

Abstract
Hemin is known to induce endocytosis of prion-protein (PrPC) from the neuronal plasma membrane, potentially limiting propagation of the disease causing PrP-scrapie (PrPSc) isoform. Hemin is therefore an attractive disease-modifying option for sporadic Creutzfeldt-Jakob disease (sCJD), a human prion disorder with no effective treatment. The hemin-PrPC interaction is also of interest in cerebral-hemorrhage (CH), a condition where potentially toxic hemin molecules come in contact with neuronal PrPC. Interestingly, PrPC is upregulated in penumbric neurons surrounding CH and is known to confer neuroprotection in a dose-dependent manner. The underlying mechanism, however, is not clear. Here, we report that hemin binds PrPC on diverse cell lines, resulting in its aggregation or degradation in a cell-type specific manner. Surprisingly, the hemin-PrPC interaction upregulates Hb synthesis in hematopoietic cells, a response reversed by deleting the hemin-binding octa-peptide repeat region of PrPC. A similar response is noted in brain organotypic cultures where exposure to hemin induces significantly more α-globin in wild-type (PrP+/+) relative to PrP-knock-out (PrP−/−) samples. Furthermore, red blood cells and brain tissue from PrP−/− mice show significantly less α-globin relative to PrP+/+ controls, indicating a positive effect of PrPC on Hb synthesis under physiological conditions as well. Surprisingly, levels of α-globin are significantly higher in sCJD brain tissue relative to controls, suggesting compensatory upregulation of Hb synthesis by surviving neurons or misregulation in diseased brains. These observations reveal a unique function of PrPC that is likely to impact the therapeutic management of CH and sCJD.
Keywords: α-Globin, hemin, neuronal hemoglobin, prion protein, sCJD
INTRODUCTION
Prion protein (PrPC) is a ubiquitously expressed plasma membrane glycoprotein most abundant on neuronal cells. Although associated with diverse physiological functions, PrPC is mostly known as the substrate for PrP-scrapie (PrPSc), a β-sheet rich isoform implicated in the pathogenesis of neurodegenerative conditions known as prion disorders. Sporadic Creutzfeldt-Jakob disease (sCJD) is the most common human prion disorder for which there is no effective treatment [1]. Search for possible therapeutic drugs led to the identification of certain porphyrins that delayed disease progression in experimental models [2]. Among these, hemin emerged as an attractive option because it induced rapid endocytosis of PrPC, thereby limiting further replication of PrPSc [3]. The interaction of hemin with PrPC was therefore thoroughly examined using recombinant PrPC and purified hemin in vitro, and in mouse neuroblastoma cells in culture where extracellular hemin induced rapid internalization of plasma membrane PrPC followed by aggregation in an intracellular compartment. Deletion of the N-terminal octa-peptide repeats of PrPC abolished this interaction, associating this region with hemin binding activity [3].
Besides sCJD, the interaction of hemin with PrPC is of clinical significance in cerebral hemorrhage (CH) where heme released from lysed red blood cells (RBCs) is rapidly oxidized to hemin and comes in contact with neuronal PrPC [4, 5]. It is believed that hemin can reach concentrations of up to 10 mM in affected brain regions [6], and is highly toxic due to its redox-active nature and the ability to intercalate within lipid membranes [6–8]. Mechanisms are therefore in place to clear free heme from the extra-cellular milieu by high affinity heme binding proteins such as hemopexin and haptoglobin [9]. Heme is also internalized by neurons, astrocytes, and macrophages by heme carrier protein 1 (HCP-1) and rapidly metabolized by the heme-oxygenase family of enzymes [10–14]. Despite these protective mechanisms, free hemin is a significant cause of neurotoxicity in CH. It is notable that PrPC is upregulated in regions affected by CH and is believed to provide neuroprotection in a dose-dependent manner [15–17]. Thus, experimentally induced cerebral hemorrhage causes significantly more neuronal death in mice lacking PrP (PrP−/−) [18] compared to wild-type controls (PrP+/+) [16, 17, 19–21], and relatively less neuronal injury in mice overexpressing PrPC [16, 22]. The underlying mechanism, however, has remained unclear.
Here, we evaluated the downstream effects of hemin-PrPC interaction using a combination of cell lines, transgenic mouse models, and brain tissue from sCJD and age-matched non-dementia controls. The main emphasis was on hemin-induced synthesis of Hb, a phenomenon studied extensively in the erythroleukemia cell line K562 [23–28] and reported recently in neuronal and glial cells [29–33]. Since neurons express modulators of hemoglobin synthesis such as the erythropoietin receptor and the hypoxia-inducible factor HIF1 [34–36], modulation of neuronal Hb by PrPC via hemin or otherwise is likely to improve neuronal viability in pathological conditions such as CH and sCJD.
In the experimental paradigms described below, expression of α-globin was considered representative of Hb concentration since this globin chain is common to both fetal Hb expressed by K562 cells and adult Hb synthesized by mouse and human RBCs and brain tissue. Under physiological conditions the expression of α-globin and β-globin chains is closely balanced since free α-globin has a tendency to aggregate and cause toxicity [37, 38].
We report that cell surface PrPC binds and internalizes extracellular hemin in hematopoietic and neuronal cell lines. This interaction is reversed by deleting the octa-peptide repeats of PrPC [3], associating this region with hemin-binding activity. More importantly, PrPC upregulates hemin-mediated synthesis of Hb in K562 cells and mouse brain organotypic cultures, indicating a positive role in hematopoietic and neuronal/glial cell Hb synthesis. Moreover, levels of α-globin and β-globin are reduced in RBCs and brain tissue of PrP−/− mice relative to PrP+/+ controls, suggesting PrPC-mediated modulation of Hb synthesis under physiological conditions as well. Unexpectedly, levels of α-globin are significantly increased in sCJD brain samples relative to non-dementia controls, indicating a compensatory response by surviving neurons or altered synthesis of Hb in diseased brains. These observations are discussed in the context of CH and sCJD where neuronal and glial cell Hb synthesis are likely to provide neuroprotection.
MATERIALS AND METHODS
Animals and ethics statement
Colonies of FVB/NJ wild type (PrP+/+) and transgenic mice lacking PrP (PrP−/−) [18] were housed in the animal facility at Case Western Reserve University (CWRU) under a 12-h day-night cycle and received ad libitum access to food and water. Animal protocols and procedures were approved by the Institutional Animal Care Committee and conformed to the recommendations of the American Veterinary Medical Association Panel on Euthanasia, United States Department of Agriculture, Public Health Service regulations and guidelines, and the Department of Health and Human Services, National Institutes of Health. The approved protocol number is 2015–0027. Standard Operating Procedures and reference materials were provided by the IACUC Office for animal use. The animal health program was directed by the Case Animal Resource Center Director, W. John Dur-fee, DVM, Diplomate ACLAM, and provided by two full-time veterinarians. Animals in each room were observed daily for signs of illness by the animal technician responsible for providing husbandry. Medical records and documentation of experimental use were maintained individually or by cage group. Veterinary technicians under the direction of the attending veterinarian provided routine veterinary medical care to all animals. Animal care and use was additionally monitored for training and compliance issues by the Training and Compliance Manager. AAALAC Accreditation, July 18, 2012 (current accreditation letter pending). USDA Registration is valid until August 23, 2017. The Case PHS Assurance number is A-3145-01, valid until 04/30/19.
Brain samples from autopsy-confirmed cases of sCJD (age 61, 70, 61, 65, 65, 73, 59, and 65 years) were obtained from the National Prion Disease Pathology Surveillance center (NPDPSC) at CWRU. Non-dementia human brain samples (age 74, 82, 77, 73, 73, 72, 77, and 79 years) were from Harvard Brain Tissue Resource Center. Pre-mortem cerebrospinal fluid (CSF) samples from autopsy-confirmed cases of sCJD (age 48, 49, and 50 years) and non-dementia controls (age 48, 48, and 49 years) were from NPDPSC.
Antibodies and reagents
Antibodies 3F4 and 8H4 specific for human and both human and mouse PrPC, respectively, were obtained from Signet Laboratories (Dedham, MA), and Abcam (cat #ab61409, Boston, MA). Antibodies specific to ferritin and β-actin were obtained from Sigma (cat# F5012, Sigma-Aldrich, St. Louis, MO) and Millipore (cat# MAB1501, Bedford, MA) respectively. Antibody to α-globin was from Abcam (cat# ab102758, Boston, MA), and for β-globin, neuroglobin, and glycophorin-A from Santa Cruz Biotechnology Inc. (cat# sc21757, sc30144, and sc19453, Dallas, Texas). Antibody specific for Tf was from GeneTex (cat# GTX21223, Irvine, CA) and for TfR from Millipore (cat# CBL47, Bedford, MA). Hemin (cat# 51280), protoporphyrin IX (cat# P8293) and FAC (cat# F5879) were purchased from Sigma (Sigma-Aldrich, St. Louis, MO). Cell culture supplies were from GIBCO (Life Technologies). All other chemicals were purchased from Sigma Aldrich (St. Louis, MO).
Cell lines and culture conditions
The hematopoietic cell line K562 and neuroblastoma cell lines SH-SY5Y and M17 were obtained from ATCC (Manassas, VA). K562 cells were cultured in RPMI medium supplemented with 10% FBS and antibiotics. SHSY5Y and M17 cells were cultured in OPTI-MEM supplemented with 10% FBS and antibiotics. Stable cell lines expressing PrPC, PrPΔ51-89 with and without the green fluorescent protein inserted between amino acids 38 and 39 of the prion protein were generated as described in previous reports [39–41]. Stock solutions of hemin and protoporphyrin IX were prepared in 50 mM NaOH (in 100 mM Tris HCl, pH-7.4) and used at a concentration of 10–30 μM and 3 μM respectively [42–44]. FAC was used at a concentration of 30 μM. Benzidine staining for the detection of Hb was performed as described [45].
SDS page and western blotting
Cells exposed to different conditions were lysed and processed for western blotting as described in previous reports [40, 46]. Antibody concentrations used were 1:5000 for 3F4, 1:1000 for α-globin, 1:750 for β-globin, 1:2500 for ferritin, 1:7000 for transferrin, 1:2000 for TfR, 1:300 for glycophorin-A, and 1:10000 for β-actin. Protein band density was quantified with UN-SCAN-IT gel 6.1 gel analysis software (Silk Scientific, Inc. Orem, UT).
Mouse brain organotypic slice culture
Brains harvested from 8 day old PrP+/+ and PrP−/− pups were sliced to a thickness of 300 μM and cultured on transwell membrane inserts (0.4 μ pore size) for 1 week in high glucose DMEM supplemented with 10% FBS and antibiotics before the addition of 20 μM hemin to experimental samples. Subsequently, brain slices were washed with PBS, incubated in RBC lysis buffer [47, 48], and processed for SDS-PAGE and western blotting. To detect α-globin expression in mice brain, 14d old mice pups (PrP+/+ and PrP−/−) were sacrificed as per approved protocol. Brain tissue was triturated, incubated in RBCs lysis buffer, washed with chilled PBS (2X), and processed for SDS-PAGE and western blotting.
Statistical analysis
Data were analyzed using GraphPad Prism5 software (GraphPad Software, Inc., La Jolla, CA) and presented as Mean ± SEM. Level of significance was calculated by unpaired t-test between the control and treatment group.
RESULTS
Hemin induces endocytosis of PrPC in neuroblastoma and hematopoietic cells
To evaluate whether hemin induces endocytosis of PrPC independent of its cellular micro-environment, neuroblastoma cell lines SH-SY5Y and the erythroleukemia cell line K562 expressing green-fluorescent-protein tagged PrPC (PrPC-GFP) or a deletion construct lacking the hemin-binding octa-peptide repeat domain (PrPΔ51-89-GFP) were exposed to 10–30 μM of hemin, and images of live cells were captured at different time-points. A representative result from 30 minutes of exposure is shown (Fig. 1). Hemin induced rapid endocytosis and intracellular accumulation of PrPC-GFP expressed on SH-SY5Y cells, but had no effect on the distribution of PrPΔ51-89-GFP (Fig. 1, panels 1–4). Similar observations were noted in M17 cells expressing PrPC-GFP and PrPΔ51-89-GFP (data not shown). In K562 cells, on the other hand, hemin reduced the expression of PrPC-GFP from the plasma membrane without significant accumulation in intracellular vesicles. The expression pattern of PrPΔ51-89-GFP was unaltered by hemin as in neuroblastoma cell lines (Fig. 1, panels 5–8). Interestingly, exposure to hemin resulted in clustering of PrPC-GFP and PrPΔ51-89-GFP-expressing K562 cells (Fig. 1, panels 6 & 8).
Fig. 1.

Hemin induces endocytosis of PrPC. SH-SY5Y and K562 cells expressing PrPC-GFP or PrPΔ51-89-GFP were exposed to vehicle (left panels) or 30 μM hemin (right panels), and images of live cells were captured after 30 minutes. Hemin induces endocytosis of PrPC-GFP in both SH-SY5Y and K562 cells (panel 1 & 5 versus 2 & 6). In SH-SY5Y cells PrPC-GFP accumulates in intracellular endocytic vesicles (panel 2), while in K562 cells expression on the plasma membrane decreases without accumulation in endosomes. A small amount of PrPC-GFP and PrPΔ51-89-GFP are detected in the perinuclear region in the Golgi compartment (panel 6). Hemin has no effect on the distribution of PrPΔ51-89-GFP in either cell line (panels 3 & 7 versus 4 & 8). Bar: 10 μm.
Exposure to hemin has been reported to cause aggregation of PrPC in N2a cells [3]. To evaluate whether this phenomenon is common to all cell types, K562, SH-SY5Y, and M17 cells expressing untagged PrPC, PrPΔ51-89, or vector were cultured in complete medium in the presence of hemin for 24 h, and lysates of untreated and hemin-exposed cells were evaluated by western blotting (Fig. 2). Probing for PrPC revealed the typical un-glycosylated, mono-glycosylated, and di-glycosylated forms migrating at 27 kDa and 30–37 kDa in control and hemin-treated cells in all three cell lines. The corresponding glycoforms of PrPΔ51-89 migrated faster on SDS-PAGE as expected (Fig. 2, 1–18). However, hemin-exposed SH-SY5Y lysates revealed an additional, slower migrating form, indicating aggregation of PrPC by hemin (Fig. 2, lane 11). In K562 and M17 cells hemin caused downregulation of PrPC, suggesting intracellular degradation (Fig. 2, lane 2 versus 5 & 14 versus 17). PrPΔ51-89 did not show a detectable change in migration pattern or expression by exposure to hemin in any cell line (Fig. 2, lanes 6, 12, 18).
Fig. 2.

Hemin-induced aggregation of PrPC is cell-type specific. A) Probing of lysates from K562, SH-SY5Y, and M17 cells for PrPC reveals the expected glycoforms of full-length and octa-peptide-repeat deleted forms of PrPΔ51-89 in all three cell lines (lanes 1–18). Exposure to hemin causes aggregation of PrPC in SH-SY5Y cells (lane 11), while in other cell lines there is a small but noticeable decrease in PrPC expression (lanes 2 versus 5 & 14 versus 17). Hemin has no effect on the expression of PrPΔ51-89 in any cell line (lanes 3, 6, 9, 12, 15, 18). Human brain sample fractionated in parallel shows the expected reaction for PrPC (lane 19). Re-probing for β-actin provides a loading control. B) Quantification by densitometry shows reduced expression of PrPC in all cell lines exposed to hemin, including SH-SY5Y cells where a fraction of PrPC is aggregated. Data represent Mean ± SEM of three independent experiments.
Thus, hemin interacts with PrPC independent of the cell line and associated plasma membrane microenvironment. This interaction results in endocytosis of the PrPC-hemin complex. In SH-SY5Y cells, PrPC undergoes aggregation, while in other cell lines it is partially degraded. Hemin has no effect on PrPΔ51-89 in any cell line, implicating this region in hemin binding [3]. Alternately, PrPC could undergo co-internalization with the low-density lipoprotein receptor related protein-1 (LRP1) that interacts with its octa-peptide repeat region and serves as a receptor for hemin-hemopexin complexes [49–52]. This possibility was checked by exposing SH-SY5Y cells expressing PrPC-GFP to heme-hemopexin or hemin, and images of live cells were captured at different time points as in Fig. 1. Rapid endocytosis of PrPC-GFP by hemin but not by heme-hemopexin indicated LRP1-independent endocytosis of PrPC by hemin (data not shown).
PrPC upregulates Hb synthesis in hematopoietic cells in vitro and in vivo
Hemin has been reported to upregulate Hb synthesis in K562 cells [26], and recently, in neuronal cells [29, 53]. To understand the role of PrPC in this process, K562 cells expressing PrPC, PrPΔ51-89, vector, and non-transfected controls were cultured in complete medium supplemented with hemin for 24–48 h, and washed cell pellets were examined visually. Surprisingly, PrPC-expressing cells showed significantly more hemoglobinization relative to cells expressing PrPΔ51-89 and controls (Fig. 3A). Reaction with benzidine, a stain used for identifying hemoglobinized cells, revealed significantly more positive cells in PrPC-expressing relative to vector controls (Fig. 3B). Culture of SH-SY5Y and M17 cells in different concentrations of hemin for 24–48 h did not induce Hb synthesis (data not shown).
Fig. 3.
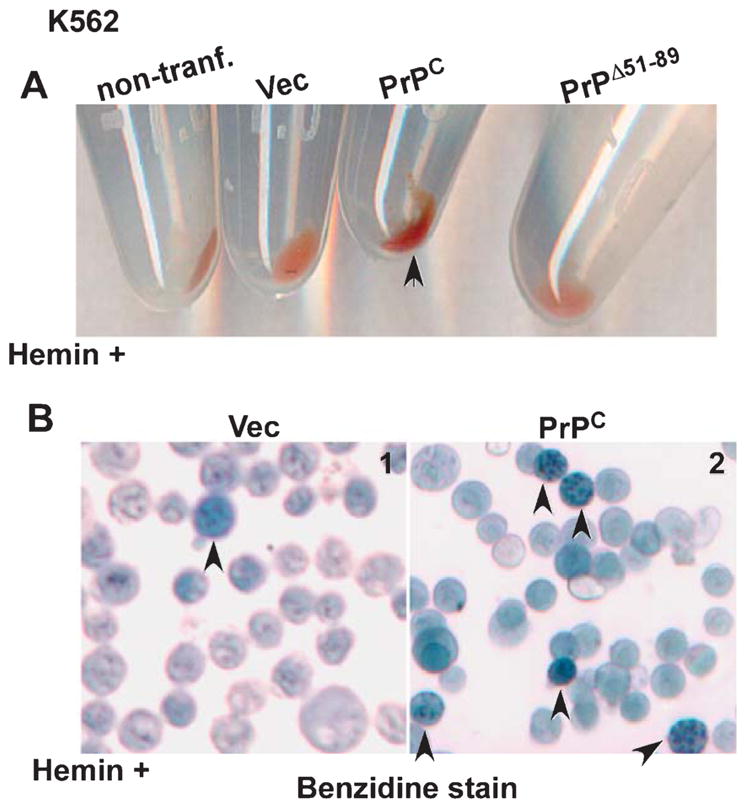
PrPC upregulates hemin-mediated synthesis of Hb in K562 cells. A) Red color of Hb is more evident in cell pellets of PrPC-expressing K562 cells (arrowhead) relative to non-transfected, vector, and PrPΔ51-89-expressing cells. B) Staining with benzidine confirms significantly more Hb in PrPC-expressing cells relative to vector controls (panel 1 versus 2, arrowhead).
To evaluate whether PrPC influences Hb synthesis in vivo, lysates of washed RBCs from age and sex-matched PrP+/+ and PrP−/− mice were fractionated on duplicate gels, and transferred proteins were probed for α-globin and β-globin (Fig. 4). Majority of α-globin and β-globin migrated as a monomer, and the levels were significantly lower in PrP−/− relative to PrP+/+ controls (Fig. 4A, B). Dimeric forms of α-globin and β-globin were visible after a longer exposure (Fig. 4A, B, right panels). Quantification of data from different mice confirmed reduced Hb expression in PrP−/− samples (Fig. 4C, D).
Fig. 4.

α- and α-globin expression is reduced in RBCs from PrP−/− mice. A, B) Lysates of washed RBCs from PrP+/+ and PrP−/− mice reveal reduced expression of α-globin (A) and β-globin (B) in PrP−/− relative to PrP+/+ samples (lanes 1–3 versus 4–6). Dimeric forms of α-globin and β-globin are evident after a longer exposure (right panels). C, D) Densitometry confirms significant reduction of α-globin and β-globin in PrP−/− samples relative to PrP+/+ controls. Reaction for β-actin provides a loading control. Data are Mean ± SEM of three independent experiments on multiple mice. *p < 0.05, **p < 0.01.
PrPC upregulates protoporphyrin IX-mediated synthesis of α-globin in K562 cells
PrPC is known to increase uptake of iron, an essential component of Hb, through its ferrireductase activity [54]. Since hemin is comprised of ferric iron enclosed in protoporphyrin IX ring, PrPC could facilitate transport of iron released from hemin across the endosomal membrane, promote protoporphyrin IX-mediated upregulation of α-globin and β-globin synthesis [55, 56], or increase de novo incorporation of hemin in newly synthesized Hb [57]. To distinguish between these possibilities, K562 cells expressing vector, PrPC, or PrPΔ51-89 were exposed to hemin or ferrous ammonium citrate (FAC) (Fig. 5), or protoporphyrin IX (Fig. 6), and expression of α-globin was evaluated by western blotting. Exposure to hemin induced a significant increase in α-globin levels relative to untreated controls (Fig. 5A, lanes 1–3 versus 4–6). However, the increase in α-globin was significantly more in PrPC-expressing cells relative to PrPΔ51-89 and vector-expressing cells (Fig. 5A, lane 5 versus 4 & 6; Fig. 5B). Re-probing for ferritin showed significant upregulation by hemin in all cell lines, indicating release of hemin-associated iron and storage in cytosolic ferritin. Consequently, expression of transferrin (Tf) and transferrin receptor (TfR) was downregulated in these samples (Fig. 5A, lanes 1–3 versus 4–6).
Fig. 5.

PrPC-mediated upregulation of α-globin is not due to increase in intracellular iron. A) K562 cells expressing vector, PrPC, or PrPΔ51-89 were cultured in the absence or presence of hemin and subjected to western blotting. Probing for α-globin reveals significant upregulation by hemin in all cell lines (lanes 1–3 versus 4–6), but most prominent in PrPC-expressing cells (lane 5 versus 4 & 6). Lysates from mouse RBCs fractionated in parallel show a strong reaction for α-globin as expected (lane 7). Ferritin is upregulated in hemin-treated samples due to the release of associated iron (lanes 1–3 versus 4–6). B) Quantification of α-globin by densitometry confirms significant upregulation by PrPC in the presence of hemin relative to vector and PrPΔ51-89-expressing cells. Data are Mean ± SEM of three independent evaluations. **p < 0.01, ***p < 0.001. C) The above experiment was repeated with in the presence of FAC (lanes 7–9). Reaction for α-globin shows upregulation by hemin as in panel A, but not by FAC (lanes 4–6 versus 7–9). Ferritin is upregulated by hemin and FAC in all cell lines as expected (lanes 4–9). Reaction for β-actin provides a loading control. D) Quantification of the data in panel C by densitometry confirms upregulation of α-globin by hemin, not by FAC, and a positive effect of PrPC on hemin-mediated increase in α-globin.
Fig. 6.

PrPC promotes protoporphyrin IX-mediated synthesis of α-globin. A) K562 cells expressing vector, PrPC, or PrPΔ51-89 were cultured in the absence or presence of protoporphyrin IX and subjected to western blotting. Probing for α-globin reveals upregulation by protoporphyrin IX in all cell lines (lanes 1–3 versus 4–6), and significantly more in PrPC-expressing cells (lane 5 versus 4 & 6). Ferritin is downregulated, and TfR upregulated by exposure to protoporphyrin IX, indicating depletion of cellular iron stores as expected (lanes 1–3 versus 4–6). Lysates from mouse RBCs provide a positive control for α-globin (lane 7). Reaction for β-actin provides a loading control. B) Quantification by densitometry confirms significantly more upregulation of α-globin by protoporphyrin IX in PrPC relative to vector and PrPΔ51-89-expressing cells. Data are Mean ± SEM of three independent experiments. **p < 0.01, ***p < 0.001.
To evaluate whether upregulation of α-globin is due to increase in intracellular iron, K562 cells expressing PrPC, PrPΔ51-89, or vector were cultured in the presence of FAC or hemin, and lysates were processed as above. Probing for α-globin revealed upregulation by hemin in all three cell lines, and significantly more in PrPC-expressing relative to other cell lines as in Fig. 5A (Fig. 5C, lanes 1–3 versus 4–6; Fig. 5D). In contrast, exposure to FAC did not induce α-globin in any of the three cell lines (Fig. 5C, lanes 7–9), though the increase in intracellular iron was significant in all three cell lines as indicated by increase in ferritin levels (Fig. 5C, lanes 1–3 versus 7–9).
The influence of porphyrin ring of hemin (without iron) on α-globin synthesis was assessed by exposing the same cell lines to protoporphyrin IX (PP IX) followed by western blotting (Fig. 6). Lysates of cells exposed to PP IX showed increased synthesis of α-globin relative to untreated controls (Fig. 6A, lanes 1–3 versus 4–6). However, the increase in α-globin was significantly more in PrPC-expressing relative to vector and PrPΔ51-89-expressing cells (Fig. 6A, lane 5 versus 4 & 6; Fig. 6B). Re-probing for ferritin, Tf, and TfR revealed downregulation of ferritin and upregulation of TfR in PP IX-treated cells, indicating depletion of cellular iron stores by this treatment due to stimulation of Hb synthesis (Fig. 6A, lanes 1–3 versus 4–6) [55, 58].
Together, the above observations suggest that PrPC facilitates protoporphyrin IX-mediated upregulation of globin genes, thereby increasing Hb synthesis. Hemin and FAC-mediated increase in intracellular iron is likely to support heme synthesis necessary for assembling tetrameric Hb, but does not influence globin chain synthesis per se, and is therefore insufficient by itself in upregulating Hb.
PrPC upregulates hemin-mediated synthesis of α-globin in mouse brain organotypic cultures
Since hemin upregulates Hb synthesis in neuronal and glial cells [53], it is likely that PrPC modulates this process as well. To explore this possibility, freshly harvested brains from 8 day post-natal PrP+/+ and PrP−/− pups were sliced and cultured in complete medium on filter supports in the absence or presence of hemin. After overnight treatment, sections were harvested, treated with RBC lysis buffer, and processed for western blotting. Lysates from human brain and mouse RBCs were fractionated in parallel as controls. Probing for α-globin revealed significant upregulation by hemin in PrP+/+ relative to PrP−/− samples (Fig. 7 lanes 2 versus 4). Surprisingly, monomeric α-globin from mouse and human brain lysates migrated slower than mouse blood α-globin (Fig. 7, lanes 1–5 versus 6). Human brain sample revealed additional α-globin forms that were absent in mouse brain samples, and migrated faster than dimeric α-globin from mouse blood (Fig. 7, lane 5 versus 6,*). Reaction for PrPC was as expected.
Fig. 7.
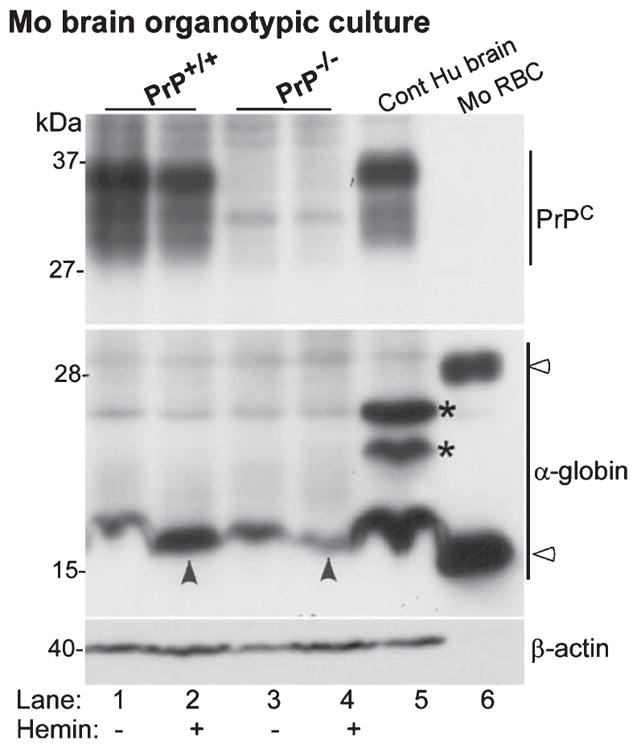
Hemin induces α-globin expression in mouse brain organotypic cultures. Lysates of organotypic brain cultures from PrP+/+ and PrP−/− mice cultured in the absence or presence of hemin were processed for western blotting. Probing for α-globin reveals significantly more α-globin in hemin treated PrP+/+ relative to PrP−/− samples (lanes 1 & 2 versus 3 & 4). Human brain sample fractionated in parallel shows significantly more α-globin relative to mouse samples (lanes 1–4 versus 5), and slower migrating forms that are absent in mouse brain and RBC lysates (lane 5, *). Expression of PrPC is as expected (lanes 1–5). Reaction for β-actin provides a loading control.
Thus, PrPC-upregulates hemin-mediated synthesis of α-globin in brain sections as well. Although the identity of specific cell types that respond to hemin is not clear, these data confirm that α-globin is quantifiable in the brain, and surprisingly, more prominent in human relative to mouse samples. The distinct migration pattern of human brain α-globin on SDS-PAGE suggests the presence of post-translational modifications specific to human brain cells. In addition, the different migration pattern of α-globin from brain and RBCs rules out contamination of brain samples with blood (also see Figs. 8 & 9).
Fig. 8.

α-globin expression is reduced in PrP−/− mouse brain. A) Processing of brain homogenates by western blotting reveals reduced expression of α-globin in PrP−/− samples relative to PrP+/+ controls (lanes 1–3 versus 4–6). Human brain sample fractionated in parallel shows monomeric α-globin that co-migrates with mouse brain samples (lane 7 versus 1–6), and slower than monomeric α-globin from mouse RBCs that show monomeric and dimeric α-globin as in Figs. 4 and 7 above (lane 7 versus 9). RPE cells that express significant amounts of neuroglobin do not show detectable reaction for α-globin (lane 8). Reaction for β-actin provides a loading control. Reaction for PrPC is as expected (lanes 1–7). B) Quantification by densitometry shows significantly less α-globin in PrP−/− relative to PrP+/+ samples. Data are Mean ± SEM of the indicated n (n = 5; *p < 0.05). C) Reaction for neuroglobin shows no difference between mouse and human brain samples and human RPE cells (lanes 1–9). Reaction for β-actin provides a loading control. D) Processing of varying amounts of total protein from mouse RBC and brain samples by Western blotting reveals a strong reaction for α-globin in RBC samples, and significantly less in brain samples despite several-fold higher total protein (lanes 1–3 versus 4). Reaction for glycophorin-A is limited to RBC samples (lanes 1–3 versus 4, lower panel).
Fig. 9.

α-globin is increased in sCJD brains. A) Probing of lysates from the frontal cortex of sCJD and non-dementia controls shows significantly higher levels for α-globin in sCJD samples relative to controls (lanes 1–4 versus 5–8). Reaction for GFAP and ferritin is higher in sCJD, while NeuN is similar in control and sCJD samples (lanes 1–4 versus 5–8). Human and mouse RBCs fractionated in parallel confirm the presence of additional α-globin forms in human brain (lanes 1–8, * versus 9 & 10, open arrow-heads), and slower migration of monomeric α-globin from brain relative to RBC samples (lanes 1–8 versus 9 & 10). Reaction for β-actin provides a loading control. B) Quantification of α-globin by densitometry shows significantly higher levels in sCJD versus controls. C) Varying amounts of total protein from control human brain and human RBCs fractionated in parallel shows a strong reaction for α-globin and β-globin in brain and RBC samples (lanes 1–3, top and middle panels). A faster migrating form of brain α-globin that does not co-migrate with dimeric β-globin from RBCs is detected as in Figs. 7 and 8 above (lane 1, top panel, arrow-head). Reaction for glycophorin-A is limited to RBC samples (lanes 2 & 3, bottom panel), ruling out contamination of brain samples with RBCs. D) Western blotting of equal volume of CSF from sCJD and control samples shows no reactivity for α-globin, though Tf is detected readily, and is reduced in sCJD samples (lanes 1–3 versus 4–6) [61]. Human brain and RBC samples react readily for α-globin as expected (lanes 7 & 8). Reaction for glycophorin-A is limited to the RBC sample (lane 8).
Expression of α-globin is reduced in PrP−/− mouse brains
To evaluate whether PrPC plays a role in Hb synthesis in the brain under physiological conditions, α-globin expression in brain samples harvested from PrP+/+ and PrP−/− was evaluated. Lysates from human brain and RBCs from PrP+/+ mice were fractioned in parallel as controls. Reaction for α-globin revealed a prominent band in both PrP+/+ and PrP−/− samples and human brain that migrated slower than α-globin from mouse RBCs as in Fig. 7 above (Fig. 8A, lanes 1–7 versus 9). Expression of α-globin was variable between mice, but was lower in PrP−/− relative to PrP+/+ controls (Fig. 8A, lanes 1–3 versus 4–6; Fig. 8B).
The α-globin antibody used in this analysis did not cross-react with neuroglobin, another oxygen carrying globin expressed in the brain, and at much higher levels in retinal pigment epithelial (RPE) cells (Fig. 8A, lane 8) [59, 60]. A separate analysis of neuroglobin expression in mouse and human brain samples and RPE cells did not show a detectable difference (Fig. 8C, lanes 1–9).
Since contamination from lysed RBCs is a concern in brain tissue samples, lysates from mouse RBCs and mouse brain (used in panel A) containing varying amounts of total protein were probed for α-globin followed by glycophorin-A, a major sialoglycoprotein of red blood cell membranes. Reactivity for α-globin was detected in RBCs and brain sample as in panel A, and was 50-fold lower in mouse brain samples relative to RBCs (Fig. 8D, lanes 1–4, upper panels). However, expression of glycophorin-A was limited to RBCs, ruling out contamination of brain samples with RBCs (Fig. 8D, lanes 1–3, lower panel).
These results indicate that PrPC modulates neuronal and glial cell Hb synthesis under physiological conditions, a function that gains increasing importance in pathological conditions where neurons are exposed to hemin.
α-Globin levels are increased in sCJD brains
Since PrPC forms non-functional PrPSc aggregates in sCJD brains, loss or altered function of PrPC is likely to alter α-globin synthesis in diseased brains, compromising the metabolic potential of affected neurons. To explore this possibility, lysates from frontal cortex of sCJD and non-dementia controls were analyzed for α-globin expression as above. Samples from human and mouse RBCs were fractionated in parallel as controls (Fig. 9). Surprisingly, α-globin expression was significantly increased in sCJD samples relative to controls (Fig. 9A, lanes 1–4 versus 5–8; Fig. 9B). Probing for the glial marker GFAP revealed a small increase in sCJD samples as expected. However, the difference in neuronal marker NeuN was minimal, indicating equivalent representation of neurons in control and sCJD samples (Fig. 9A, lanes 1–4 versus 5–8). Ferritin was upregulated in sCJD samples as reported earlier (Fig. 9A, lanes 5–8), and expression of PrP was as expected (Fig. 9A, lanes 1–8). As noted above, α-globin from brain samples migrated slower than its counterpart from RBCs (Fig. 9, lanes 1–8 versus 9 & 10). Additional forms of α-globin were detected in control and sCJD samples, probably representing modified isoforms whose identify requires further exploration (lanes 1–8,*).
Since contamination of brain tissue with RBCs is a major concern, representative samples from human brain and RBCs were fractionated in duplicate, and re-analyzed by western blotting as above (Fig. 9C). Probing for α-globin and β-globin revealed slower migrating bands in the brain sample relative to corresponding bands from RBCs (Fig. 9C, lane 1 versus 2 & 3; upper and middle panels). However, reaction with glycophorin-A was limited to RBC samples (Fig. 9C, lane 1 versus 2 & 3, bottom panel). These observations rule out any contamination of brain samples with RBCs. No reactivity for α-globin or β-globin was detected in the CSF of control and sCJD samples (Fig. 9D, lanes 1–6), though the expected bands were detected in brain and RBC samples (Fig. 9D, lanes 7 & 8). Levels of CSF Tf were lower in sCJD samples relative to controls as reported earlier (Fig. 9D, lanes 1–3 versus 4–6) [61].
DISCUSSION
This report confirms and extends previous observations on the interaction of hemin with PrPC [3]. We demonstrate that PrPC binds and internalizes extra-cellular hemin regardless of the plasma membrane micro-environment. This interaction causes aggregation or degradation of PrPC in a cell-type specific manner, while internalized hemin is degraded by all cell lines and the released iron stored in cytosolic ferritin. More importantly, PrPC upregulates hemin-induced synthesis of fetal Hb in K562 cells and adult Hb in mouse brain organotypic cultures, and modulates the synthesis of adult Hb in hematopoietic, neuronal, and glial cells under physiological conditions. Surprisingly, levels of α-globin are increased in sCJD brains, suggesting a compensatory response by surviving neurons or proliferating glial cells, or dys-regulation of Hb synthesis due to sCJD-associated pathology. These observations are discussed in the context of CH and sCJD, pathological conditions where interaction of hemin with PrPC and altered functional activity of PrPSc respectively are likely to play a significant role.
The neuroprotective role of PrPC in CH is well-documented, but poorly understood [15–17, 62]. Our observations suggest that PrPC protects neurons by promoting the endocytosis and degradation of extracellular hemin, helping in its clearance from the neuronal micro-environment. Exposure to hemin concentrations comparable to brain regions affected by CH [6] led to rapid internalization of PrPC and hemin in both hematopoietic and neuronal cell lines. Internalized PrPC was partially degraded in most cell lines and organotypic brain cultures. In SH-SY5Y cells, however, PrPC formed detergent-insoluble aggregates that accumulated in intracellular vesicles as reported for N2a cells [3]. The differential fate of PrPC in specific cell lines probably reflects the efficiency with which the hemin-PrPC complex is degraded, influenced in part by factors that modify the secondary structure of PrPC and resistance to proteolytic enzymes [3]. However, absence of such aggregates in hemin-exposed K562 cells and organotypic brain cultures indicates that hemin-induced aggregation of PrPC is not a common phenomenon, and unlikely to occur in vivo.
The positive effect of PrPC on hemin-induced synthesis of Hb is surprising and of clinical significance because of its potential to improve neuronal viability. PrPC upregulated hemin-mediated synthesis of Hb 2-fold in K562 cells, and ~3-fold in mouse organotypic brain slice cultures. PrPC also amplified the effect of protoporphyrin IX that lacks iron [56, 63] on α-globin synthesis, while extracellular iron had no effect. Since deletion of the octa-peptide repeat region abolished this effect, it is likely that PrPC increases the uptake and possibly transport of hemin and protoporphyrin IX through heme transporters across the endosomal membrane [64], facilitating their influence on α-globin gene expression [23, 24, 65] or direct incorporation in Hb [66]. These observations explain expression of PrPC on proerythroblasts in bone marrow niches where surrounding macrophages are likely to export hemin recycled from senescent RBCs for incorporation in newly synthesized Hb [66–69]. Decreased levels of α-globin and β-globin in PrP−/− RBCs support an active role of PrPC in this pathway. A hemin-independent role of PrPC in hematopoiesis has also been described, though the biochemical pathways involved in this process are not entirely clear [67, 70, 71].
Upregulation of hemin-mediated synthesis of α-globin by PrPC in organotypic brain cultures was surprising, and indicated common pathway(s) of Hb synthesis in hematopoietic precursors and brain cells. These observations are of significance in CH where endocytosis of hemin by PrPC followed by upregulation of Hb is likely to improve neuronal viability by the dual mechanism of clearing extracellular hemin and increasing their respiratory potential. Upregulation of Hb in neurons and astrocytes has been reported in primary cells exposed to hemin [29] and in areas surrounding CH [33, 53], supporting our observations. Although it is difficult to conclude whether neurons, glia, or both cell types respond to hemin, significant upregulation of Hb in PrP+/+ relative to PrP−/− samples suggests participation of neurons that express higher levels of PrPC. A small but significant reduction of α-globin in cortical brain tissue from PrP−/− brains suggests a positive role of PrPC in Hb synthesis under physiological conditions as well, though the underlying mechanism is not clear. Considering the high metabolic rate of neurons at steady state and their susceptibility to oxygen deprivation, a positive role of PrPC in neuronal Hb synthesis has clinical implications.
A significant increase in α-globin in the brain tissue of autopsy-confirmed cases of sCJD was surprising especially since a significant amount of PrPC is nonfunctional in diseased brains due to conversion to the PrPSc isoform. Lack of α-globin reactivity in CSF samples despite significant upregulation in the brain tissue indicates upregulation of Hb, not α-globin per se. The increase in α-globin did not correlate with the glial marker GFAP or the neuronal marker NeuN, making it difficult to attribute this change to gliosis or neuronal loss, two main attributes of sCJD. It is likely that surviving neurons upregulate Hb synthesis in response to sCJD-associated stress, increasing the levels of α-globin in diseased tissue. Upregulation of ferritin in sCJD brains has been reported [72], but increased availability of iron in the absence of globin chain upregulation is unlikely to upregulate Hb synthesis. Interestingly, increased levels of α-Hb stabilizing protein (AHSP) have been reported in the serum of sCJD cases [73], suggesting upregulation of α-globin in the peripheral blood of sCJD cases. However, absence of α-globin in the CSF of both control and sCJD cases argues against this conclusion which was refuted in a later report [74], leaving the matter unsettled. Altered levels of neuronal Hb have been reported in multiple sclerosis [75], Alzheimer’s disease, Parkinson’s disease, and dementia with Lewy bodies [76, 77], suggesting a complex interplay of disease pathology and neuronal Hb synthesis.
In conclusion, this report highlights the dual role of PrPC in clearing hemin from the neuronal micro-environment and upregulating neuronal Hb, thereby promoting neuronal survival in CH. A positive role of PrPC in Hb synthesis under physiological conditions reveals a novel function of this protein in hematopoietic and neuronal Hb synthesis that requires further exploration. Upregulation of Hb in sCJD brains may reflect a compensatory mechanism by surviving neurons, an observation with significant therapeutic implications. Further investigations are necessary to understand the mechanism underlying disease-specific changes in neuronal Hb in sCJD and other neurodegenerative conditions.
Acknowledgments
This study was supported by NIH grants NS 092145, NS 076139, and NS 077438 to N.S.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/15-1039).
References
- 1.Head MW, Ironside JW. Review: Creutzfeldt-Jakob disease: Prion protein type, disease phenotype and agent strain. Neuropathol Appl Neurobiol. 2012;38:296–310. doi: 10.1111/j.1365-2990.2012.01265.x. [DOI] [PubMed] [Google Scholar]
- 2.Kocisko DA, Caughey WS, Race RE, Roper G, Caughey B, Morrey JD. A porphyrin increases survival time of mice after intracerebral prion infection. Antimicrob Agents Chemother. 2006;50:759–761. doi: 10.1128/AAC.50.2.759-761.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee KS, Raymond LD, Schoen B, Raymond GJ, Kett L, Moore RA, Johnson LM, Taubner L, Speare JO, Onwubiko HA, Baron GS, Caughey WS, Caughey B. Hemin interactions and alterations of the subcellular localization of prion protein. J Biol Chem. 2007;282:36525–36533. doi: 10.1074/jbc.M705620200. [DOI] [PubMed] [Google Scholar]
- 4.Li RC, Saleem S, Zhen G, Cao W, Zhuang H, Lee J, Smith A, Altruda F, Tolosano E, Dore S. Heme-hemopexin complex attenuates neuronal cell death and stroke damage. J Cereb Blood Flow Metab. 2009;29:953–964. doi: 10.1038/jcbfm.2009.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wagner KR, Sharp FR, Ardizzone TD, Lu A, Clark JF. Heme and iron metabolism: Role in cerebral hemorrhage. J Cereb Blood Flow Metab. 2003;23:629–652. doi: 10.1097/01.WCB.0000073905.87928.6D. [DOI] [PubMed] [Google Scholar]
- 6.Robinson SR, Dang TN, Dringen R, Bishop GM. Hemin toxicity: A preventable source of brain damage following hemorrhagic stroke. Redox Rep. 2009;14:228–235. doi: 10.1179/135100009X12525712409931. [DOI] [PubMed] [Google Scholar]
- 7.Dang TN, Robinson SR, Dringen R, Bishop GM. Uptake metabolism and toxicity of hemin in cultured neurons. Neurochem Int. 2011;58:804–811. doi: 10.1016/j.neuint.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 8.Gutteridge JM, Smith A. Antioxidant protection by haemopexin of haem-stimulated lipid peroxidation. Biochem J. 1988;256:861–865. doi: 10.1042/bj2560861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schaer DJ, Buehler PW, Alayash AI, Belcher JD, Vercellotti GM. Hemolysis and free hemoglobin revisited: Exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood. 2013;121:1276–1284. doi: 10.1182/blood-2012-11-451229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dang TN, Bishop GM, Dringen R, Robinson SR. The putative heme transporter HCP1 is expressed in cultured astrocytes and contributes to the uptake of hemin. Glia. 2010;58:55–65. doi: 10.1002/glia.20901. [DOI] [PubMed] [Google Scholar]
- 11.Latunde-Dada GO, Simpson RJ, McKie AT. Recent advances in mammalian haem transport. Trends Biochem Sci. 2006;31:182–188. doi: 10.1016/j.tibs.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 12.Chen-Roetling J, Cai Y, Lu X, Regan RF. Hemin uptake and release by neurons and glia. Free Radic Res. 2014;48:200–205. doi: 10.3109/10715762.2013.859386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen-Roetling J, Regan RF. Effect of heme oxygenase-1 on the vulnerability of astrocytes and neurons to hemoglobin. Biochem Biophys Res Commun. 2006;350:233–237. doi: 10.1016/j.bbrc.2006.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen L, Zhang X, Chen-Roetling J, Regan RF. Increased striatal injury and behavioral deficits after intracerebral hemorrhage in hemopexin knockout mice. J Neurosurg. 2011;114:1159–1167. doi: 10.3171/2010.10.JNS10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McLennan NF, Brennan PM, McNeill A, Davies I, Fotheringham A, Rennison KA, Ritchie D, Brannan F, Head MW, Ironside JW, Williams A, Bell JE. Prion protein accumulation and neuroprotection in hypoxic brain damage. Am J Pathol. 2004;165:227–235. doi: 10.1016/S0002-9440(10)63291-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shyu WC, Lin SZ, Chiang MF, Ding DC, Li KW, Chen SF, Yang HI, Li H. Overexpression of PrPC by adenovirus-mediated gene targeting reduces ischemic injury in a stroke rat model. J Neurosci. 2005;25:8967–8977. doi: 10.1523/JNEUROSCI.1115-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weise J, Sandau R, Schwarting S, Crome O, Wrede A, Schulz-Schaeffer W, Zerr I, Bahr M. Deletion of cellular prion protein results in reduced Akt activation, enhanced postischemic caspase-3 activation, and exacerbation of ischemic brain injury. Stroke. 2006;37:1296–1300. doi: 10.1161/01.STR.0000217262.03192.d4. [DOI] [PubMed] [Google Scholar]
- 18.Bueler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, De Armond SJ, Prusiner SB, Aguet M, Weissmann C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- 19.Black SA, Stys PK, Zamponi GW, Tsutsui S. Cellular prion protein and NMDA receptor modulation: Protecting against excitotoxicity. Front Cell Dev Biol. 2014;2:45. doi: 10.3389/fcell.2014.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitsios N, Saka M, Krupinski J, Pennucci R, Sanfeliu C, Miguel Turu M, Gaffney J, Kumar P, Kumar S, Sullivan M, Slevin M. Cellular prion protein is increased in the plasma and peri-infarcted brain tissue after acute stroke. J Neurosci Res. 2007;85:602–611. doi: 10.1002/jnr.21142. [DOI] [PubMed] [Google Scholar]
- 21.Spudich A, Frigg R, Kilic E, Kilic U, Oesch B, Raeber A, Bassetti CL, Hermann DM. Aggravation of ischemic brain injury by prion protein deficiency: Role of ERK-1/-2 and STAT-1. Neurobiol Dis. 2005;20:442–449. doi: 10.1016/j.nbd.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 22.Weise J, Doeppner TR, Muller T, Wrede A, Schulz-Schaeffer W, Zerr I, Witte OW, Bahr M. Overexpression of cellular prion protein alters postischemic Erk1/2 phosphorylation but not Akt phosphorylation and protects against focal cerebral ischemia. Restor Neurol Neurosci. 2008;26:57–64. [PubMed] [Google Scholar]
- 23.Tahara T, Sun J, Igarashi K, Taketani S. Heme-dependent up-regulation of the alpha-globin gene expression by transcriptional repressor Bach1 in erythroid cells. Biochem Biophys Res Commun. 2004;324:77–85. doi: 10.1016/j.bbrc.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 24.Tahara T, Sun J, Nakanishi K, Yamamoto M, Mori H, Saito T, Fujita H, Igarashi K, Taketani S. Heme positively regulates the expression of beta-globin at the locus control region via the transcriptional factor Bach1 in erythroid cells. J Biol Chem. 2004;279:5480–5487. doi: 10.1074/jbc.M302733200. [DOI] [PubMed] [Google Scholar]
- 25.Tsiftsoglou AS, Tsamadou AI, Papadopoulou LC. Heme as key regulator of major mammalian cellular functions: Molecular, cellular, and pharmacological aspects. Pharmacol Ther. 2006;111:327–345. doi: 10.1016/j.pharmthera.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 26.Fibach E, Kollia P, Schechter AN, Noguchi CT, Rodgers GP. Hemin-induced acceleration of hemoglobin production in immature cultured erythroid cells: Preferential enhancement of fetal hemoglobin. Blood. 1995;85:2967–2974. [PubMed] [Google Scholar]
- 27.Rutherford TR, Clegg JB, Weatherall DJ. K562 human leukaemic cells synthesise embryonic haemoglobin in response to haemin. Nature. 1979;280:164–165. doi: 10.1038/280164a0. [DOI] [PubMed] [Google Scholar]
- 28.Moore A, Merad Boudia M, Lehalle D, Massrieh W, Derjuga A, Blank V. Regulation of globin gene transcription by heme in erythroleukemia cells: Analysis of putative heme regulatory motifs in the p45 NF-E2 transcription factor. Antioxid Redox Signal. 2006;8:68–75. doi: 10.1089/ars.2006.8.68. [DOI] [PubMed] [Google Scholar]
- 29.He Y, Hua Y, Keep RF, Liu W, Wang MM, Xi G. Hemoglobin expression in neurons and glia after intracerebral hemorrhage. Acta Neurochir Suppl. 2011;111:133–137. doi: 10.1007/978-3-7091-0693-8_22. [DOI] [PubMed] [Google Scholar]
- 30.Richter F, Meurers BH, Zhu C, Medvedeva VP, Chesselet MF. Neurons express hemoglobin alpha- and beta-chains in rat and human brains. J Comp Neurol. 2009;515:538–547. doi: 10.1002/cne.22062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schelshorn DW, Schneider A, Kuschinsky W, Weber D, Kruger C, Dittgen T, Burgers HF, Sabouri F, Gassler N, Bach A, Maurer MH. Expression of hemoglobin in rodent neurons. J Cereb Blood Flow Metab. 2009;29:585–595. doi: 10.1038/jcbfm.2008.152. [DOI] [PubMed] [Google Scholar]
- 32.Biagioli M, Pinto M, Cesselli D, Zaninello M, Lazarevic D, Roncaglia P, Simone R, Vlachouli C, Plessy C, Bertin N, Beltrami A, Kobayashi K, Gallo V, Santoro C, Ferrer I, Rivella S, Beltrami CA, Carninci P, Raviola E, Gustincich S. Unexpected expression of alpha- and beta-globin in mesencephalic dopaminergic neurons and glial cells. Proc Natl Acad Sci U S A. 2009;106:15454–15459. doi: 10.1073/pnas.0813216106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He Y, Hua Y, Liu W, Hu H, Keep RF, Xi G. Effects of cerebral ischemia on neuronal hemoglobin. J Cereb Blood Flow Metab. 2009;29:596–605. doi: 10.1038/jcbfm.2008.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Digicaylioglu M, Bichet S, Marti HH, Wenger RH, Rivas LA, Bauer C, Gassmann M. Localization of specific erythropoietin binding sites in defined areas of the mouse brain. Proc Natl Acad Sci U S A. 1995;92:3717–3720. doi: 10.1073/pnas.92.9.3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Masuda S, Okano M, Yamagishi K, Nagao M, Ueda M, Sasaki R. A novel site of erythropoietin production. Oxygen-dependent production in cultured rat astrocytes. J Biol Chem. 1994;269:19488–19493. [PubMed] [Google Scholar]
- 36.Stroka DM, Burkhardt T, Desbaillets I, Wenger RH, Neil DA, Bauer C, Gassmann M, Candinas D. HIF-1 is expressed in normoxic tissue and displays an organ-specific regulation under systemic hypoxia. FASEB J. 2001;15:2445–2453. doi: 10.1096/fj.01-0125com. [DOI] [PubMed] [Google Scholar]
- 37.Kong Y, Zhou S, Kihm AJ, Katein AM, Yu X, Gell DA, Mackay JP, Adachi K, Foster-Brown L, Louden CS, Gow AJ, Weiss MJ. Loss of alpha-hemoglobin-stabilizing protein impairs erythropoiesis and exacerbates beta-thalassemia. J Clin Invest. 2004;114:1457–1466. doi: 10.1172/JCI21982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brunori A, Falcioni G, Fioretti E, Giardina B, Rotilio G. Formation of superoxide in the autoxidation of the isolated α and β chains of human hemoglobin and its involvement in hemichrome precipitation. Eur J Biochem. 1975;53:99–104. [Google Scholar]
- 39.Gu Y, Verghese S, Mishra RS, Xu X, Shi Y, Singh N. Mutant prion protein-mediated aggregation of normal prion protein in the endoplasmic reticulum: Implications for prion propagation and neurotoxicity. J Neurochem. 2003;84:10–22. doi: 10.1046/j.1471-4159.2003.01255.x. [DOI] [PubMed] [Google Scholar]
- 40.Haldar S, Tripathi A, Qian J, Beserra A, Suda S, McElwee M, Turner J, Hopfer U, Singh N. Prion protein promotes kidney iron uptake via its ferrireductase activity. J Biol Chem. 2015;290:5512–5522. doi: 10.1074/jbc.M114.607507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tripathi AK, Haldar S, Qian J, Beserra A, Suda S, Singh A, Hopfer U, Chen SG, Garrick MD, Turner JR, Knutson MD, Singh N. Prion protein functions as a ferrireductase partner for ZIP14 and DMT1. Free Radic Biol Med. 2015;84:322–330. doi: 10.1016/j.freeradbiomed.2015.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Addya S, Keller MA, Delgrosso K, Ponte CM, Vadigepalli R, Gonye GE, Surrey S. Erythroid-induced commitment of K562 cells results in clusters of differentially expressed genes enriched for specific transcription regulatory elements. Physiol Genomics. 2004;19:117–130. doi: 10.1152/physiolgenomics.00028.2004. [DOI] [PubMed] [Google Scholar]
- 43.Rowley PT, Ohlsson-Wilhelm BM, Rudolph NS, Farley BA, Kosciolek B, LaBella S. Hemin preferentially stimulates synthesis of alpha-globin in K562 human erythroleukemia cells. Blood. 1982;59:1098–1102. [PubMed] [Google Scholar]
- 44.Wanda PE, Lee LT, Howe C. A spectrophotometric method for measuring hemoglobin in erythroleukemic cells (K562) J Histochem Cytochem. 1981;29:1442–1444. doi: 10.1177/29.12.6948040. [DOI] [PubMed] [Google Scholar]
- 45.Pettiford SM, Herbst R. The protein tyrosine phosphatase HePTP regulates nuclear translocation of ERK2 and can modulate megakaryocytic differentiation of K562 cells. Leukemia. 2003;17:366–378. doi: 10.1038/sj.leu.2402767. [DOI] [PubMed] [Google Scholar]
- 46.Singh A, Qing L, Kong Q, Singh N. Change in the characteristics of ferritin induces iron imbalance in prion disease affected brains. Neurobiol Dis. 2012;45:930–938. doi: 10.1016/j.nbd.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alonso MM, Jiang H, Gomez-Manzano C, Fueyo J. Targeting brain tumor stem cells with oncolytic adenoviruses. Methods Mol Biol. 2012;797:111–125. doi: 10.1007/978-1-61779-340-0_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim HA, Whittle SC, Lee S, Chu HX, Zhang SR, Wei Z, Arumugam TV, Vinh A, Drummond GR, Sobey CG. Brain immune cell composition and functional outcome after cerebral ischemia: Comparison of two mouse strains. Front Cell Neurosci. 2014;8:365. doi: 10.3389/fncel.2014.00365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parkyn CJ, Vermeulen EG, Mootoosamy RC, Sunyach C, Jacobsen C, Oxvig C, Moestrup S, Liu Q, Bu G, Jen A, Morris RJ. LRP1 controls biosynthetic and endocytic trafficking of neuronal prion protein. J Cell Sci. 2008;121:773–783. doi: 10.1242/jcs.021816. [DOI] [PubMed] [Google Scholar]
- 50.Hvidberg V, Maniecki MB, Jacobsen C, Hojrup P, Moller HJ, Moestrup SK. Identification of the receptor scavenging hemopexinheme complexes. Blood. 2005;106:2572–2579. doi: 10.1182/blood-2005-03-1185. [DOI] [PubMed] [Google Scholar]
- 51.Jen A, Parkyn CJ, Mootoosamy RC, Ford MJ, Warley A, Liu Q, Bu G, Baskakov IV, Moestrup S, McGuinness L, Emptage N, Morris RJ. Neuronal low-density lipoprotein receptor-related protein 1 binds and endocytoses prion fibrils via receptor cluster 4. J Cell Sci. 2010;123:246–255. doi: 10.1242/jcs.058099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Taylor DR, Hooper NM. The low-density lipoprotein receptor-related protein 1 (LRP1) mediates the endocytosis of the cellular prion protein. Biochem J. 2007;402:17–23. doi: 10.1042/BJ20061736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.He Y, Hua Y, Lee JY, Liu W, Keep RF, Wang MM, Xi G. Brain alpha- and beta-globin expression after intracerebral hemorrhage. Transl Stroke Res. 2010;1:48–56. doi: 10.1007/s12975-009-0004-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Singh A, Haldar S, Horback K, Tom C, Zhou L, Meyerson H, Singh N. Prion protein regulates iron transport by functioning as a ferrireductase. J Alzheimers Dis. 2013;35:541–552. doi: 10.3233/JAD-130218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Coccia EM, Perrotti E, Stellacci E, Orsatti R, Del Russo N, Marziali G, Testa U, Battistini A. Regulation of expression of ferritin H-chain and transferrin receptor by protoporphyrin IX. Eur J Biochem. 1997;250:764–772. doi: 10.1111/j.1432-1033.1997.00764.x. [DOI] [PubMed] [Google Scholar]
- 56.Palma JF, Gao X, Lin CH, Wu S, Solomon WB. Iron protoporphyrin IX (hemin) but not tin or zinc protoporphyrin IX can stimulate gene expression in K562 cells from enhancer elements containing binding sites for NF-E2. Blood. 1994;84:1288–1297. [PubMed] [Google Scholar]
- 57.Fibach E, Aker M. Hemin augments growth and hemoglobinization of erythroid precursors from patients with diamond-blackfan anemia. Anemia. 2012;2012:940260. doi: 10.1155/2012/940260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Coccia EM, Profita V, Fiorucci G, Romeo G, Affabris E, Testa U, Hentze MW, Battistini A. Modulation of ferritin H-chain expression in Friend erythroleukemia cells: Transcriptional and translational regulation by hemin. Mol Cell Biol. 1992;12:3015–3022. doi: 10.1128/mcb.12.7.3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brittain T. The anti-apoptotic role of neuroglobin. Cells. 2012;1:1133–1155. doi: 10.3390/cells1041133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Palladino P, Scaglione GL, Arcovito A, Maria Vitale R, Amodeo P, Vallone B, Brunori M, Benedetti E, Rossi F. Neuroglobin-prion protein interaction: What’s the function? J Pept Sci. 2011;17:387–391. doi: 10.1002/psc.1333. [DOI] [PubMed] [Google Scholar]
- 61.Singh A, Beveridge AJ, Singh N. Decreased CSF transferrin in sCJD: A potential pre-mortem diagnostic test for prion disorders. PLoS One. 2011;6:e16804. doi: 10.1371/journal.pone.0016804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Steele AD, Zhou Z, Jackson WS, Zhu C, Auluck P, Moskowitz MA, Chesselet MF, Lindquist S. Context dependent neuroprotective properties of prion protein (PrP) Prion. 2009;3:240–249. doi: 10.4161/pri.3.4.10135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nakajima O, Kagechika H, Shudo K, Hashimoto Y, Iwasaki S. Possible involvement of retinoid-like cofactor in serum in hemin/protoporphyrin-IX-induced differentiation of human leukemia K562 cells. Biochem Biophys Res Commun. 1995;206:1003–1010. doi: 10.1006/bbrc.1995.1142. [DOI] [PubMed] [Google Scholar]
- 64.White C, Yuan X, Schmidt PJ, Bresciani E, Samuel TK, Campagna D, Hall C, Bishop K, Calicchio ML, Lapierre A, Ward DM, Liu P, Fleming MD, Hamza I. HRG1 is essential for heme transport from the phagolysosome of macrophages during erythrophagocytosis. Cell Metab. 2013;17:261–270. doi: 10.1016/j.cmet.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Higgs DR, Garrick D, Anguita E, De Gobbi M, Hughes J, Muers M, Vernimmen D, Lower K, Law M, Argentaro A, Deville MA, Gibbons R. Understanding alpha-globin gene regulation: Aiming to improve the management of thalassemia. Ann N Y Acad Sci. 2005;1054:92–102. doi: 10.1196/annals.1345.012. [DOI] [PubMed] [Google Scholar]
- 66.Schultz IJ, Chen C, Paw BH, Hamza I. Iron and porphyrin trafficking in heme biogenesis. J Biol Chem. 2010;285:26753–26759. doi: 10.1074/jbc.R110.119503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Griffiths RE, Heesom KJ, Anstee DJ. Normal prion protein trafficking in cultured human erythroblasts. Blood. 2007;110:4518–4525. doi: 10.1182/blood-2007-04-085183. [DOI] [PubMed] [Google Scholar]
- 68.Keel SB, Doty RT, Yang Z, Quigley JG, Chen J, Knoblaugh S, Kingsley PD, De Domenico I, Vaughn MB, Kaplan J, Palis J, Abkowitz JL. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science. 2008;319:825–828. doi: 10.1126/science.1151133. [DOI] [PubMed] [Google Scholar]
- 69.Yuan X, Fleming MD, Hamza I. Heme transport and erythropoiesis. Curr Opin Chem Biol. 2013;17:204–211. doi: 10.1016/j.cbpa.2013.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Panigaj M, Glier H, Wildova M, Holada K. Expression of prion protein in mouse erythroid progenitors and differentiating murine erythroleukemia cells. PLoS One. 2011;6:e24599. doi: 10.1371/journal.pone.0024599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang CC, Steele AD, Lindquist S, Lodish HF. Prion protein is expressed on long-term repopulating hematopoietic stem cells and is important for their self-renewal. Proc Natl Acad Sci U S A. 2006;103:2184–2189. doi: 10.1073/pnas.0510577103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Singh A, Isaac AO, Luo X, Mohan ML, Cohen ML, Chen F, Kong Q, Bartz J, Singh N. Abnormal brain iron homeostasis in human and animal prion disorders. PLoS Pathog. 2009;5:e1000336. doi: 10.1371/journal.ppat.1000336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Miele G, Manson J, Clinton M. A novel erythroid-specific marker of transmissible spongiform encephalopathies. Nat Med. 2001;7:361–364. doi: 10.1038/85515. [DOI] [PubMed] [Google Scholar]
- 74.Appleford NE, Wilson K, Houston F, Bruce LJ, Morrison A, Bishop M, Chalmers K, Miele G, Massey E, Prowse C, Manson J, Will RG, Clinton M, MacGregor I, Anstee DJ. alpha-Hemoglobin stabilizing protein is not a suitable marker for a screening test for variant Creutzfeldt-Jakob disease. Transfusion. 2008;48:1616–1626. doi: 10.1111/j.1537-2995.2008.01759.x. [DOI] [PubMed] [Google Scholar]
- 75.Broadwater L, Pandit A, Clements R, Azzam S, Vadnal J, Sulak M, Yong VW, Freeman EJ, Gregory RB, McDonough J. Analysis of the mitochondrial proteome in multiple sclerosis cortex. Biochim Biophys Acta. 2011;1812:630–641. doi: 10.1016/j.bbadis.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ferrer I, Gomez A, Carmona M, Huesa G, Porta S, Riera-Codina M, Biagioli M, Gustincich S, Aso E. Neuronal hemoglobin is reduced in Alzheimer’s disease, argyrophilic grain disease, Parkinson’s disease, and dementia with Lewy bodies. J Alzheimers Dis. 2011;23:537–550. doi: 10.3233/JAD-2010-101485. [DOI] [PubMed] [Google Scholar]
- 77.Shephard F, Greville-Heygate O, Marsh O, Anderson S, Chakrabarti L. A mitochondrial location for haemoglobins–dynamic distribution in ageing and Parkinson’s disease. Mitochondrion. 2014;14:64–72. doi: 10.1016/j.mito.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]