reported as described previously.5,13,14,38
2.8. Quantification of the per cent lesion-associated lipid within macrophages
The lesion area staining positive for macrophages and for neutral lipid was quantified using Image-Pro Plus. An imprint was then made for each area and superimposed using Adobe Photoshop CS2 (Version 9.0.2). Both imprints are still distinguishable at this point, only the overlapping areas appeared darker. The superimposed image is then imported to Image-Pro Plus to measure the amount of overlap and the percentage of lipid associated within macrophages is calculated by dividing the overlapping area with the total area of lesion-associated lipid. It is important to note that each section used for the overlay of lesion-associated lipid and macrophage areas was only separated by 10 µm.
2.9. Laser capture microdissection and RNA extraction
Using laser capture microdissection (LCM), we were able to extract the atherosclerotic lesions from our control and experimental mice. The PixCell IIe LCM system was set at the following parameters: 30 µm laser spot size, 50 mW power, and 10 ms duration. Total RNA was extracted from dissected tissue using the Qiagen RNeasy Micro Kit (Qiagen) as per manufacturer's instructions.
2.10. Quantitative real-time RT–PCR
Real-time RT–PCR was used to quantify transcription levels of IL-4, IL-10, IFN-γ, and β-actin in total RNA isolated from lesion tissue collected by LCM. A standard curve of each cytokine mRNA was constructed using serial dilutions of stock mouse IFN-γ, IL-4, IL-10, and β-actin cDNA. The real-time RT–PCRs were performed using the reaction conditions, nested TaqMan probes, and a forward and reverse set of cytokine-specific PCR primers as described by Giulietti et al.,42 for IFN-γ, IL-4, and IL-10 and as provided by Qiagen Life Sciences for β-actin (QuantiTect Mm β-actin Assay). Resulting values were normalized to the β-actin values.
2.11. Cytokine production assay
Splenocytes (2 × 105) isolated from Ldlr−/− mice that were either Jα18+/+ vs. −/− were incubated for 72 h in the presence or absence of 50 ng/mL of α-GalCer (Cedarlane Laboratories, Burlington, ON, Canada) in RPMI 1640 medium supplemented with 10% FCS, 50 µM 2-mercaptoethanol, 2 mM glutamine, and antibiotics. Levels of IFN-γ and IL-4 in the supernatant were determined by ELISA (R&D Systems).
2.12. Statistics
Data analysis was performed using SigmaStat 2.03 software (SPSS Inc., Chicago, IL, USA). For each parameter, the mean and standard error of mean (SEM) were calculated. Statistical analysis between groups was by a One Way Analysis of Variance on Ranks with all pairwise multiple comparison procedures performed using the Dunn Method, after testing that the data complied with the constraints of parametric analysis. Values with P ≤ 0.05 were considered statistically significant.
3. Results
Compared with Jα18+/+ × Ldlr−/− mice, deficiency of Vα14 NKT cells in Ldlr−/− mice did not affect total serum-cholesterol concentrations (Table 1) or -cholesterol distribution between various fractions of lipoproteins (Figure 1).
Table 1.
Serum cholesterol values, quantification of lesion-associated cells expressing CD3, MHC class II, and lesion-associated IFN-γ, IL-4, and IL-10 mRNA concentrations
| Gender | Jα18 genotype of Ldlr−/− mice | Lesion-associated CD3 positively stained cells (n) | Lesion-associated MHC class II positively stained cells (n) | Final serum cholesterol values (n) | IL-4/β-actin mRNA ratio (n) ×10−3 | IL-10/β-actin mRNA ratio (n) ×10−3 | IFN-γ/β-actin mRNA ratio (n) ×10−3 |
|---|---|---|---|---|---|---|---|
| Male | +/+ | 13.3 ± 1.6 (8) | 16.1 ± 1.8 (8) | 1748 ± 169 (9) | ND (6) | ND (6) | 15.1 ± 1.4 (6) |
| −/− | 15.8 ± 2.0 (8) | 12.4 ± 1.3 (8) | 2202 ± 204 (9) | ND (7) | ND (7) | 2.0 ± 1.3* (7) | |
| Female | +/+ | 9.0 ± 1.3 (9) | 15.6 ± 2.2 (9) | 1742 ± 60 (9) | ND (5) | ND (5) | 28.3 ± 1.6 (5) |
| −/− | 10.0 ± 2.0 (11) | 16.5 ± 2.3 (11) | 1981 ± 112 (11) | ND (8) | ND (8) | 2.0 ± 0.61† (8) |
ND, not detected.
*P = 0.05 vs. Jα18 +/+ males.
† P = 0.05 vs. Jα18 +/+ females.
When compared with Jα18+/+ mice, loss of Vα14 NKT cells decreased lesion size by ∼20 and 28% within the ascending aorta of both male (0.103 ± 0.009 mm2 vs. 0.078 ± 0.007 mm2, respectively, n = 9 per group; P = 0.021) and female (0.110 ± 0.008 mm2 (n = 9) vs. 0.087 ± 0.006 mm2 (n = 11), respectively; P = 0.026) mice (Figure 2A), and by 28 and 37% in the aortic arch of both males (5.9 ± 0.5% vs. 4.2 ± 0.6%, respectively, n = 9 per group; P = 0.037;) and females (7.1 ± 1.0% (n = 11) vs. 4.4 ± 0.6% (n = 9), respectively; P = 0.038) mice (Figure 2B). When lesions of the ascending aorta were measured for the area occupied by Sudan IV staining and the area occupied by macrophage staining, loss of Vα14 NKT cells resulted in a significant reduction in the size of lipid staining and macrophage staining of said lesions (Table 2).
Figure 2.
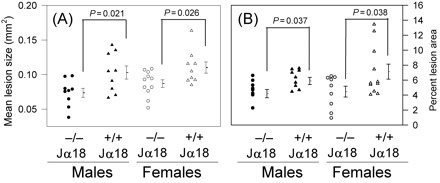
Figures show (A) mean lesion size and (B) per cent lesion area. The extent of atherosclerotic lesion development in the (A) ascending aorta and the (B) aortic arch of male and female Ldlr−/− mice either +/+ or −/− for the Jα18 gene was determined as described under Section 2. Values of individual mice are represented as circles (Jα18−/−) and triangles (Jα18+/+), whereas the mean lesion size of each group of mice is presented as a single horizontal line (to the right of each symbol grouping) with error bars denoting SEM. *P-values showing significant differences between groups are noted.
Table 2.
Lipid-positive stained area values, macrophage-positive stained area, and % overlap of lipid and macrophage stained areas
| Gender | Jα18 genotype of Ldlr−/− mice | Lipid-positive stained area (mean value mm2) (n) | Macrophage-positive stained area (mean value mm2) (n) | % overlap of lipid and macrophage areas (n) |
|---|---|---|---|---|
| Male | +/+ | 0.0997 ± 0.0104 (9) | 0.1028 ± 0.0127 (9) | 93.3 ± 3.2 (9) |
| −/− | 0.0722 ± 0.0045* (9) | 0.0750 ± 0.0048† (9) | 95.9 ± 1.2 (9) | |
| Female | +/+ | 0.0997 ± 0.0109 (9) | 0.1012 ± 0.0116 (9) | 93.4 ± 4.2 (9) |
| −/− | 0.0707 ± 0.0047** (11) | 0.0733 ± 0.0049‡ (11) | 96.2 ± 1.7 (11) |
*P = 0.0264 vs. Jα18 +/+ males.
**P = 0.0251 vs. Jα18 +/+ females.
† P = 0.0471 vs. Jα18 +/+ males.
‡ P = 0.0465 vs. Jα18 +/+ females.
As a marker of lesion-associated foam cell development, we have devised a computer-assisted way of calculating the percentage of lesion-associated lipid (Sudan IV positive) contained within the corresponding area that stained positive for macrophages. By performing this type of analysis, we discovered no significant difference in the percentage of neutral lipid associated within macrophages located in the ascending aortic lesions of both male and female Jα18+/+ vs. −/− mice (Table 2).
Detailed histological analysis indicated that all lesions from both male and female Jα18+/+ and −/− mice were at the early-stage of development, consisting mainly of macrophage-derived foam cells (Figure 3). Absent from these lesions was the presence of a definable necrotic core or fibrous cap, which would indicate the existence of complex atherosclerotic plaques.
Figure 3.
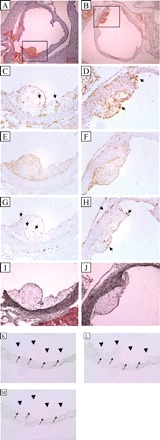
Representative histological sections from a region where the aortic sinus becomes the ascending aorta of a female (A, C, E, G, I) Jα18−/−× Ldlr−/− and a female (B, D, F, H, J) Jα18+/+ × Ldlr−/− mouse. (A, B) Sudan IV for neutral lipids, (C, D) monoclonal antimouse MHC class II, (E, F) rabbit antisera to mouse macrophages, (G, H) purified hamster antimouse CD3+E molecular complex antibody, and (I, J) Gomori trichrome to detect collagen. Magnification: (A, B) ×40; (C–M) ×200. (A, inset) and (B, inset) area contained in C, E, G, I, and D, F, H, J, respectively. Arrows indicate individual cells that are staining positive for: (C, D) MHC class II; and (G, H) CD3+. Control (negative) slides were performed on serial sections from the ascending aorta of an Ldlr−/− mouse (K–M): (K) Staining was performed in the absence of the biotinylated primary antibody against MHC class II. (L) Staining was performed in the absence of the primary antibody against macrophages and the presence of the biotinylated secondary antibody, goat anti-rat. (M) Staining was performed in the absence of the primary antibody against CD3 and the presence of the biotinylated secondary goat anti-hamster antibody. (K–M) Arrowheads are used to outline the luminal boundary of the atherosclerotic lesion and small arrows are used to outline the internal elastic lamina.
By coupling the techniques of LCM and quantitative real-time RT–PCR, we noted a significant reduction (85–93%) in the amount of IFN-γ mRNA expressed in atherosclerotic lesions extracted from serial sections of the aortic root of Jα18+/+ vs. −/− mice (Table 1). However, using the same technique, we could not detect either IL-4 or IL-10 (Table 1). Using Immunohistochemistry, we also detected a greater staining pattern for IFN-γ protein in the lesions of Jα18+/+ vs. −/− mice (Figure 4). Despite the noted decrease in lesion-associated IFN-γ expression (at both the mRNA and protein levels), the mean number of lesion-associated CD3+ T cells and the activation status of APC (those expressing MHC class II) within the lesions of Jα18+/+ vs. −/− mice were not significantly different (Table 1).
Figure 4.
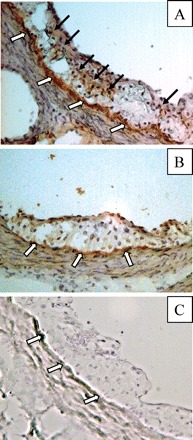
Representative immunohistological sections staining specifically for IFN-γ using atherosclerotic lesions of a female (A) Jα18+/+ × Ldlr−/− mouse and a female (B) Jα18−/− × Ldlr−/− mouse. (C) Representative control lesion, where staining was performed in the absence of the primary antibody against IFN-γ and the presence of the biotinylated secondary goat anti-rat antibody. Black arrows indicate areas of positive staining for IFN-γ. White arrows indicate areas of minor background staining which appear to be held to the internal elastic lamina.
Treatment of splenocytes isolated from Jα18+/+ × Ldlr−/− mice with α-GalCer caused a significant increase in the secretion of both IFN-γ and IL-4 by these cells (Figure 5). However, α-GalCer treatment of splenocytes from Jα18−/− mice did not cause a significant increase in the secretion of either IFN-γ or IL-4, and that the levels of both cytokines in the supernatant of splenocytes from Jα18−/− mice were not significantly different from control cells (spenocytes isolated from Jα18+/+ mice) not exposed to α-GalCer.
Figure 5.
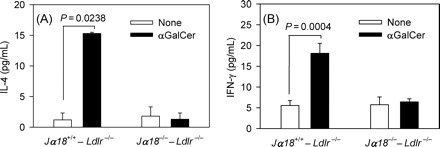
Spleen cells from Jα18+/+ × Ldlr−/− and Jα18−/− × Ldlr−/− mice were cultured in vitro in the presence or absence of α-GalCer (50 ng/mL). Three days later, IL-4 (A) and IFN-γ (B) levels were measured by ELISA.
4. Discussion
Invariant CD1d-restricted NKT cells function as a ‘bridge’ between innate and adaptive immunity.43 In this capacity, NKT cells have been shown to regulate numerous immune responses involving conditions such as autoimmune disease, tumour surveillance, and infection caused by microbial pathogens.44–49 Evidence gathered from the analysis of both human and mouse atherosclerotic lesions suggests the presence of NKT cells in this disease as well. First, nested RT–PCR has detected the invariant Vα14Jα18 TCR α-chain rearrangement in atherosclerotic lesions from Apoe−/− mice.31 Secondly, all four CD1 proteins (a, b, c, and d) have been detected in human atherosclerotic lesions associated with both macrophage-derived foam cells34 and dendritic cells.50 Finally, a number of groups have recently shown that CD1d deficiency reduces lesion development in both Apoe−/−30,31 and Ldlr−/−33 mice. Together, this evidence strongly implicates the participation of NKT cells in the disease process, but it does not identify the specific subpopulation of NKT cell(s) that directly promote lesion formation. By superimposing the deficiency of the Jα18 gene onto mice that are already susceptible to atherosclerosis, we show in this current study that loss of functionally active Vα14 NKT cells significantly reduces the formation of early-stage lesions in both genders of mice, without affecting either serum total-cholesterol values or lipoprotein-cholesterol distributions. Furthermore, by coupling the techniques of LCM with quantitative real-time RT–PCR and by performing immunohistochemistry, we also made the important finding that expression of the proatherogenic cytokine IFN-γ is significantly reduced in lesions from Jα18−/− mice.
By using Cd1d−/− mice, which effectively depletes the mouse of all functional CD1d-restricted NKT cells, Nakai et al.,31 Tupin et al.,30 Major et al.,32 and Aslanian et al.,33 explored the collective role of CD1d-restricted NKT cells in the development of atherosclerosis in both the Apoe−/− and the LDLr−/− murine backgrounds. The Vα14Jα18 NKT cells are not the only CD1d-restricted NKT cells that are rendered functionally deficient in the CD1d−/− mouse. However, in C57BL/6 mice, targeted deletion of the Jα18 gene results in the selective depletion of Vα14 NKT cells without affecting the population of other CD1d-dependent and CD1d-independent NKT cells, or the populations of NK cells and conventional T and B cells.26,36 By making Ldlr−/− mice deficient of Vα14Jα18 NKT cells, we saw a significant reduction in lesion formation in this model of atherosclerosis, which highlights the significant role this subpopulation of NKT cells plays in lesion formation. If we focus our attention on the study by Aslanian et al.,33 who chose to study CD1d deficiency on the Ldlr−/− background, we find that the results of our study differ from those of Aslanian et al.33 where we found that compared with CD1d deficiency, deficiency of Jα18 resulted in a significant, yet much smaller reduction in atherosclerosis (20–28% in our study vs. 47% in the study by Aslanian et al.,33 based on analysis within the ascending aorta). These are very important differences between our study and that of Aslanian et al.33 and, as such, point out that Vα14Jα18 NKT cells are most likely not the only CD1d-restricted NKT cells contributing to lesion formation as at least one other cell type, which has yet to be identified, is contributing an equal share to the promotion of lesion development. This comparison highlights that our current study has shown a specific subpopulation of NKT cells to be proatherogenic, and it is precisely this unique finding that sets our current work apart from these earlier cited studies. As other subpopulations of NKT cells are identified, such as that described by Skold et al.,51 targeted deletion of these unique NKT cells within a mouse model of atherosclerosis will allow for the examination of other subpopulations of NKT cell with respect to their own contribution to lesion progression. Although this type of systematic determination of the per cent contribution of other CD1d-restricted NKT cells to lesion development is greatly encouraged, especially given our findings compared with those of Aslanian et al.,33 such a task still remains infinitely too complex since the existing criteria for the populations of other NKT cells are still only broadly defined and therefore the possible number of specific NKT cell types too great. For example, other CD1d-dependent NKT cells, the non-Vα14 CD1d-dependent NKT, includes Vα3.2-Jα9/Vβ8, Vα8/Vβ8, and other species, whereas the CD1d-independent NKT cells include those that are CD49B+ or those that are NK1.1+ and have diverse TCRs and can either be CD8+, CD4+, or CD8−CD4−.
In order to truly define the participatory role of Vα14 NKT cells in promoting atherosclerosis, it is important to know whether this particular immune cell resides within atherosclerotic lesions. Two independent groups have in fact confirmed the presence of Vα14-Jα18 transcripts within atherosclerotic lesions of two different mouse models of the disease. Nakai et al.31 showed that the development of atherosclerosis in wild type or Apoe−/− mice fed an atherogenic diet was associated with the presence of Vα14-Jα18 transcripts within the developing atheroma. In the second study, Aslanian et al.33 detected Vα14-Jα18 TCR mRNA in lesion-containing segments of the aortic arch from Ldlr−/− mice. Taken together, these independent findings are able to place Vα14 NKT cells at the site of lesion formation.
The noted reduction in both splenocyte-secreted and lesion-associated IFN-γ concentrations in this study upon Vα14 NKT cell deficiency provides strong mechanistic evidence that activated Vα14 NKT cells promote atherosclerosis by producing and/or by mediating the production of IFN-γ by other immune cells, such as NK cells at the site of a developing lesion. In vitro, IFN-γ has been shown to be proatherogenic by promoting the processes of lipoprotein oxidation, macrophage-derived foam cell formation, and immune cell activation.52–61 We and others have previously shown that administration of exogenous IFN-γ13 will accelerate early-stage atherosclerotic lesion development in Apoe−/− mice and that endogenous IFN-γ promotes lesion development in both Apoe−/− mice14 and Ldlr−/− mice.9 The identification of Vα14 NKT cells within atherosclerotic lesions of mice also validates our IFN-γ data as it relates to Vα14Jα18 NKT cells as being the most likely mediator for the production of this pro-inflammatory cytokine and that IFN-γ, produced locally at the site of lesion development, is a key contributor to lesion formation as noted in earlier studies that used IFN-γ null mice placed on both the Ldlr−/−9 and Apoe−/−14 backgrounds. Future studies will also need to examine whether Vα14 NKT cells promote not only early-stage, but also late-stage lesion development, and whether Vα14 NKT cells accelerate disease by affecting the activation status of additional populations of lesion-associated immune cells such as conventional T cells,62 NK cells,5 and/or the process of macrophage-derived foam cell formation by interrupting the ability of foam cells to efflux cholesterol to HDL via ABCA1.63,64 Interestingly, we discovered no significant difference in the percentage of neutral lipid associated within macrophages located in the ascending aortic lesions of Jα18+/+ vs. −/− mice, despite the fact that the overall size of atherosclerotic lesions in the same area of the ascending aorta of the Jα18−/− mice were significantly smaller than their Jα18+/+ littermates. These findings could possibly suggest that Vα14 NKT cell deficiency reduced atherosclerotic burden by causing a net decrease in the population of lesion-associated macrophages, rather than a decrease in the ability of macrophages to undergo lipid-loading. The cellular target of IFN-γ expressed by Vα14 NKT cells is currently an area of investigation in our laboratory as the identification of these target cells would be seen as an important step in the elucidation of this particular proatherogenic pathway.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
Heart and Stroke Foundation of Ontario Grant-in-Aid (#T 5691 to S.C.W.). S.C.W is the recipient of a Great-West Life and London Life New Investigator Award from the Heart and Stroke Foundation of Canada.
Supplementary Material
Acknowledgements
We would like to thank Valerie Barrette for her help with maintaining the mouse colony used in this study and Karen Hoke for her constant moral support.
Conflict of interest: none declared.
References
- 1.Hansson GK. Immune mechanisms in atherosclerosis. Arterioscler Thromb Vasc Biol. 2001;21:1876–1890. doi: 10.1161/hq1201.100220. [DOI] [PubMed] [Google Scholar]
- 2.Binder CJ, Chang MK, Shaw PX, Miller YI, Hartvigsen K, Dewan A, et al. Innate and acquired immunity in atherogenesis. Nat Med. 2002;8:1218–1226. doi: 10.1038/nm1102-1218. [DOI] [PubMed] [Google Scholar]
- 3.Hansson GK, Libby P, Schonbeck U, Yan ZQ. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ Res. 2002;91:281–291. doi: 10.1161/01.res.0000029784.15893.10. [DOI] [PubMed] [Google Scholar]
- 4.Linton MF, Major AS, Fazio S. Proatherogenic role for NK cells revealed. Arterioscler Thromb Vasc Biol. 2004;24:992–994. doi: 10.1161/01.ATV.0000128896.45976.f0. [DOI] [PubMed] [Google Scholar]
- 5.Whitman SC, Rateri DL, Szilvassy SJ, Yokoyama W, Daugherty A. Depletion of natural killer cell function decreases atherosclerosis in low-density lipoprotein receptor null mice. Arterioscler Thromb Vasc Biol. 2004;26:1049–1054. doi: 10.1161/01.ATV.0000124923.95545.2c. [DOI] [PubMed] [Google Scholar]
- 6.Zhou X, Nicoletti A, Elhage R, Hansson GK. Transfer of CD4(+) T cells aggravates atherosclerosis in immunodeficient apolipoprotein E knockout mice. Circulation. 2000;102:2919–2922. doi: 10.1161/01.cir.102.24.2919. [DOI] [PubMed] [Google Scholar]
- 7.Laurat E, Poirier B, Tupin E, Caligiuri G, Hansson GK, Bariety J, et al. In vivo downregulation of T helper cell 1 immune responses reduces atherogenesis in apolipoprotein E-knockout mice. Circulation. 2001;104:197–202. doi: 10.1161/01.cir.104.2.197. [DOI] [PubMed] [Google Scholar]
- 8.Huber SA, Sakkinen P, David C, Newell MK, Tracy RP. T helper-cell phenotype regulates atherosclerosis in mice under conditions of mild hypercholesterolemia. Circulation. 2001;103:2610–2616. doi: 10.1161/01.cir.103.21.2610. [DOI] [PubMed] [Google Scholar]
- 9.Buono C, Come CE, Stavrakis G, Maguire GF, Connelly PW, Lichtman AH. Influence of interferon-gamma on the extent and phenotype of diet-induced atherosclerosis in the LDLR-deficient mouse. Arterioscler Thromb Vasc Biol. 2003;23:454–460. doi: 10.1161/01.ATV.0000059419.11002.6E. [DOI] [PubMed] [Google Scholar]
- 10.Caligiuri G, Nicoletti A, Poirier B, Hansson GK. Protective immunity against atherosclerosis carried by B cells of hypercholesterolemic mice. J Clin Invest. 2002;109:745–753. doi: 10.1172/JCI07272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Major AS, Fazio S, Linton MF. B-lymphocyte deficiency increases atherosclerosis in LDL receptor-null mice. Arterioscler Thromb Vasc Biol. 2002;22:1892–1898. doi: 10.1161/01.atv.0000039169.47943.ee. [DOI] [PubMed] [Google Scholar]
- 12.Gupta S, Pablo AM, Jiang X, Wang N, Tall AR, Schindler C. IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. J Clin Invest. 1997;99:2752–2761. doi: 10.1172/JCI119465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whitman SC, Ravisankar P, Elam H, Daugherty A. Exogenous interferon-gamma enhances atherosclerosis in apolipoprotein E-/- mice. Am J Pathol. 2000;157:1819–1824. doi: 10.1016/s0002-9440(10)64820-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whitman SC, Ravisankar P, Daugherty A. IFN-gamma deficiency exerts gender-specific effects on atherogenesis in apolipoprotein E-/- mice. J Interferon Cytokine Res. 2002;22:661–670. doi: 10.1089/10799900260100141. [DOI] [PubMed] [Google Scholar]
- 15.Elhage R, Clamens S, Besnard S, Mallat Z, Tedgui A, Arnal J, et al. Involvement of interleukin-6 in atherosclerosis but not in the prevention of fatty streak formation by 17beta-estradiol in apolipoprotein E-deficient mice. Atherosclerosis. 2001;156:315–320. doi: 10.1016/s0021-9150(00)00682-1. [DOI] [PubMed] [Google Scholar]
- 16.Huber SA, Sakkinen P, Conze D, Hardin N, Tracy R. Interleukin-6 exacerbates early atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 1999;19:2364–2367. doi: 10.1161/01.atv.19.10.2364. [DOI] [PubMed] [Google Scholar]
- 17.Pinderski LJ, Fischbein MP, Subbanagounder G, Fishbein MC, Kubo N, Cheroutre H, et al. Overexpression of interleukin-10 by activated T lymphocytes inhibits atherosclerosis in LDL receptor-deficient Mice by altering lymphocyte and macrophage phenotypes. Circ Res. 2002;90:1064–1071. doi: 10.1161/01.res.0000018941.10726.fa. [DOI] [PubMed] [Google Scholar]
- 18.Pinderski Oslund LJ, Hedrick CC, Olvera T, Hagenbaugh A, Territo M, Berliner JA, et al. Interleukin-10 blocks atherosclerotic events in vitro and in vivo. Arterioscler Thromb Vasc Biol. 1999;19:2847–2853. doi: 10.1161/01.atv.19.12.2847. [DOI] [PubMed] [Google Scholar]
- 19.Namiki M, Kawashima S, Yamashita T, Ozaki M, Sakoda T, Inoue N, et al. Intramuscular gene transfer of interleukin-10 cDNA reduces atherosclerosis in apolipoprotein E-knockout mice. Atherosclerosis. 2004;172:21–29. doi: 10.1016/j.atherosclerosis.2003.08.032. [DOI] [PubMed] [Google Scholar]
- 20.Mallat Z, Besnard S, Duriez M, Deleuze V, Emmanuel F, Bureau MF, et al. Protective role of interleukin-10 in atherosclerosis. Circ Res. 1999;85:e17–e24. doi: 10.1161/01.res.85.8.e17. [DOI] [PubMed] [Google Scholar]
- 21.Caligiuri G, Rudling M, Ollivier V, Jacob MP, Michel JB, Hansson GK, et al. Interleukin-10 deficiency increases atherosclerosis, thrombosis, and low-density lipoproteins in apolipoprotein E knockout mice. Mol Med. 2003;9:10–17. [PMC free article] [PubMed] [Google Scholar]
- 22.Joyce S, Van Kaer L. CD1-restricted antigen presentation: an oily matter. Curr Opin Immunol. 2003;15:95–104. doi: 10.1016/s0952-7915(02)00012-2. [DOI] [PubMed] [Google Scholar]
- 23.Bendelac A, Bonneville M, Kearney JF. Autoreactivity by design: innate B and T lymphocytes. Nat Rev Immunol. 2001;1:177–186. doi: 10.1038/35105052. [DOI] [PubMed] [Google Scholar]
- 24.Wilson SB, Delovitch TL. Janus-like role of regulatory iNKT cells in autoimmune disease and tumour immunity. Nat Rev Immunol. 2003;3:211–222. doi: 10.1038/nri1028. [DOI] [PubMed] [Google Scholar]
- 25.Kronenberg M, Gapin L. The unconventional lifestyle of NKT cells. Nat Rev Immunol. 2002;2:557–568. doi: 10.1038/nri854. [DOI] [PubMed] [Google Scholar]
- 26.Taniguchi M, Harada M, Kojo S, Nakayama T, Wakao H. The regulatory role of Valpha14 NKT cells in innate and acquired immune response. Annu Rev Immunol. 2003;21:483–513. doi: 10.1146/annurev.immunol.21.120601.141057. [DOI] [PubMed] [Google Scholar]
- 27.Godfrey DI, Hammond KJ, Poulton LD, Smyth MJ, Baxter AG. NKT cells: facts, functions and fallacies. Immunol Today. 2000;21:573–583. doi: 10.1016/s0167-5699(00)01735-7. [DOI] [PubMed] [Google Scholar]
- 28.Fujii S, Shimizu K, Smith C, Bonifaz L, Steinman RM. Activation of natural killer T cells by alpha-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J Exp Med. 2003;198:267–279. doi: 10.1084/jem.20030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, et al. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science. 1997;278:1626–1629. doi: 10.1126/science.278.5343.1626. [DOI] [PubMed] [Google Scholar]
- 30.Tupin E, Nicoletti A, Elhage R, Rudling M, Ljunggren HG, Hansson GK, et al. CD1d-dependent activation of NKT cells aggravates atherosclerosis. J Exp Med. 2004;199:417–422. doi: 10.1084/jem.20030997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakai Y, Iwabuchi K, Fujii S, Ishimori N, Dashtsoodol N, Watano K, et al. Natural killer T cells accelerate atherogenesis in mice. Blood. 2004;104:2051–2059. doi: 10.1182/blood-2003-10-3485. [DOI] [PubMed] [Google Scholar]
- 32.Major AS, Wilson MT, McCaleb JL, Ru SY, Stanic AK, Joyce S, et al. Quantitative and qualitative differences in proatherogenic NKT cells in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2004;24:2351–2357. doi: 10.1161/01.ATV.0000147112.84168.87. [DOI] [PubMed] [Google Scholar]
- 33.Aslanian AM, Chapman HA, Charo IF. Transient role for CD1d-restricted natural killer T cells in the formation of atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2005;25:628–632. doi: 10.1161/01.ATV.0000153046.59370.13. [DOI] [PubMed] [Google Scholar]
- 34.Melian A, Geng YJ, Sukhova GK, Libby P, Porcelli SA. CD1 expression in human atherosclerosis. A potential mechanism for T cell activation by foam cells. Am J Pathol. 1999;155:775–786. doi: 10.1016/S0002-9440(10)65176-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park SH, Weiss A, Benlagha K, Kyin T, Teyton L, Bendelac A. The mouse CD1d-restricted repertoire is dominated by a few autoreactive T cell receptor families. J Exp Med. 2001;193:893–904. doi: 10.1084/jem.193.8.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cui J, Shin T, Kawano T, Sato H, Kondo E, Toura I, et al. Requirement for Valpha14 NKT cells in IL-12-mediated rejection of tumors. Science. 1997;278:1623–1626. doi: 10.1126/science.278.5343.1623. [DOI] [PubMed] [Google Scholar]
- 37.Whitman SC. A practical approach to using mice in atherosclerosis research. Clin Biochem Rev. 2004;25:81–93. [PMC free article] [PubMed] [Google Scholar]
- 38.Whitman SC, Ravisankar P, Daugherty A. Interleukin-18 enhances atherosclerosis in apolipoprotein E(-/-) mice through release of interferon-gamma. Circ Res. 2002;90:E34–E38. doi: 10.1161/hh0202.105292. [DOI] [PubMed] [Google Scholar]
- 39.Daugherty A, Whitman SC. Quantification of atherosclerosis in mice. Methods Mol Biol. 2003;209:293–309. doi: 10.1385/1-59259-340-2:293. [DOI] [PubMed] [Google Scholar]
- 40.Daugherty A, Pure E, Delfel-Butteiger D, Chen S, Leferovich J, Roselaar SE, et al. The effects of total lymphocyte deficiency on the extent of atherosclerosis in apolipoprotein E-/- mice. J Clin Invest. 1997;100:1575–1580. doi: 10.1172/JCI119681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105:1605–1612. doi: 10.1172/JCI7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Giulietti A, Overbergh L, Valckx D, Decallonne B, Bouillon R, Mathieu C. An overview of real-time quantitative PCR: applications to quantify cytokine gene expression. Methods. 2001;25:386–401. doi: 10.1006/meth.2001.1261. [DOI] [PubMed] [Google Scholar]
- 43.Taniguchi M, Seino K, Nakayama T. The NKT cell system: bridging innate and acquired immunity. Nat Immunol. 2003;4:1164–1165. doi: 10.1038/ni1203-1164. [DOI] [PubMed] [Google Scholar]
- 44.Koseki H, Imai K, Ichikawa T, Hayata I, Taniguchi M. Predominant use of a particular alpha-chain in suppressor T cell hybridomas specific for keyhole limpet hemocyanin. Int Immunol. 1989;1:557–564. doi: 10.1093/intimm/1.6.557. [DOI] [PubMed] [Google Scholar]
- 45.Balk SP, Ebert EC, Blumenthal RL, McDermott FV, Wucherpfennig KW, Landau SB, et al. Oligoclonal expansion and CD1 recognition by human intestinal intraepithelial lymphocytes. Science. 1991;253:1411–1415. doi: 10.1126/science.1716785. [DOI] [PubMed] [Google Scholar]
- 46.Porcelli S, Yockey CE, Brenner MB, Balk SP. Analysis of T cell antigen receptor (TCR) expression by human peripheral blood CD4-8- alpha/beta T cells demonstrates preferential use of several V beta genes and an invariant TCR alpha chain. J Exp Med. 1993;178:1–16. doi: 10.1084/jem.178.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singh N, Hong S, Scherer DC, Serizawa I, Burdin N, Kronenberg M, et al. Cutting edge: activation of NK T cells by CD1d and alpha-galactosylceramide directs conventional T cells to the acquisition of a Th2 phenotype. J Immunol. 1999;163:2373–2377. [PubMed] [Google Scholar]
- 48.Sharif S, Arreaza GA, Zucker P, Mi QS, Sondhi J, Naidenko OV, et al. Activation of natural killer T cells by alpha-galactosylceramide treatment prevents the onset and recurrence of autoimmune Type 1 diabetes. Nat Med. 2001;7:1057–1062. doi: 10.1038/nm0901-1057. [DOI] [PubMed] [Google Scholar]
- 49.Hong S, Wilson MT, Serizawa I, Wu L, Singh N, Naidenko OV, et al. The natural killer T-cell ligand alpha-galactosylceramide prevents autoimmune diabetes in non-obese diabetic mice. Nat Med. 2001;7:1052–1056. doi: 10.1038/nm0901-1052. [DOI] [PubMed] [Google Scholar]
- 50.Bobryshev YV, Lord RS. CD1 expression and the nature of CD1-expressing cells in human atherosclerotic plaques. Am J Pathol. 2000;156:1477–1478. doi: 10.1016/s0002-9440(10)65016-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Skold M, Faizunnessa NN, Wang CR, Cardell S. CD1d-specific NK1.1+ T cells with a transgenic variant TCR. J Immunol. 2000;165:168–174. doi: 10.4049/jimmunol.165.1.168. [DOI] [PubMed] [Google Scholar]
- 52.Geng YJ, Hansson GK. Interferon-gamma inhibits scavenger receptor expression and foam cell formation in human monocyte-derived macrophages. J Clin Invest. 1992;89:1322–1330. doi: 10.1172/JCI115718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fong LG, Fong TA, Cooper AD. Inhibition of mouse macrophage degradation of acetyl-low density lipoprotein by interferon-gamma. J Biol Chem. 1990;265:11751–11760. [PubMed] [Google Scholar]
- 54.Cornicelli JA, Butteiger D, Rateri DL, Welch K. Daugherty A. Interleukin-4 augments acetylated LDL-induced cholesterol esterification in macrophages. J Lipid Res. 2000;41:376–383. [PubMed] [Google Scholar]
- 55.LaMarre J, Wolf BB, Kittler EL, Quesenberry PJ, Gonias SL. Regulation of macrophage alpha 2-macroglobulin receptor/low density lipoprotein receptor-related protein by lipopolysaccharide and interferon-gamma. J Clin Invest. 1993;91:1219–1224. doi: 10.1172/JCI116283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Conrad DJ, Kuhn H, Mulkins M, Highland E, Sigal E. Specific inflammatory cytokines regulate the expression of human monocyte 15-lipoxygenase. Proc Natl Acad Sci USA. 1992;89:217–221. doi: 10.1073/pnas.89.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Christen S, Thomas SR, Garner B, Stocker R. Inhibition by interferon-gamma of human mononuclear cell-mediated low density lipoprotein oxidation. Participation of tryptophan metabolism along the kynurenine pathway. J Clin Invest. 1994;93:2149–2158. doi: 10.1172/JCI117211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jonasson L, Hansson GK, Bondjers G, Noe L, Etienne J. Interferon-gamma inhibits lipoprotein lipase in human monocyte-derived macrophages. Biochim Biophys Acta. 1990;1053:43–48. doi: 10.1016/0167-4889(90)90024-8. [DOI] [PubMed] [Google Scholar]
- 59.Sempowski GD, Derdak S, Phipps RP. Interleukin-4 and interferon-gamma discordantly regulate collagen biosynthesis by functionally distinct lung fibroblast subsets. J Cell Physiol. 1996;167:290–296. doi: 10.1002/(SICI)1097-4652(199605)167:2<290::AID-JCP13>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 60.Li H, Cybulsky MI, Gimbrone MA, Jr, Libby P. Inducible expression of vascular cell adhesion molecule-1 by vascular smooth muscle cells in vitro and within rabbit atheroma. Am J Pathol. 1993;143:1551–1559. [PMC free article] [PubMed] [Google Scholar]
- 61.Hansson GK, Jonasson L, Holm J, Clowes MM, Clowes AW. Gamma-interferon regulates vascular smooth muscle proliferation and Ia antigen expression in vivo and in vitro. Circ Res. 1988;63:712–719. doi: 10.1161/01.res.63.4.712. [DOI] [PubMed] [Google Scholar]
- 62.Roselaar SE, Kakkanathu PX, Daugherty A. Lymphocyte populations in atherosclerotic lesions of apoE -/- and LDL receptor -/- mice. Decreasing density with disease progression. Arterioscler Thromb Vasc Biol. 1996;16:1013–1018. doi: 10.1161/01.atv.16.8.1013. [DOI] [PubMed] [Google Scholar]
- 63.Wang XQ, Panousis CG, Alfaro ML, Evans GF, Zuckerman SH. Interferon-gamma-mediated downregulation of cholesterol efflux and ABC1 expression is by the Stat1 pathway. Arterioscler Thromb Vasc Biol. 2002;22:e5–e9. doi: 10.1161/01.atv.0000018287.03856.dd. [DOI] [PubMed] [Google Scholar]
- 64.Panousis CG, Zuckerman SH. Regulation of cholesterol distribution in macrophage-derived foam cells by interferon-gamma. J Lipid Res. 2000;41:75–83. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.