

Abstract
Mdm2 and Mdm4 are negative regulators of the tumour suppressor p53; hence, this relationship is the focus of many cancer-related studies. A multitude of experiments across various developmental stages have been conducted to explore the tissue-specific roles of these proteins in the mouse. When Mdm2 or Mdm4 are deleted in the germline or specific tissues, they display different phenotypic defects, some of which lead to embryonic lethality. Mdm2 loss is often more deleterious than loss of its homologue Mdm4. All tissues experience activation of p53 target genes upon loss of Mdm2 or Mdm4; however, the degree to which the p53 pathway is perturbed is highly tissue-specific and does not correlate to the severity of the morphological phenotypes. Therefore, a need for further understanding of how these proteins regulate p53 activity is warranted, as therapeutic targeting of the p53 pathway is rapidly evolving and gaining attention in the field of cancer research. In this review, we discuss the tissue-specificity of Mdm proteins in regulating p53 and expose the need for investigation at the cell-specific level.
Keywords: Mdm2, Mdm4, MdmX, development, embryogenesis, tissue-specific, p53
Mdm2-null and Mdm4-null mice are embryonic lethal
Mdm2 (murine double minute 2) and its homologue, Mdm4 (also referred to as MdmX), are two major negative regulators of the tumour suppressor p53 (Figure 1)(Pant et al., 2013). Both proteins contain RING finger domains at their respective C-termini. The RING domain of Mdm2 harbours E3 ligase activity, giving it the ability to initiate p53 degradation, Mdm4 degradation, and degradation of itself. Although the RING domain of Mdm4 is not enzymatically active, both Mdm2 and Mdm4 bind to the N-terminal transcriptional activation domain of p53 and repress the transcriptional activity of p53 in homoeostasis. Furthermore, p53 and Mdm2 are involved in a negative feedback loop; p53 transactivates Mdm2 and Mdm2 in turn targets p53 for proteasomal degradation. Lastly, Mdm2 and Mdm4 form a heterodimer that maximizes inhibition of p53 (Wu et al., 1993; Tanimura et al., 1999).
Figure 1.
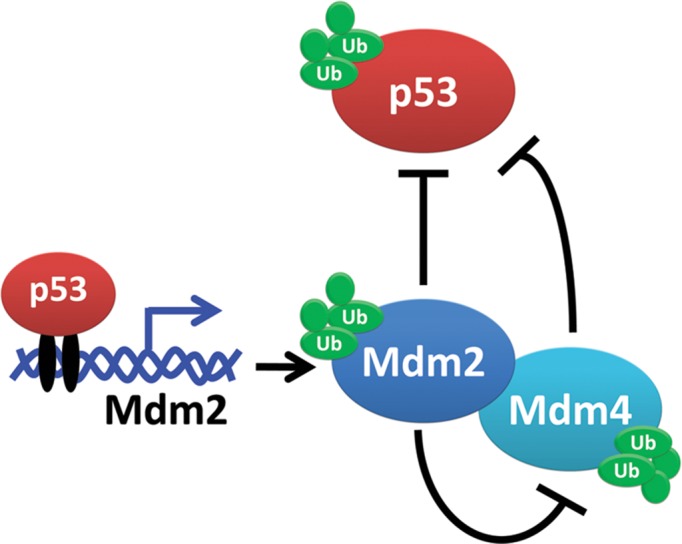
Schematic of the Mdm2, Mdm4, and p53 relationships described in this review. Mdm2 and Mdm4 are negative regulators of p53. Mdm2 also has the ability to ubiquitinate p53, itself, and Mdm4 proteins to target them for proteasomal degradation. p53 signals for the transcription of Mdm2 when it is needed in the cell. Mdm2 and Mdm4 have the ability to form and function as a heterodimer.
Mdm2-null and Mdm4-null mice are embryonic lethal, indicating that Mdm proteins are vital in development. Moreover, these phenotypes can be rescued with concomitant loss of p53 (Mdm2−/−p53−/− and Mdm4−/−p53−/− mice are viable), indicating that the lethality is due to activation of p53. The loss of Mdm2 in an embryo results in death due to a p53-mediated apoptotic mechanism (Jones et al., 1995; Chavez-Reyes et al., 2003; Montes de Oca Luna et al., 1995; Parant et al., 2001). This occurs prior to implantation of the embryos at embryonic day 3.5 (E3.5). In contrast, embryos lacking Mdm4 initiate cell cycle arrest and occasionally apoptosis, but they do so at later developmental timepoints—normally around gastrulation of the embryo (E7.5) (Parant et al., 2001; Migliorini et al., 2002). The progression of mouse development is shown in Figure 2.
Figure 2.

Illustration of mouse embryos at important embryonic timepoints that are highlighted throughout this review. The shaded area at E9.5 represents the location of the heart, at E14.5 the location of the kidney, and at E17.5 the location of the brain and CNS.
Due to limitations of studying development in embryonic lethal mouse models, conditional alleles were generated to evaluate the functions of Mdm2 and Mdm4 at later developmental stages and in various tissues of adult mice. In this review, we will explore the research that aims at understanding the roles of Mdm2 and Mdm4 during organogenesis and organ development.
Alterations in intrinsic functions of Mdm proteins lead to accumulation and/or hyperactivity of p53
In attempts to gain a better understanding of the interactions between Mdm2, Mdm4, and p53, multiple mouse models were created that alter the intrinsic functions of Mdm2 or Mdm4 in order to examine their physiological importance. The Mdm2 gene contains two promoters; a p53-independent promoter (P1), located upstream from the first exon that is constitutively active under basal conditions, and a p53-dependent promoter (P2) residing in the first intron that is only activated in response to DNA damage and stress conditions (Barak et al., 1994). When DNA is damaged, p53 binds to the P2 promoter of Mdm2, which leads to increased Mdm2 levels and subsequent decreased p53 levels. Pant et al. (2013) generated point mutations in the P2 promoter that disrupt interactions with p53 (Mdm2P2/P2 mice) to examine the significance of the Mdm2-p53 feedback loop (Figure 3A) (Pant et al., 2013). Abrogation of p53 binding at the Mdm2-P2 promoter does not alter basal levels of Mdm2 or p53, but mice are extremely sensitive to low-dose ionizing radiation (IR). The inability of p53 to upregulate Mdm2 results in extended p53 activity and a p53-dependent death. Another mouse, Mdm2Y487A/Y487A, was generated to specifically examine the importance of Mdm2 E3 ligase activity. A point mutation at amino acid 487 in the C-terminus of Mdm2 disrupts E3 ligase activity yet allows Mdm2 to inhibit p53 and thus results in viable mice. However, as Mdm2P2/P2 mice, Mdm2Y487A/Y487A mice are sensitive to IR (Tollini et al., 2014). These differences in viability showcase the lower levels of Mdm2 and/or Mdm4 activity required for homoeostasis. A mouse with a mutation at amino acid 462 of Mdm2 that disrupts both E3 ligase function and interactions with Mdm4, Mdm2C462A/C462A, is lethal because of too much p53 activity (Itahana et al., 2007). Other studies with specific deletion of the Mdm4 RING domain that removes interactions with Mdm2 (Mdm4ΔRING/ΔRING) and a point mutation in Mdm4 that also disrupts interactions with Mdm2 (Mdm4C462A/C462A) are embryonic lethal (Figure 3B) (Huang et al., 2011; Pant et al., 2011). Again these phenotypes are rescued by p53 loss (Mdm4ΔRING/ΔRINGp53−/− and Mdm4C462A/C462Ap53−/−, respectively). Thus, interactions between the RING domains of Mdm2 and Mdm4 are vital for sufficient Mdm activity to inhibit p53 during early development.
Figure 3.

Generated alleles of Mdm2 or Mdm4 (MdmX). Exons are depicted by black boxes marked with the appropriate number. (A) The wild-type (WT) allele of Mdm2 is shown. The null allele contains a neomycin cassette inserted in place of exons 10–12. The Mdmflox allele creates a conditionally null allele by flanking exons 7 through 9 with loxP sites. These exons will be removed after Cre-mediated recombination of the loxP sites. Mdm2FM is generated using loxP sites surrounding exons 5 and 6, which encodes the p53 interaction domain. The Mdm2puro/Δ7–12 allele retains the puromycin cassette in intron 6. The PND (mutant P2 promoter, neomycin cassette, and 184-bp deletion) allele has a 184-bp deletion in intron 3 in conjunction with retention of the neomycin cassette and deletion of the p53 response elements present in the Mdm2P2/P2, a knock-in mouse harbouring mutation of the p53-binding sequences in the Mdm2-P2 promoter, allowing for basal Mdm2 expression. The Mdm2Y487A allele contains a point mutation at amino acid number 487. This knock-in mutation converts a Tyrosine to an Alanine thus disrupting the E3 ligase activity of the Mdm2 RING domain but still allowing it to bind to Mdm4. The Mdm2C462A allele contains a point mutation at amino acid number 462. This knock-in mutation converts a Cysteine to an Alanine thus removing E3 ligase and disrupting interactions with Mdm4. (B) The WT allele of Mdm4 is shown. The original Mdm4-null allele has a neomycin cassette in place of exons 3 and 4 removing the majority of the p53-binding domain. This allele creates a small C-terminal protein that retains interactions with Mdm2. The Mdm4Δ2 deletes exon 2. The Mdm4Δ2 allele is a true null. The Mdm4FX allele is generated using a floxed exon 2. Through Cre recombination, this exon can be conditionally removed. Exon 2 of Mdm4 (MdmX) is the first coding exon and its loss results in a functionally null allele. The Mdm4ΔRING allele contains an in-frame deletion of the RING domain within exon 11 composed of 49 amino acids. The Mdm4C462A allele contains a neomycin-resistant gene cassette in intron 10 and a point mutation at amino acid number 462 within exon 11, making it unable to form a heterodimer with Mdm2.
Loss of Mdm2 and Mdm4 in the central nervous system
Given that radiation of embryos displays a very strong p53-dependent effect within the central nervous system (CNS), Mdm2 and Mdm4 are also implicated in nervous system development. Thus, Xiong et al. (2006) used a neuron-specific transgenic Cre, Nestin-Cre (Nes-Cre), to delete Mdm2 and Mdm4 within the CNS to explore any redundancies that these two genes may play in the development of the CNS (Table 1). Mdm2FM/FM or Mdm4FX/FX (conditional alleles of Mdm2 or Mdm4, respectively) mice were crossed to Mdm2FM/+Nes-Cre or Mdm4FX/+Nes-Cre mice, respectively (Figure 3) (Grier et al., 2006). The neuron-specific enhancer of the Nestin promoter drives Cre expression and recombination in the neurons and glia begins at E10.5 (Graus-Porta et al., 2001). At weaning, no Mdm2FM/FMNes-Cre mice are obtained. Occasionally, mice are born with abnormally shaped heads; however, they die very soon after birth. Phenotypic defects due to the loss of Mdm2 include a domed-head phenotype, decreased neuronal tissue, thinner cerebral cortexes, and excess cerebrospinal fluid in the brain observed as early as E12.5. At E18.5, Mdm2FM/FMNes-Cre embryos have virtually no neuronal tissue remaining and are unable to survive until time of birth.
Table 1.
Synopsis of the different drivers of the Cre-recombinase system utilized within the discussed works.
| Type | Driver | Site of expression | Expression initiation | Cited works | |
|---|---|---|---|---|---|
| αHoxb7-Cre | Transgenic | Homeobox B7 | Ureteric bud and mesonephric duct | E9.5 | Hilliard et al. (2011) |
| αMyHC-Cre | Transgenic | α-myosin heavy chain | Cardiomyocytes | E8.5 | Ng et al. (1991), Agah et al. (1997), Xiong et al. (2007) |
| CAG-CreER | Transgenic | CMV enhancer/chicken β-actin | Ubiquitous expression | Tamoxifen-inducible | Zhang et al. (2014a) |
| Le-Cre | Transgenic | Pax6 | Surface ectoderm, retinal, and pancreatic primordial cells | E9 | Zhang et al. (2014b) |
| Nes-Cre | Transgenic | Nestin | Central and peripheral nervous systems | E10.5 | Graus-Porta et al. (2001), Francoz et al. (2006), Grier et al. (2006), Xiong et al. (2006) |
| SM22-Cre | Knock-in | Smooth muscle protein 22-α gene | Vascular and visceral smooth muscle cells of adult mice | Tamoxifen-inducible | Kuhbandner et al. (2000), Boesten et al. (2006) |
| Tie2-Cre (Tek-Cre) | Transgenic | Tek receptor Tyrosine kinase | Endothelial cells | E7.5 | Kisanuki et al. (2001), Zhang et al. (2012) |
| Villin-Cre | Transgenic | Villin 1 | Epithelial cells of small and large intestines | E9 | Creamer et al. (1961), Madison et al. (2002), Valentin-Vega et al. (2008, 2009) |
| Zp3-Cre | Transgenic | Zona Pellucida 3 | Growing oocytes | Before first meiotic division | de Vries et al. (2000), Livera et al. (2016) |
Data include the Cre recombinase, whether a knock-in or transgenic model, the gene driver of the recombinase, tissue- or cell-specific site of expression, and initiation of expression. Additional information on these specific Cre-recombinase systems can be found in the attached cited works.
The same experiments are performed on the Mdm4 CNS-specific deletion mice (Xiong et al., 2006). No Mdm4FX/FXNes-Cre mice are observed at birth. Embryonically, they do not begin to show any phenotypic defects until E17.5, at which time, they display a flat head resulting from a large cavity that developed within the brain. This phenotype increases in severity through E18.5. Deletion of both Mdm2 and Mdm4 led to more severe phenotypes. These results indicate that Mdm2 and Mdm4 are essential for the development of the CNS, although the phenotypes of Mdm2 loss are more dramatic and present at an earlier embryonic timepoint.
Further experiments utilizing these mice indicate that Mdm2 as well as Mdm4 CNS-specific deletions ultimately lead to both apoptosis and cell cycle arrest within these tissues. However, Mdm2FM/FMNes-Cre mice show an increase in p53 4 days before Mdm4FX/FXNes-Cre mice do so (E10.5 and E14.5, respectively) resulting in death. Additionally, crosses with p53−/− mice show that these lethal phenotypes are p53-dependent, verifying that Mdm2 and Mdm4 are indispensable regulators of p53 during the development of the CNS.
Francoz et al. (2006) conducted a similar study in which they used Mdm2 and Mdm4-null mice in conjunction with Nes-Cre and the p53LSL/LSL allele. Upstream of the normal coding region of p53, the p53LSL/LSL allele has a transcriptional stop element surrounded by loxP sites. In the absence of tamoxifen, no p53 protein is produced from this allele. However, upon administration of tamoxifen, the loxP sites recombine and remove the stop element, allowing for the production of a functional wild-type (WT) p53 protein. Analyses of individual deletions (p53LSL/−Mdm2−/−Nes-Cre and p53LSL/−Mdm4−/−Nes-Cre mice) show analogous results to the aforementioned study. Moreover, this group also examined Mdm2;Mdm4 double null (p53LSL/−Mdm2−/−Mdm4−/−Nes-Cre) mice. Surprisingly, all observed phenotypes are intensified in these double knockout mice and are p53-dependent. Morphologically, these mice have the smallest heads and the least amount of neuronal tissue at E14.5. Additionally, these p53LSL/−Mdm2−/−Mdm4−/−Nes-Cre mice exhibit less cycling cells coupled with extremely high apoptosis and p53 stabilization. Since the phenotype of double knockout mice is worse than either single knockout, these data suggest that Mdm2 and Mdm4 play non-overlapping and important roles when regulating p53 in the CNS.
Mdm2 and Mdm4 trade off to support cardiomyocyte maintenance
Due to the fact that apoptosis and proliferation are key processes in heart development, tissue-specific Cre systems were used to delete Mdm2 and Mdm4 in the heart (Jones et al., 1995; Xiong et al., 2007). The αMyHC-Cre transgene is under control of the α-myosin heavy chain promoter, which is specific to cardiomyocytes and is expressed early in cardiogenesis at E8.5 (cardiogenesis is initiated at E7.5) (Ng et al., 1991; Agah et al., 1997). No mice survived with complete loss of Mdm2 in the heart. Results indicate that all Mdm2FM/−αMyHC-Cre mice die by E13.5. Defects can be seen starting at E9.5, an embryonic day later than the initiation of Cre expression. At this time, the walls of the heart are much thinner than normal and, if able to develop further, at E13.5 the heart does not function properly resulting in blood leakage from the ventricles. The deficiencies seen within these embryos ultimately lead to heart failure and death prior to birth. Further analysis reveals that these phenotypic changes are due to apoptosis arising in conjunction with low levels of proliferation among the few remaining cardiomyocytes. The loss of Mdm2 specifically in the heart cells (Mdm2FM/−αMyHC-Cre) results in embryonic death due to the activation of p53, disrupting the delicate pattern of proliferation and apoptosis necessary for the developing heart. Concomitant loss of p53 leads to viable p53−/−Mdm2FM/−αMyHC-Cre mice with the same lifespan as p53−/− and p53−/−Mdm2−/− mice. Thus, the lethal phenotype is p53-dependent.
The same study found that loss of Mdm4 (Mdm4FX/Δ2αMyHC-Cre—Mdm4Δ2 is a null allele) specifically in cardiomyocytes does not result in embryonic lethality (Grier et al., 2006). Mdm4FX/Δ2αMyHC-Cre mice appear to be phenotypically and developmentally normal. Thus, Mdm2 but not Mdm4 is required for the proper development of the heart. However, abnormalities in the Mdm4FX/Δ2αMyHC-Cre mice become noticeable as early as 3 months. By 8 months of age, the functional defects of the heart are apparent. Mice are swollen, have decreased mobility, and display shortness of breath. By 8 months, Mdm4FX/Δ2αMyHC-Cre mice have half the number of cardiomyocytes. TUNEL staining reveals that this is a result of apoptosis coupled with an expected lack of proliferation. All of these mice die within a year, giving them a significantly decreased lifespan compared to controls. Xiong et al. (2007) noted that Mdm4FX/Δ2αMyHC-Cre mice die of dilated cardiomyopathy, a leading cause of heart failure with few genes being implicated in its heredity. Further analyses reveal that all chambers of the heart are dilated with thinner ventricle walls compared with controls. Additionally, fibrosis is present in the Mdm4FX/Δ2αMyHC-Cre hearts, which ultimately leads to heart failure. Moreover, Mdm4FX/Δ2αMyHC-Cre male mice die significantly earlier than females, which is consistent with human cardiomyopathy data. All of these phenotypes are rescued by loss of p53. These experiments indicate that Mdm2 and Mdm4 have critical roles to play in the development and homoeostasis of murine hearts. While Mdm2 trumps organogenesis of this tissue, Mdm4 is fundamental in maintaining regulation of p53 in adult cardiomyocytes.
Zhang et al. (2012) orchestrated a similar study in which they used Tie2-Cre to evaluate Mdm loss specifically during cardiac endocardial cushion morphogenesis. The Tie2-Cre is under the control of the mouse endothelial-specific receptor tyrosine kinase promoter (Tie2 or Tek) and is expressed beginning at E7.5 (Kisanuki et al., 2001). Much like the aforementioned study, the Mdm2FM/FMTie2-Cre mice die before E10.5 due to a misshapen developing heart and severe vascular defects throughout their bodies. The p53 target genes p21 and Puma are significantly upregulated in the hearts of these mice. This group also evaluated Mdm2FM/+Mdm4+/Δ2Tie2-Cre mice in order to determine whether loss of only one allele of Mdm2 in conjunction with loss of one allele of Mdm4 would produce different results than loss of both copies of Mdm2 (Mdm2FM/FMTie2-Cre). Mdm2FM/+Mdm4+/Δ2Tie2-Cre mice are viable yet the majority die within the first 2 weeks after birth with all mice dying by 8 weeks. The majority of Mdm2FM/+Mdm4+/Δ2Tie2-Cre mice did not achieve closure of their atrial or ventricular septum leading to significant decreases in heart function and arrhythmias. Thus, haploinsufficiency of Mdm2 and Mdm4 in the endothelial cells of mice leads to heart malfunctions detrimental enough to lead to postnatal death. Additionally, the phenotypic defects of these Mdm2FM/+Mdm4+/Δ2Tie2-Cre mice can be rescued with deletion of one copy of p53 (p53−/−Mdm2FM/+Mdm4+/Δ2Tie2-Cre).
Mdm2 is vital for the development of fully functioning kidneys
The majority of kidney failure cases are linked to developmental defects in proper ureteric bud formation. This process is directly affected by choreographed cell death and proliferation. Studies in mice with low amounts of Mdm2 or Mdm4 reveal small and poorly functioning kidneys (Terzian et al., 2007), suggesting that p53 levels may impact normal kidney function. An intricate spatial relationship between Mdm2 and p53 exists during kidney development, with both genes being highly expressed in the kidney early in embryogenesis and then slowly declining during the remainder of development. Mdm2 was deleted specifically in the ureteric bud lineage using αHoxb7-Cre transgene in conjunction with the Mdm2flox allele, resulting in a functionally null Mdm2 allele (Mendrysa et al., 2003; Zhao et al., 2004; Hilliard et al., 2011). Significant differences between the Mdm2flox/+αHoxb7-Cre and control kidneys are seen during embryogenesis as early as E11.5. The phenotypic and morphological defects observed correlate with an increase in apoptosis in the ureteric bud branches at E14.5. The embryonic Mdm2flox/+αHoxb7-Cre kidney is riddled with apoptosis by E16.5. In addition to this, the mutant kidneys experience a decrease in cell proliferation, further explaining why they are underdeveloped. At postnatal day 1 (P1), Mdm2flox/+αHoxb7-Cre kidneys are visibly smaller and misshapen when compared to controls, and mice do not survive past weaning. The cortical and medullary areas of the mutant kidneys are indiscernible. Furthermore, the ureteric bud branches fail to extend the entire length of the kidney. All phenotypes are p53-dependent and can be rescued with concomitant loss of p53. More details on the roles of Mdm2 and Mdm4 specifically in the context of kidney development are published in an accompanying review by El-Dahr et al. (2017) in this special issue.
Eyeless mice
Eye development is a precise process relying on tight regulation of multiple transcription factors. The ectoderm slowly thickens starting at E9.5 to form the lens of the eye. At this time, p53-mediated apoptosis, differentiation, and proliferation play crucial roles, implicating Mdm2 and Mdm4 in morphogenesis of the eye (Zhang et al., 2014b). The Lens-Cre (Le-Cre) transgene is used in combination with conditional alleles of Mdm2 and Mdm4 in order to remove these genes in the eye lens. The transgene begins expressing in the surface ectoderm around E9 and recombination is seen at E9.5. At E10, there are clear signs that the lens is unable to develop, progressing to E12.5 at which time a misshapen retina is all that remains in Mdm2FM/FMLe-Cre mice. Loss of Mdm2 in the lens thus results in an eyeless Mdm2FM/FMLe-Cre mouse and neonatal lethality of unknown causes. Using the same genetic approach, loss of Mdm4 also results in a viable eyeless Mdm4FX/FXLe-Cre mouse. The delay of the Mdm4FX/FXLe-Cre eyeless phenotype can be seen at E10.5, approximately an embryonic day later than Mdm2FM/FMLe-Cre mice. These data indicate that Mdm2 and Mdm4 play independent roles in development and maintenance of the lens structure.
The loss of Mdm2 and Mdm4 by Le-Cre increases p53 levels. TUNEL staining and BrdU incorporation are both notably increased signifying the increase in p53 function in Mdm2 and Mdm4-deficient eye tissue. Notably, Pax6, a transcription factor highly expressed in eye development, does not show any significant differences following loss of Mdm2 or Mdm4 and thus, functions separately from the p53 pathway during lens organogenesis. Not surprisingly, all phenotypes of Mdm2FM/FMLe-Cre or Mdm4FX/FXLe-Cre mice are completely absolved through concomitant loss of p53.
Mdm2, but not Mdm4, is necessary for smooth muscle cell homoeostasis
Boesten et al. (2006) wanted to determine whether non-cycling cells are dependent on p53 similar to rapidly proliferating cell types. They used a tamoxifen-inducible Cre-recombinase system in conjunction with either the floxed Mdm2 or Mdm4 allele in order to inactivate these genes specifically within the terminally differentiated smooth muscle cells (SMCs) of the intestine. This Cre is under the control of the SM22 gene promoter, restricting its activity to the vascular and visceral SMCs of adult mice (Kuhbandner et al., 2000). SM-CreERT2Mdm2FM/FM have significantly decreased body weights, are less responsive to stimuli, and develop hunched backs. Necropsies reveal a shortening and dilation of the small intestine. It is likely that the SM-CreERT2Mdm2FM/FM mice die too quickly for additional significant pathologies to arise within these tissues—all mice are moribund between 8 and 11 days after the start of tamoxifen injections. Additionally, loss of Mdm2 causes an elevation in the protein levels of p53 and its transcriptional activity in the affected tissues. Transcriptional targets, such as p21, are increased at the mRNA and protein level. Additionally, there is no evidence of increased apoptosis in the affected cells, showcasing that cell cycle regulation, but not cell death, is the key p53 stress pathway of SMCs after Mdm2 loss. The phenotypes observed in SM-CreERT2Mdm2FM/FM mice can be fully rescued through loss of p53. Unlike Mdm2, Mdm4 is not essential for normal SMC homoeostasis in mice. Mice that lose Mdm4 (SM-CreERT2Mdm4FX/FX) appear phenotypically normal compared to controls and show no signs of decreased functionality or increased p53 levels and activity.
Intestines bypass Mdm2 loss
The epithelium of the intestines has the fastest cellular turnover taking between 24 and 60 h to repopulate the entire organ (Creamer et al., 1961). Radiation of the intestine reveals that p53-dependent cell fate is determined by cell type. Undifferentiated cells are more sensitive to damage and thus apoptose, while differentiated epithelium has the ability to arrest.
To determine the significance of Mdm2 in regulating p53 in the intestinal epithelium, Villin-Cre was used (Valentin-Vega et al., 2008). This gene is first active at E9 (Madison et al., 2002). Surprisingly, loss of Mdm2 in the intestine leads to a viable mouse (Mdm2FM/FMVil-Cre or Mdm2FM/−Vil-Cre). However, many early adverse phenotypes exist. These Mdm2FM/FMVil-Cre mice display significantly lower body weights than their control littermates despite the fact that they are properly feeding and obtaining milk. This difference is apparent as early as P3. In addition, the intestines of these Mdm2FM/FMVil-Cre mice show signs of inflammation as well as an increase in the intervillus pocket, indicating decreased absorption ability and abnormal tissue structure. Loss of Mdm2 in the intestine leads to increased stabilization and activity of p53. In fact, the more severe the phenotype (smallest mice), the more p53-positive cells are present within the epithelium. Unexpectedly, no cell cycle arrest occurs despite an increase in mRNA levels of known cell cycle arrest-related p53 target genes. Surprisingly, with time, the intestinal epithelium of Mdm2FM/FMVil-Cre mice selects against cells expressing Cre and regenerates the entire organ with cells that lack Cre and retain functional Mdm2. The intestinal epithelium increases proliferation of Mdm2-expressing cells and increases crypt fission events. This is dependent on intestinal stem cells, which also increase in number. As with other Mdm2 loss-of-function phenotypes, this too is rescued by p53 loss.
The same Cre was used to explore loss of Mdm4 in the intestinal epithelium (Mdm4FX/FXVil-Cre or Mdm4FX/Δ2Vil-Cre) (Valentin-Vega et al., 2009). This leads to a viable mouse with no obvious phenotypic differences throughout development and adulthood. However, there is an increase in p53 activity in the intestinal epithelium of these Mdm4FX/FXVil-Cre mice, but it is confined to the highly proliferative areas. Similarly, staining reveals that apoptotic response in the intestine of these Mdm4FX/FXVil-Cre mice is also confined to the highly proliferative cells after Mdm4 loss. Additionally, p21 is increased throughout the intestine despite the fact that the proliferation rate of these cells is not affected by the loss of Mdm4. In this cellular context, Mdm2 regulates p53 activity throughout the intestines, whereas Mdm4 only shows signs of p53 regulation specifically in the highly proliferative compartments.
Loss of Mdm2 in oocytes results in infertility
To determine whether Mdm2 and Mdm4 are implicated in the survival of oocytes during their development, a Cre gene driven by the Zona Pellucida Glycoprotein 3 promoter (Zp3-Cre), which is exclusively expressed in growing oocytes, is used in conjunction with the conditional Mdm2FM and Mdm4FX alleles (de Vries et al., 2000; Livera et al., 2016). Loss of Mdm2 in the oocytes (Mdm2FM/−Zp3-Cre) leads to significantly decreased fertility, irregular menstrual cycling, and small ovaries. However, not all of the oocytes display the same phenotypes after loss of Mdm2. The authors hypothesized that the abnormal hormonal environment (high plasma follicle-stimulating hormone and luteinizing hormone coupled with low anti-mullerian hormone levels) and the resulted reproductive tract defects in the Mdm2FM/−Zp3-Cre mouse cause the fertility defects, not loss of Mdm2 in oocytes. Crosses to p53-null mice rescue the phenotypes observed in the Mdm2 knockout oocytes and restore fertility. Loss of Mdm4 in oocytes (Mdm4FX/−Zp3-Cre) has no effect on fertility, hormone levels, nor show any morphological differences in the genital tract.
Global Mdm2 loss destroys normal tissues in adult mice
Since Mdm2 is the major negative regulator of p53, it has become an alluring therapeutic target for cancers that overexpress Mdm2 and remain WT for p53. However, these therapies are not tumour-specific and thus adverse effects of these drugs on normal tissues must also be explored. Zhang et al. (2014a) addressed this question by conditionally removing Mdm2 in a fully developed mouse (2–4 months old) using a tamoxifen-inducible CAG-CreER to recombine the Mdm2FM allele in all tissue types. Tamoxifen injections were administered once a day for three consecutive days, and all Mdm2FM/−CAG-CreER mice in the study died within 2 days after the last tamoxifen injection. It is important to note that although all tissues experience recombination of the Mdm2 allele, the degree of recombination is tissue-specific and does not directly correlate with the severity of the resulting phenotype.
Results indicate that both radio-sensitive and radio-insensitive tissues, such as the spleen and kidney, respectively, are negatively affected by the loss of Mdm2. Functional and morphological defects vary by age of the Mdm2FM/−CAG-CreER mouse and tissue type, yet all tissues experience some degree of perturbation of the p53 pathway shown through RT-PCR analysis of downstream targets. For example, after global Mdm2 loss, there is an increase in protein casts and tubule dilation in kidneys of 4-month-old mice, whilst maintaining normal blood urine nitrogen (BUN) concentration. Normal BUN concentration signifies that the Mdm2FM/−CAG-CreER kidneys are still functioning properly despite significant morphological differences. Additionally, the kidney harbours the highest levels of p53 target genes, p21 and Puma. Immunohistochemistry (IHC) reveals that p53 is stabilized in the kidney after Mdm2 loss and apoptosis is evident. Conversely, the Mdm2FM/−CAG-CreER liver appears phenotypically normal compared to control tissues. However, the level of aspartate aminotransferase increases substantially, indicating that the livers of these mice are not functioning properly. The liver also stained positively for p53 and apoptosis after interrupted tamoxifen injections despite no dramatic increase in the mRNA levels of p53 target genes. Interestingly, the Mdm2FM/−CAG-CreER heart has no obvious morphological defects after acute Mdm2 loss yet still shows significant increases in the mRNA levels of p53 target genes p21 and Puma. Additionally, IHC does reveal changes in p53 protein levels. However, after prolonged exposure to Mdm2 loss (through interrupted tamoxifen injections), fibrosis starts to show up in the heart. Increased exposure to Mdm2 loss causes stabilized p53 in addition to the morphological defects. Additionally, the same tamoxifen injections were given to year old mice. This study revealed that p53 activity was not as robust in older Mdm2-deleted mice.
Similarly, another group conducted analogous studies in which they globally explored p53 reactivation in Mdm2-null or Mdm4-null backgrounds (Ringshausen et al., 2006; Garcia et al., 2011). Both studies utilize the p53KI/− mouse model that contains a p53-ER fusion. In the absence of tamoxifen, the p53KI allele is functionally null (it should be noted however that p53KI/KI mice do not rescue the Mdm2-null phenotype but p53KI/− mice do). However, upon tamoxifen treatment, the WT p53 activity of this allele is restored. Thus, Mdm proteins are never present within these mice, and p53 activity is being toggled on and off by the presence or absence of tamoxifen injections. Mdm2−/−p53KI/− mice exhibit similar defects to those found in the study of Zhang et al. (2014a). After the reactivation of p53 through daily tamoxifen injections, these Mdm2−/−p53KI/− mice only survive for up to 6 days. At that time, tissues were harvested for analysis. This group found that defects are seen in radio-sensitive tissues as early as 6 h after reactivation of WT p53. Not surprisingly, these Mdm2−/−p53KI/− mice experience dramatic atrophy in radio-sensitive tissues such as the bone marrow, spleen (which lacks white pulp), thymus, small intestine, and colon (which has shortened villi). In addition, all of these tissues show dramatic upregulation in p21 and Puma expression, and have numerous apoptotic cells. These mice also exhibit haematopoietic defects evident by an extreme reduction in all blood cell types.
Contrary to the Zhang study, radio-insensitive tissues experience fewer defects when utilizing the p53KI allele. The Mdm2−/−p53KI/− heart, liver, kidney, and brain all remain morphologically normal with no signs of increased proliferation or apoptosis. However, all tissues, especially the kidney and the brain, display elevated mRNA levels of p21 and Puma, indicating that they are still affected by reactivation of p53.
Mdm4 −/− p53 KI/− mice surprisingly survive long past 1 week after p53 reactivation. Even with a continuation of tamoxifen injections, the median time of death is 29 days. This study reveals that lack of Mdm4 is deleterious only in radio-sensitive tissues, not radio-insensitive. However, although the morphological defects and upregulation of Puma mRNA are limited to the bone marrow, spleen, thymus, and intestinal epithelium, all Mdm4−/−p53KI/− tissues show upregulated p21 mRNA. Thus, the only phenotypic differences seen are increased apoptosis in all radio-sensitive tissues as well as severe anaemia that is thought to lead to bone marrow failure resulting in the demise of these mice. The Mdm4−/−p53KI/− liver, heart, kidney, and lung do not display any signs of apoptosis.
These studies exemplify the fact that radio-sensitive and radio-insensitive tissues are vulnerable to the loss of Mdm2 (Mdm2−/−p53KI/−), while only radio-sensitive ones are vulnerable to Mdm4 (Mdm4−/−p53KI/−), yet both result in the demise of mice due to unrestricted activity of p53. However, it is important to note that the results from the p53KI allele are different from the genetic ablation of Mdm2 due to the presence of only one copy of p53, which must be kept active by continuous tamoxifen injections. The tissue-specificity of unrestricted p53 activity must be explored further to better understand the differences observed in these studies.
Alterations in Mdm2 levels alone are sufficient to achieve deleterious unrestricted p53 affects
Haploinsufficiency (50% of normal expression) of Mdm2 or Mdm4 leads to increased radiosensitivity due to defects in the haematopoietic system in a p53-dependent manner (Grier et al., 2006; Terzian et al., 2007). The Mdm2/Mdm4 double heterozygotes (Mdm2+/−Mdm4+/−) show defects in neural tube closure during development and have multiple organ defects. Furthermore, all blood cells are depleted in the double heterozygotes, though there is no sign of this in single heterozygotes (Mdm2+/− or Mdm4+/−). Importantly, all of the double heterozygotes (Mdm2+/−Mdm4+/−) die within 20 days of birth, signifying the importance of the levels of these proteins in survival and proper development. All, however, are rescued by concurrent loss of one allele of p53.
Mendrysa et al. (2003) generated a hypomorphic allele for Mdm2 (not Mdm4), which only expresses 30% of WT levels. These Mdm2puro/Δ7–12 mice are viable with defects in homoeostatic tissues. These mice have smaller body weights accompanied by small organs with those having the most significant decrease in size being the spleen and thymus. Not surprisingly, these Mdm2puro/Δ7–12 mice also have a poorly functioning haematopoietic system. They exhibit significant reduction of red blood cells, white blood cells, lymphocytes, T cells, and B cells compared to WT animals. Additionally, the lymphatic system of Mdm2puro/Δ7–12 mice shows twice the normal amount of apoptosis and an increase in p53 transcriptional targets, with p21 experiencing the greatest upregulation. As expected, these mice also exhibit a p53-dependent increased sensitivity to IR.
Recently, another Mdm2 hypomorphic mouse (Mdm2PND/PND) was generated and identified due to increased pigmentation of its ears, tail, and paws (Pant et al., 2016). These mice express levels of Mdm2 close to the threshold needed for survival; Mdm2PND/PND mice are viable whilst Mdm2PND/− mice are not. This mouse harbours a mutation in the P2 promoter in addition to a large deletion in intron 3 and retains a neomycin cassette. Thus, minimal levels of Mdm2 are being encoded solely from the P1 promoter. Characterization of these mice reveals high basal levels of canonical p53 targets expressed in spleens that increase upon radiation. Furthermore, Mdm2PND/PND mice are extremely sensitive to radiation and die after exposure to 3-gray IR despite the fact that this is not immediately lethal for WT mice. Even without induced stresses such as IR, these Mdm2PND/PND mice have shorter lifespans than WT controls, signifying that the prolonged increase in p53 is harmful.
Further phenotypic defects include weight differences in the Mdm2PND/PND mice compared to WT controls at 1 year of age. Not only do the Mdm2PND/PND mice weigh less in general, but they also have smaller hearts and testicles at 6 weeks of age. The small heart phenotype resolves itself by 1 year of age at which time they weigh the same as WT hearts. The reasons for the differences in heart weight are unknown, but the Mdm2PND/PND mice do not seem to be negatively affected by the reduced heart weight during development. Despite some mice being fertile, the litter numbers are lower than expected, and the reproductive tracts of these mice are significantly smaller throughout their life and contain less germ cells resulting in breeding difficulties.
Not surprisingly, the haematopoietic system of the Mdm2PND/PND mice is also altered. Blood count analyses reveal that both white and red blood cell counts in these mice are significantly lower than the WT age and sex-matched controls. The obvious dark pigmentation in these mice is coupled with spindled melanocytes and higher than normal melanin content. Furthermore, the pigmentation changes are specific to the epidermal layer; the dermis is indistinguishable from WT. This signifies clear tissue-specificity of Mdm2 loss and p53 hyperactivity, though the reasons for these phenotypic differences have not yet been determined.
Loss of one copy of p53 is sufficient to alleviate the melanin hyperpigmentation and completely rescues the differences in weight and reproductive tract deficiencies of the Mdm2PND/PND mice (Pant et al., 2016). Complete loss of p53 was able to fully restore normal skin pigmentation. Moreover, to explore the tissue-specificity of downstream p53 targets, p21-null, Puma-null, and p21;Puma double null mice were crossed with Mdm2PND/PND mice as attempted rescues. p21-loss decreases the severity of testes shrinkage, but does not rescue fertility nor does it alleviate any reproductive issues seen in Mdm2PND/PND females, suggesting gender differences in sensitivity to hypomorphic Mdm2. Puma-loss completely rescues all reproductive shortcomings, resulting in normal litter numbers and sizes. The Mdm2PND/PNDp21−/−Puma−/− mice were generated in attempts to rescue the hyperpigmentation phenotype, after Puma-loss and p21-loss individually were unsuccessful. However, the double knockout also does not resolve the pigmentation alterations. Kit ligand (Kitl) is a downstream target of p53 that is involved in melanocyte migration, and Kitl mRNA levels are significantly upregulated in the Mdm2PND/PND animals. However, removal of Kitl to determine whether melanocytes are able to return to normal is not possible due to the fact that Kitl inhibitors only affect melanocytes that have not yet migrated. Thus, inhibitors would have no effect on existing hyperpigmentation. It is important to note, however, that crossing the Mdm2PND/PND mouse to a Kitl knockout mouse may successfully alleviate the skin pigmentation phenotype, but this has not been done.
Perspective and conclusions
A large number of studies, summarized in this review, have probed the importance of Mdm2 and Mdm4 in a cell and tissue-specific manner. Regardless of the Cre driving deletion of Mdm2, Mdm2 loss results in a cell lethal phenotype. For example, deletion in the heart results in cardiac defects that are lethal, deletion in the intestine results in compensatory repopulation of the intestinal epithelium with cells that express Mdm2, and global deletion of Mdm2 in two different models results in a myriad of detrimental phenotypes that are lethal. All of these phenotypes are p53-dependent, further fortifying the role of Mdm2 as a major inhibitor of p53 activity.
Mdm4 loss, on the other hand, is tolerated by many cell types but not all. During the highly proliferative embryogenesis process, Mdm4 is required in the CNS during development, but not the heart for example. Interestingly, Mdm4 loss is relatively well tolerated in many adult tissues. In all of these cases, the phenotypes are p53-dependent, emphasizing that too much p53 is detrimental to cell viability. It is the highly proliferative cells that appear to need both Mdm2 and Mdm4; perhaps because these cells need to dampen p53 levels quickly to allow developmental processes to proceed in a timely manner. The observation that disruption of the Mdm2–Mdm4 interaction is embryonic lethal further supports the idea that the Mdm2/Mdm4 heterodimer is the best p53 inhibitor during the highly proliferative embryogenesis process.
Studies of haploinsufficient and hypomorphic mice have further highlighted the need for Mdm2 in adult tissues. The haematopoietic system is a highly proliferative one that shows defects in many cases with slight decreases in Mdm2 levels. These studies, as well, have emphasized the need for increased levels of Mdm2 in response to damage. DNA damage increases p53 levels, and the basal levels of Mdm2 that exist in these mice are insufficient to return p53 to normal quickly enough, thus resulting in lethal phenotypes. The feedback loop-deficient mice are a perfect example of the need to make more Mdm2 to dampen p53 activities after DNA damage. These studies suggest that the relationship between Mdm2 and p53 is dynamic and changes with the needs of the cell.
Lacking is a real understanding of the p53 downstream effectors in a cell and tissue-specific manner. Kitl appears to be an important target in melanocytes and Lif in the uterus for implantation (Hu et al., 2007; Pant et al., 2016). p21 is elevated 200-fold in kidneys upon Mdm2 loss, the consequences of which are unknown. Puma deletion bypasses the fertility defects in Mdm2 hypomorphic mice. An understanding of the factors implementing p53 function in a cell-specific manner may yield new therapeutic targets for reactivation of the p53 pathway in tumour development.
Since many cancers have high Mdm2 levels and WT p53, Mdm2 inhibitors have been developed and are now in clinical trials. Mdm2 inhibitors have yielded toxicities—the observed haematopoietic toxicities were predicted from mouse models as far back as 2003 (Mendrysa et al., 2003). Another interesting observation that needs further exploration is the fact that Mdm2 loss results in less p53 activity in older mice as compared to young mice. This difference may be exploited in treatment of older patients. While current therapies target Mdm2, Mdm4 loss often displays less dramatic or deleterious effects, suggesting that Mdm4 may be a better therapeutic target due to its decreased toxicity. Clearly, additional studies are needed to understand the p53 signalling pathways upregulated upon Mdm2 and Mdm4 loss in a cell and tissue-specific manner.
Acknowledgements
We thank Vinod Pant, Amanda Wasylishen, and Yun Zhang (Department of Genetics, The University of Texas MD Anderson Cancer Center) for their helpful discussion and comments on this review. We would also like to acknowledge all the other studies in this field that were not discussed or cited solely due to space limitations.
Funding
This work is supported by NIH grant CA47296 to G.L.
Conflict of interest: none declared.
References
- Agah R., Frenkel P.A., French B.A., et al. (1997). Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J. Clin. Invest. 100, 169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak Y., Gottlieb E., Juven-Gershon T., et al. (1994). Regulation of mdm2 expression by p53: alternative promoters produce transcripts with nonidentical translation potential. Genes Dev. 8, 1739–1749. [DOI] [PubMed] [Google Scholar]
- Boesten L.S., Zadelaar S.M., De Clercq S., et al. (2006). Mdm2, but not Mdm4, protects terminally differentiated smooth muscle cells from p53-mediated caspase-3-independent cell death. Cell Death Differ. 13, 2089–2098. [DOI] [PubMed] [Google Scholar]
- Chavez-Reyes A., Parant J.M., Amelse L.L., et al. (2003). Switching mechanisms of cell death in mdm2- and mdm4-null mice by deletion of p53 downstream targets. Cancer Res. 63, 8664–8669. [PubMed] [Google Scholar]
- Creamer B., Shorter R.G., and Bamforth J (1961). The turnover and shedding of epithelial cells. I. The turnover in the gastro-intestinal tract. Gut 2, 110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries W.N., Binns L.T., Fancher K.S., et al. (2000). Expression of Cre recombinase in mouse oocytes: a means to study maternal effect genes. Genesis 26, 110–112. [PubMed] [Google Scholar]
- El-Dahr S., Hilliard S., and Saifudeen Z (2017). Regulation of kidney development by the Mdm2/Mdm4–p53 axis. J. Mol. Cell Biol. doi: 10.1093/jmcb/mjx005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francoz S., Froment P., Bogaerts S., et al. (2006). Mdm4 and Mdm2 cooperate to inhibit p53 activity in proliferating and quiescent cells in vivo. Proc. Natl Acad. Sci. USA 103, 3232–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia D., Warr M.R., Martins C.P., et al. (2011). Validation of MdmX as a therapeutic target for reactivating p53 in tumors. Genes Dev. 25, 1746–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graus-Porta D., Blaess S., Senften M., et al. (2001). β1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron 31, 367–379. [DOI] [PubMed] [Google Scholar]
- Grier J.D., Xiong S., Elizondo-Fraire A.C., et al. (2006). Tissue-specific differences of p53 inhibition by Mdm2 and Mdm4. Mol. Cell. Biol. 26, 192–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilliard S., Aboudehen K., Yao X., et al. (2011). Tight regulation of p53 activity by Mdm2 is required for ureteric bud growth and branching. Dev. Biol. 353, 354–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W., Feng Z., Teresky A.K., et al. (2007). p53 regulates maternal reproduction through LIF. Nature 450, 721–724. [DOI] [PubMed] [Google Scholar]
- Huang L., Yan Z., Liao X., et al. (2011). The p53 inhibitors MDM2/MDMX complex is required for control of p53 activity in vivo. Proc. Natl Acad. Sci. USA 108, 12001–12006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itahana K., Mao H., Jin A., et al. (2007). Targeted inactivation of Mdm2 RING finger E3 ubiquitin ligase activity in the mouse reveals mechanistic insights into p53 regulation. Cancer Cell 12, 355–366. [DOI] [PubMed] [Google Scholar]
- Jones S.N., Roe A.E., Donehower L.A., et al. (1995). Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 378, 206–208. [DOI] [PubMed] [Google Scholar]
- Kisanuki Y.Y., Hammer R.E., Miyazaki J., et al. (2001). Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev. Biol. 230, 230–242. [DOI] [PubMed] [Google Scholar]
- Kuhbandner S., Brummer S., Metzger D., et al. (2000). Temporally controlled somatic mutagenesis in smooth muscle. Genesis 28, 15–22. [DOI] [PubMed] [Google Scholar]
- Livera G., Uzbekov R., Jarrier P., et al. (2016). Loss of oocytes due to conditional ablation of Murine double minute 2 (Mdm2) gene is p53-dependent and results in female sterility. FEBS Lett. 590, 2566–2574. [DOI] [PubMed] [Google Scholar]
- Madison B.B., Dunbar L., Qiao X.T., et al. (2002). Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J. Biol. Chem. 277, 33275–33283. [DOI] [PubMed] [Google Scholar]
- Mendrysa S.M., McElwee M.K., Michalowski J., et al. (2003). mdm2 Is critical for inhibition of p53 during lymphopoiesis and the response to ionizing irradiation. Mol. Cell. Biol. 23, 462–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliorini D., Lazzerini Denchi E., Danovi D., et al. (2002). Mdm4 (Mdmx) regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Mol. Cell. Biol. 22, 5527–5538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montes de Oca Luna R., Wagner D.S., and Lozano G (1995). Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 378, 203–206. [DOI] [PubMed] [Google Scholar]
- Ng W.A., Grupp I.L., Subramaniam A., et al. (1991). Cardiac myosin heavy chain mRNA expression and myocardial function in the mouse heart. Circ. Res. 68, 1742–1750. [DOI] [PubMed] [Google Scholar]
- Pant V., Xiong S., Chau G., et al. (2016). Distinct downstream targets manifest p53-dependent pathologies in mice. Oncogene 35, 5713–5721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant V., Xiong S., Iwakuma T., et al. (2011). Heterodimerization of Mdm2 and Mdm4 is critical for regulating p53 activity during embryogenesis but dispensable for p53 and Mdm2 stability. Proc. Natl Acad. Sci. USA 108, 11995–12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant V., Xiong S., Jackson J.G., et al. (2013). The p53-Mdm2 feedback loop protects against DNA damage by inhibiting p53 activity but is dispensable for p53 stability, development, and longevity. Genes Dev. 27, 1857–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parant J., Chavez-Reyes A., Little N.A., et al. (2001). Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat. Genet. 29, 92–95. [DOI] [PubMed] [Google Scholar]
- Ringshausen I., O'Shea C.C., Finch A.J., et al. (2006). Mdm2 is critically and continuously required to suppress lethal p53 activity in vivo. Cancer Cell 10, 501–514. [DOI] [PubMed] [Google Scholar]
- Tanimura S., Ohtsuka S., Mitsui K., et al. (1999). MDM2 interacts with MDMX through their RING finger domains. FEBS Lett. 447, 5–9. [DOI] [PubMed] [Google Scholar]
- Terzian T., Wang Y., Van Pelt C.S., et al. (2007). Haploinsufficiency of Mdm2 and Mdm4 in tumorigenesis and development. Mol. Cell. Biol. 27, 5479–5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollini L.A., Jin A., Park J., et al. (2014). Regulation of p53 by Mdm2 E3 ligase function is dispensable in embryogenesis and development, but essential in response to DNA damage. Cancer Cell 26, 235–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentin-Vega Y.A., Box N., Terzian T., et al. (2009). Mdm4 loss in the intestinal epithelium leads to compartmentalized cell death but no tissue abnormalities. Differentiation 77, 442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentin-Vega Y.A., Okano H., and Lozano G (2008). The intestinal epithelium compensates for p53-mediated cell death and guarantees organismal survival. Cell Death Differ. 15, 1772–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Bayle J.H., Olson D., et al. (1993). The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 7, 1126–1132. [DOI] [PubMed] [Google Scholar]
- Xiong S., Van Pelt C.S., Elizondo-Fraire A.C., et al. (2007). Loss of Mdm4 results in p53-dependent dilated cardiomyopathy. Circulation 115, 2925–2930. [DOI] [PubMed] [Google Scholar]
- Xiong S., Van Pelt C.S., Elizondo-Fraire A.C., et al. (2006). Synergistic roles of Mdm2 and Mdm4 for p53 inhibition in central nervous system development. Proc. Natl Acad. Sci. USA 103, 3226–3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., He X., Chen L., et al. (2012). Synergistic regulation of p53 by Mdm2 and Mdm4 is critical in cardiac endocardial cushion morphogenesis during heart development. J. Pathol. 228, 416–428. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Xiong S., Li Q., et al. (2014. a). Tissue-specific and age-dependent effects of global Mdm2 loss. J. Pathol. 233, 380–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Zhang X., and Lu H (2014. b). Aberrant activation of p53 due to loss of MDM2 or MDMX causes early lens dysmorphogenesis. Dev. Biol. 396, 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H., Kegg H., Grady S., et al. (2004). Role of fibroblast growth factor receptors 1 and 2 in the ureteric bud. Dev. Biol. 276, 403–415. [DOI] [PMC free article] [PubMed] [Google Scholar]