

Abstract
2-Amino-9H-pyrido[2,3-b]indole (AαC) is the most abundant carcinogenic heterocyclic aromatic amine (HAA) formed in mainstream tobacco smoke. AαC is a liver carcinogen in rodents, but its carcinogenic potential in humans is not known. To obtain a better understanding of the genotoxicity of AαC in humans, we have investigated its metabolism and its ability to form DNA adducts in human hepatocytes. Primary human hepatocytes were treated with AαC at doses ranging from 0.1 to 50 μM and the metabolites were characterized by UPLC/ ion trap multistage mass spectrometry (UPLC/MSn). Six major metabolites were identified: a ring-oxidized doubly conjugated metabolite, N2-acetyl-2-amino-9H-pyrido[2,3-b]indole-6-yl-oxo-(β-D-glucuronic acid) (N2-acetyl-AαC-6-O-Gluc); two ring-oxidized glucuronide (Gluc) conjugates: 2-amino-9H-pyrido[2,3-b]indol-3-yl-oxo-(β-D-glucuronic acid) (AαC-3-O-Gluc) and 2-amino-9H-pyrido[2,3-b]indol-6-yl-oxo-(β-D-glucuronic acid) (AαC-6-O-Gluc); two sulfate conjugates, 2-amino-9H-pyrido[2,3-b]indol-3-yl sulfate (AαC-3-O-SO3H) and 2-amino-9H-pyrido[2,3-b]indol-6-yl sulfate (AαC-6-O-SO3H); and the Gluc conjugate, N2-(β-D-glucosidurony1)-2-amino-9H-pyrido[2,3-b]indole (AαC-N2-Gluc). In addition, four minor metabolites were identified: N2-acetyl-9H-pyrido[2,3-b]indol-3-yl sulfate (N2-acetyl-AαC-3-O-SO3H); N2-acetyl-9H-pyrido[2,3-b]indol-6-yl sulfate (N2-acetyl-AαC-6-O-SO3H), N2-acetyl-2-amino-9H-pyrido[2,3-b]indol-3-yl-oxo-(β-D-glucuronic acid) (N2-acetyl-AαC-3-O-Gluc), and O-(β-D-glucosidurony1)-2-hydroxyamino-9H-pyrido[2,3-b]indole (AαC-HN2-O-Gluc). The latter metabolite, AαC-HN2-O-Gluc is a reactive intermediate which binds to DNA to form the covalent adduct N-(2′-deoxyguanosin-8-yl)-2-amino-9H-pyrido[2,3-b]indole (dG-C8-AαC). Pre-incubation of hepatocytes with furafylline, a selective mechanism-based inhibitor of P450 1A2, resulted in a strong decrease in the formation of AαC-HN2-O-Gluc and a concomitant decrease in DNA adduct formation. Our findings describe the major pathways of metabolism of AαC in primary human hepatocytes and reveal the importance of N-acetylation and glucuronidation in metabolism of AαC. P450 1A2 is a major isoform involved in the bioactivation of AαC to form the reactive AαC-HN2-O-Gluc conjugate and AαC-DNA adducts.
Keywords: 2-amino-9H-pyrido[2,3-b]indole (AαC); primary human hepatocytes; metabolism; P450 1A2; DNA adduct; UDP-glucuronosyltransferase
Graphical abstract
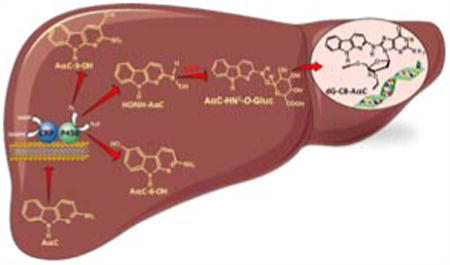
Introduction
Epidemiologic studies conducted over the past two decades have consistently shown that smoking is a risk factor for liver and gastrointestinal tract cancer.1-3 Cigarette smoking is a prominent source of exposure to a number of genotoxicants including nitrosamines, aromatic amines, polycyclic aromatic hydrocarbons, and heterocyclic aromatic amines (HAA).4 HAA are also formed in well-done cooked meat, poultry and fish,5 and some occur in diesel gas exhaust.6,7 2-Amino-9H-pyrido[2,3-b]indole (AαC) is by far the most abundant HAA formed in tobacco smoke with a level range between 25 to 260 ng per cigarette.8-11 These amounts are comparable to those of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), and 25-100 times higher than those of 4-aminobiphenyl (4-ABP) and benzo[a]pyrene (B[a]P);9,12,13 these three chemicals are recognized as human carcinogens.3,14
AαC has been detected in urine of smokers of the Shanghai Cohort study.15 The urinary levels were positively correlated to the number of cigarettes smoked per day. A similar finding was reported in Zengshu, China, where AαC was present at higher levels in the urine of smokers than nonsmokers.16 Recently, a tobacco smoking cessation study conducted in the United States revealed that AαC was present in urine during the smoking phase in greater than 90% of the subjects, and the geometric mean urinary level of AαC decreased by 87% six weeks after cessation of tobacco usage.17 These urinary biomarker data demonstrate that tobacco smoking is a significant source of AαC exposure. The data reported in the literature on urinary level of AαC in human is restricted to few reports where the mean level of AαC in urine of subjects who smoked greater than 20 cigarette per day range between 11.9 and 3511.9 pg/mg creatinine.15-17
AαC induces liver and blood vessel tumors in CDF1 mice,5,18 it induces lacI transgene mutations in the colon of C57BL/6 mice,19 and aberrant crypt foci, an early biomarker of colon neoplasia, in Big Blue mice.20 However, the genotoxicity of AαC remains unknown in humans and only few reports have been reported on the genotoxicity of AαC in vitro in human cells. AαC is genotoxic in human lymphoblastoid cells (MLC-5)21 and in peripheral blood lymphocytes cells, when assessed the by the comet and micronucleus assays.22 We also showed that AαC forms high and persistent levels of DNA adducts in primary human hepatocytes.23,24 The major DNA adduct formed by AαC is N-(deoxyguanosin-8-yl)-2-amino-9H-pyrido[2,3-b]indole (dG-C8-AαC)25,26 which is regarded as a mutagenic lesion.27 The levels of dG-C8-AαC formed in primary human hepatocytes were greater than those dG-C8 adducts formed with other HAA, including 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP), 2-amino-3,8-dimethylmidazo[4,5-f]quinoxaline (MeIQx), 2-amino-3-methylimidazo[4,5-f]quinoline (IQ) or the structurally related arylamine 4-aminobiphenyl (4-ABP).24
AαC requires metabolism to exert its genotoxic effects. AαC undergoes metabolic activation, by P450 1A2-catalyzed N-oxidation of the exocyclic amine group to form 2-hydroxyamino-9H-pyrido[2,3-b]indole (HONH-AαC).28,29 Conjugation enzymes such as N-acetyltransferases (NAT) or sulfotransferases (SULT), catalyze the conversion of HONH-AαC to unstable esters, which undergo heterolytic cleavage to form the presumed short-lived nitrenium ion, which covalently adducts to DNA.30 In contrast to the NAT and SULT enzymatic pathways of bioactivation, the UDP-Glucuronosyltransferases (UGT) are largely viewed as conjugation pathways that lead to detoxication.31 UGT-mediated conjugation of glucuronic acid (Gluc) to the exocyclic amino groups of HAA or N-hydroxylated HAA is a detoxification pathway for HAA.31-33 However, we discovered that the UGT also catalyze the O-glucuronidation of HONH-AαC to form the AαC-HN2-O-Gluc conjugate, a reactive metabolite that contributes to the genotoxicity of AαC.34
The abundance of AαC in tobacco smoke and its propensity to undergo bioactivation by hepatic enzymes provides a plausible biochemical mechanism that may explain how AαC can induce DNA damage and play a role in the development of liver and digestive tract cancers in smokers.1,35,36 However stable biomarkers must be developed and implemented in molecular epidemiological studies designed to assess the role of chemical exposures to AαC in cancer risk. For this purpose, a better understanding of the metabolism of AαC is required.
Studies have been devoted to the metabolism of AαC in vitro with hepatic human liver microsomes or recombinant human P450s29,30,37-39 and in vivo mainly in rats26,37,40 and mice.41 However, knowledge about the major metabolic pathways of AαC and the key enzymes involved in bioactivation of this carcinogen in liver and extrahepatic tissues of humans are limited. There is only one study reported the metabolism of AαC in human hepatocarcinoma cell line HepG2.40 This cell line is not fully metabolically competent since it does not express some important metabolism enzymes such as UGT.40 In contrast, primary human hepatocytes are the gold standard to investigate different pathways of carcinogen metabolism, since this cell model retains liver function for at least seven days and expresses phase I and phase II enzymes as well as cofactors at physiological concentrations.42
The aim of our study was to characterize the major metabolites of AαC formed in human liver using primary cultured human hepatocytes. The metabolites were characterized by UPLC/MSn in conjunction with HPLC and ultraviolet detection, and by treatment with the deconjugation enzymes arylsulfatase and β-glucuronidase. Our findings show that AαC undergoes multiple pathways of metabolism that include N2-acetylation, N2-glucuronidation, as well as ring-oxidation at the C-3 and C-6 atoms of the heterocyclic ring of AαC. The metabolic activation of AαC through N-oxidation was shown to occur by formation of AαC-HN2-O-Gluc, a genotoxic metabolite that reacts with DNA.34 Preincubation of hepatocytes with furafylline, a selective inhibitor of P450 1A243 resulted in a strong decrease in the formation of AαC-HN2-O-Gluc with a concomitant decrease in DNA adducts. Our data describes for the first time, the metabolism of AαC in human liver, and the importance of P450 1A2 in formation of AαC-HN2-O-Gluc and DNA adducts.
Materials and Methods
Caution
AαC and its derivatives are potential human carcinogens. These chemicals must be handled in a well-ventilated fume hood with proper use of gloves and protective clothing.
Chemicals
AαC was purchased from the Toronto Research Chemicals (Toronto, ON, Canada). [4b,5,6,7,8,8a-13C6]AαC was a gift from Dr. Daniel Doerge, National Center for Toxicological Research (Jefferson, AR). Human liver microsomes were obtained from the Tennessee Donor Services, Nashville, TN, and kindly provided by Prof. F. P. Guengerich, Vanderbilt University. DMSO, ethoxyresorufin, methoxyresorufin, methanol, acetonitrile, ascorbic acid, hydrochloric acid, ammonium acetate, sulfatase from Helix pomatia (≥10,000 units/g solid) and β-Glucuronidase from Helix pomatia (≥30,000 units/g solid) were purchased from Sigma Aldrich (St. Louis, MO, USA). AαC-3-OH and AαC-6-OH were prepared with human liver microsomes or rat liver microsomes and spectroscopically characterized as previously reported.17,34 N-(deoxyguanosin-8-yl)-2-amino-9H-pyrido[2,3-b]indole (dG-C8-AαC) and [13C10]-dG-C8-AαC were prepared as previously described.27
Cell Isolation, Culture, and Treatment
Human liver samples were obtained from patients undergoing liver resection for primary or secondary hepatomas through the Centre de Resources Biologiques (CRB)-Santé of Rennes (http://www.crbsante-rennes.com). The demographics information is provided in supporting information (Table S1). The research protocol was conducted under French legal guidelines and fulfilled the requirements of the local institutional ethics committee. Hepatocytes were isolated by a two-step collagenase perfusion procedure and seeded in petri dishes at a density of 3×106 viable cells/19.5 cm2 dish, in William's medium (Gibco, Life technologies, Carlsbad, CA, USA), supplemented by bovine serum albumin (1 g/L) (Life technologies), glutamine (2 mM) (Life technologies), bovine insulin (5 μg/mL) (Life technologies), penicillin (10 U/mL) (Life technologies), streptomycin (10 μg/mL) (Life technologies) and 10% fetal calf serum (v/v) (Life technologies). After 18 h of cell seeding, the media was replaced with cell media lacking fetal calf serum and containing hydrocortisone hemisuccinate (54 μM) (Laboratoire SERB, Paris, France). After 36 h of culture, the differentiated cells were incubated with AαC in DMSO (0.1% v/v) for 24 h. At the end of treatment, the supernatants were collected and the cells were washed with PBS. Cell pellets were collected by centrifugation at 3500 g for 10 min at 4 °C. Both supernatants and cell pellets were stored at -80 °C until further use. To assess the role of P450 1A2 in the metabolism of AαC, the cells were pre-treated with furafylline (5 μM) or 0.1% DMSO (v/v) for 24 h. The media was then renewed with furafylline (5 μM) or 0.1% DMSO (v/v), and the cells were incubated with AαC (0.1, 1, 10 or 50 μM) for an additional 24 h. At the end of each time point, the cell media was collected for metabolite analysis and the cellular pellets were collected after PBS washing for DNA adducts measurements.
EROD/MROD Activity
Ethoxyresorufin O-deethylase (EROD) and methoxyresorufin O-demethylase (MROD) activities associated with P450 1A1/244 and P450 1A2,45 respectively, were measured in all primary cultured hepatocytes used in this study as described previously.24 The reaction rates were linear over the reaction time and proportional to protein concentration estimated by the Bradford procedure.46
Samples preparation and HPLC analysis of the major metabolites of AαC
Cell media was treated by 3 volumes of cold methanol and incubated on ice for 30 min, followed by a centrifugation at 20 000 g for 10 min at 4 °C. The supernatant was collected and evaporated to dryness, and the extracts were dissolved in HPLC grade water. The metabolites were separated and collected with an Agilent model 1100 HPLC Chemstation (Palo Alto, CA) equipped with UV/V is detector. The metabolites were separated with Aquasil C18 column (4.6 × 250 mm, 5 μm particle size) from Thermo Scientific (Bellefonte, PA). The chromatography commenced isocratically at 95% A solvent (20 mM ammonium acetate and 5% acetonitrile) for 10 min, followed by a linear gradient over 20 min to 50% B (acetonitrile) at a flow rate of 1 mL/min. The column was then washed with 100% acetonitrile and re-equilibrated at starting solvent conditions. Once collected, the metabolites were evaporated to dryness and stored at -80 °C until LC/MS analysis.
UV Spectral Characterization of AαC metabolites by treatment with deconjugating enzymes or acid hydrolysis
The proposed ring oxidized sulfate conjugates (AαC-3-O-SO3H and AαC-6-O-SO3H) and the ring oxidized Gluc conjugates (AαC-3-O-Gluc and AαC-6-O-Gluc) were hydrolyzed, respectively with sulfatase or β-glucuronidase. The metabolites purified by HPLC (∼25 ng) were concentrated to dryness by vacuum centrifugation, resuspended in 25 μL of 50 mM potassium phosphate buffer (pH 7.0) and incubated with 25 μL of sulfatase (200 units/mL) or 25 μL of β-glucuronidase (300 units/mL) for 1 h at 37 °C. The enzymes solutions and buffers were purged with argon before use, and the incubations were conducted in tightly closed Eppendorf tubes to minimize oxidation of the deconjugated metabolites. The hydrolysis was terminated by the addition of 1 volume of ice-cold methanol. The precipitated proteins were removed by centrifugation and the supernatants were analyzed by HPLC as described above. The proposed N-acetylated ring-oxidized AαC Gluc conjugate (N2-acetyl-AαC-6-O-Gluc) was subjected to enzymatic hydrolysis by β-glucuronidase under the same conditions as described above, followed by acid hydrolysis (0.1 M HCl) for 3 h at 60 °C. After addition of 1 volume of ice-cold methanol and incubation for 5 min on ice, the precipitated protein was eliminated by centrifugation. The supernatant was dried and re-suspended in HPLC grade water. The hydrolysis products were analyzed by HPLC as described above. The UV spectra of each hydrolysis products was compared to those spectra of AαC-6-OH and AαC-3-OH, which were produced using human liver microsome as previously reported.34,41
Solid-phase extraction (SPE) of hepatocyte metabolites for UPLC-ESI-MSn
The cell media containing AαC metabolites were processed by SPE using Oasis HLB 1 cc (30 mg) cartridge (Waters, Milford, MA, USA), prior to UPLC-ESI-MSn analysis. After the conditioning the cartridge with 1 mL of methanol, followed by 1 mL of LC/MS grade water, the cell media (100-500 μL) diluted in 2 mM ammonium acetate (added up to 1 mL) were loaded in the cartridge. The cartridge was washed with 1 mL of 2 mM ammonium acetate, and metabolites were eluted with 1 mL of methanol. The eluates were evaporated to dryness by vacuum centrifugation at 42 °C. The residues were dissolved in mobile phase buffer (2 mM ammonium acetate).
UPLC-ESI-MSn analysis
Ultrapeformance liquid chromatography electrospray ionization mass spectrometry (UPLC-ESI-MS) analyses was performed with NanoAcquity UPLC system (Waters Corp., New Milford, MA) equipped with a Waters Symmetry C18 Trap Column (180 μm × 20 mm, 5 μm particle size) (Waters Corp., New Milford, MA) and an Advance CaptiveSpray ion source (Michrom Bioresources, Auburn, CA) interfaced with a linear quadrupole ion trap mass spectrometer (LTQ Velos, Thermo Fisher, San Jose, CA). Solvent A was 2 mM ammonium acetate and solvent B was 95% acetonitrile and 5% H2O. After 3 min at 5% of B, a linear gradient was employed, starting at 5% B, arriving at 50% B in 20 min and ended at 99% B at 25 min at a flow rate of 5 μL/min. The transitions were as follow: AαC-O-SO3H ([M+H]+ at m/z 280.1 > 200.1 >); AαC-O-Gluc ([M+H]+ at m/z 376.1 > 200.1 >); N2-acetyl-AαC-O-SO3H ([M+H]+ at m/z 322.1 > 241.1 >); N2-acetyl-AαC-O-Gluc ([M+H]+ at m/z 418.1 > 242.1 >); AαC-N2-Gluc ([M+H]+ at m/z 360.1 > 183.1 >); AαC-HN2-O-Gluc ([M+H]+ at m/z 376.1 > 200.1 >); AαC ([M+H]+ at m/z 184.1 > 167.1 >).
Isolation and Digestion of DNA for Adduct Measurements
Cell pellets containing 3×106 primary human hepatocytes were homogenized in 400 μL of TE buffer at pH 8.0 (50 mM Tris-HCl and 10 mM EDTA) and incubated with RNase T1 (319 U) and RNase A (20 μg) for 30 min at 37 °C. Then proteinase K (200 μg) and SDS (0.05% final concentration) were added and the mixture was incubated for 1 h at 37 °C. The DNA was purified by the phenol/chloroform extraction, followed by precipitation with ethanol.47 DNA was resuspended in 200 μL of sterile water and quantified with a NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific). Each DNA sample was spiked with isotopically labeled internal standards at a level of 1 adduct per 107 bases. DNA digestion was performed in 5 mM Bis-Tris-HCl buffer (pH 7.1). DNA was incubated for 3.5 h with DNase I and Nuclease P1 at 37 °C followed by 18 h incubation with alkaline phosphatase and phosphodiesterase at 37 °C.48 After vacuum centrifugation to the dryness, digested DNA was resuspended in 30 μL of 1:1 water/DMSO (v/v) and sonicated for 5 min. Samples were then centrifuge for 5 min at 21 000 g and the supernatant was transferred to LC vials.
UPLC/MS3 measurement of DNA adducts
DNA adduct measurements were performed with the NanoAcquity UPLC system (Waters Corp, New Milford, MA) equipped with a Waters Symmetry trap column (180 μm × 20 mm, 5 μm particle size), a Michrom C18 AQ column (0.3 mm×150 mm, 3 μm particle size), and a Michrom Captive Spray source interfaced with LTQ Velos. Chromatographic conditions were described previously.24 The ions were monitored at the MS3 scan stage as follows: dG-C8-AαC (m/z 449.1 > 333.1 > 209.2, 291.4, 316.4); [13C10]-dG-C8-AαC (m/z 459.1 > 338.1 > 210.2, 295.5, 321.5).
Results
Identification of the major metabolites of AαC formed in human hepatocytes
Six major metabolites were produced by incubating primary human hepatocytes with of AαC (50 μM) and arbitrarily named M1 to M6 as function of the order of their elution time by HPLC (Figure 1). All of the metabolites retained the characteristic UV chromophore of AαC with slight changes in the spectral properties. The metabolites were collected and infused into the LTQ Velos ion trap MS for characterization.
Figure 1.
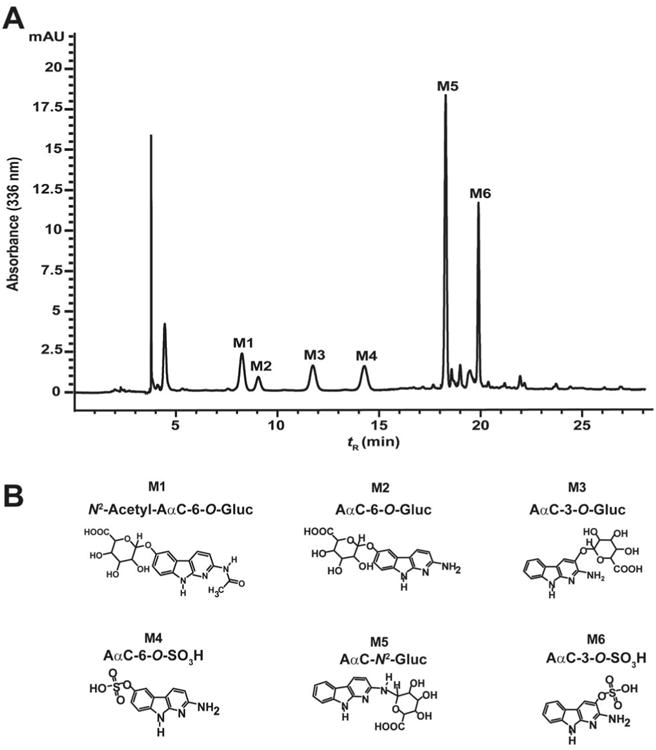
(A) HPLC-UV profile and (B) proposed chemical structures of AαC metabolites formed in primary human hepatocytes incubated with 50 μM of AαC over 24 h.
ESI-MS product ion and UV spectra of AαC metabolites
The product ion spectrum of AαC-N2-Gluc ([M+H]+ at m/z 360.1) (M5) at MS2 stage, shown in Figure 2A, displays fragment ions at m/z 280.1 ([M+H-2H2O-CO2]+), m/z 226.1 ([M+H-C4H6O5]+) and at m/z 184.1 ([M+H-C6H8O6]+). The latter two fragment ions are attributed to the positively charged acetyl derivative of AαC and the protonated AαC. These results are consistent with a previous study.49
Figure 2.
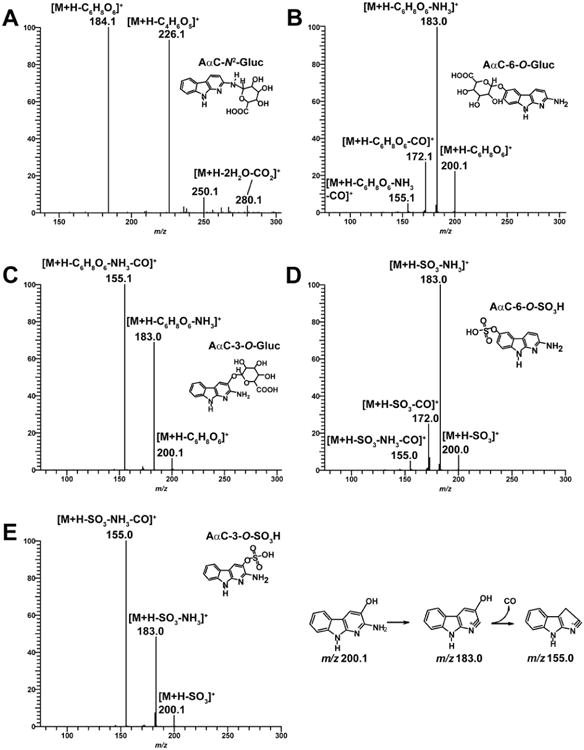
(A) LC/MS2 product ion mass spectra of AαC-N2-Gluc (m/z 360.1 >) and LC/MS3 product ion mass spectra of (B) AαC-6-O-Gluc (m/z 376.1 > 200.1 >), (C) AαC-3-O-Gluc (m/z 376.1 > 200.1 >), (D) AαC-6-O-SO3H (m/z 280.1 > 200.1 >), (E) AαC-3-O-SO3H (m/z 280.1 > 200.1 >) in positive ionization mode. The proposed mechanism of formation of the prominent fragment ion of AαC-3-OH at m/z 155.0 is presented.
The two AαC-O-Gluc isomers (M2 and M3) are readily distinguished by their product ion spectra at MS3 scan stage. The product ion spectrum of AαC-6-O-Gluc ([M+H]+ at m/z 376.1) (M2) at MS3 scan stage displays two fragments ions: one ion at m/z 200.1 ([M+H-C6H8O6]+) attributed to the positively charged AαC-6-OH, which formed by the cleavage of the Gluc moiety, and the second ion at m/z 183.0 ([M+H-C6H8O6-NH3]+) is attributed to the loss of NH3 from AαC-6-OH (Figure 2B). In the case of AαC-3-O-Gluc, ([M+H]+ at m/z 376.1) (M3) the fragment ions at m/z 200.1 and m/z 183.0 are also observed, in addition to a prominent fragment ion at m/z 155.1 ([M+H-C6H8O6-NH3-CO]+), which is proposed to occur by the successive loss of NH3 and CO from AαC-3-OH (Figure 2C). The ion at m/z 155.1 is a prominent product ion of AαC-3-OH, but not for AαC-6-OH, when these hydroxylated AαC derivatives undergo CID with a triple quadrupole MS, or by ion trap at the MS3 scan stage (unpublished results, R. Turesky). The differences in the pattern of fragmentation of AαC-O-Gluc isomers lead us to tentatively assign M3 as AαC-3-O-Gluc and M2 as AαC-6-O-Gluc.
The hydrolysis products of M2 and M3 obtained by treatment with β-glucuronidase showed that M2 co-eluted with AαC-6-OH and displayed an UV spectrum identical to that of AαC-6-OH, whereas the hydrolysis product of M3 co-eluted with AαC-3-OH and exhibited the same UV spectrum as AαC-3-OH (Figure S1). The AαC-6-OH and AαC-3-OH reference compounds produced by human liver microsomes were characterized by 1H-NMR, which unambiguously identified the sites of ring-oxidation of AαC.17,34 Taken together these results lead us to identify M2 as AαC-6-O-Gluc and M3 as AαC-3-O-Gluc.
As was observed for the O-Gluc conjugates, the isomeric sulfate conjugates can be distinguished by their product ion spectra. The product ion spectrum of AαC-6-O-SO3H (M6) ([M+H]+ at m/z 280.0) at MS3 stage displays two fragments ions at m/z 200.0 ([M+H-SO3]+) and m/z 183.0 ([M+H-SO3-NH3]+) (Figure 2D). The product ion spectrum of AαC-3-O-SO3H (M4) ([MH]+ at m/z 280.0) at MS3 stage also displays the fragment ions at m/z 200.1 and 183.0, and a major fragment ion at m/z 155.0 ([M+H-SO3-NH3-CO]+) attributed to the loss of NH3 and CO from AαC-3-OH (Figure 2E). These results lead to the tentative identification of M4 as AαC-6-O-SO3H and M6 as AαC-3-O-SO3H. The sulfatase hydrolysis product of M4 and M6 co-eluted by HPLC and exhibited the characteristic UV spectra of AαC-3-OH and AαC-6-OH, respectively support the assignment of M4 as AαC-6-O-SO3H and M6 as AαC-3-O-SO3H (Figure S1).
The product ion spectrum of N2-acetyl-AαC-6-O-Gluc (M1) ([M+H]+ at m/z 418.1) at MS3 scan stage is shown in Figure 3A. A major fragment ion is observed at m/z 200.1 ([M+H-C6H8O6-C2H3O]+) attributed to the cleavage of the Gluc and the acetyl linkages. Fragment ions at m/z 183.1 ([M+H-C6H8O6-C2H3O-NH3]+) and m/z 172.1 ([M+H-C6H8O6-C2H3O-CO]+) are also observed. A fragment ion at m/z 155.1 attributed to the successive loss of NH3 and CO from protonated AαC-6-OH was also observed. However, this ion is relatively minor in abundance. The position of oxidation and identification of the metabolite as N2-acetyl-AαC-6-O-Gluc was determined by comparing the UV spectrum of hydrolysis product to those UV spectra of AαC-6-OH and AαC-3-OH. The N2-acetyl-AαC-6-O-Gluc hydrolysis product (enzymatic hydrolysis using β-glucuronidase followed by acid hydrolysis) co-eluted and exhibited an UV spectrum identical to that of AαC-6-OH, whereas the UV spectrum and retention time of AαC-3-OH were different. These data confirm the identification of M1 as N2-acetyl-AαC-6-O-Gluc (Figure 3B).
Figure 3.
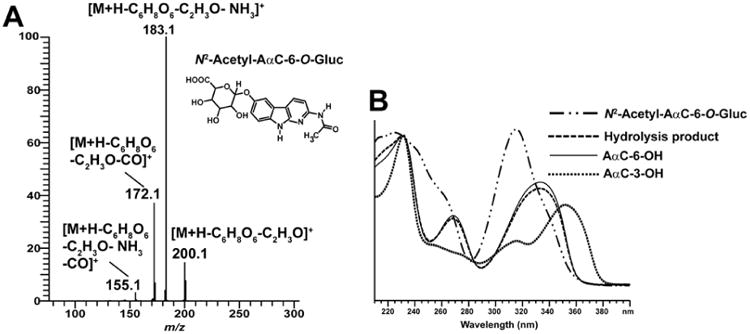
Identification and characterization of N2-Acetyl-AαC-6-O-Gluc. (A) LC/MS3 product ion mass spectra of N2-Acetyl-AαC-6-O-Gluc (m/z 418.1 > 242.1 >) formed in primary human hepatocytes. N2-Acetyl-AαC-6-O-Gluc was purified by HPLC and characterized by mass spectrometry. (B) UV spectra of AαC-6-OH, AαC-3-OH, N2-acetyl-AαC-O-Gluc and its hydrolysis product obtained after β-glucuronidase followed by acid treatment of N2-acetyl-AαC-O-Gluc as described in material and method.
Characterization of minor metabolites of AαC in human hepatocytes
We assessed the metabolite formation in primary human hepatocytes treated with various concentrations of AαC (0.1, 1, 10 and 50 μM) over 24 h followed by UPLC/MSn analysis. In addition to the previously described metabolites, several minor metabolites were identified. There were two N-acetylated ring-oxidized AαC sulphate conjugates (N2-acetyl-AαC-6-O-SO3H, N2-acetyl-AαC-3-O-SO3H), one N-acetylated ring-oxidized AαC Gluc conjugate (N2-acetyl-AαC-3-O-Gluc) and the genotoxic metabolite, AαC-HN2-O-Gluc (Figure 4).
Figure 4.
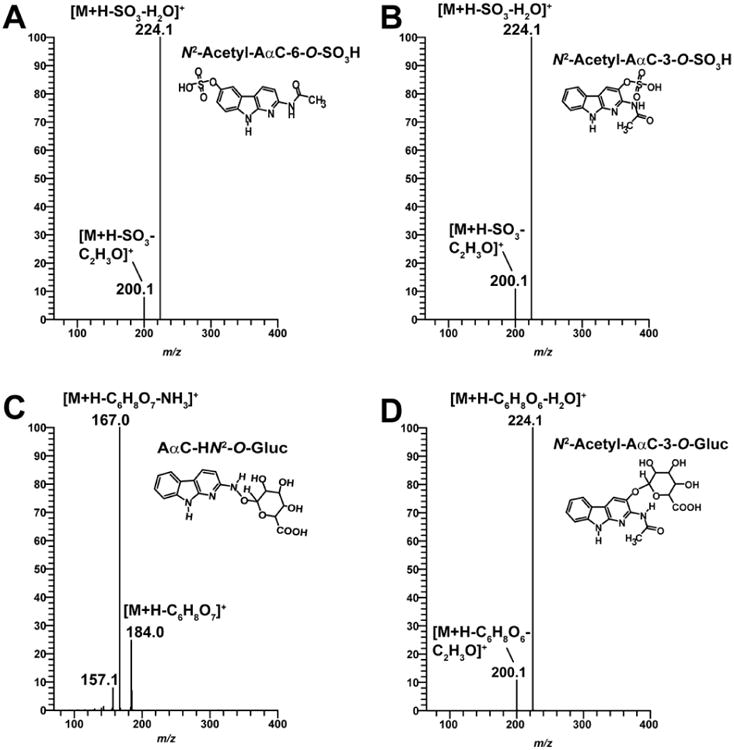
LC/MS3 product ion mass spectra of N2-acetyl-AαC-6-O-SO3H (m/z 322.1 > 241.1 >) (A), N2-acetyl-AαC-3-O-SO3H (m/z 322.1 > 241.1 >) (B), AαC-HN2-O-Gluc (m/z 376.1 > 200.1 >) (C) and N2-acetyl-AαC-3-O-Gluc (m/z 418.1 > 242.1 >) (D).
The product ion spectra of the two N2-acetyl-AαC-O-SO3H conjugates ([M+H]+ at m/z 322.1) at the MS3 scan stage revealed a prominent fragment ion at m/z 224.1 ([M+H-H2SO4]+) which occurs by cleavage of the sulfate linkage. In addition, fragment ions were observed at m/z 200.1 ([M+H-SO3-C2H3O]+) attributed to the loss of SO3 and the acetyl linkage to form a protonated AαC-OH species (Figure 4A and 4B). We were unable to distinguish the identities of these two isomers based on mass spectral data; however, based on their polarity and the fact that the retention time for the AαC-6-O-SO3H metabolite was characteristically shorter than that of AαC-3-O-SO3H, the metabolite observed at tR 15.2 min was tentatively assigned as N2-acetyl-AαC-6-O-SO3H and the metabolite observed at tR 16.9 min assigned as N2-acetyl-AαC-3-O-SO3H (Figure 5).
Figure 5.
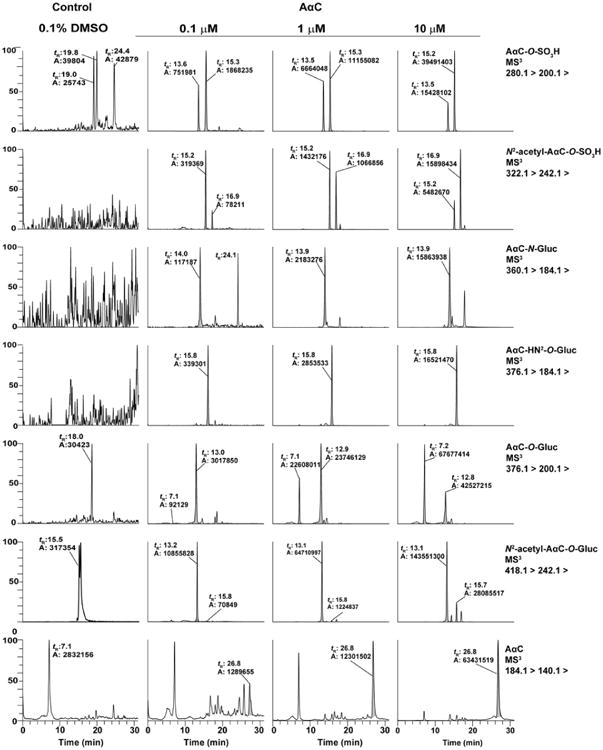
Mass chromatograms of AαC metabolites formed during 24h in primary human hepatocytes incubated 0.1, 1 and 10 μM of AαC. The retention time (tR) are AαC-6-O-SO3H (13.5), AαC-3-O-SO3H (15.2), N2-acetyl-AαC-6-O-SO3H (15.2), N2-acetyl-AαC-3-O-SO3H (16.9), AαC-6-O-Gluc (7.2), AαC-3-O-Gluc (12.8), N2-acetyl-AαC-6-O-Gluc (13.1), N2-acetyl-AαC-3-O-Gluc (15.7) min.
In the case of N2-acetyl-AαC-3-O-Gluc ([M+H]+ at m/z 418.1), The product ion spectra et MS3 stage shows a fragment ion at m/z 224.1 ([M+H-C6H8O6-H2O]+), which is attributed to the cleavage of the Gluc linkage and the loss of water. In addition, we observed a fragment ion at m/z 200.1 attributed to the protonated AαC-3-OH resulting from the concomitant cleavages of the Gluc and the acetyl moieties (Figure 4D). Based on its polarity and its longer retention time in comparison to its N2-acetyl-AαC-6-O-Gluc isomer described above, we tentatively identified this metabolite as N2-acetyl-AαC-3-O-Gluc (Figure 5).
The product ion spectrum of the previously reported AαC-HN2-O-Gluc ([M+H]+ at m/z 376.0) at MS3 stage shown in Figure 4C displays two fragment ions: the ion at m/z 184.0 ([M+H-C6H8O7]+) is attributed to the positively charged AαC, which is formed by the cleavage of the Gluc linkage. The ion at m/z 167.0 ([M+H-C6H8O7-NH3]+) is attributed to the loss of NH3 from the protonated AαC. The online LC-ESI-MSn consecutive reaction product ion spectra acquired in the negative mode of AαC-HN2-O-Gluc [M-H]- at m/z 374.1 at the MS2 and MS3 scan stages support the proposed structure with an O-linkage formed between the HONH-AαC and Gluc. The base peak ion in the product ion spectrum at the MS2 scan stage was observed at m/z 193.0 [M-H-AαC]-. The second generation product ion spectrum of AαC-HN2-O-Gluc acquired on m/z 193.0 shows the typical CID fragmentation pattern previously reported for the glucoronate,50 and proved that the linkage formed between the glucuronic acid and HONH-AαC occurred through the oxygen atom of HONH-AαC (Figure S2). These spectral data are consistent with the published data on the metabolite produced by recombinant UGTs.34
In addition to these fully characterized metabolites, we observed the formation of several other minor metabolites, which are isomers of those characterized above and include two N2-acetyl-AαC-O-Gluc, one N2-acetyl-AαC-O-SO3H, and one AαC-N-Gluc.
AαC metabolism at low concentrations in human hepatocytes
The biotransformation of AαC was examined as a function of concentration in primary human hepatocytes treated with 0.1, 1 and 10 μM of AαC. As shown in Figure 6, the concentration of AαC did not greatly impact its metabolism; indeed the patterns of metabolites formed were comparable at all of the concentrations studied. Based on the total ion counts of each metabolite in positive ion mode, the N2-acetyl-AαC-6-O-Gluc is the most prominent metabolite, followed by the two ring-oxidized AαC glucuronide conjugates (AαC-3-O-Gluc and AαC-6-O-Gluc) and the two ring-oxidized AαC sulfate conjugates (AαC-3-O-SO3H and AαC-6-O-SO3H). However, these estimates are approximate as the ionization efficiencies, particularly those of the sulfate conjugates in the positive ion mode, may result in underestimation of product formation. The findings show that the ring oxidation of C3 and C6 position constitutes a major pathway of metabolism of AαC. It is noteworthy that the formation of the genotoxic metabolite AαC-HN2-O-Gluc occurred at all the concentrations studied.
Figure 6.
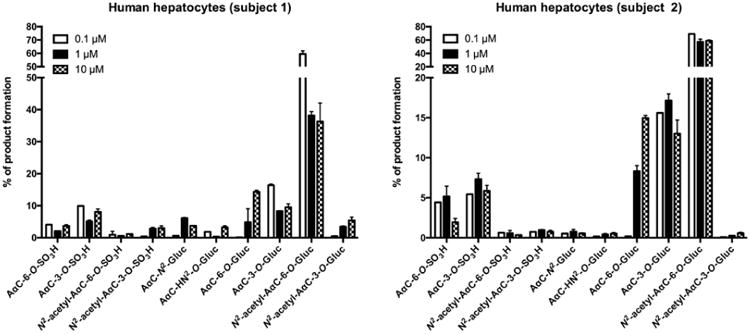
Distribution of AαC metabolites formed as a function of dose in two human primary hepatocyte preparations after 24 h of incubation. Primary human hepatocytes were incubated with various concentrations of AαC (0.1, 1 and 10 μM) during 24 h and the relative ion abundance of each metabolites derived from AαC formed were determined by mass spectrometry based on total ion counts in positive ion mode.
Role of P450 1A2 in AαC metabolism and DNA adduct formation in human hepatocytes
Human P450 1A2 is well recognized as the major isoform responsible for bioactivation of a number of HAA.29,51-53 The role of P450 1A2 in the formation of AαC metabolites in hepatocytes was assessed with furafylline, a selective, mechanism-based P450 1A2 inhibitor.43 In our experimental conditions, we observe that 24 h of treatment with 5 μM of furafylline leads to more than 80% decrease of P450 1A2 activity (date not shown). As shown in Figure 7, the pre-incubation of human hepatocytes with furafylline resulted in a strong decrease in the levels of AαC-HN2-O-Gluc with a concomitant increase in the amount of unmetabolized AαC and AαC-N2-Gluc. These results demonstrate that P450 1A2 is the major isoform involved in the metabolism of AαC and formation of AαC-HN2-O-Gluc. In addition, we observed that P450 1A2 inhibition lead to a decrease in the levels of the two ring-oxidized AαC sulfate conjugates (AαC-3-O-SO3H and AαC-6-O-SO3H), the two ring-oxidized AαC glucuronide conjugates (AαC-3-O-Gluc and AαC-6-O-Gluc) and the two N-acetylated AαC-6-OH conjugates (N2-acetyl-AαC-6-O-Gluc and N2-acetyl-AαC-6-O-SO3H). In contrast, the relative abundance of the N2-acetyl-AαC-3-O-SO3H and N2-acetyl-AαC-3-O-Gluc were increased by the inhibition of P450 1A2. Thus, other P450s may have catalysed the C-3 oxidation N2-acetyl-AαC. The potency of furafylline P450 1A2 mediated inhibition was most pronounced when AαC was incubated at low substrate concentrations, indicating that other P450s29 or oxidases17 contributed to the metabolism of AαC at higher substrate concentrations.
Figure 7.
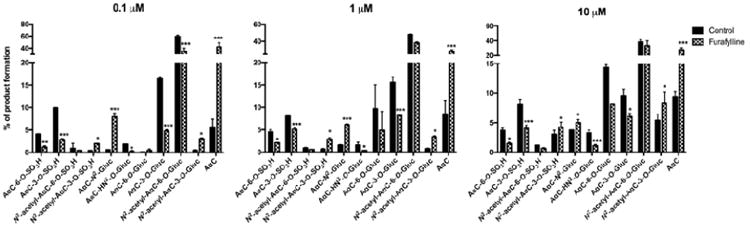
Role of P450 1A2 in AαC metabolism in human hepatocytes. Cells were pre-treated with furafylline (5 μM) for 24h prior to treatment with AαC (0.1, 1 and 10 μM) during 24h. The relative abundance of each AαC derived metabolites was determined by mass spectrometry based on total ion counts in positive ion mode. (Student's t-test, * P<0.05; **P<0.01, ***P<0.005 versus control).
The levels of dG-C8-AαC were measured in conjunction with the amounts of AαC-HN2-O-Gluc formed in human hepatocytes treated with several doses of AαC (0.1, 1 and 10 μM) in the presence or absence of furafylline. As shown in Figure 8, AαC-HN2-O-Gluc and dG-C8-AαC were formed in a dose-dependent manner. The inhibition of P450 1A2 lead to a 70 - 80% decrease in the amount of AαC-HN2-O-Gluc and dG-C8-AαC adduct levels. These results signify that P450 1A2 is the major isoform involved in the bioactivation of AαC in human hepatocytes, to form genotoxic metabolites, including AαC-HN2-O-Gluc, and dG-C8-AαC adduct formation. A representative UPLC-ESI/MS3 chromatogram and product ion spectra of DNA adduct are shown in supplementary data (Figure S3). The product ion spectrum is in excellent agreement to our previously published data27.
Figure 8.
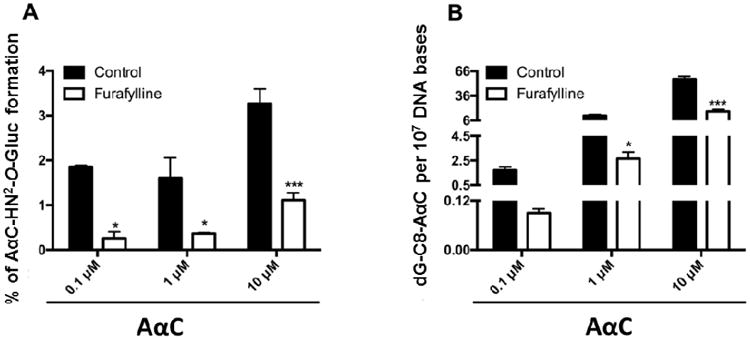
Correlation between DNA adducts levels derived from AαC and the relative abundance of AαC-HN2-O-Gluc formed in human hepatocytes. Relative abundance of AαC-HN2-O-Gluc (A) and dG-C8-AαC (B) formed in human hepatocytes. Cells were pre-treated with furafylline (5 μM) for 24h prior to incubation with 0.1, 1, 10 and 50 μM of AαC for 24h and the relative abundance of AαC-HN2-O-Gluc and dG-C8-AαC levels derived from AαC formed were estimated by mass spectrometry. (Student's t-test, * P<0.05; **P<0.01, ***P<0.005 versus control).
Discussion
The aim of this study was to characterize the major metabolites of AαC formed in primary human hepatocytes using UPLC/MSn. Our data demonstrate that AαC is subjected to direct conjugation reactions such as N-glucuronidation, but also double and triple biotransformation reactions by the combination of N-oxidation or ring-oxidation followed by O-sulfonation, O-glucuronidation, as well as N-acetylation. The amounts of N-acetyl-AαC were extremely low in hepatocytes, indicating that this metabolite undergoes further metabolism (unpublished data, R. Turesky). We detected 10 stable metabolites including six major metabolites: AαC-3-O-SO3H, AαC-6-O-SO3H, AαC-3-O-Gluc, AαC-6-O-Gluc, N2-acetyl-AαC-6-O-Gluc and AαC-N2-Gluc, and four minor products: N2-acetyl-AαC-6-O-Gluc, N2-acetyl-AαC-6-O-SO3H, N2-acetyl-AαC-3-O-SO3H and AαC-HN2-O-Gluc.
Ring oxidation of AαC at the C-3 and C-6 atoms of the heterocylic ring are the major pathways of AαC metabolism in human hepatocytes. These oxidation pathways are similar to the data reported by Raza et al. using human liver microsomes, where three oxidized products of AαC were identified: one N-oxidized product (HONH-AαC) and two ring-oxidized products (AαC-3-OH and AαC-6-OH).29 The ratio of ring-oxidized products to N-oxidized product was estimated to be 85:15 and was relatively constant across the several human liver microsomal preparations.29 Since AαC-3-OH and AαC-6-OH possess no mutagenic activities,28,29 the predominant routes for AαC metabolism in human hepatocytes appear as detoxication pathways. However, AαC-3-O-SO3H also can be produced by the rearrangement of the genotoxic N-sulfate ester of HONH-AαC.17 Moreover, AαC undergoes metabolism to produce higher levels of DNA adducts than 4-ABP, PhIP or MeIQx in human hepatocytes.23,24 Therefore, DNA adducts are important biomarkers to measure when characterizing procarcinogen metabolism in cell systems. Although AαC undergoes extensive metabolism by N2-acetylation and oxidation, we have not yet detected DNA adducts of AαC that possesses an N-acetyl group (unpublished observations, R. Turesky) as was reported for the structurally related aromatic amine, 2-aminofluorene.54,55
There are large interspecies differences in metabolism of AαC in human hepatocytes compared to those previously reported in vivo in rats.40 The four major metabolites of AαC identified in the bile and urine of adult male Sprague-Dawley rats treated intravenously with AαC were: AαC-3-O-SO3H, N2-acetyl-AαC-3-O-SO3H, N2-acetyl-AαC-6-O-SO3H, N2-acetyl-AαC-3-O-Gluc.40 The latter three metabolites were produced in relatively minor amounts in primary human hepatocytes. This discrepancy may be explained, in part by the different catalytic activities and regioselectivity of human and rat enzymes orthologues toward this procarcinogen. We previously reported interspecies differences in metabolism of two other HAA, PhIP and MeIQx in rodent and human hepatocytes.52,53 Indeed, the catalytic activity of human P450 1A2 was 9 to 11 fold higher than rat P450 1A2 in the N-oxidation of MeIQx and PhIP,53,56 and human P450 1A2 catalyzed the N-oxidation as the major pathway of metabolism whereas ring oxidation of these HAA was negligible.52,53,57,58 The biotransformation of AαC was also previously investigated in the human liver HepG2 cell line, where four major metabolites were identified: AαC-3-O-SO3H, AαC-6-O-SO3H, N2-acetyl-AαC-6-O-SO3H and N2-acetyl-AαC.40 The absence of Gluc metabolites is not surprising since this cell line is devoid of UGT.40,59 Taken together, these metabolic data show that rats and HepG2 cell lines do not fully capture the metabolism of AαC that occurs in human hepatocytes.
The genotoxicity of AαC is dependent upon N-oxidation of AαC. In vitro studies conducted with human liver microsomes or recombinant human P450s reveal that P450 1A2 is the primary isoform involved in N-oxidation and bioactivation of AαC.26,29 We previously reported that P450 1A2 is the primary isoform involved in N-oxidation of AαC in primary human hepatocytes and that the catalytic activity of P450 1A2 is critical for AαC derived DNA adduct formation.23 In this current study, we demonstrate that P4501A2 is also the major isoform involved in metabolism of AαC in human hepatocytes, by use of furafylline, a selective inhibitor of P450 1A2.43 There is a strong correlation among P450 1A2 activity, AαC-HN2-O-Gluc and dG-C8-AαC formation in human hepatocytes. AαC-HN2-O-Gluc is a biologically reactive metabolite that binds to DNA,34,60 however, other reactive intermediates such as the N-sulfooxy- or N-acetoxy esters of AαC also bind to DNA. In contrast to the N-sulfooxy- or N-acetoxy esters of AαC, which are labile, AαC-HN2-O-Gluc is sufficiently stable and can be isolated and measured.61,62 NAT1 and NAT2 and SULT1A2 do catalyse the DNA binding of HONH-AαC to DNA in vitro.30 The relative contributions of UGT, NAT, SULT or other conjugating enzymes in the metabolic activation of AαC in humans remain to be determined. Pretreatment of hepatocytes with furafylline also strongly decreased the levels of sulfate and Gluc conjugates of AαC-3-OH and AαC-6-OH, but the N-acetylated derivatives of these metabolites increased when cells were pretreated with furafylline. These findings suggest that N-acetyl-AαC undergoes and C-6 oxidation by P450s other than P4501A2.
Recently, some oxidative products of AαC, including AαC-3-O-SO3H, were detected in plasma of liver specific P-450 reductase null mice, suggesting that a non-hepatic P450 enzymes catalysed oxidation of AαC in the mouse model.41 The level of DNA adduct formation of AαC in liver was also unaffected in the liver specific P-450 reductase null mice treated with high doses of AαC.43 Our data show that the contribution of non-P450 oxidases to the metabolism of AαC and bioactivation in human hepatocytes is minor.
In summary, human hepatocytes extensively metabolize AαC into more than ten products and also produce high levels of DNA adducts. Some of the metabolites of AαC may be used as biomarkers of exposure to AαC in molecular epidemiological studies since AαC is extensively metabolized in humans.17,63 AαC-3-OSO3H has already been employed as a urinary biomarker and frequently detected in the urine of smokers.17 The development of AαC-HN2-O-Gluc as a urinary biomarker could provide valuable information about the extent of bioactivation of AαC in humans. Given the high levels of AαC in mainstream tobacco smoke,8-11 and the relatively high levels of AαC DNA adducts formed in human hepatocytes compared to other HAA or aromatic amines,23,24 further studies on the potential role of AαC in tobacco-associated liver and gastrointestinal cancers are warranted.
Supplementary Material
Acknowledgments
MB was a recipient of Anses and UFR Pharmacie of University of Rennes 1 as well as travel fellowships from Université Européenne de Bretagne
Funding Sources: This work was supported by Inserm, la Ligue contre le Cancer, the PNREST Anses, Cancer TMOI AVIESAN, 2013/1/166 (SL), and in part by National Cancer Institute of the National Institutes of Health grant RO1CA134700 (R.J.T) and R01CA134700-S (R.J.T) of the Family Smoking Prevention and Tobacco Control Act, and in part by National Cancer Institute Cancer Center Support Grant No. CA077598 (R.J.T).
Abbreviations
- AαC
2-Amino-9H-pyrido[2,3-b]indole
- NNK
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
- 4-ABP
4-aminobiphenyl
- B[a]P
benzo[a]pyrene
- PhIP
2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine
- MeIQx
2-amino-3,8-dimethylmidazo[4,5-f]quinoxaline
- IQ
2-amino-3-methylimidazo[4,5-f]quinolone
- dG-C8-AαC
N-(deoxyguanosin-8-yl)-2-amino-9H-pyrido[2,3-b]indole
- NAT
N-acetyltransferases
- SULT
Sulfotransferases
- UGT
UDP-Glucuronosyltrasferases
- UPLC/MSn
UPLC/ion trap multistage mass spectrometry
- HONH-AαC
2-hydroxyamino-9H-pyrido[2,3-b]indole
- AαC-3-OH
2-amino-3-hydroxy-9H-pyrido[2,3-b]indole
- AαC-6-OH
2-amino-6-hydroxy-9H-pyrido[2,3-b]indole
- AαC-3-O-SO3H
2-amino-9H-pyrido[2,3-b]indol-3-yl-sulfate
- AαC-6-O-SO3H
2-amino-9H-pyrido[2,3-b]indol-6-yl-sulfate
- AαC-3-O-Gluc
2-amino-9H-pyrido[2,3-b]indol-3-yl-oxo-(β-D-glucuronic acid)
- AαC-6-O-Gluc
2-amino-9H-pyrido[2,3-b]indol-6-yl-oxo-(β-D-glucuronic acid)
- N2-acetyl-AαC-3-O-SO3H
N2-acetyl-9H-pyrido[2,3-b]indol-3-yl sulfate
- N2-acetyl-AαC-6-O-SO3H
N2-acetyl-9H-pyrido[2,3-b]indol-6-yl sulfate
- N2-acetyl-AαC-3-O-Gluc
N2-acetyl-2-amino-9H-pyrido[2,3-b]indol-3-yl-oxo-(β-D-glucuronic acid)
- N2-acetyl-AαC-6-O-Gluc
N2-acetyl-2-amino-9H-pyrido[2,3-b]indol-6-yl-oxo-(β-D-glucuronic acid)
- AαC-N2-Gluc
N2-(β-D-glucosidurony1)-2-amino-9H-pyrido[2,3-b]indole
- AαC-HN2-O-Gluc
O-(β-D-glucosidurony1)-2-hydroxyamino-9H-pyrido[2,3-b]indole
- SPE
Solid-phase extraction
Footnotes
Supporting Information: Regioisomer characterization of AαC-6-O-Gluc, AαC-3-O-Gluc, AαC-6-O-SO3H and AαC-3-O-SO3H. LC/MS2 product ion mass spectra of AαC-HN2-O-Gluc in negative ion mode. Second generation product ion spectrum of AαC-HN2-O-Gluc acquired on m/z 193 in negative ion mode. UPLC-ESI/MS3 chromatograms of dG-C8-AαC formed in primary human hepatocytes with or without inhibition of P4501A2 with furafylline. Primary human hepatocytes were pre-treated for 24 h with 0.1 % DMSO, orfurafylline (5 μM) followed by 24 h of treatment with 0.1 % DMSO (Ctrl), or AαC (0.1, 1 or 10 μM), Donor information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Giovannucci E. An updated review of the epidemiological evidence that cigarette smoking increases risk of colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2001;10:725–731. [PubMed] [Google Scholar]
- 2.IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. IARC. Tobacco smoke and involuntary smoking. 2004;83:1–1438. [PMC free article] [PubMed] [Google Scholar]
- 3.IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Personal habits and indoor combustions. 2012;100:1–538. [PMC free article] [PubMed] [Google Scholar]
- 4.Hoffmann D, Hoffmann I, El-Bayoumy K. The less harmful cigarette: a controversial issue. a tribute to Ernst L. Wynder. Chem Res Toxicol. 2001;14:767–790. doi: 10.1021/tx000260u. [DOI] [PubMed] [Google Scholar]
- 5.Sugimura T, Wakabayashi K, Nakagama H, Nagao M. Heterocyclic amines: Mutagens/carcinogens produced during cooking of meat and fish. Cancer Sci. 2004;95:290–299. doi: 10.1111/j.1349-7006.2004.tb03205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manabe S, Izumikawa S, Asakuno K, Wada O, Kanai Y. Detection of carcinogenic amino-alpha-carbolines and amino-gamma-carbolines in diesel-exhaust particles. Environ Pollut. 1991;70:255–265. doi: 10.1016/0269-7491(91)90013-m. [DOI] [PubMed] [Google Scholar]
- 7.Manabe S, Kurihara N, Wada O, Izumikawa S, Asakuno K, Morita M. Detection of a carcinogen, 2-amino-1-methyl-6-phenylimidazo [4,5-b]pyridine, in airborne particles and diesel-exhaust particles. Environ Pollut. 1993;80:281–286. doi: 10.1016/0269-7491(93)90049-t. [DOI] [PubMed] [Google Scholar]
- 8.Zhang L, Ashley DL, Watson CH. Quantitative analysis of six heterocyclic aromatic amines in mainstream cigarette smoke condensate using isotope dilution liquid chromatography-electrospray ionization tandem mass spectrometry. Nicotine Tob Res. 2011;13:120–126. doi: 10.1093/ntr/ntq219. [DOI] [PubMed] [Google Scholar]
- 9.Hoffmann DHI. Letters to the editor, tobacco smoke components. Beiträge zur Tabakforschung International/Contributions to Tobacco Research. 1998;18 [Google Scholar]
- 10.Smith CJ, Qian X, Zha Q, Moldoveanu SC. Analysis of alpha- and beta-carbolines in mainstream smoke of reference cigarettes by gas chromatography-mass spectrometry. J Chromatogr A. 2004;1046:211–216. doi: 10.1016/j.chroma.2004.06.082. [DOI] [PubMed] [Google Scholar]
- 11.Yoshida D, Matsumoto T. Amino-alpha-carbolines as mutagenic agents in cigarette smoke condensate. Cancer Lett. 1980;10:141–149. doi: 10.1016/0304-3835(80)90037-3. [DOI] [PubMed] [Google Scholar]
- 12.Patrianakos C, Hoffmann D. Chemical Studies on Tobacco-Smoke. 64 Analysis of Aromatic-Amines in Cigarette-Smoke. J of Ana Toxicol. 1979;3:150–154. [Google Scholar]
- 13.Zha Q, Qian NX, Moldoveanu SC. Analysis of polycyclic aromatic hydrocarbons in the particulate phase of cigarette smoke using a gas chromatographic-high-resolution mass spectrometric technique. J Chromatogr Sci. 2002;40:403–408. doi: 10.1093/chromsci/40.7.403. [DOI] [PubMed] [Google Scholar]
- 14.Hecht SS. Human urinary carcinogen metabolites: biomarkers for investigating tobacco and cancer. Carcinogenesis. 2002;23:907–922. doi: 10.1093/carcin/23.6.907. [DOI] [PubMed] [Google Scholar]
- 15.Turesky RJ, Yuan JM, Wang R, Peterson S, Yu MC. Tobacco smoking and urinary levels of 2-amino-9H-pyrido[2,3-b]indole in men of Shanghai, China. Cancer Epidemiol Biomarkers Prev. 2007;16:1554–1560. doi: 10.1158/1055-9965.EPI-07-0132. [DOI] [PubMed] [Google Scholar]
- 16.Fu Y, Zhao G, Wang S, Yu J, Xie F, Wang H, Xie J. Simultaneous determination of fifteen heterocyclic aromatic amines in the urine of smokers and nonsmokers using ultra-high performance liquid chromatography-tandem mass spectrometry. J Chromatogr A. 2014;1333:45–53. doi: 10.1016/j.chroma.2014.01.057. [DOI] [PubMed] [Google Scholar]
- 17.Konorev D, Koopmeiners JS, Tang Y, Franck Thompson EA, Jensen JA, Hatsukami DK, Turesky RJ. Measurement of the Heterocyclic Amines 2-Amino-9H-pyrido[2,3-b]indole and 2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyridine in Urine: Effects of Cigarette Smoking. Chem Res Toxicol. 2015;28:2390–2399. doi: 10.1021/acs.chemrestox.5b00401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ohgaki H, Matsukura N, Morino K, Kawachi T, Sugimura T, Takayama S. Carcinogenicity in mice of mutagenic compounds from glutamic acid and soybean globulin pyrolysates. Carcinogenesis. 1984;5:815–819. doi: 10.1093/carcin/5.6.815. [DOI] [PubMed] [Google Scholar]
- 19.Zhang XB, Felton JS, Tucker JD, Urlando C, Heddle JA. Intestinal mutagenicity of two carcinogenic food mutagens in transgenic mice: 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine and amino(alpha)carboline. Carcinogenesis. 1996;17:2259–2265. doi: 10.1093/carcin/17.10.2259. [DOI] [PubMed] [Google Scholar]
- 20.Okonogi H, Ushijima T, Zhang XB, Heddle JA, Suzuki T, Sofuni T, Felton JS, Tucker JD, Sugimura T, Nagao M. Agreement of mutational characteristics of heterocyclic amines in lacI of the Big Blue mouse with those in tumor related genes in rodents. Carcinogenesis. 1997;18:745–748. doi: 10.1093/carcin/18.4.745. [DOI] [PubMed] [Google Scholar]
- 21.Pfau W, Martin FL, Cole KJ, Venitt S, Phillips DH, Grover PL, Marquardt H. Heterocyclic aromatic amines induce DNA strand breaks and cell transformation. Carcinogenesis. 1999;20:545–551. doi: 10.1093/carcin/20.4.545. [DOI] [PubMed] [Google Scholar]
- 22.Majer BJ, Kassie F, Sasaki Y, Pfau W, Glatt H, Meinl W, Darroudi F, Knasmuller S. Investigation of the genotoxic effects of 2-amino-9H-pyrido[2,3-b]indole in different organs of rodents and in human derived cells. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;802:167–173. doi: 10.1016/j.jchromb.2003.10.042. [DOI] [PubMed] [Google Scholar]
- 23.Nauwelaers G, Bellamri M, Fessard V, Turesky RJ, Langouet S. DNA adducts of the tobacco carcinogens 2-amino-9H-pyrido[2,3-b]indole and 4-aminobiphenyl are formed at environmental exposure levels and persist in human hepatocytes. Chem Res Toxicol. 2013;26:1367–1377. doi: 10.1021/tx4002226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nauwelaers G, Bessette EE, Gu D, Tang Y, Rageul J, Fessard V, Yuan JM, Yu MC, Langouet S, Turesky RJ. DNA adduct formation of 4-aminobiphenyl and heterocyclic aromatic amines in human hepatocytes. Chem Res Toxicol. 2011;24:913–925. doi: 10.1021/tx200091y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pfau W, Schulze C, Shirai T, Hasegawa R, Brockstedt U. Identification of the major hepatic DNA adduct formed by the food mutagen 2-amino-9H-pyrido[2,3-b]indole (A alpha C) Chem Res Toxicol. 1997;10:1192–1197. doi: 10.1021/tx9701182. [DOI] [PubMed] [Google Scholar]
- 26.Frederiksen H, Frandsen H. Excretion of metabolites in urine and faeces from rats dosed with the heterocyclic amine, 2-amino-9H-pyrido[2,3-b]indole (AalphaC) Food Chem Toxicol. 2004;42:879–885. doi: 10.1016/j.fct.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 27.Turesky RJ, Bendaly J, Yasa I, Doll MA, Hein DW. The impact of NAT2 acetylator genotype on mutagenesis and DNA adducts from 2-amino-9H-pyrido[2,3-b]indole. Chem Res Toxicol. 2009;22:726–733. doi: 10.1021/tx800473w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niwa T, Yamazoe Y, Kato R. Metabolic activation of 2-amino-9H-pyrido[2,3-b]indole by rat-liver microsomes. Mutat Res. 1982;95:159–170. doi: 10.1016/0027-5107(82)90254-8. [DOI] [PubMed] [Google Scholar]
- 29.Raza H, King RS, Squires RB, Guengerich FP, Miller DW, Freeman JP, Lang NP, Kadlubar FF. Metabolism of 2-amino-alpha-carboline. A food-borne heterocyclic amine mutagen and carcinogen by human and rodent liver microsomes and by human cytochrome P4501A2. Drug Metab Dispos. 1996;24:395–400. [PubMed] [Google Scholar]
- 30.King RS, Teitel CH, Kadlubar FF. In vitro bioactivation of N-hydroxy-2-amino-alpha-carboline. Carcinogenesis. 2000;21:1347–1354. [PubMed] [Google Scholar]
- 31.Turesky RJ, Le Marchand L. Metabolism and biomarkers of heterocyclic aromatic amines in molecular epidemiology studies: lessons learned from aromatic amines. Chem Res Toxicol. 2011;24:1169–1214. doi: 10.1021/tx200135s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nowell SA, Massengill JS, Williams S, Radominska-Pandya A, Tephly TR, Cheng Z, Strassburg CP, Tukey RH, MacLeod SL, Lang NP, Kadlubar FF. Glucuronidation of 2-hydroxyamino-1-methyl-6-phenylimidazo[4, 5-b]pyridine by human microsomal UDP-glucuronosyltransferases: identification of specific UGT1A family isoforms involved. Carcinogenesis. 1999;20:1107–1114. doi: 10.1093/carcin/20.6.1107. [DOI] [PubMed] [Google Scholar]
- 33.Malfatti MA, Felton JS. Human UDP-glucuronosyltransferase 1A1 is the primary enzyme responsible for the N-glucuronidation of N-hydroxy-PhIP in vitro. Chem Res Toxicol. 2004;17:1137–1144. doi: 10.1021/tx049898m. [DOI] [PubMed] [Google Scholar]
- 34.Tang Y, LeMaster DM, Nauwelaers G, Gu D, Langouet S, Turesky RJ. UDP-glucuronosyltransferase-mediated metabolic activation of the tobacco carcinogen 2-amino-9H-pyrido[2,3-b]indole. J Biol Chem. 2012;287:14960–14972. doi: 10.1074/jbc.M111.320093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee YC, Cohet C, Yang YC, Stayner L, Hashibe M, Straif K. Meta-analysis of epidemiologic studies on cigarette smoking and liver cancer. Int J Epidemiol. 2009;38:1497–1511. doi: 10.1093/ije/dyp280. [DOI] [PubMed] [Google Scholar]
- 36.Vineis P, Alavanja M, Buffler P, Fontham E, Franceschi S, Gao YT, Gupta PC, Hackshaw A, Matos E, Samet J, Sitas F, Smith J, Stayner L, Straif K, Thun MJ, Wichmann HE, Wu AH, Zaridze D, Peto R, Doll R. Tobacco and cancer: recent epidemiological evidence. J Natl Cancer Inst. 2004;96:99–106. doi: 10.1093/jnci/djh014. [DOI] [PubMed] [Google Scholar]
- 37.Frederiksen H. Two food-borne heterocyclic amines: metabolism and DNA adduct formation of amino-alpha-carbolines. Mol Nutr Food Res. 2005;49:263–273. doi: 10.1002/mnfr.200400061. [DOI] [PubMed] [Google Scholar]
- 38.Frederiksen H, Frandsen H. In vitro metabolism of two heterocyclic amines, 2-amino-9H-pyrido[2,3-b]indole (A(alpha)C) and 2-amino-3-methyl-9H-pyridol2,3-b]indole (MeA(alpha)C) in human and rat hepatic microsomes. Pharmacol Toxicol. 2002;90:127–134. doi: 10.1034/j.1600-0773.2002.900303.x. [DOI] [PubMed] [Google Scholar]
- 39.King RS, Teitel CH, Shaddock JG, Casciano DA, Kadlubar FF. Detoxification of carcinogenic aromatic and heterocyclic amines by enzymatic reduction of the N-hydroxy derivative. Cancer Lett. 1999;143:167–171. doi: 10.1016/s0304-3835(99)00119-6. [DOI] [PubMed] [Google Scholar]
- 40.Yuan ZX, Jha G, McGregor MA, King RS. Metabolites of the carcinogen 2-amino-alpha-carboline formed in male Sprague-Dawley rats in vivo and in rat hepatocyte and human HepG2 cell incubates. Chem Res Toxicol. 2007;20:497–503. doi: 10.1021/tx600303d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Turesky RJ, Konorev D, Fan X, Tang Y, Yao L, Ding X, Xie F, Zhu Y, Zhang QY. Effect of cytochrome P450 reductase deficiency on 2-amino-9H-pyrido[2,3-b]indole metabolism and DNA adduct formation in liver and extrahepatic tissues of mice. Chem Res Toxicol. 2015;28:2400–2410. doi: 10.1021/acs.chemrestox.5b00405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guillouzo A, Morel F, Langouet S, Maheo K, Rissel M. Use of hepatocyte cultures for the study of hepatotoxic compounds. J Hepatol. 1997;26(2):73–80. doi: 10.1016/s0168-8278(97)80499-0. [DOI] [PubMed] [Google Scholar]
- 43.Kunze KL, Trager WF. Isoform-selective mechanism-based inhibition of human cytochrome P450 1A2 by furafylline. Chem Res Toxicol. 1993;6:649–656. doi: 10.1021/tx00035a009. [DOI] [PubMed] [Google Scholar]
- 44.Burke MD, Mayer RT. Differential effects of phenobarbitone and 3-methylcholanthrene induction on the hepatic microsomal metabolism and cytochrome P-450-binding of phenoxazone and a homologous series of its n-alkyl ethers (alkoxyresorufins) Chem Biol Interact. 1983;45:243–258. doi: 10.1016/0009-2797(83)90072-8. [DOI] [PubMed] [Google Scholar]
- 45.Eugster HP, Probst M, Wurgler FE, Sengstag C. Caffeine, estradiol, and progesterone interact with human CYP1A1 and CYP1A2. Evidence from cDNA-directed expression in Saccharomyces cerevisiae. Drug Metab Dispos. 1993;21:43–49. [PubMed] [Google Scholar]
- 46.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 47.Gupta RC. 32P-postlabelling analysis of bulky aromatic adducts. IARC Sci Publ. 1993:11–23. [PubMed] [Google Scholar]
- 48.Goodenough AK, Schut HA, Turesky RJ. Novel LC-ESI/MS/MS(n) method for the characterization and quantification of 2′-deoxyguanosine adducts of the dietary carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine by 2-D linear quadrupole ion trap mass spectrometry. Chem Res Toxicol. 2007;20:263–276. doi: 10.1021/tx0601713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chiu YT, Liu J, Tang K, Wong YC, Khanna KK, Ling MT. Inactivation of ATM/ATR DNA damage checkpoint promotes androgen induced chromosomal instability in prostate epithelial cells. PLoS One. 2012;7:e51108. doi: 10.1371/journal.pone.0051108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Levsen K, Schiebel HM, Behnke B, Dotzer R, Dreher W, Elend M, Thiele H. Structure elucidation of phase II metabolites by tandem mass spectrometry: an overview. J Chromatogr A. 2005;1067:55–72. doi: 10.1016/j.chroma.2004.08.165. [DOI] [PubMed] [Google Scholar]
- 51.Butler MA, Iwasaki M, Guengerich FP, Kadlubar FF. Human cytochrome P-450PA (P-450IA2), the phenacetin O-deethylase, is primarily responsible for the hepatic 3-demethylation of caffeine and N-oxidation of carcinogenic arylamines. Proc Natl Acad Sci. 1989;86:7696–7700. doi: 10.1073/pnas.86.20.7696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Langouet S, Welti DH, Kerriguy N, Fay LB, Huynh-Ba T, Markovic J, Guengerich FP, Guillouzo A, Turesky RJ. Metabolism of 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline in human hepatocytes: 2-amino-3-methylimidazo[4,5-f]quinoxaline-8-carboxylic acid is a major detoxification pathway catalyzed by cytochrome P450 1A2. Chem Res Toxicol. 2001;14:211–221. doi: 10.1021/tx000176e. [DOI] [PubMed] [Google Scholar]
- 53.Langouet S, Paehler A, Welti DH, Kerriguy N, Guillouzo A, Turesky RJ. Differential metabolism of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine in rat and human hepatocytes. Carcinogenesis. 2002;23:115–122. doi: 10.1093/carcin/23.1.115. [DOI] [PubMed] [Google Scholar]
- 54.Kriek E. Fifty years of research on N-acetyl-2-aminofluorene, one of the most versatile compounds in experimental cancer research. J Cancer Res Clin Oncol. 1992;118:481–489. doi: 10.1007/BF01225261. [DOI] [PubMed] [Google Scholar]
- 55.Kriek E. Persistent binding of a new reaction product of the carcinogen N-hydroxy-N-2-acetylaminofluorene with guanine in rat liver DNA in vivo. Cancer Res. 1972;32:2042–2048. [PubMed] [Google Scholar]
- 56.Turesky RJ, Guengerich FP, Guillouzo A, Langouet S. Metabolism of heterocyclic aromatic amines by human hepatocytes and cytochrome P4501A2. Mutat Res. 2002;506-507:187–195. doi: 10.1016/s0027-5107(02)00165-3. [DOI] [PubMed] [Google Scholar]
- 57.Turesky RJ, Constable A, Richoz J, Varga N, Markovic J, Martin MV, Guengerich FP. Activation of heterocyclic aromatic amines by rat and human liver microsomes and by purified rat and human cytochrome P450 1A2. Chem Res Toxicol. 1998;11:925–936. doi: 10.1021/tx980022n. [DOI] [PubMed] [Google Scholar]
- 58.Wallin H, Mikalsen A, Guengerich FP, Ingelman-Sundberg M, Solberg KE, Rossland OJ, Alexander J. Differential rates of metabolic activation and detoxication of the food mutagen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine by different cytochrome P450 enzymes. Carcinogenesis. 1990;11:489–492. doi: 10.1093/carcin/11.3.489. [DOI] [PubMed] [Google Scholar]
- 59.Aninat C, Piton A, Glaise D, Le Charpentier T, Langouet S, Morel F, Guguen-Guillouzo C, Guillouzo A. Expression of cytochromes P450, conjugating enzymes and nuclear receptors in human hepatoma HepaRG cells. Drug Metab Dispos. 2006;34:75–83. doi: 10.1124/dmd.105.006759. [DOI] [PubMed] [Google Scholar]
- 60.Cai T, Yao L, Turesky RJ. Bioactivation of Heterocyclic Aromatic Amines by UDP Glucuronosyltransferases. Chem Res Toxicol. 2016;29:879–891. doi: 10.1021/acs.chemrestox.6b00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pathak KV, Bellamri M, Wang Y, Langouet S, Turesky RJ. 2-Amino-9H-pyrido[2,3-b]indole (AalphaC) Adducts and Thiol Oxidation of Serum Albumin as Potential Biomarkers of Tobacco Smoke. J Biol Chem. 2015;290:16304–16318. doi: 10.1074/jbc.M115.646539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Novak M, Nguyen TM. Unusual reactions of the model carcinogen N-acetoxy-N-acetyl-2-amino-alpha-carboline. J Org Chem. 2003;68:9875–9881. doi: 10.1021/jo034505u. [DOI] [PubMed] [Google Scholar]
- 63.Holland RD, Taylor J, Schoenbachler L, Jones RC, Freeman JP, Miller DW, Lake BG, Gooderham NJ, Turesky RJ. Rapid biomonitoring of heterocyclic aromatic amines in human urine by tandem solvent solid phase extraction liquid chromatography electrospray ionization mass spectrometry. Chem Res Toxicol. 2004;17:1121–1136. doi: 10.1021/tx049910a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.