

Abstract
Increased de novo synthesis of fatty acids is a distinctive feature of prostate cancer, which continues to be a leading cause of cancer-related deaths among American men. Therefore, inhibition of de novo fatty acid synthesis represents an attractive strategy for chemoprevention of prostate cancer. We have shown previously that dietary feeding of phenethyl isothiocyanate (PEITC), a phytochemical derived from edible cruciferous vegetables such as watercress, inhibits incidence and burden of poorly-differentiated prostate cancer in Transgenic Adenocarcinoma of Mouse Prostate (TRAMP) model. The present study was designed to test the hypothesis of whether fatty acid intermediate(s) can serve as noninvasive biomarker(s) of prostate cancer chemoprevention by PEITC using archived plasma and tumor specimens from the TRAMP study as well as cellular models of prostate cancer. Exposure of prostate cancer cells (LNCaP and 22Rv1) to pharmacological concentrations of PEITC resulted in downregulation of key fatty acid metabolism proteins, including acetyl-CoA carboxylase 1 (ACC1), fatty acid synthase (FASN), and carnitine palmitoyltransferase 1A (CPT1A). The mRNA expression of FASN and CPT1A as well as acetyl-CoA levels were decreased by PEITC treatment in both cell lines. PEITC administration to TRAMP mice also resulted in a significant decrease in tumor expression of FASN protein. Consistent with these findings, the levels of total free fatty acids, total phospholipids, triglyceride, and ATP were significantly lower in the plasma and/or prostate tumors of PEITC-treated TRAMP mice compared with controls. The present study is the first to implicate inhibition of fatty acid synthesis in prostate cancer chemoprevention by PEITC.
Keywords: Prostate Cancer, Fatty Acids, Phenethyl Isothiocyanates, Chemoprevention
Introduction
More than 26,000 American men are expected to die from prostate cancer in 2016 alone despite our increasingly broader understanding of the risk factors, genomic landscape, and biology of this neoplasm (1–5). Chemoprevention represents a powerful yet clinically under explored strategy for reducing the death and suffering from prostate cancer (6, 7). Feasibility of prostate cancer chemoprevention has been explored clinically with pioneering investigations of inhibitors of 5α-reductase (finasteride and dutasterise), which is responsible for the conversion of testosterone to dihydrotestosterone (8, 9). Even though this strategy resulted in about 23–25% decrease in the relative risk, there were high-grade tumors in the treatment arm (8, 9). After 18 years of follow-up, high-grade prostate cancer was still more common in the finasteride (PCPT trial) arm when compared to controls, but the overall survival rate or survival after diagnosis of prostate cancer was not significantly different between the groups (10). Similarly, the SELECT trial was disappointing and did not show any preventative benefit of vitamin E and selenium supplementation (11, 12). Therefore, a safe and inexpensive intervention with broad acceptance for chemoprevention of prostate cancer is still lacking.
Multiple observations implicate increased de novo synthesis of fatty acids in prostate cancer development (13–15). Moreover, fatty acid oxidation is a prevailing bioenergetic pathway in prostate cancer (16). Fatty acid synthesis is elevated upon exposure to androgens in LNCaP prostate cancer cell line (17). Pharmacological or genetic suppression of key fatty acid synthesis proteins, including ATP citrate lyase (ACLY), acetyl-CoA carboxylase (ACC), and fatty acid synthase (FASN), elicit anticancer effects in cultured and xenografted prostate cancer cell models (18–21). Blood levels of saturated and monounsaturated fatty acids are markers of de novo lipogenesis and the risk of prostate cancer (22). Therefore, inhibition of fatty acid metabolism represents an attractive alternative for chemoprevention of prostate cancer.
Phytochemicals are appealing for cancer chemoprevention based on epidemiological observations and preclinical experimental evidence (23, 24). The isothiocyanate family of small-molecule phytochemicals (e.g., phenethyl isothiocyanate; hereafter abbreviated as PEITC) abundant in cruciferous vegetables is particularly promising for cancer chemoprevention because of multifaceted effects, including inhibition of cancer initiation in carcinogen-exposed rodent models (25, 26), prevention of post-initiation cancer development in transgenic mouse models (27, 28), and suppression of angiogenesis and oncogenic signaling in cancer cells (29–31). Some of the isothiocyanates, including PEITC, have also been studied clinically for their biological activity (32, 33). In cruciferous vegetables, PEITC is stored as a thioglucoside conjugate (gluconasturtiin) and generated through catalytic mediation of myrosinase (Fig. 1A).
Figure 1.

PEITC treatment decreases ACC1 and FASN protein level in human prostate cancer cells (A) A simplified cartoon depicting metabolic conversion of gluconasturtiin to PEITC and their chemical structures. Immunocytochemical images showing expression of ACLY (B) ACC1 (C), and FASN protein (D) in LNCaP and 22Rv1 cells treated for 24 hours with DMSO (control) or 5 μmol/L PEITC. Similar results were observed in replicate experiments except for some morphological changes in ACLY staining especially in the LNCaP cell line for unclear reasons.
We showed previously that feeding of PEITC-supplemented diet (3 μmol PEITC/g diet) to Transgenic Adenocarcinoma of Mouse Prostate (TRAMP) mice resulted in a significant decrease in the incidence and burden (area) of poorly-differentiated prostate cancer (27). Identification of noninvasive biomarkers is particularly desirable for clinical development of chemopreventive interventions. The present study was undertaken to test the hypothesis of whether fatty acid synthesis intermediate(s) represent such noninvasive biomarker(s) of prostate cancer chemoprevention by PEITC. Both cellular models of prostate cancer and archived plasma and tumor tissues/sections from the TRAMP study were utilized to test this hypothesis.
Materials and Methods
Ethics statement
Fresh frozen plasma and tumor tissues as well as paraffin-embedded prostate adenocarcinoma sections from the previously published TRAMP study (27) were used to determine the in vivo effect of PEITC administration on levels of fatty acids and lipids and expression of fatty acid metabolism proteins. Use of mice was approved by the University of Pittsburgh Institutional Animal Care and Use Committee.
Reagents
PEITC (purity ≥98%) was purchased from LKT Laboratories (St. Paul, MN). RPMI1640 medium was from Cellgro-Mediatech (Manassas, VA), whereas other cell culture reagents, fetal bovine serum, and BODIPY® 493/503 (D3922) were purchased from Life Technologies-Thermo Fisher Scientific (Waltham MA). Anti-FASN (3180) and anti-ACC1 (4190) antibodies were from Cell Signaling Technology (Danvers, MA), whereas anti-ACLY (ab40793) and anti-carnitine palmitoyltransferase 1A (CPT1A) (ab128568) antibodies were from Abcam (Cambridge, MA). A kit for colorimetric measurement of triglyceride (10010303) was from Cayman Chemical (Ann Arbor, MI). Total phospholipid (MAK122) and total free fatty acid (MAK044) quantification kits were from Sigma-Aldrich (St. Louis, MO). A fluorometric kit for acetyl-CoA (K317-100) and a colorimetric kit for ATP (K354-100) were purchased from BioVision (Milpitas, CA).
Cell lines and culture condition
LNCaP and 22Rv1 cells were obtained from the American Type Culture Collection (Manassas, VA) and last authenticated by us in 2012 (22Rv1) or 2015 (LNCaP). Authenticated stocks of these cell lines were used for the in vitro experiments. Cell line authentication was done using short tandem repeat profiling at the IDEXX BioResearch. The cell lines were cultured as suggested by the supplier.
Confocal microscopy
Cells were plated on coverslips in 24-well plates. After overnight incubation to allow attachment of the cells, they were treated with dimethyl sulfoxide (DMSO) or desired doses of PEITC for 24 hours and then fixed and permeabilized with 2% paraformaldehyde and 0.5% Triton X-100, respectively. After blocking, cells were incubated with anti-ACLY (1:2000 dilution), anti-ACC1 (1:1000 dilution) or anti-FASN (1:5000 dilution) antibody overnight at 4°C. Cells were then washed with phosphate-buffered saline (PBS) and incubated with Alexa Fluor 488-conjugated goat anti-rabbit secondary antibody (1:2000 dilution) for 1 hour at room temperature in the dark. Cells were counterstained with DRAQ5 (nuclear stain) and then mounted and examined under a Confocal TCSSL microscope.
For microscopic analysis of CPT1A, cells were treated with DMSO or PEITC as described above and stained with 100 nmol/L MitoTracker red in complete medium at 37°C for 1 hour followed by washing and fixation of cells with 4% formaldehyde in complete medium at 37°C for 15 minutes. The cells were permeabilized with 0.5% Triton X-100 for 5 minutes and incubated for 1 hour with blocking solution consisting of PBS, 0.5% bovine serum albumin, and 0.15% glycine. After blocking, cells were incubated with anti-CPT1A antibody (1:3000 dilution) at 4°C overnight and then with Alexa Fluor 488-conjugated goat anti-mouse secondary antibody for 1 hour at room temperature. The cells were examined under a Confocal TCSSL microscope.
Western blotting
LNCaP (3.5×105) and 22Rv1 (5×105) cells were plated in media supplemented with 10% fetal bovine serum and allowed to attach overnight. Cells were then treated with DMSO or desired concentrations of PEITC for different time points. Cell lysates were prepared as described by us previously (34). Western blotting was performed as described by us previously (34).
Quantitative polymerase chain reaction (PCR)
Expression of FASN and CPT1A were determined in LNCaP and 22Rv1 cells by real time PCR. Total RNA from DMSO or PEITC treated cells was isolated using RNeasy kit. 2 μg RNA was used for cDNA synthesis with the use of SuperScript III reverse transcriptase and oligo (dT)20 primer. Quantitative PCR was performed using 2x SYBR Green Master Mix under same conditions, with the number of cycles changed to 40. Primers for FASN, CPT1A, and GAPDH were as follows. Forward (FASN): 5′-CTGGCTCAGCACCTCTATCC-3′; Reverse: (FASN) 5′-CAGGTTGTCCCTGTGATCCT-3′; Forward (CPT1A): 5′-CAAGGACATGGGCAAGTTTT-3′; Reverse: (CPT1A) 5′-AAAGGCAGAAGAGGTGACGA-3′; Forward (GAPDH): 5′-GGACCTGACCTGCCGTCTAGAA-3′; Reverse (GAPDH): 5′-GGTGTCGCTGTTGAAGTCAGAG-3′. The PCR conditions were as follows: 95°C for 10 minutes followed by 40 cycles of 95°C for 15 seconds, 60°C for 1 minute, and 72°C for 30 seconds. Relative gene expression was calculated using the 2−ΔΔCT method (35).
Immunohistochemistry
Immunohistochemistry was performed as described by us previously (27, 28) with some modifications. Briefly, prostate tumor sections (4–5 μm thick) were de-paraffinized, hydrated in graded alcohol, and then washed with PBS. Sections were immersed in 0.3% H2O2 in 100% methanol for 20 minutes at room temperature. Sections were then incubated with blocking buffer for 1 hour followed by incubation with primary antibody (FASN- 1:500; CPT1A- 1:1000) for overnight in humid chambers at room temperature. After washing, sections were incubated with Alexa Fluor 488-conjugated secondary antibody (1:1000) for 1 hour at room temperature. After washing with PBS, sections were mounted with an anti-fade mounting media containing 4′,6-diamidino-2-phenylindole. Stained sections were examined under Nikon A1 (s216.6) confocal microscope. At least five non-overlapping and non-necrotic images were captured from each section and analyzed with NIS element 4.0 software.
Neutral lipid droplets in prostate tumor sections were visualized by BODIPY493/503 staining. Sections were incubated with 25 μg/mL BODIPY 493/503 for 1 hour at room temperature. After washing with PBS, sections were mounted with an anti-fade mounting media containing 4′,6-diamidino-2-phenylindole and examined under Nikon A1 (s216.6) confocal microscope. At least five non-overlapping and non-necrotic images were captured from each section and number of lipid droplets/cell was quantified using FIJI ImageJ (NIH) software. Number of lipid droplets/cell was determined in at least 50 cells per high-power field.
Determination of metabolites
A fluorometric assay kit was used to measure acetyl-CoA levels in human prostate cancer (LNCaP and 22Rv1) cells, and tumor lysates and plasma from TRAMP mice. LNCaP (3.5×105) and 22Rv1 (5×105) cells were seeded in 60 mm plates and treated with DMSO or the indicated doses of PEITC for 24 hours. Human prostate cancer cells and tumor tissues were sonicated in acetyl-CoA assay buffer and centrifuged at 10,000g for 10 minutes. The fluorescence was measured at excitation and emission wavelengths of 535 and 587 nm, respectively, using a fluorimeter (Biotek, VT).
For measurement of total free fatty acids (C8 and longer), 10–20 mg tumor tissues were sonicated in 200 μl of 1% (w/v) Triton X-100 in chloroform. The homogenates were centrifuged at 14,000 rpm for 10 minutes, and organic phase was collected from the supernatant and air dried to remove chloroform. The dried lipids were dissolved in fatty acid assay buffer and used for the assay. Plasma was diluted in fatty acid assay buffer and used for the assay. Assay was performed according to manufacturer’s instructions. Absorbance was measured at 570 nm.
A kit was used to measure total phospholipids in the tumor and plasma according to the supplier’s instructions. The tumor tissue was sonicated in phospholipid assay buffer and centrifuged at 10,000g for 10 minutes. The supernatant was used for phospholipid assay and protein estimation. Plasma was directly used for the assay. Assay was performed according to manufacturer’s instructions. Absorbance was measured at 570 nm.
For the measurement of triglyceride, 10–20 mg tumor tissue was homogenized in manufacture supplied solution followed by centrifugation at 10,000g for 10 minutes. The supernatant was used for triglyceride assay and protein estimation. Plasma was directly used for the assay. Assay was performed as per manufacturer’s protocol. Absorbance was measured at 550 nm.
A colorimetric assay kit was used to measure ATP levels in the tumor. Tumor tissue was sonicated in ATP assay buffer, centrifuged at 10,000g for 10 minutes, and the supernatant was filtered using 10 kDa membrane. Absorbance was measured at 570 nm.
Statistical analysis
GraphPad Prism (version 7.02) was used for statistical analysis. Statistical significance of difference was determined by one-way analysis of variance (ANOVA) followed by Dunnett’s test or unpaired Student’s t test.
Results
Fatty acid synthesis in the cytosol begins with ACLY-mediated conversion of citrate, which is derived from the tricarboxylic acid cycle in the mitochondia, to acetyl-CoA. Expression of ACLY was not affected by PEITC treatment in either LNCaP or 22Rv1 cell line (Fig. 1B). The next step in the de novo synthesis of fatty acids is conversion of acetyl-CoA to malonyl-CoA through catalytic mediation of ACC, which is a rate liming step. FASN complex subsequently utilizes one molecule of acetyl-CoA and seven molecules of malonyl-CoA to produce fatty acids through a series of catalytic domains (14,15). Protein levels of both ACC1 (Fig. 1C) and FASN (Fig. 1D) were markedly suppressed by PEITC treatment in both LNCaP and 22Rv1 cells. It is important to mention that LNCaP is an androgen-responsive cell line with expression of mutant androgen receptor (T877A) and mutant phosphatase and tensin homolog. On the other hand, 22Rv1 cell line expresses wild-type phosphatase and tensin homolog but mutant androgen receptor and its splice variants including AR-V7. These results indicated that PEITC-mediated downregulation of ACC1 and FASN was maintained in prostate cancer cell lines with different genetic alterations.
Inhibitory effect of PEITC on ACC1 and FASN was confirmed by western blotting. Levels of both ACC1 and FASN proteins were decreased markedly after PEITC treatment and this downregulation was evident as early as 8 hours post-treatment (Fig. 2A). Real-time quantitative PCR revealed downregulation of FASN mRNA expression in PEITC-treated LNCaP and 22Rv1 cells relative to corresponding DMSO-treated control cells (Fig. 2B). Fig. 2C depicts immunohistochemical images for FASN protein expression in a representative prostate adenocarcinoma from the control and the PEITC-treated TRAMP mouse. The semi-quantitative H-score for FASN protein expression was lower by about 50% (P= 0.005 by unpaired Student’s t test) in the poorly-differentiated adenocarcinoma sections of the PEITC-treated TRAMP mice when compared with controls (Fig. 2D). These results indicated in vitro and in vivo downregulation of key fatty acid synthesis proteins after PEITC treatment in prostate cancer cells.
Figure 2.

PEITC downregulates ACC1 and FASN expression in human prostate cancer cells. (A) Immunoblotting for ACC1 and FASN proteins using lysates from control and PEITC-treated LNCaP and 22Rv1 cells. (B) Real-time quantitative PCR of FASN in LNCaP and 22Rv1 cells after treatment with DMSO or PEITC. Results shown are mean ± SD (n= 3). *Significantly different (P < 0.05) compared with corresponding DMSO-treated control cells by one way ANOVA followed by Dunnett’s test. (C) Representative immunohistochemistry images showing expression of FASN protein in prostate adenocarcinoma of control and PEITC treated TRAMP mice. Lower panel shows hematoxylin and eosin-staining. (D) Quantitation of FASN expression (40x magnification, scale bar = 50 μm). Results shown are mean ± SD (n=5 for both groups). P value was determined by unpaired Student’s t test. Each experiment (A and B) was repeated at least twice and the results were generally consistent.
CPT1A is another protein with critical role in the overall metabolism of fatty acids (14, 15). CPT1A, also known as carnitine acyltransferase I, is a mitochondria-localized enzyme responsible for generation of acyl carnitines by catalyzing the transfer of the acyl group of a long-chain fatty acyl-CoA from coenzyme A to carnitine. Western blotting revealed downregulation of CPT1A protein in both LNCaP and 22Rv1 cell lines at least at 16- and 24-hour time points (Fig. 3A). Due to faint expression of CPT1A in western blot especially in the LNCaP cell line, immunocytochemistry was also performed. As expected, CPT1A was localized in the mitochondria as indicated by yellow-orange fluorescence in DMSO-treated control cells due to the merging of CPT1A-associated green fluorescence and red color signal from MitoTracker red (Fig. 3B). The yellow-orange fluorescence was markedly suppressed by PEITC treatment in both cell lines (Fig. 3B). PEITC treatment caused transcriptional repression of CPT1A in both LNCaP and 22Rv1 cells as revealed by real-time quantitative PCR (Fig. 3C). Similar to FASN (Fig. 2C), PEITC administration to TRAMP mice resulted in suppression of CPT1A protein level in the poorly-differentiated prostate cancer relative to control but the difference was not statistically significant with a P value of 0.07 (Fig. 3D, E).
Figure 3.

PEITC decrease CPT1A expression. (A) Immunoblotting for CPT1A protein using lysates from control and PEITC-treated LNCaP and 22Rv1 cells. (B) Immunocytochemical images depicting CPT1A expression in control and PEITC treated LNCaP and 22Rv1 cells (24 hour treatment). (C) Real-time quantitative PCR for CPT1A in LNCaP and 22Rv1 cells after 8, 16, and 24 hours of treatment with DMSO or PEITC. Results shown are mean ± SD (n= 3 for each column). *Significantly different (P < 0.05) compared with corresponding DMSO-treated control cells by one way ANOVA followed by Dunnett’s test. (D) Representative immunohistochemistry images showing expression of CPT1A protein in prostate adenocarcinoma of a control and a PEITC-treated TRAMP mouse. (E) Quantitation of CPT1A protein expression (40x magnification, scale bar = 50 μm). Results shown are mean ± SD. Statistical significance was determined by unpaired Student’s t test. Each experiment (A to C) was repeated at least twice and the results were generally consistent.
Because PEITC treatment downregulated CPT1A protein, it was of interest to determine whether this effect was accompanied by a decrease in the levels of acetyl-CoA. Intracellular level of acetyl-CoA was indeed lower in PEITC-treated cells compared with control and the difference was significant at the 5 μmol/L dose (Fig. 4A). Plasma level of acetyl-CoA (Fig. 4B), but not the tumor acetyl-CoA level (Fig. 4C), was significantly lower in the TRAMP mice of PEITC treatment group compared with control. Lack of statistical significance for tumor acetyl-CoA could be due to small sample size especially in the PEITC arm.
Figure 4.

PEITC treatment decreases acetyl-CoA level in human prostate cancer cells and in plasma of TRAMP mice. (A) Acetyl-CoA levels in LNCaP and 22Rv1 cells treated for 24 hours with DMSO or PEITC. Results shown are mean ± SD (n= 3 for each column). *Significantly different (P < 0.05) compared with corresponding DMSO-treated control by one way ANOVA followed by Dunnett’s test. Acetyl-CoA levels in the plasma (B) and prostate adenocarcinoma of (C) TRAMP mice. Results shown are mean ± SD. Plasma (n = 17 for control group and n = 16 for PEITC treatment group) and tumor samples (n = 8 for control group and n = 4 for PEITC treatment group) from different mice of each group were used for determination of Acetyl-CoA levels. Statistical significance was determined by unpaired Student’s t test.
Some fatty acids are esterified to phospholipids, which play an important role in signal transduction and intracellular trafficking (36). PEITC feeding to TRAMP mice resulted in a significant decrease in plasma levels of total free fatty acids (Fig. 5A), total phospholipids (Fig. 5B), and triglyceride (Fig. 5C).
Figure 5.
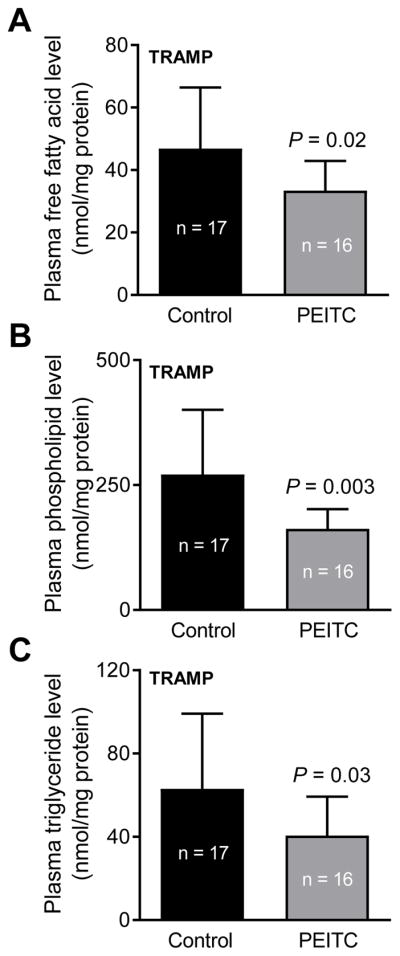
Effect of PEITC administration on fatty acid metabolites in the plasma of TRAMP mice. Levels of total free fatty acids (A), total phospholipids (B) and triglyceride (C) in the plasma of control and PEITC-treated TRAMP mice. Results shown are mean ± SD (n = 17 for control group and n = 16 for PEITC treatment group). Plasma samples from different mice of each group were used for the assays. Statistical significance was determined by unpaired Student’s t test.
Mean total free fatty acid level in the tumors of PEITC-treated TRAMP mice was lower by about 25% when compared to control group (Fig. 6A), but the difference was not significant (P= 0.06). On the other hand, PEITC administration to TRAMP mice resulted in a significant decrease in prostate tumor levels of total phospholipids (Fig. 6A). The difference was not significant for prostate tumor level of triglyceride between control and PEITC treatment groups (Fig. 6A; P= 0.09). Nevertheless, tumor ATP level was lower by 32% (P= 0.03) in the PEITC treatment group compared to control (Fig. 6A).
Figure 6.

PEITC administration decreases fatty acid metabolite levels in the prostate adenocarcinoma of TRAMP mice. (A) Levels of total free fatty acids, total phospholipids, triglyceride, and ATP in prostate tumors of control and PEITC-treated TRAMP mice. Results shown are mean ± SD (n = 8 for control and n = 4 for PEITC treatment group). Tumor tissues from different mice of each group were used for the assays. Statistical significance was determined by unpaired Student’s t test. (B) Representative images for BODIPY493/503 staining showing lipid droplets in prostate adenocarcinoma of a control and a PEITC-treated TRAMP mouse. (C) Quantitation of number of lipid droplets/cell (60x magnification, scale bar = 5 μm). Results shown are mean ± SD. Statistical significance was determined by unpaired Student’s t test. (D) A simplified cartoon for fatty acid synthesis. PEITC-mediated suppression of fatty acid metabolism proteins and intermediates is also summarized. Abbreviations: FACS- fatty acyl-CoA synthase; ACLY- ATP citrate lyase; ACC- acetyl-CoA carboxylase; FASN- fatty acid synthase; and CPT- carnitine palmitoyltransferase.
Figure 6B depicts BODIPY staining (neutral lipids) in a representative prostate tumor section of a control and a PEITC-treated TRAMP mouse (white dots in the images represent neutral lipid droplets). The mean number of neutral lipid droplets/cell was significantly lower in the prostate tumor sections of PEITC-treated mice when compared with control (Fig. 6C). A cartoon summarizing fatty acid metabolism as well as PEITC-mediated suppression of fatty acid synthesis enzyme proteins and intermediates is shown in Fig. 6D.
Discussion
The present study demonstrates, for the first time, significant downregulation of FASN protein and mRNA expression upon PEITC treatment in prostate cancer cells in vitro and in vivo leading to a decrease in levels of total free fatty acids and total phospholipids. Evidence is quite strong for an oncogenic role of FASN in prostate cancer. For example, immunohistochemistry and fluorescence in situ hybridization analysis in paraffin-embedded tissue microarrays revealed FASN gene copy gain as well as protein overexpression in prostate adenocarcinoma specimens (37). Increased FASN gene expression was documented in nearly 50% of all prostate tissues with elevated FASN protein levels (37). Overexpression of FASN in an immortalized normal human prostate epithelial cell line (iPrEC) resulted in palmitoylation of Wnt1 as well as cytoplasmic stabilization of β-catenin (38). Consistent with these results, overexpression of FASN in iPrECs and prostate cancer cells (LNCaP) increased cell proliferation and soft agar growth (39). More importantly, transgenic expression of FASN in mice produced prostatic intraepithelial neoplasia at 7 months of age (39). In radical prostatectomy needle biopsy cores, elevated fatty acid synthase expression was suggested to predict upgraded Gleason score (40). Even though FASN is a legitimate therapeutic target in prostate cancer, clinical development of natural or synthetic FASN inhibitor has been challenging due to a variety of reasons, including stability (Cerulenin), bioavailability (epigallocatechin- 3-gallate and Orlistat), and side effects (appetite suppression and weight loss through direct activation of CPT1 (41,42). PEITC, on the other hand, has excellent oral bioavailability and is well-tolerated in mice after chronic administration (27, 28).
In comparison to FASN, ACC is relatively less studied in the context of prostate cancer despite its rate limiting role in conversion of acetyl-CoA to malonyl-CoA. Nevertheless, pharmacological inhibition of ACC was shown to inhibit PC-3 prostate cancer cell migration (43). Chemical inhibition of ACC by 5-tetradecyl-oxy-2-furoic acid was accompanied by caspase-dependent cell death in prostate cancer cells regardless of the p53 status (44). RNA interference of ACC-α also resulted in inhibition of cell proliferation and caspase-dependent apoptosis in LNCaP prostate cancer cells (19). We have shown previously that PEITC treatment results in caspase-dependent apoptosis in cultured human prostate cancer cells (45, 46). Surprisingly, prostate cancer chemoprevention by PEITC in the TRAMP model was not associated with increased tumor cell apoptosis (27). We conclude that ACC1 downregulation by PEITC may not be important for apoptosis induction, at least for this phytochemical.
This study shows a decrease in intracellular levels of acetyl-CoA in vitro by PEITC treatment in human prostate cancer cells. PEITC administration to TRAMP mice also results in lowering of plasma acetyl-CoA levels (present study). This intermediate not only is the building block of fatty acid synthesis but is also generated during β-oxidation of fatty acids. Because ACLY protein expression is not altered by PEITC treatment, at least in LNCaP and 22Rv1 cells (present study), it is reasonable to postulate that PEITC-mediated decrease in acetyl-CoA level is most likely due to inhibition of fatty acid β-oxidation. However, further work is necessary to experimentally test this possibility.
In conclusion, the present study provides in vitro and in vivo evidence for PEITC-mediated downregulation of FASN expression that is accompanied by a decrease in plasma and/or tumor levels of total free fatty acids, phospholipids, and ATP. We propose that plasma free fatty acids and/or total phospholipids may serve as noninvasive biomarkers of PEITC in future clinical trials.
Acknowledgments
Grant Support
This work was supported by the grant RO1 CA101753-11 awarded by the National Cancer Institute (S.V. Singh). This research used the Animal Facility and the Tissue and Research Pathology Facility supported in part by a grant from the National Cancer Institute at the National Institutes of Health (P30 CA047904; Dr. Edward Chu- Principal Investigator).
Footnotes
Conflict of Interest: None of the authors has any conflict of interest.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Key T. Risk factors for prostate cancer. Cancer Surv. 1995;23:63–77. [PubMed] [Google Scholar]
- 3.Roobol MJ, Carlsson SV. Risk stratification in prostate cancer screening. Nat Rev Urol. 2013;10:38–48. doi: 10.1038/nrurol.2012.225. [DOI] [PubMed] [Google Scholar]
- 4.Drudge-Coates L, Turner B. Prostate cancer overview. Part 1: non-metastatic disease. Br J Nurs. 2012;21:S23–8. [PubMed] [Google Scholar]
- 5.Mitchell T, Neal DE. The genomic evolution of human prostate cancer. Br J Cancer. 2015;113:193–8. doi: 10.1038/bjc.2015.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ting H, Deep G, Agarwal C, Agarwal R. The strategies to control prostate cancer by chemoprevention approaches. Mutat Res. 2014;760:1–15. doi: 10.1016/j.mrfmmm.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jandial DD, Blair CA, Zhang S, Krill LS, Zhang YB, Zi X. Molecular targeted approaches to cancer therapy and prevention using chalcones. Curr Cancer Drug Targets. 2014;14:181–200. doi: 10.2174/1568009614666140122160515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thompson IM, Goodman PJ, Tangen CM, Lucia MS, Miller GJ, Ford LG, et al. The influence of finasteride on the development of prostate cancer. N Engl J Med. 2003;349:215–224. doi: 10.1056/NEJMoa030660. [DOI] [PubMed] [Google Scholar]
- 9.Andriole GL, Bostwick DG, Brawley OW, Gomella LG, Marberger M, Montorsi F, et al. Effect of dutasteride on the risk of prostate cancer. N Engl J Med. 2010;362:1192–202. doi: 10.1056/NEJMoa0908127. [DOI] [PubMed] [Google Scholar]
- 10.Thompson IM, Goodman PJ, Tangen CM, Parnes HL, Minasian LM, Godley PA, et al. Long-term survival of participants in the prostate cancer prevention trial. N Engl J Med. 2013;369:603–10. doi: 10.1056/NEJMoa1215932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lippman SM, Klein EA, Goodman PJ, Lucia MS, Thompson IM, Ford LG, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT) JAMA. 2009;301:39–51. doi: 10.1001/jama.2008.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klein EA, Thompson IM, Tangen CM, Crowley JJ, Lucia MS, Goodman PJ, et al. Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT) JAMA. 2011;306:1549–56. doi: 10.1001/jama.2011.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suburu J, Chen YQ. Lipids and prostate cancer. Prostaglandins Other Lipid Mediators. 2012;98:1–10. doi: 10.1016/j.prostaglandins.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zadra G, Photopoulos C, Loda M. The fat side of prostate cancer. Biochim Biophys Acta. 2013;1831:1518–32. doi: 10.1016/j.bbalip.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu X, Daniels G, Lee P, Monaco ME. Lipid metabolism in prostate cancer. Am J Clin Exp Urol. 2014;2:111–20. [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis. 2006;9:230–4. doi: 10.1038/sj.pcan.4500879. [DOI] [PubMed] [Google Scholar]
- 17.Swinnen JV, Esquenet M, Goossens K, Heyns W, Verhoeven G. Androgens stimulate fatty acid synthase in the human prostate cancer cell line LNCaP. Cancer Res. 1997;57:1086–90. [PubMed] [Google Scholar]
- 18.Kridel SJ, Axelrod F, Rozenkrantz N, Smith JW. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res. 2004;64:2070–5. doi: 10.1158/0008-5472.can-03-3645. [DOI] [PubMed] [Google Scholar]
- 19.Brusselmans K, De Schrijver E, Verhoeven G, Swinnen JV. RNA interference-mediated silencing of the acetyl-CoA-carboxylase-α gene induces growth inhibition and apoptosis of prostate cancer cells. Cancer Res. 2005;65:6719–25. doi: 10.1158/0008-5472.CAN-05-0571. [DOI] [PubMed] [Google Scholar]
- 20.Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, et al. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. 2005;8:311–21. doi: 10.1016/j.ccr.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 21.Beckers A, Organe S, Timmermans L, Scheys K, Peeters A, Brusselmans K, et al. Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res. 2007;67:8180–7. doi: 10.1158/0008-5472.CAN-07-0389. [DOI] [PubMed] [Google Scholar]
- 22.Chavarro JE, Kenfield SA, Stampfer MJ, Loda M, Campos H, Sesso HD, et al. Blood levels of saturated and monounsaturated fatty acids as markers of de novo lipogenesis and risk of prostate cancer. Am J Epidemiol. 2013;178:1246–55. doi: 10.1093/aje/kwt136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stan SD, Kar S, Stoner GD, Singh SV. Bioactive food components and cancer risk reduction. J Cell Biochem. 2008;104:339–56. doi: 10.1002/jcb.21623. [DOI] [PubMed] [Google Scholar]
- 24.Lee KW, Bode AM, Dong Z. Molecular targets of phytochemicals for cancer prevention. Nat Rev Cancer. 2011;11:211–8. doi: 10.1038/nrc3017. [DOI] [PubMed] [Google Scholar]
- 25.Hecht SS. Chemoprevention by isothiocyanates. J Cell Biochem. 1995;22(suppl):195–209. doi: 10.1002/jcb.240590825. [DOI] [PubMed] [Google Scholar]
- 26.Stoner GD, Morse MA. Isothiocyanates and plant polyphenols as inhibitors of lung and esophageal cancer. Cancer Lett. 1997;114:113–9. doi: 10.1016/s0304-3835(97)04639-9. [DOI] [PubMed] [Google Scholar]
- 27.Powolny AA, Bommareddy A, Hahm ER, Normolle DP, Beumer JH, Nelson JB, et al. Chemopreventative potential of the cruciferous vegetable constituent phenethyl isothiocyanate in a mouse model of prostate cancer. J Natl Cancer Inst. 2011;103:571–84. doi: 10.1093/jnci/djr029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh SV, Kim SH, Sehrawat A, Arlotti JA, Hahm ER, Sakao K, et al. Biomarkers of phenethyl isothiocyanate-mediated mammary cancer chemoprevention in a clinically relevant mouse model. J Natl Cancer Inst. 2012;104:1228–39. doi: 10.1093/jnci/djs321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keum YS, Jeong WS, Kong AN. Chemoprevention by isothiocyanates and their underlying molecular signaling mechanisms. Mutat Res. 2004;555:191–202. doi: 10.1016/j.mrfmmm.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 30.Xiao D, Singh SV. Phenethyl isothiocyanate inhibits angiogenesis in vitro and ex vivo. Cancer Res. 2007;67:2239–46. doi: 10.1158/0008-5472.CAN-06-3645. [DOI] [PubMed] [Google Scholar]
- 31.Singh SV, Singh K. Cancer chemoprevention with dietary isothiocyanates mature for clinical translational research. Carcinogenesis. 2012;33:1833–42. doi: 10.1093/carcin/bgs216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuan JM, Stepanov I, Murphy SE, Wang R, Allen S, Jensen J, et al. Clinical trial of 2-phenethyl isothiocyanate as an inhibitor of metabolic activation of a tobacco-specific lung carcinogen in cigarette smokers. Cancer Prev Res (Phila) 2016;9:396–405. doi: 10.1158/1940-6207.CAPR-15-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amjad AI, Parikh RA, Appleman LJ, Hahm ER, Singh K, Singh SV. Broccoli-derived sulforaphane and chemoprevention of prostate cancer: From bench to bedside. Curr Pharmacol Rep. 2015;1:382–90. doi: 10.1007/s40495-015-0034-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiao D, Srivastava SK, Lew KL, Zeng Y, Hershberger P, Johnson CS, et al. Allyl isothiocyanate, a constituent of cruciferous vegetables, inhibits proliferation of human prostate cancer cells by causing G2/M arrest and inducing apoptosis. Carcinogenesis. 2003;24:891–7. doi: 10.1093/carcin/bgg023. [DOI] [PubMed] [Google Scholar]
- 35.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(ΔΔ-CT) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 36.Swinnen JV, Brusselmans K, Verhoeven G. Increased lipogenesis in cancer cells: new players, novel targets. Curr Opin Clin Nutr Metab Care. 2006;9:358–65. doi: 10.1097/01.mco.0000232894.28674.30. [DOI] [PubMed] [Google Scholar]
- 37.Shah US, Dhir R, Gollin SM, Chandran UR, Lewis D, Acquafondata M, et al. Fatty acid synthase gene overexpression and copy number gain in prostate adenocarcinoma. Hum Pathol. 2006;37:401–9. doi: 10.1016/j.humpath.2005.11.022. [DOI] [PubMed] [Google Scholar]
- 38.Fiorentino M, Zadra G, Palescandolo E, Fedele G, Bailey D, Fiore C, et al. Overexpression of fatty acid synthase is associated with palmitoylation of Wnt1 and cytoplasmic stabilization of β-catenin in prostate cancer. Lab Invest. 2008;88:1340–8. doi: 10.1038/labinvest.2008.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Migita T, Ruiz S, Fornari A, Fiorentino M, Priolo C, Zadra G, et al. Fatty acid synthase: a metabolic enzyme and candidate oncogene in prostate cancer. J Natl Cancer Inst. 2009;101:519–32. doi: 10.1093/jnci/djp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hamada S, Horiguchi A, Kuroda K, Ito K, Asano T, Miyai K, et al. Elevated fatty acid synthase expression in prostate needle biopsy cores predicts upgraded Gleason score in radical prostatectomy specimens. Prostate. 2014;74:90–6. doi: 10.1002/pros.22732. [DOI] [PubMed] [Google Scholar]
- 41.Kuhajda FP, Pizer ES, Li JN, Mani NS, Frehywot GL, Townsend CA. Synthesis and antitumor activity of an inhibitor of fatty acid synthase. Proc Natl Acad Sci USA. 2000;97:3450–4. doi: 10.1073/pnas.050582897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lambert JD, Yang CS. Cancer chemopreventive activity and bioavailability of tea and tea polyphenols. Mutat Res. 2003;523–524:201–8. doi: 10.1016/s0027-5107(02)00336-6. [DOI] [PubMed] [Google Scholar]
- 43.Scott KE, Wheeler FB, Davis AL, Thomas MJ, Ntambi JM, Seals DF, et al. Metabolic regulation of invadopodia and invasion by acetyl-CoA carboxylase 1 and de novo lipogenesis. PLoS One. 2012;7:e29761. doi: 10.1371/journal.pone.0029761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guseva NV, Rokhlin OW, Glover RA, Cohen MB. TOFA (5-tetradecyl-oxy-2-furoic acid) reduces fatty acid synthesis, inhibits expression of AR, neuropilin-1 and Mcl-1 and kills prostate cancer cells independent of p53 status. Cancer Biol Ther. 2011;12:80–5. doi: 10.4161/cbt.12.1.15721. [DOI] [PubMed] [Google Scholar]
- 45.Xiao D, Singh SV. Phenethyl isothiocyanate-induced apoptosis in p53-deficient PC-3 human prostate cancer cell line is mediated by extracellular signal-regulated kinases. Cancer Res. 2002;62:3615–9. [PubMed] [Google Scholar]
- 46.Xiao D, Zeng Y, Choi S, Lew KL, Nelson JB, Singh SV. Caspase-dependent apoptosis induction by phenethyl isothiocyanate, a cruciferous vegetable-derived cancer chemopreventive agent, is mediated by Bak and Bax. Clin Cancer Res. 2005;11:2670–9. doi: 10.1158/1078-0432.CCR-04-1545. [DOI] [PubMed] [Google Scholar]