

Abstract
Production of NO by the endothelial nitric oxide synthase (eNOS) has a major role in blood pressure control and suppression of atherosclerosis. In a previous study, we presented evidence implicating the Pin1 prolyl isomerase in negative modulation of eNOS activity in bovine aortic endothelial cells (BAECs). Pin1 recognizes phosphoserine/phosphothreonine–proline motifs in target proteins and catalyzes prolyl isomerization at the peptide bond. In the present study, we show, first, with purified proteins, that Pin1 binds to eNOS directly via the Pin1 WW domain. Binding is enhanced by mimicking phosphorylation of eNOS at S116. Interaction of Pin1 with eNOS markedly reduces eNOS enzymatic activity. Second, in BAECs, we show that TNFα induces ERK 1/2-mediated S116 phosphorylation of eNOS, accompanied by Pin1 binding. TNFα treatment of BAECs results in a reduction in NO release from the cells in a manner that depends on the activities of both Pin1 and ERK 1/2. Evidence is also presented that this mechanism of eNOS regulation cannot occur in rat and mouse cells because there is no proline residue in the mouse and rat amino acid sequences adjacent to the putative phosphorylation site. Moreover, we find that phosphorylation of this site is not detectable in mouse eNOS.
Keywords: Endothelial nitric oxide synthase (eNOS), Pin1, TNFα
1. Introduction
Two important mechanisms of biochemical regulation of the endothelial nitric oxide synthase (eNOS) are eNOS phosphorylation and eNOS protein–protein interactions. In many instances, phosphorylation and protein–protein interactions act in concert or in sequence in regulation of eNOS activity. One example is the phosphorylation-dependent binding of the Pin1 prolyl isomerase to eNOS. Phosphoserine–proline and phosphothreonine–proline motifs in a large number of different proteins are specifically recognized and bound by the N-terminal domain of Pin1 known as the WW domain. The C-terminal isomerase domain then catalyzes the cis to trans isomerization of the phosphoserine/phosphothreonine–proline peptide bond. In a previous study, we showed that eNOS is subject under basal conditions in cultured bovine aortic endothelial cells (BAECs) to phosphorylation by ERK 1/2 protein kinases at the eNOS inhibitory phosphorylation site at S116, adjacent to P117 (bovine sequence numbering of eNOS amino acids will be used in this manuscript, except where otherwise noted). We also showed that eNOS and Pin1 are co-immunoprecipitated from BAECs to a greater extent when S116 is phosphorylated. We showed further that inhibition of Pin1 in BAECs increases the amount of NO release from the cells while overexpression of Pin1 reduces NO production. Additionally, we demonstrated that overexpression of Pin1 in intact aortae isolated from mice reduces acetylcholine-induced relaxation of aortic rings [16]. Prolyl isomerization can have profound effects on protein conformation and, consequently, on protein function. We, therefore, interpreted the results of our earlier study as evidence of a previously unrecognized form of inhibitory regulation of eNOS in endothelial cells and blood vessels through S116 phosphorylation-dependent interaction of eNOS with Pin1. This interaction likely promotes prolyl isomerization of eNOS at S116–P117, which we hypothesized as having an inhibitory effect on eNOS activity.
More recently, and in contrast to our conclusions, Chaisson et al. [4] presented evidence that purported to show that Pin1 binding and Pin1-catalyzed prolyl isomerization of eNOS at phosphorylated S116 enables dephosphorylation at this site of inhibitory phosphorylation. These authors thus concluded that the phospho-S116–P117 eNOS– Pin1 interaction reverses the inhibitory effect of S116 phosphorylation and stimulates, rather than inhibits, eNOS activity. To support their hypothesis, Chaisson et al. also showed that Pin1 knockout mice exhibited endothelial dysfunction and hypertension. All of the experimental data reported by Chaisson et al., however, came from studies with cultured rat aortic endothelial cells or from ex vivo or in vivo studies performed in mice. Consequently, it is very important to note that a comparison of the amino acid sequences from bovine, human, rat, and mouse eNOS [7] shows that there is a glutamine residue rather than a proline residue in the mouse and rat sequences at the position that corresponds to P117 in the bovine sequence (equivalent to P115 in the human sequence). Without a proline at this position, Pin1 binding and prolyl isomerization cannot occur.
Most recently, Paneni et al. [14] reported results that differ from those of Chaisson et al. and that are more consistent with our conclusions. Paneni et al. showed that Pin1 inhibits eNOS activity in human aortic endothelial cells through recognition of phospho-S116 in eNOS. They showed further that Pin1 knockout mice do not exhibit any endothelial dysfunction or hypertension. Rather, genetic deletion of Pin1 in their study had a protective effect against the endothelial dysfunction induced by vascular inflammation in diabetic mice. They also reported an increase in Pin1 activity in peripheral blood monocytes in diabetic human patients.
Vascular inflammation is associated with accelerated atherosclerosis and cardiovascular disease. A major factor in the process of inflammation is the pro-inflammatory cytokine, TNFα [10, 17]. In the vasculature, TNFα binds to endothelial cell surface receptors and suppresses NO production by modifying the expression or activity of the eNOS enzyme. TNFα also decreases NO bioavailability by producing a reduction in availability of the eNOS substrate, L-arginine, by accumulation of the endogenous eNOS inhibitor, asymmetric dimethylarginine, and by increased scavenging of NO due to TNFα-induced superoxide production [10,17]. The biochemical mechanisms by which TNFα negatively modulates eNOS enzymatic activity, however, are not fully understood. Therefore, in the present study, we have investigated whether Pin1 interaction with eNOS at phospho-S116–P117 might be involved in the process by which TNFα reduces eNOS activity in intact endothelial cells. Also, to provide support for our original hypothesis, we have carried out additional studies with purified proteins. These studies demonstrate that the Pin1 WW domain binds directly to phospho-S116–P117 in eNOS and that this interaction has an inhibitory, rather than a stimulatory, effect on eNOS activity.
2. Materials and methods
2.1. Materials
Anti-eNOS antibody and all materials for protein expression in the baculovirus system were obtained from BD Biosciences. Anti-phospho-S116 eNOS antibody, anti-ERK 1/2 antibody, and anti-phospho-ERK 1/2 antibody were purchased from Upstate-Cell Signaling Solutions. Anti-Pin1 antibody was obtained from Santa Cruz Biotechnology. TNFα, active human ERK2, and all chemical compounds (except where otherwise noted) were obtained from Sigma–Aldrich. Tetrahydrobiopterin was purchased from Cayman Chemical. L-[14C]Arginine was from Perkin Elmer and AG-50W-X8 was from Bio-Rad. 2′,5′-ADP Sepharose came from GE Healthcare. Glutathione–Sepharose and the GST-fusion cloning vector, pGEX-4T-1, were purchased from Amersham Pharmacia Biotech.
2.2. Expression and purification of eNOS proteins
Bovine wild-type and S116D eNOS and murine wild-type eNOS were expressed and purified in a baculovirus/Sf9 insect cell system as described in detail previously [18]. Briefly, baculovirus expression of the two forms of eNOS enzyme was carried out in Sf9 insect cells. Cells were lysed and eNOS was purified to N95% homogeneity.
2.3. In vitro binding assays with GST fusion proteins and Baculovirus-expressed eNOS proteins
Glutathione-S-transferase (GST) and GST fusion proteins were expressed in E. coli and purified by affinity binding to glutathione–Sepharose. Proteins (100 pmol of each), prebound to glutathione–Sepharose beads, were incubated overnight with shaking at 4 °C in 1 ml of buffer containing 50 mM Tris-HCl, pH 7.4, 20% glycerol, 1% PMSF, and 100 pmol of bovine wild-type or S116D eNOS, purified from the baculovirus expression system. Following the overnight binding reaction, beads were washed six times in 1 ml of 50 mM Hepes, pH 7.5, 120 mM NaCl, 1 mM EDTA, 0.5% CHAPS, and 1% PMSF. Bound proteins were eluted with SDS sample buffer and boiling for 5 min. Eluted proteins were then subjected to immunoblotting with anti-eNOS antibody.
2.4. Assay of eNOS enzymatic activity
Purified eNOS activity was measured by the method of Bredt and Snyder [3] which determines the rate of formation of L-[14C]citrulline from L-[14C]arginine (100 μM) in the presence of excess cofactors including Ca2+ (2 mM), calmodulin (200 units), NADPH (1 mM), FAD (4 μM), FMN (4 μM), and tetrahydrobiopterin (40 μM). Product (L-[14C]citrulline) was separated from substrate (L-[14C]arginine) on Bio-Rad AG 50W-X8 cation exchange columns.
2.5. Cell culture
Primary cultures of bovine aortic endothelial cells (BAECs) were purchased from VEC Technologies Inc. All endothelial cell experiments were performed on BAECs in passages 2–6. Cells were maintained in medium 199 supplemented with 10% FBS, 5% iron-supplemented calf serum, 0.6 μg/ml thymidine, 2.2 mg/ml sodium bicarbonate, 500 IU/ml penicillin, and 500 μg/ml streptomycin. COS-7 cells were maintained in DMEM supplemented with 10% FBS, 500 IU penicillin, and 500 μg/ml streptomycin. Sf9 insect cells were maintained in Grace’s medium supplemented with 10% FBS, 500 IU penicillin, and 500 μg/ml streptomycin.
2.6. Immunoprecipitation and immunoblotting
Cell lysates were prepared by lysis of cells in ice-cold lysis buffer containing 50 mM Tris-HCl, pH 7.4, 100 mM NaF, 15 mM Na4P2O7, 1 mM Na3VO4, 1% Triton X-100, and 1 mM PMSF. Immunoprecipitation and immunoblotting were then carried out as described previously [6].
2.7. In vitro phosphorylation
Purified wild-type mouse or bovine eNOS (1 μg) was incubated with purified constitutively active ERK2 (1 μg) for 1 h at 30 °C in buffer containing 50 mM Hepes, pH 7.5, 10 mM MgCl2, 1 mM EGTA, 0.001% TWEEN 20, in the absence and presence of 3.3 mM ATP. The reaction samples were placed on ice before being analyzed by immunoblotting.
2.8. Measurement of NO release
NO release was determined by the chemiluminescence assay that measures nitrite levels in conditioned media as described previously [16].
3. Results
3.1. Pin1 binds to eNOS directly through its WW domain and binding is increased by mimicking of phosphorylation of eNOS at S116
In order to determine whether eNOS interacts directly with Pin1 and to determine whether mimicking of phosphorylation of eNOS at S116 results in an increase in the eNOS–Pin1 interaction, we expressed wild-type (WT) bovine eNOS and a phospho-mimetic S116D form of bovine eNOS (in which serine 116 was changed to an aspartate) in a baculovirus expression system. Both of the proteins were purified as active enzymes to >95% homogeneity (as judged by Coomassie staining of SDS polyacrylamide gels) by affinity binding to 2′,5′-ADP-Sepharose. In addition, we expressed (in E. coli) a GST non-fusion protein and GST fusion proteins of human full-length (FL) Pin1 (residues 1–163) and a ΔWW Pin1 (residues 43–163), which contains the prolyl isomerase domain but lacks the WW domain. The GST fusion proteins and the GST non-fusion protein were purified by affinity binding to glutathione–Sepharose and the proteins, prebound to Sepharose beads, were used in in vitro binding assays with equal quantities of WT eNOS and S116D eNOS. As shown in the representative blot in Fig. 1A and in the densitometric analysis of 3 blots from 3 separate experiments shown in Fig. 1B, full-length Pin1 bound WT eNOS and S116D eNOS in these assays and consistently bound more than twice as much of the phospho-mimetic S116D form of eNOS as compared to WT eNOS. By comparison, GST alone or ΔWW Pin1 showed either no detectable binding or greatly diminished binding in these in vitro binding assays.
Fig. 1.
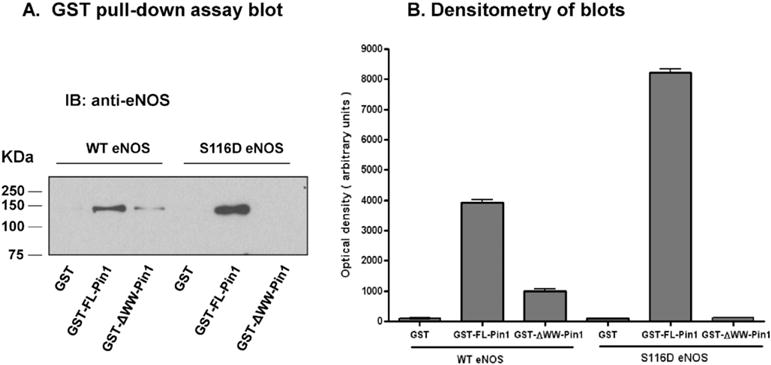
In vitro binding of wild-type and phospho-mimetic eNOS to GST-Pin1 fusion proteins. GST fusion proteins representing full-length (FL) human Pin1 (residues 1–163), Δ WW Pin1 (residues 43–163), and a GST non-fusion protein were expressed in E. coli and purified by affinity binding to glutathione–Sepharose. Proteins, prebound to beads, were incubated with purified baculovirus-expressed bovine wild-type (WT) and S116D eNOS. Following binding, extensive washing, and boiling in SDS sample buffer, proteins were immunoblotted (IB) with anti-eNOS antibody. A, Representative blot. B, Densitometric analysis of 3 blots from 3 separate experiments (means ± S.E.).
3.2. The enzymatic activity of purified S116D eNOS is significantly reduced by incubation with purified Pin1
In order to assess whether direct binding of phospho-S116 eNOS by Pin1 affects eNOS enzymatic activity, and to determine whether the observed effect, if any, is positive or negative, we carried out experiments with purified bovine S116D eNOS and purified full-length human Pin1. Pin1 was expressed as a 6×-His-tagged protein in a baculovirus system to allow for its purification (as an active enzyme) to >95% homogeneity (as judged by Coomassie staining of SDS polyacrylamide gels) by affinity binding to Ni-Sepharose. Freshly isolated (never frozen) S116D eNOS (1 μg) was incubated at 37 °C for 30 min without calmodulin or with calmodulin (100 units) and either Pin1 (1 μg) or bovine serum albumin (BSA, 1 μg). Enzyme activity during the incubation was measured by arginine-to-citrulline conversion assay in the presence of excess cofactors including Ca2+, NADPH, FAD, FMN, and tetrahydrobiopterin. As shown in Fig. 2, eNOS was completely dependent on calmodulin for activity in this assay. Activity was markedly suppressed by co-incubation with Pin1 but was not affected by co-incubation with an equal quantity of BSA. No effect of BSA in these experiments demonstrated that the effect of Pin1 on eNOS activity was not simply due to a non-specific effect of additional protein in the assay solution.
Fig. 2.
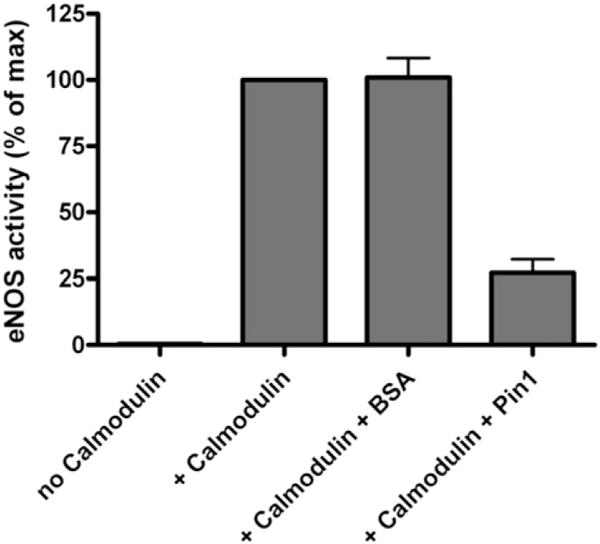
Effect of incubation of purified Pin1 with purified S116D eNOS on eNOS enzymatic activity. Bovine S116D eNOS and human Pin1 were each expressed and purified from a baculovirus expression system. Purified eNOS was incubated for 30 min at 37 °C either by itself or with the additions indicated and total eNOS enzyme activity during the incubation period was measured by arginine-to-citrulline conversion assay. Results shown represent means ± S.E. from 3 separate purifications of eNOS.
3.3. TNFα a induces sustained phosphorylation of eNOS at S116 in endothelial cells accompanied by activation of the ERK 1/2 MAP kinases
We next extended our investigation from experiments with purified proteins to experiments with intact endothelial cells. To determine whether TNFα induces phosphorylation of eNOS at S116 in BAECs, we treated cultured cells with TNFα (50 ng/ml) for 0, 1, 2, 3, 4, 5, and 6 h. Cells were lysed and lysates were subjected to immunoblotting with phospho-S116-specific and nonphospho-specific antibodies to the eNOS protein. The representative blot of phospho-S116 eNOS shown in Fig. 2A and the densitometric analysis of blots from 3 separate experiments shown in Fig. 2B confirmed that TNFα stimulates a sustained phosphorylation of eNOS at S116 and that phosphorylation is maximal by 3 h. Blotting with nonphospho-specific anti-eNOS antibody showed that there was no TNFα-induced change in total eNOS protein during the 6 h treatment period. We have shown previously that ERK 1/2 MAP kinases are responsible for eNOS phosphorylation at S116 in BAECs under basal conditions [16]. In an effort to determine whether ERK 1/2 might also be responsible for the TNFα-stimulated phosphorylation of S116, we treated BAECs with TNFα for the times indicated and probed for ERK 1/2 activation with an antibody that recognizes only the activated, phosphorylated forms of ERK 1/2 (p-ERK 1/2). As shown in the representative blot in Fig. 3C and the densitometric analysis of 3 separate blots from 3 different experiments shown in Fig. 3D, TNFα induced the activation of ERK 1/2 with a time course of increasing activation out to at least 5 h. Immunoblotting to quantify total ERK 1/2 showed that there was no change in the amount of total ERK 1/2 protein during the incubation period.
Fig. 3.
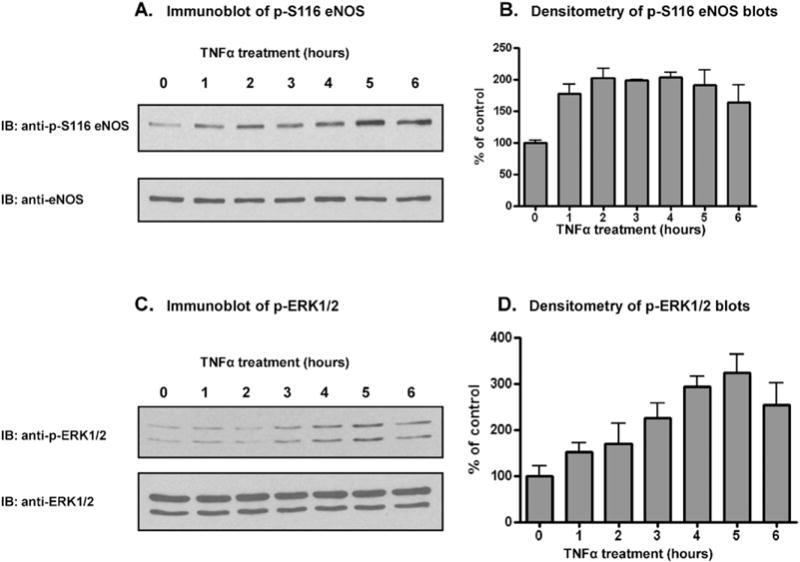
TNFα-induced phosphorylation of eNOS at S116 and TNFα-induced activation of ERK 1/2 in BAECs. BAECS were incubated for the times indicated, cells were lysed, and lysates were immunoblotted (IB) with phospho-S116-specific and nonphospho-specific antibodies for eNOS and phospho-specific and nonphospho-specific antibodies for ERK 1/2. A and C, Representative immunoblots. B and D, Densitometric analysis of 3 blots from 3 separate experiments (means ± S.E.).
3.4. TNFα-induced phosphorylation of eNOS at S116 in endothelial cells is blocked by specific inhibitors of ERK 1/2 and is accompanied by Pin1 binding to eNOS
To confirm that ERK 1/2 mediates the TNFα-stimulated phosphorylation of eNOS at S116 in endothelial cells, BAECs were either not exposed or exposed to TNFα (50 ng/ml) for 4 h in the absence or presence of two structurally distinct, selective MEK 1/2 (and thus ERK 1/2) inhibitors termed PD98059 [1] and U0126 [5]. Cells were lysed at the end of 4 h and lysates were immunoblotted with phospho-S116-specific and nonphospho-specific antibodies to eNOS. As shown in the representative blot of Fig. 4A and in the densitometric analysis of blots from 4 separate experiments shown in Fig. 4B, both of the ERK 1/2 kinase inhibitors blocked the cytokine-stimulated phosphorylation event in question. ERK 1/2 is thus identified as the kinase responsible for catalyzing TNFα-induced S116 phosphorylation of eNOS in endothelial cells. In order to confirm that TNFα-induced phosphorylation of S116 in eNOS promotes Pin1 binding to eNOS, we also carried out co-immunoprecipitation experiments. BAECs were either not treated or treated with TNFα (50 ng/ml) for 4 h, cells were lysed, and lysates were subjected to immunoprecipitation with anti-Pin1 antibody. Anti-Pin1 immunoprecipates were then immunoblotted with anti-eNOS and anti-Pin1 antibodies. Fig. 3C and D show a representative immunoblot and densitometry of 3 blots from 3 separate co-immunoprecipitation experiments. Analysis of the results shows a clear increase in association of eNOS with Pin1 that is induced by TNFα. Blotting for Pin1 also confirmed that equal amounts of Pin1 were immunoprecipitated in each condition.
Fig. 4.
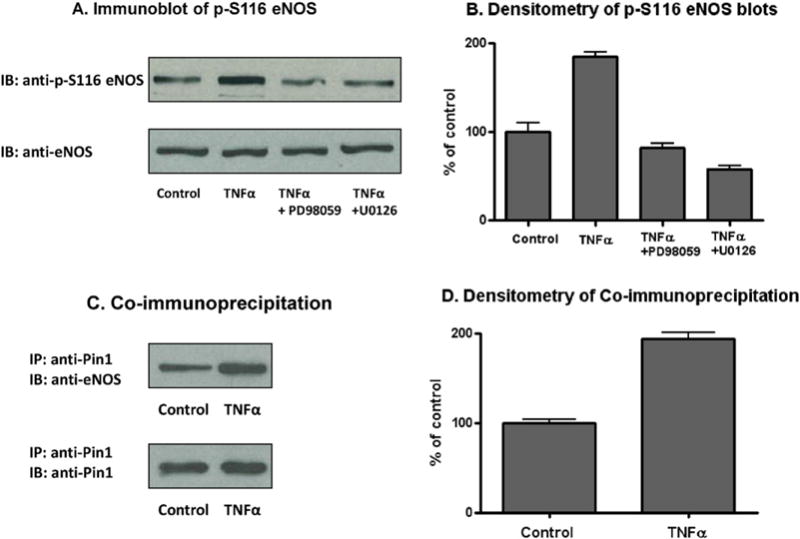
Effects of ERK 1/2 inhibitors on TNFα-induced phosphorylation of eNOS at S116 and effects of TNFα on eNOS–Pin1 complex formation in BAECs. A, Representative blot from experiments in which BAECs were incubated for 4 h without any additions (Control) or with the additions indicated, cells were lysed, and lysates were immunoblotted (IB) with phospho-S116-specific and nonphospho-specific antibodies for eNOS. B, Densitometric analysis of 4 blots from 4 separate experiments described in A (means ± S.E.). C, Representative blot from experiments in which BAECs were incubated for 4 h without (Control) and with TNFα, cells were lysed, Pin1 was immunoprecipitated (IP) with anti-Pin1 antibody, and immunoprecipitates were immunoblotted (IB) with anti-eNOS and anti-Pin1 antibodies. D, Densitometric analysis of the co-immunoprecipitation results from 3 separate experiments (means ± S.E.).
3.5. TNFα induces a reduction in NO release from endothelial cells in a manner that depends on the activities of both Pin1 and ERK 1/2
We next examined whether a 4 h treatment of BAECs with TNFα which, as shown above, results in ERK 1/2-mediated phosphorylation of eNOS at S116 and increased association of eNOS with Pin1 (Fig. 4), alters the activity of eNOS in these cells in a manner that depends on the activities of Pin1 and ERK 1/2. BAECs were incubated in culture medium either containing or not containing TNFα (50 ng/ml) for 4 h with the addition or lack of addition of various inhibitors. Cell culture medium was then replaced with fresh medium containing the same additions and cells were incubated for an additional 60 min. At the end of the 60 min, culture medium was removed from the cells and the accumulation of nitrite (the stable breakdown product of NO) in the conditioned medium was measured by using a NO-specific chemiluminescence analyzer as described previously [6]. In a first set of experiments, we tested the effect of a specific pharmacological inhibitor of Pin1 activity termed juglone. Juglone forms a covalent bond with the thiol groups of cysteines 41 and 69 of human Pin1 and thereby irreversibly inhibits Pin1 activity [9]. As shown in Fig. 5A, TNFα treatment reduced the amount of NO released by the cells by an average of about 40% compared to the control in 3 separate experiments. The TNFα-induced reduction in NO release was completely blocked by juglone in these experiments. We next tested the effect of inhibition of Pin1 activity using a different approach, namely, by using the adenovirus expressing a dominant negative form of Pin1 (Ad-DN Pin1) that was used in our previous investigation to inhibit Pin1 activity [16]. In the control condition, cells were transduced with a ß-galactosidase adenovirus (Ad-ß-gal). Fig. 5B shows the results of this set of experiments. TNFα treatment of Ad-ß-gal-transduced BAECs resulted in about a 50% reduction in NO release compared to the control in 3 separate experiments. This reduction was completely abolished by transduction with the adenovirus expressing the dominant negative form of Pin1. We next tested the effects of the two different pharmacological ERK 1/2 inhibitors, PD98059 and U0126. These two sets of experiments, each repeated 3 times, again showed about a 50% reduction in NO release induced by TNFα that was completely or almost completely blocked by both of the ERK 1/2 inhibitors (Fig. 5C and D).
Fig. 5.
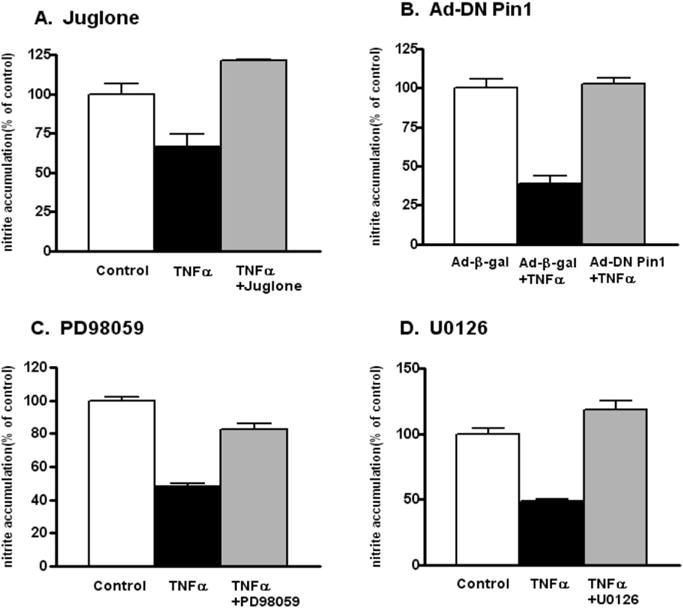
Effects of TNFα and various inhibitors on NO release from BAECs. BAECs were incubated for 4 h without (Control) and with TNFα and with the addition or lack of addition of the indicated inhibitors. Culture medium was then replaced with fresh medium containing the same additions. After a further 1 h incubation, nitrite accumulation in the conditioned medium was measured with an NO-specific chemiluminescence analyzer. Results shown are means ± S.E. for 3 separate experiments performed with each inhibitor.
3.6. Evidence that the commercially available phospho-specific antibody that detects Phospho-S116 eNOS in bovine endothelial cells may not detect a possible equivalent Phospho-T113 eNOS in mouse or rat endothelial cells
Numerous published studies have examined eNOS phosphorylation at S116. In all of these studies, including the one by Chaisson et al. [4], the antibody that was used to detect phosphorylation was a phospho-specific polyclonal antibody that targets a specific human eNOS phosphopeptide sequence. This antibody (which is available from several different commercial vendors) was raised in rabbits injected with a phosphopeptide corresponding to the human eNOS 106–118 sequence PRKLQGRPS(p)PGPP. This sequence is identical at 12 out of 13 positions to the bovine eNOS 108–120 sequence PRKLQT*RPS(p)PGPP (where an asterisk identifies the single mismatch). The antibody appears to recognize phospho-S116 eNOS in bovine cells and phospho-S114 in human cells. We have attempted to use this antibody to probe for phosphorylation of eNOS at S116 in aortic homogenates from wild-type and atherosclerotic mice (ApoE knockout mice fed a high fat diet). These studies did not yield definitive results in that there was no band detected with the phospho-specific antibody that matched a band detected with the nonphospho-specific anti-eNOS antibody. This prompted us to look carefully at the mouse eNOS sequence [7] and compare it to the human and bovine sequences. The eNOS sequence in the mouse (which is identical in the rat) that corresponds to the human 106–118 sequence is numbered 105–117. Mouse/rat eNOS 105–117 has a sequence of amino acids of PRKLQS*RPT(p)*Q*GPS* (where an asterisk identifies the mismatches with the human sequence, and (p) identifies the putative phosphorylation site). The mouse (and rat) 13-mer sequence contains 4 mismatches with the human sequence, one of which is the putatively phosphorylated residue itself (T113). The other 3 mismatches are located very close to the putative phosphorylation site at T113. The most important insight that is gained from this analysis, however, is that a glutamine residue (rather than a proline residue) is found in the mouse/rat sequence at the +1 position relative to the putative phosphorylation site (T113, equivalent to S116 in the bovine sequence). Thus, even if T113 is phosphorylated in mouse or rat eNOS, the protein lacks a proline at amino acid number 114. Therefore, this site cannot function as either a binding site for Pin1 or as a site of enzyme catalysis by the Pin1 prolyl isomerase. Furthermore, based on the lack of homology between the rat/mouse and human eNOS sequences at and around the potential rat/mouse phosphorylation site at T113, we speculated that the phospho-S116-specific anti-eNOS antibody, which was raised against the human phosphopeptide sequence, might not be suitable for use with mouse and rat cells. In order to obtain evidence in support of this hypothesis, we expressed and purified the full-length mouse eNOS protein in a baculovirus system using the same protocol used for bovine eNOS. As shown in the Coomassie-stained gel in Fig. 6A, mouse and bovine eNOS proteins were both equally and highly purified by this method. The two proteins (mouse and bovine) were then used as substrates in a cell-free phosphorylation system for commercially purchased purified constitutively active ERK2. The phosphorylation reaction was carried out in a reaction mixture in the absence (phosphorylation −) or presence (phosphorylation +) of ATP, in order to discriminate between non-phosphorylated and phosphorylated bands after immunoblotting of proteins. Immunoblotting with the nonphospho-specific commercially available anti-eNOS antibody detected bands for both the non-phosphorylation and phosphorylation reaction conditions for both mouse and bovine eNOS (Fig. 6B). Immunoblotting was also performed with the commercially available anti-phospho-S116 eNOS antibody. As shown in Fig. 6C, a clear band was detected for ERK2-phosphorylated (but not non-phosphorylated) bovine eNOS. No band was detected for mouse eNOS, either in the non-phosphorylation or phosphorylation reaction conditions.
Fig. 6.
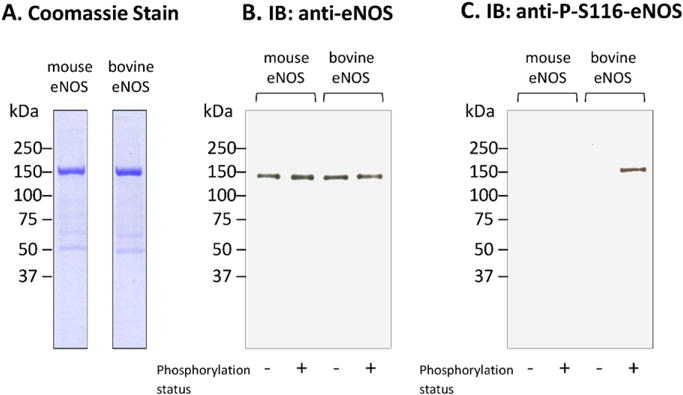
Reactivity of the anti-phospho-S116 eNOS antibody with ERK2-phosphorylated and nonphosphorylated mouse and bovine eNOS proteins. Purified wild-type mouse or bovine eNOS was incubated with purified constitutively active ERK2 for 1 h at 30 °C under the conditions described in Materials and methods. Results shown are representative of 3 separate experiments.
4. Discussion
Controversy currently exists regarding whether Pin1 binding upon phosphorylation of eNOS at S116 results in increased or decreased eNOS activity. We have shown in a previous study that complex formation of eNOS with Pin1 in endothelial cells is dependent on eNOS phosphorylation at S116 and that inhibition of the activity of Pin1 increases NO release from the cells while overexpression of Pin1 decreases NO release [16]. It has also been established in earlier studies from several laboratories that S116 phosphorylation of eNOS per se has an inhibitory effect on eNOS activity [2,11,12,15], although the possible involvement of Pin1 in this inhibition was not known at the time these studies were published. We have thus proposed the hypothesis that Pin1 interaction with S116-phosphorylated eNOS inhibits eNOS activity. This hypothesis has since been supported by the results of Paneni et al. [14] but has been contradicted by the results of Chaisson et al., who have concluded that Pin1 interaction with eNOS leads to an increase in eNOS activity [4]. The results reported here, showing a clear and substantial inhibition of the activity of purified S116D eNOS by purified Pin1, strongly support our original hypothesis that S116 phosphorylation-dependent interaction of eNOS with Pin1 results in prolyl isomerization of eNOS with a consequent conformational change in eNOS that decreases activity. This hypothesis is further supported by the results of our in vitro binding assays with GST fusion proteins. These “pull-down” assays demonstrate at least 3 different aspects of the eNOS–Pin1 interaction. First, that complex formation between eNOS and Pin1 is direct and does not require an intermediary or adaptor protein, such as Hsp90, that acts as an adaptor in the interaction between eNOS and soluble guanylate cyclase [19]. Second, binding of Pin1 to eNOS is mediated by the Pin1 WW domain. This domain is known to act as a modular domain in Pin1, as well as in certain other proteins, that specifically recognizes a phosphoserine/phosphothreonine–proline pair of adjacent amino acids [13]. Third, binding of eNOS by Pin1 in this in vitro binding assay is increased by more than 100% by mimicking eNOS phosphorylation at S116, demonstrating an important contribution of S116 phosphorylation. Significant binding also occurs in the absence of phospho-mimicking, however, suggesting that the adjacent proline residue and likely other additional amino acids participate in the eNOS–Pin1 binding interaction.
Our original hypothesis is further supported by novel results that demonstrate the involvement of this specific mechanism of eNOS regulation in the TNFα signaling pathway of endothelial cells that leads to reduced eNOS activity. First, we have shown that TNFα induces eNOS phosphorylation at S116 in endothelial cells and that this phosphorylation can be blocked by the two structurally distinct ERK 1/2 inhibitors, PD98059 and U0126. Second, we have demonstrated by coimmunoprecipition that TNFα treatment of endothelial cells induces complex formation between eNOS and Pin1. Third, we have shown that TNFα reduces eNOS activity in endothelial cells by a mechanism that depends on both ERK 1/2 activity and Pin1 activity. Taken together with the results of our studies with purified proteins, our investigation in intact endothelial cells provides strong support for an interpretation that TNFα signaling in endothelial cells proceeds in part through a cascade of ERK 1/2 activation, eNOS phosphorylation at S116, Pin1 binding to phosphorylated S116, and inhibition of eNOS in a manner that depends on Pin1 activity. This is a mechanism of TNFα signaling that has not been previously reported, and one that could contribute to vascular inflammation and vascular lesion formation in human disease due to a reduction in the anti-atherogenic actions of NO. A final conclusion from this study that has broad implications is that species differences need to be taken into consideration when choosing animal models to study biochemical and cellular mechanisms with relevance to human biology. A search by NCBI BLAST as well as the published results of Gnanapandithen et al. [7] shows that the mouse and rat eNOS sequences have a threonine–glutamine pair of amino acids, rather than a serine–proline pair, at positions equivalent to S116–P117 in the bovine sequence and S114–P115 in the human sequence. Consequently, rat and mouse cells and tissues are not appropriate models for study of the physiological role of eNOS phosphorylation at S116 or of Pin1-catalyzed prolyl isomerization at S116–P117. It remains possible that Pin1 does act on eNOS, but it cannot occur at this particular site. This possibility is suggested by our previously published results showing that overexpression of Pin1 by adenoviral transduction of intact mouse aortae reduces the amount of relaxation of vascular rings that is induced by acetylcholine treatment [16]. Further support for this possibility has also been provided by the experiments in mouse carried out by Paneni et al. [14]. A second related observation is that it is important to take species differences into account when using phospho-specific antibodies. Our results suggest that the commercially available anti-phospho-S116 eNOS antibody may not be suitable for use in rat and mouse. This follows from the fact that a 13-amino-acid phosphopeptide based on the human amino acid sequence was used to raise the polyclonal antibody. The corresponding mouse/rat eNOS sequence has 4 mismatches to the human sequence, one of which is the putative phosphorylation site itself (threonine instead of serine) and the other 3 of which are located in very close proximity to this site.
TNFα signaling and VEGF signaling both involve ERK 1/2 activation. VEGF signaling, however, is associated with stimulation, rather than suppression, of endothelial NO release [20]. We have shown previously that VEGF-stimulated NO release from BAECs is rapid and transient, occurring entirely within 30 min. We showed further that NO production induced by VEGF is completely dependent on rapid and transient VEGF-stimulated intracellular Ca2+ mobilization [8]. In contrast, TNFα-dependent ERK 1/2 activation and phosphorylation of eNOS at S116, which is accompanied by suppression of NO release, occurs over a much longer, more gradual, and sustained time-frame, out to at least 5–6 h (Fig. 3). Consequently, the measurements of NO release that we have reported here were taken after 4 h of exposure of BAECs to TNFα (Fig. 5). NO production by eNOS in this context is likely to be modulated very differently from that seen in response to VEGF. Unlike VEGF-induced positive modulation of eNOS, TNFα-induced negative modulation of eNOS would not, for example, depend on a transient elevation of intracellular Ca2+ and activation of eNOS by binding of Ca2+–calmodulin. In addition, elevation of intracellular Ca2+ induced by VEGF will produce a dephosphorylation of eNOS at S116 [15], an opposite effect on the phosphorylation status of S116 to that produced in response to TNFα.
Acknowledgments
This work was supported by the National Institutes of Health National Heart, Lung, and Blood Institute [grant RO1 HL108719].
Abbreviations
- eNOS
Endothelial nitric oxide synthase
- BAECs
Bovine aortic endothelial cells
- FL
Full-length
- BSA
Bovine serum albumin
- Ad
Adenovirus
- WT
Wild-type
- ß-gal
ß-galactosidase
References
- 1.Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 2.Bauer PM, Fulton D, Boo YC, Sorescu GP, Kemp BE, Jo H, et al. Compensatory Phosphorylation and protein–protein interactions revealed by loss of function and gain of function mutants of multiple serine phosphorylation sites in endothelial nitric-oxide synthase. J Biol Chem. 2003;278:14841–14849. doi: 10.1074/jbc.M211926200. [DOI] [PubMed] [Google Scholar]
- 3.Bredt DS, Snyder SH. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc Natl Acad Sci U S A. 1990;87:682–685. doi: 10.1073/pnas.87.2.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiasson VL, Munshi N, Chatterjee P, Young KJ, Mitchell BM. Pin1 deficiency causes endothelial dysfunction and hypertension. Hypertension. 2011;58:431–438. doi: 10.1161/HYPERTENSIONAHA.111.172338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 6.Fulton D, Church JE, Ruan L, Li C, Sood SG, Kemp BE, et al. Src Kinase Activates Endothelial Nitric-Oxide Synthase by Phosphorylating Tyr-83. J Biol Chem. 2005;280:35943–35952. doi: 10.1074/jbc.M504606200. [DOI] [PubMed] [Google Scholar]
- 7.Gnanapandithen K, Chen Z, Kau CL, Gorczynski RM, Marsden PA. Cloning and characterization of murine endothelial constitutive nitric oxide synthase. Biochem Biophys Acta. 1996;1308:103–106. doi: 10.1016/0167-4781(96)00098-x. [DOI] [PubMed] [Google Scholar]
- 8.He H, Venema VJ, Venema RC, Gu X, Marrero M, Caldwell RB. Vascular endothelial growth factor signals endothelial cell production of nitric oxide and prostacyclin through Flk-1/KDR activation of c-Src. J Biol Chem. 1999;274:25130–25135. doi: 10.1074/jbc.274.35.25130. [DOI] [PubMed] [Google Scholar]
- 9.Hennig L, Christner C, Kipping M, Schelbert B, Rücknagel KP, Grabley S, et al. Selective inactivation of parvulin-like peptidyl-prolyl Cis/Trans isomerases by juglone. Biochemistry. 1998;37:5953–5960. doi: 10.1021/bi973162p. [DOI] [PubMed] [Google Scholar]
- 10.Kleinbongard P, Heusch G, Schulz R. Tnf-alpha in atherosclerosis, myocardial ischemia/reperfusion and heart failure. Pharmacol Ther. 2010;127:295–314. doi: 10.1016/j.pharmthera.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 11.Kou R, Greif D, Michel T. Dephosphorylation of endothelial nitric oxide synthase by vascular endothelial growth factor: implications for the vascular responses to cyclosporin A. J Biol Chem. 2002;277:29669–29673. doi: 10.1074/jbc.M204519200. [DOI] [PubMed] [Google Scholar]
- 12.Li C, Ruan L, Sood SG, Papapetropoulos A, Fulton D, Venema RC. Role of eNOS Phosphorylation at Ser-116 in Regulation of eNOS Activity in Endothelial Cells. Vasc Pharmacol. 2007;47:257–264. doi: 10.1016/j.vph.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu PJ, Zhou XZ, Shen M, Lu KP. Function of WW domains as phosphoserine-or phosphothreonine-binding modules. Science. 1999;283:1325–1328. doi: 10.1126/science.283.5406.1325. [DOI] [PubMed] [Google Scholar]
- 14.Paneni F, Costantino S, Castello L, Battista R, Capretti G, Chiandotto D, et al. Targeting prolyl-isomerase Pin1 prevents mitochondrial oxidative stress and vascular dysfunction: insights in patients with diabetes. Eur Heart J. 2014;36:817–828. doi: 10.1093/eurheartj/ehu179. [DOI] [PubMed] [Google Scholar]
- 15.Ruan L, Torres CM, Buffett RJ, Kennard S, Fulton D, Venema RC. Calcineurin-mediated dephosphorylation of eNOS at serine 116 affects eNOS enzymatic activity indirectly by facilitating c-Src binding and tyrosine 83 phosphorylation. Vasc Pharmacol. 2013;59:27–35. doi: 10.1016/j.vph.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruan L, Torres CM, Qian J, Chen F, Mintz JD, Stepp DW, et al. Pin1 Prolyl isomerase regulates endothelial nitric oxide synthase. Arterioscler, Thromb Vasc Biol. 2011;31:392–398. doi: 10.1161/ATVBAHA.110.213181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steyers CM, Miller FJ., Jr Endothelial dysfunction in chronic inflammatory diseases. Int J Mol Sci. 2014;15:11324–11349. doi: 10.3390/ijms150711324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Venema RC, Sayegh HS, Arnal JF, Harrison DG. Role of the enzyme calmodulin-binding domain in membrane association and phospholipid inhibition of endothelial nitric oxide synthase. J Biol Chem. 1995;270:14705–14711. doi: 10.1074/jbc.270.24.14705. [DOI] [PubMed] [Google Scholar]
- 19.Venema RC, Venema VJ, Ju H, Harris MB, Snead C, Jilling T, et al. Novel complexes of guanylate cyclase with heat shock protein 90 and nitric oxide synthase. Am J Physiol Heart Circ Physiol. 2003;285:H669–H678. doi: 10.1152/ajpheart.01025.2002. [DOI] [PubMed] [Google Scholar]
- 20.Zachary I. VEGF signaling: integration and multi-tasking in endothelial cell biology. Biochem Soc Trans. 2003;6:1171–1177. doi: 10.1042/bst0311171. [DOI] [PubMed] [Google Scholar]