

Summary
Iron is an essential metal involved in several major cellular processes required to maintain life. Because of iron’s ability to cause oxidative damage, its transport, metabolism, and storage is strictly controlled in the body, especially in the small intestine, liver, and kidney. Iron plays a major role in acute kidney injury and has been a target for therapeutic intervention. However, the therapies that have been effective in animal models of acute kidney injury have not been successful in human beings. Targeting iron trafficking via ferritin, ferroportin, or hepcidin may offer new insights. This review focuses on the biology of iron, particularly in the kidney, and its implications in acute kidney injury.
Keywords: Iron, kidney, injury, ferroportin, hepcidin, ferritin
Iron is an essential element that is necessary for life. The primary function of iron in the body is the transportation of oxygen. Iron is bound to the heme group in hemoglobin, which allows red bloods cells to supply oxygen to the tissues. Hemoglobin also facilitates the transfer of carbon dioxide from the tissues to the lungs for removal from the body. Myoglobin is another heme protein that uses iron to store and transport oxygen in muscle cells. More than 70% of the body’s iron is contained within hemoglobin and myoglobin. Another 12% can be found in iron storage proteins such as ferritin and transferrin. The remaining 15% of the body’s iron is in heme-containing proteins (such as cytochromes, respiratory burst enzymes, catalase, nitric oxide synthase, myeloperoxidase, and others) that are ubiquitous and imperative to the maintenance of proper functioning of cells and tissues.
Heme is a highly conjugated heterocyclic organic ring with ferrous iron in the center, which is a necessary component of several proteins in the body involved in respiration and energy metabolism. Cytochromes, such as cytochrome c, contain a heme group and are responsible for the production of adenosine triphosphate in the electron transport chain. Other heme-containing enzymes such as cytochrome P450s are monooxygenases that catalyze the metabolism of a wide variety of endogenous and exogenous compounds for either synthesis or detoxification. Although peroxidases such as catalase are protective and catalyze the reduction of hydrogen peroxide to water, cyclooxygenases convert fatty acids into vasoactive prostaglandins. In addition, iron is an essential component of several enzymes involved in the synthesis of collagen and neurotransmitters. Iron also is necessary for proper immune function.
Although iron is essential for life, it is extremely labile in certain forms and, as a result, can be highly reactive and toxic. Iron can be found in two different redox states: the ferrous state (Fe2+) and the ferric state (Fe3+). The fact that iron can bind six different ligands simultaneously accounts for the high reactivity levels of this metal and its ability to undergo Fenton chemistry. When the body insufficiently reduces oxygen to water, a superoxide radical is formed, but it is subsequently converted to hydrogen peroxide by superoxide dismutases. During the Haber-Weiss reaction, the ferrous form of iron can catalyze a reaction with the seemingly innocuous superoxide and hydrogen peroxide to form the toxic hydroxyl radical. Consequently, the superoxide radical can remove iron from iron-sulfur clusters in proteins, which can lead to certain diseases.1 On the other hand, the deleterious effects of the hydroxyl radical are well established. Several researchers have studied the ability of the hydroxyl radical to cause oxidative DNA damage, ultimately leading to disease.2 Because of the potential reactivity of iron, its movement throughout the body is strictly controlled. Table 1 shows some of the major proteins involved in regulating iron homeostasis.
Table 1.
Some Key Proteins Involved in Iron Homeostasis
| Protein | Function |
|---|---|
| DMT1 | The divalent metal transporter 1 imports Fe2+ into the cell |
| Ferritin | Iron storage protein with heavy (H)- and light (L)-chain subtypes H-ferritin has ferroxidase activity that allows for the safe incorporation of iron into ferritin for storage |
| Ferroportin | An iron transporter that exports iron from the cell |
| Hepcidin | A peptide synthesized in the liver that controls iron absorption by regulating ferroportin expression |
| Hephaestin | A ferroxidase localized mostly in the small intestine that is responsible for converting Fe2+ to Fe3+ |
| NGAL | Neutrophil gelatinase- associated lipocalin (also known as lipocalin-2) sequesters iron to inhibit bacterial growth It is also a biomarker for acute kidney injury |
| Transferrin | A glycoprotein that can bind up to two atoms of iron in biological fluids. Binding is reversible and the iron pool in transferrin has a high turnover rate |
IRON TRAFFICKING IN SPECIFIC ORGANS
The body obtains iron from the diet. Some iron-rich foods include meats, dark green leafy vegetables, beans, dried fruits, and other iron-fortified foods such as cereal. Dietary iron is absorbed in the intestine and metabolized and stored in other tissues, including the liver and the kidneys. Iron is eliminated from the body through blood loss, urinary excretion, and by discarding mucosal and skin cells. The small intestine, particularly the duodenum, is the organ responsible for dietary iron absorption (Fig. 1). The acidic environment of the stomach increases the solubility of the iron acquired from the diet, usually as Fe3+, to aid with passage through the digestive tract. In the intestinal lumen, ascorbic acid and ferric reductases such as duodenal cytochrome b, a plasma membrane protein located in the brush-border membrane of enterocytes, reduces ferric iron to the ferrous form (Fe2+).3 Iron can enter enterocytes at the apical surface using the proton gradient powered divalent metal transporter-1 (DMT1).4 It is thought that iron uses transcytosis or a chaperone to travel from the apical membrane to the basolateral membrane, but the exact mechanism has not yet been elucidated.5 The efflux of iron from the basolateral side of the enterocyte is facilitated by ferroportin (FPN)6; interestingly, there also is evidence to suggest that FPN may modulate the activity of DMT1 at the apical membrane.7 Although it is unclear how iron navigates from the enterocyte through the intestinal mucosa to the plasma, the ferroxidases hephaestin and/or ceruloplasmin are involved, depending on the physiological state.8 After being oxidized to the ferric form, iron binds to transferrin in the plasma, which disseminates it to the bone marrow and all tissues and organs. In addition to inorganic iron, heme iron also is absorbed by the intestinal mucosa.9 Either the heme carrier protein-1 or the folate transporter is responsible for heme transport across the brush-border membrane of duodenal enterocytes.10,11 Heme oxygenases (HOs) degrade heme to release free iron into the internal cellular iron pool. When iron is not being used actively, it can be bound to transferrin in the plasma or ferritin in the tissues.
Figure 1.
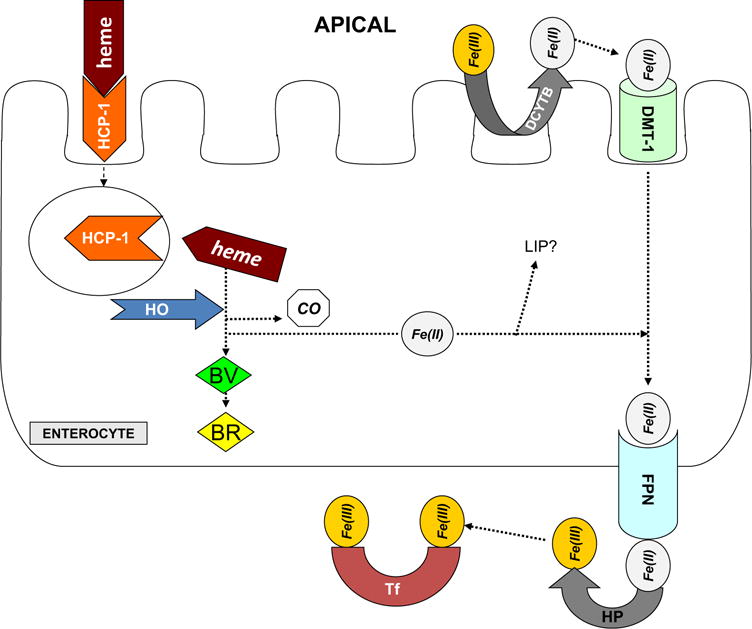
Intestinal iron absorption. This figure shows iron absorption in an enterocyte. BR, bilirubin; BV, biliverdin; CO, carbon monoxide; DCYTB, duodenal cytochrome b; HCP-1, heme carrier protein-1; HP, hephaestin; Tf, transferrin. Reprinted with permission from Zarjou et al.100
The liver is primarily responsible for the storage of the body’s iron surplus, but macrophages participate as well. The liver has the capacity to store 10 times the normal amount of iron.12 Because of its storage potential, there are several opportunities for iron uptake by the liver, including transferrin-bound iron (TBI) uptake, non–transferrin bound iron (NTBI) uptake, and the clearance of iron-containing complexes from the circulation.13 Environmental pH seems to be the driving force behind TBI uptake. The neutral pH of the extracellular fluid facilitates the binding of diferric transferrin, a transferrin group carrying two iron molecules, to the transferrin receptor 1 (TFR1).14 An endosome forms to encapsulate the diferric transferrin bound to TFR1 and the acidic environment of the endosome facilitates the dissociation of the iron atoms from transferrin.15 The newly minted apotransferrin, transferrin without iron, binds tightly to the TFR1 in the acidic environment of the endosome; however, when the endosome merges with the plasma membrane, apotransferrin readily dissociates from the TFR1 in the neutral extracellular environment. The TFR2 uses a similar mechanism, except the TFR2-mediated uptake becomes more relevant in the liver when serum iron transcends the transferrin binding capacity, similar to that of NTBI uptake.9
The exact mechanism of NTBI uptake in the liver has not been elucidated. DMT1 is the preferred method of NTBI in the liver.16 Because DMT1 has reduced activity at a neutral pH, other methods of iron absorption must exist. One potential candidate is the zinc transporter 14.17 The liver also can absorb heme iron from hemoglobin when a heme-hemoglobin complex binds to a heme-hemopexin complex.18,19 Although ferritin and lactoferrin bound to their respective receptors can be internalized by the liver and selected for lysosomal degradation after the release of the iron, ferritin is still a major storage option for iron in the liver. Iron also can be stored in hemosiderin.9,18 Iron is exported from hepatocytes in the liver using the FPN transporter.20 The rate of iron exportation is controlled by ferroxidase ceruloplasmin.19 The liver regulates the body’s iron content by producing the peptide hepcidin, which can manipulate both the distribution and the absorption of iron in other tissues. When hepcidin binds to FPN, janus kinase 2 (Jak2) is activated and subsequently phosphorylates one of two adjacent tyrosine residues.21 After FPN is phosphorylated it is internalized and undergoes ubiquitination.22
KIDNEY
There are a limited number of mechanisms for iron to be eliminated from the body and one of those routes is through urinary excretion. Iron is filtered in the glomerulus and reabsorbed in the renal tubules.13,23 Similar to the liver, the kidney can absorb iron using TBI and NTBI uptake. In fact, the glomerular filtrate contains transferrin, especially in individuals with proximal tubular dysfunction.24 The kidney can absorb transferrin-bound iron using the TFR19; however, the kidney can internalize ferritin when it binds to Scara5 to transport iron.25 Transferrin also can undergo endocytosis in the renal proximal tubules via cubilin and megalin receptors for the uptake of iron.26
In addition to TBI, there are several mechanisms that do not involve transferrin for iron absorption in the kidney. With its expression localized to the cortex, the kidney has the highest reported levels of DMT1 messenger RNA, suggesting a role in the reabsorption of iron from the lumen.27 Most iron reabsorption in the kidney occurs in the loop of Henle and the collecting duct system.28 Research has suggested that iron competes with copper and/or manganese for reabsorption, which is evidence for the involvement of zinc and zinclike iron-like (ZIP) transporters in kidney-mediated iron uptake.29 ZIP8 and ZIP14 are the two zinc transporters that are expressed in proximal tubules.30 There are several other receptors and transporters that are believed to facilitate iron uptake in the kidney including the lactoferrin receptor, neutrophil gelatinase-associated lipocalin (NGAL) receptor, proton-coupled folate transporter, and mucolipin 1 and 2.31–34 CD163 and pro-low-density lipoprotein receptor-related protein 1 may be responsible for the uptake of heme-hemopexin complexes and haptoglobin-hemopexin complexes in the kidney, respectively.35–38 Both heavy- and light-chain ferritins are expressed in the kidney to mediate iron storage.23,39 Proteins expressed in renal tubules potentially handle all iron export from the kidney. These proteins include ferroportin, feline leukemia virus subgroup c receptor, and hephaestin.23,40,41 Considering the vast machinery available to handle iron in the kidney, it is conceivable to think that altering some of these pathways may prove useful in acute kidney injury.
IRON HANDLING IN ACUTE KIDNEY INJURY
Acute kidney injury (AKI) is associated with increased mortality, length of stay, and health care costs for hospitalized patients.42,43 The severity of the disease can range from a small decrement in glomerular filtration to complete kidney failure.44 AKI is particularly common in critically ill patients with septic shock and in patients undergoing major cardiac surgery.42,43,45,46 Iron may play a major role in the damage caused during AKI. Several studies have reported an increase in tissue iron content in the kidney after injury. Bleomycin detection was used to determine the increase in kidney tissue catalytic iron after glycerol-induced AKI47 and ischemia-reperfusion injury.45 Introduction of an iron-deficient diet decreased, but did not eliminate, the increased iron levels in the kidney after ischemia-reperfusion injury.45 Iron also has been implicated in the pathogenesis of cisplatin-induced nephrotoxicity.48,49 In addition to the increased free iron in kidney tissue, there is also more iron excreted in the urine with AKI. This observation is independent of the origin of the injury because it has been observed in ischemia-reperfusion injury,45,50 transplant ischemia,51 chemotherapy-induced nephrotoxicity,48,52 and hemoglobin/myoglobin-induced kidney injury.47
Although the source of catalytic iron in AKI has not been shown clearly, several possibilities have been suggested. In 1993, Baliga et al45 reported that the excess iron that accumulated in the kidney after AKI may originate from degraded red blood cells. Some researchers have pointed to ferritin as the culprit responsible for iron release,53–55 whereas others have suggested the iron may originate from mitochondria rich in heme and nonheme iron.56,57 In a model of AKI initiated by administration of the antibiotic gentamycin, only catalase was protective despite the use of a superoxide anion scavenger, hydroxyl scavengers, and iron chelators.56 This lead Ueda et al56 in 1993 to conclude that the iron was derived from mitochondria and resulted in oxidant-induced damage. Another potential candidate is cytochrome P450. In the kidney, in vitro and in vivo cisplatin treatment causes a reduction in P450 expression and an increase in bleomycin-detectable iron.49 Administration of the P450 inhibitor, piperonyl butoxide, reversed these effects, in addition to conferring functional and histologic protection.49 Even though the precise source of the iron is not completely known, the role of reactive oxygen species (ROS) in iron-induced kidney injury has been well documented in animal models.
Multiple studies have shown that iron plays a major role in ROS-induced nephrotoxicity.45,47,48,58 Oxygen free radicals cause lipid peroxidation–induced renal injury in a rat model of ischemic AKI.59 Moreover, in 1988 Paller60 observed that free iron and heme iron caused lipid peroxidation in several rat models of AKI, including glycerol-, hemoglobin-, and ischemia-induced renal injury. Kirschner and Fantini61 and others45,62 have reported that iron and its production of ROS plays a major role in the pathology of renal ischemia-reperfusion injury. In another form of AKI induced by glycerol, Shah and Walker,63 in 1988, suggested that iron generates toxic hydroxyl radicals.
TARGETING IRON AS THERAPY IN AKI
Iron Removal
If the damage sustained during AKI is initiated by iron, then altering iron trafficking in the kidney should be potentially helpful. Because of the protective effect of iron chelators in several models of kidney injury, it is believed that iron causes the production of the hydroxyl radicals responsible for renal damage.64 Several researchers have used iron chelators such as deferoxamine (DFO) to treat AKI. After administration of DFO, glycerol-induced kidney injury was attenuated significantly as determined by reduced blood urea nitrogen and creatinine levels in rats.63 DFO also caused a pronounced decrease in histologic renal damage.63 Paller60 showed that DFO was beneficial in hemoglobin- and myolgobin-induced AKI. Likewise, infusion of DFO during reperfusion improved renal function and curtailed lipid peroxidation in a rat model of postischemic renal injury, whereas addition of iron exacerbated the injury.65 These studies led to a phase 2 randomized controlled clinical trial to test the efficacy of CRMD-001 (a unique formulation of the iron chelator, deferiprone) in CKD patients with a high risk of developing AKI owing to contrast exposure during coronary angiography (clinicaltrials.gov, NCT01146925). The result of this clinical trial was inconclusive.
Pharmacologic therapy with apotransferrin inhibits oxidative stress, inflammation, and loss of function associated with renal ischemia-reperfusion injury.66 Consequently, the L-type calcium channel blocker nifedipine regulates DMT1 activity to eliminate iron overload by increasing the amount of iron excreted in the urine.67 In addition to free iron, free circulating heme is increased after AKI.68 As a result, increasing circulating levels of hemopexin may decrease kidney damage because it can remove free heme.68
As an alternative to using therapies that target the removal of iron, several free radical scavengers have proved beneficial in animal models of AKI. Cisplatin is an efficacious chemotherapy drug, but its use is limited by the significant renal toxicity it causes by direct injury to proximal tubules.69 Hydroxyl radical scavengers attenuate the histologic and functional damage in models of cisplatin-induced nephrotoxicity.48 To determine the level of protection afforded by hydroxyl radical scavengers, Shah and Walker63 used dimethylthiourea and sodium benzoate and showed significant renal protection. Rats pretreated with dimethylthiourea along with superoxide dismutase, or the xanthine oxidase inhibitor allopurinol, also were protected in a model of AKI induced by renal artery occlusion.59 Despite the success of these treatments in animal models of AKI, translation of these therapies to human beings has not been as fruitful as expected.70
Because of the minimal therapy options for individuals with AKI, a focus on therapies that could be administered within the first hours of symptom manifestation would be ideal.71,72 Implementation of such strategies has been hampered by the ability to diagnose AKI early enough before damage already has occurred. Currently, creatinine is used as a biomarker of AKI, but is a poor choice because of the time it takes for serum creatinine levels to increase after AKI.73 Several biomarkers have been developed to address this limitation and this is a subject of great interest. These include NGAL, kidney injury molecule-1, interleukin-18, liver-type fatty acid-binding protein, α-1 microglobulin, N-acetyl-β-D-glucosaminidase, and others.74,75 Recent studies have validated the use of urinary levels of insulin-like growth factor binding protein-7 and tissue inhibitor of metalloproteinase-2, both inducers of G1 cell-cycle arrest, in human AKI.76
Iron Regulators
Although hemojuvelin, hepicidin, and NGAL are excellent biomarkers of AKI, they potentially can act as therapeutic targets for the treatment of AKI.77–79 Hemojuvelin regulates hepcidin expression,80,81 and its soluble form is a potential early biomarker for AKI.79 Furin protease inhibitors block the conversion of membrane-bound hemojuvelin to soluble hemojuvelin. Membrane-bound hemojuvelin is associated with reducing iron content in the kidney, hepcidin secretion, and ferroportin degradation in AKI.79 As a result, increasing the expression levels of membrane-bound hemojuvelin may be a potential therapeutic option for AKI.
Urine hepcidin levels may be useful as a predictive biomarker of AKI, especially after cardiopulmonary bypass.82 A clinical study in patients undergoing cardiopulmonary bypass reported that postoperative urinary hepcidin levels increase when compared with preoperative levels in cardiac patients who did not develop AKI, but for patients who were diagnosed with AKI, their urinary hepcidin levels remain relatively unchanged preoperatively and postoperatively.82 Hepatic overexpression of hepcidin occurs in situations of iron overload.83 Renal ischemia-reperfusion injury caused an increase in serum and nonheme iron levels.78 Because AKI increases the levels of iron, one potential treatment or pretreatment may be to increase circulating hepcidin levels. Hepcidin reduces renal oxidative stress, apoptosis, inflammatory cell infiltration, and ischemia-reperfusion–induced renal injury.78 Likewise, the damage caused by ischemia-reperfusion injury was magnified in mice deficient of hepcidin.78 In fact, administration of exogenous hepcidin to the hepcidin-deficient mice attenuated kidney injury by inducing iron sequestration in the liver and increasing the expression of heavy-chain ferritin in the kidney.78
Heavy chain ferritin (FtH) is thought to be cytoprotective, especially in the kidney. In 2015, Bolisetty et al84 reported that FtH plays a crucial role in tubular-macrophage cross-talk during renal injury. The conditional deletion of FtH in proximal tubules (FtHPT−/−) caused a substantial increase in proinflammatory macrophages in the kidneys of mice subjected to unilateral ureteral obstruction, a model of renal inflammation and fibrosis.84 Moreover, these transgenic mice experienced a pronounced increase in the levels of inflammatory cytokines and fibrosis after kidney injury compared with a macrophage-specific knockout of FtH. In addition, FtHPT−/− mice showed a decrease in renal structure and function after rhabdomyolysis and cisplatin-induced kidney injury (Fig. 2A).39 After injury, these mice also experienced increased mortality compared with wild-type mice (Fig. 2B).39 The proximal tubule-specific deletion of FtH increased the urinary levels of several iron acceptor proteins, including hemopexin, transferrin, and, most notably, NGAL.39
Figure 2.
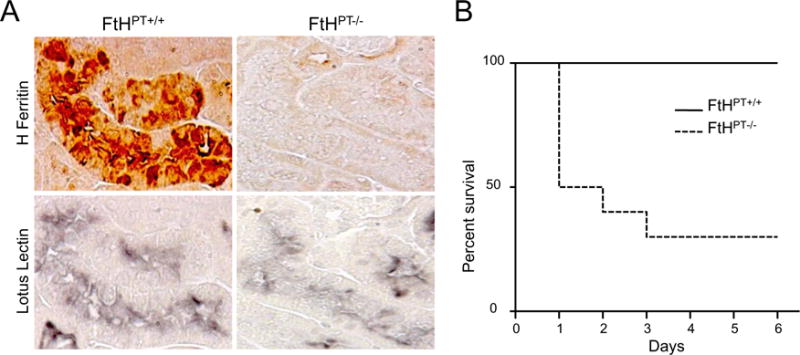
Deletion of heavy-chain ferritin in the proximal tubules exacerbates AKI. (A) Immunohistochemical staining of heavy-chain ferritin and the proximal tubule marker lotus lectin on serial kidney sections from FtHPT+/+ and FtHPT−/− mice (n = 4–8 per group). (B) Glycerol was administered to FtHPT+/+ and FtHPT−/− mice and their survival was monitored for up to 6 days (n = 10 per group). Reproduced with permission from Zarjou et al.39
NGAL is a protein that binds small iron-carrying molecules called siderophores that act as iron chelators/transporters in several diseases.71 Similar to other types of kidney injury, urine and serum levels of NGAL are a predictive early indicator of AKI after cardiopulmonary bypass surgery.85 Moreover, microarray data show that NGAL becomes overexpressed in kidney tissues soon after ischemic injury.86 Data suggest that increasing NGAL concentrations in the circulation before kidney injury may attenuate or potentially eliminate functional and structural damage.86,87 Purified NGAL administered intravenously is taken up in renal proximal tubules and conserves histologic integrity, function, and cell viability in proximal tubules after an ischemic insult.86 Interestingly, the renal protective effects of NGAL are at least in part dependent on heme oxygenase enzyme activity.88
HO-1 breaks down heme released from heme proteins to form biliverdin, iron, and carbon monoxide. The iron released from this reaction is sequestered by ferritin. Several lines of evidence support an important role for the induction of HO-1 in the pathophysiology of AKI,89,90 as follows: (1) HO-1 is strongly induced in the kidney in both animal models and human AKI90,91; (2) genetic and pharmacologic manipulation of HO-1 in animal models determines the course of AKI, deficiency or inhibition worsens renal structure and function, and increased expression is protective (reviewed by Nath90); (3) plasma and urine levels of HO-1 have been implicated as biomarkers of AKI in both animal models and human beings92; (4) polymorphisms in the human HO-1 promoter correlate with HO-1 expression and outcomes in AKI93,94; (5) many drugs/interventions that have been tested in preclinical models of AKI (eg, α-melanocyte-stimulating hormone, erythropoietin, interleukin-10, NGAL) are potent inducers of HO-1 and mediate their effects, at least in part, through HO-1 induction95–99; (6) each of the products of the HO-1-catalyzed reaction has shown renal protective effects in animal models of AKI; and (7) several clinical trials targeting the HO-1 pathway in the kidney, heart, and other organ systems have been initiated (Clinicaltrials.gov, NCT01430156, NCT00483587, NCT02142699, and NCT00531856).
In conclusion, iron is an important element in the body. Because of the labile nature of iron, its transport, metabolism, and storage is tightly controlled. Evidence suggests that there is an excess of iron after AKI. There are several therapies that have proven to be effective in animal models to attempt to reduce the iron levels and/or protect the kidney from ROS produced from iron-catalyzed reactions. Such treatments have not yet been effective in human beings. Approaches to target iron trafficking via ferritin, ferroportin, or hepcidin may offer new insights. Because of the complex nature of AKI, it is likely that a combination of different therapies discussed in this review may be required to ascertain the desired therapeutic outcome.
Acknowledgments
Supported in part by National Institutes of Health grant R01 DK059600 and University of Alabama at Birmingham-University of California at San Diego O’Brien Center grant P30 DK079337 (A.A.), and a MERIT (Mentored Experiences in Research, Instruction, and Teaching) postdoctoral fellowship award K12GM088010 (V.J.W.).
Footnotes
Conflict of interest statement: none.
References
- 1.Rouault TA. Biogenesis of iron-sulfur clusters in mammalian cells: new insights and relevance to human disease. Dis Model Mech. 2012;5:155–64. doi: 10.1242/dmm.009019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kell DB. Iron behaving badly: inappropriate iron chelation as a major contributor to the aetiology of vascular and other progressive inflammatory and degenerative diseases. BMC Med Genomics. 2009;2:2. doi: 10.1186/1755-8794-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gunshin H, Starr CN, Direnzo C, Fleming MD, Jin J, Greer EL, et al. Cybrd1 (duodenal cytochrome b) is not necessary for dietary iron absorption in mice. Blood. 2005;106:2879–83. doi: 10.1182/blood-2005-02-0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mackenzie B, Garrick MD. Iron imports. II. Iron uptake at the apical membrane in the intestine. Am J Physiol Gastrointest Liver Physiol. 2005;289:G981–6. doi: 10.1152/ajpgi.00363.2005. [DOI] [PubMed] [Google Scholar]
- 5.Ma Y, Yeh M, Yeh KY, Glass J. Iron imports. V. Transport of iron through the intestinal epithelium. Am J Physiol Gastrointest Liver Physiol. 2006;290:G417–22. doi: 10.1152/ajpgi.00489.2005. [DOI] [PubMed] [Google Scholar]
- 6.McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K, Barrow D, et al. A novel duodenal iron-regulated transporter, ireg1, implicated in the basolateral transfer of iron to the circulation. Mol Cell. 2000;5:299–309. doi: 10.1016/s1097-2765(00)80425-6. [DOI] [PubMed] [Google Scholar]
- 7.Thomas C, Oates PS. Ferroportin/ireg-1/mtp-1/slc40a1 modulates the uptake of iron at the apical membrane of enterocytes. Gut. 2004;53:44–9. doi: 10.1136/gut.53.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen H, Attieh ZK, Su T, Syed BA, Gao H, Alaeddine RM, et al. Hephaestin is a ferroxidase that maintains partial activity in sex-linked anemia mice. Blood. 2004;103:3933–9. doi: 10.1182/blood-2003-09-3139. [DOI] [PubMed] [Google Scholar]
- 9.Muckenthaler MU, Galy B, Hentze MW. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (ire/irp) regulatory network. Annu Rev Nutr. 2008;28:197–213. doi: 10.1146/annurev.nutr.28.061807.155521. [DOI] [PubMed] [Google Scholar]
- 10.Le Blanc S, Garrick MD, Arredondo M. Heme carrier protein 1 transports heme and is involved in heme-fe metabolism. Am J Physiol Cell Physiol. 2012;302:C1780–5. doi: 10.1152/ajpcell.00080.2012. [DOI] [PubMed] [Google Scholar]
- 11.Qiu A, Jansen M, Sakaris A, Min SH, Chattopadhyay S, Tsai E, et al. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell. 2006;127:917–28. doi: 10.1016/j.cell.2006.09.041. [DOI] [PubMed] [Google Scholar]
- 12.Ganz T, Nemeth E. Regulation of iron acquisition and iron distribution in mammals. Biochim Biophys Acta. 2006;1763:690–9. doi: 10.1016/j.bbamcr.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 13.Garrick MD, Garrick LM. Cellular iron transport. Biochim Biophys Acta. 2009;1790:309–25. doi: 10.1016/j.bbagen.2009.03.018. [DOI] [PubMed] [Google Scholar]
- 14.Dautry-Varsat A, Ciechanover A, Lodish HF. pH and the recycling of transferrin during receptor-mediated endocytosis. Proc Natl Acad Sci USA. 1983;80:2258–62. doi: 10.1073/pnas.80.8.2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bali PK, Aisen P. Receptor-modulated iron release from transferrin: differential effects on n- and c-terminal sites. Biochemistry. 1991;30:9947–52. doi: 10.1021/bi00105a019. [DOI] [PubMed] [Google Scholar]
- 16.Shindo M, Torimoto Y, Saito H, Motomura W, Ikuta K, Sato K, et al. Functional role of dmt1 in transferrin-independent iron uptake by human hepatocyte and hepatocellular carcinoma cell, hlf. Hepatol Res. 2006;35:152–62. doi: 10.1016/j.hepres.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 17.Liuzzi JP, Aydemir F, Nam H, Knutson MD, Cousins RJ. Zip14 (slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc Natl Acad Sci USA. 2006;103:13612–7. doi: 10.1073/pnas.0606424103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Graham RM, Chua AC, Herbison CE, Olynyk JK, Trinder D. Liver iron transport. World J Gastroenterol. 2007;13:4725–36. doi: 10.3748/wjg.v13.i35.4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith A, Hunt RC. Hemopexin joins transferrin as representative members of a distinct class of receptor-mediated endocytic transport systems. Eur J Cell Biol. 1990;53:234–45. [PubMed] [Google Scholar]
- 20.Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, et al. The iron exporter ferroportin/slc40a1 is essential for iron homeostasis. Cell Metab. 2005;1:191–200. doi: 10.1016/j.cmet.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 21.De Domenico I, Lo E, Ward DM, Kaplan J. Hepcidin-induced internalization of ferroportin requires binding and cooperative interaction with jak2. Proc Natl Acad Sci USA. 2009;106:3800–5. doi: 10.1073/pnas.0900453106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–3. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 23.Smith CP, Thevenod F. Iron transport and the kidney. Biochim Biophys Acta. 2009;1790:724–30. doi: 10.1016/j.bbagen.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 24.Norden AG, Lapsley M, Lee PJ, Pusey CD, Scheinman SJ, Tam FW, et al. Glomerular protein sieving and implications for renal failure in Fanconi syndrome. Kidney Int. 2001;60:1885–92. doi: 10.1046/j.1523-1755.2001.00016.x. [DOI] [PubMed] [Google Scholar]
- 25.Li JY, Paragas N, Ned RM, Qiu A, Viltard M, Leete T, et al. Scara5 is a ferritin receptor mediating non-transferrin iron delivery. Dev Cell. 2009;16:35–46. doi: 10.1016/j.devcel.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kozyraki R, Fyfe J, Verroust PJ, Jacobsen C, Dautry-Varsat A, Gburek J, et al. Megalin-dependent cubilin-mediated endocytosis is a major pathway for the apical uptake of transferrin in polarized epithelia. Proc Natl Acad Sci U S A. 2001;98:12491–6. doi: 10.1073/pnas.211291398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tchernitchko D, Bourgeois M, Martin ME, Beaumont C. Expression of the two mRNA isoforms of the iron transporter Nramp2/DMTI in mice and function of the iron responsive element. Biochem J. 2002;363:449–55. doi: 10.1042/0264-6021:3630449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wareing M, Ferguson CJ, Green R, Riccardi D, Smith CP. In vivo characterization of renal iron transport in the anaesthetized rat. J Physiol. 2000;524:581–6. doi: 10.1111/j.1469-7793.2000.00581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jorge-Nebert LF, Galvez-Peralta M, Landero Figueroa J, Somarathna M, Hojyo S, Fukada T, et al. Comparing gene expression during cadmium uptake and distribution: untreated versus oral Cd-treated wild-type and ZIP14 knockout mice. Toxicol Sci. 2015;143:26–35. doi: 10.1093/toxsci/kfu204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jenkitkasemwong S, Wang CY, Mackenzie B, Knutson MD. Physiologic implications of metal-ion transport by ZIP14 and ZIP8. Biometals. 2012;25:643–55. doi: 10.1007/s10534-012-9526-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khan AA, Quigley JG. Control of intracellular heme levels: heme transporters and heme oxygenases. Biochim Biophys Acta. 2011;1813:668–82. doi: 10.1016/j.bbamcr.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suzuki YA, Lonnerdal B. Baculovirns expression of mouse lactoferrin receptor and tissue distribution in the mouse. Biometals. 2004;17:301–9. doi: 10.1023/b:biom.0000027709.42733.e4. [DOI] [PubMed] [Google Scholar]
- 33.Nissenson AR. Should medical directors assume responsibility for facility-specific outcomes? Semin Dial. 2012;25:284–5. doi: 10.1111/j.1525-139X.2012.01073.x. [DOI] [PubMed] [Google Scholar]
- 34.Cheng X, Shen D, Samie M, Xu H. Mucolipins: intracellular trpml1-3 channels. FEBS Lett. 2010;584:2013–21. doi: 10.1016/j.febslet.2009.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gutierrez E, Egido J, Rubio-Navarro A, Buendia I, Blanco Colio LM, Toldos O, et al. Oxidative stress, macrophage infiltration and CD163 expression are determinants of longterm renal outcome in macrohematuria-induced acute kidney injury of IgA nephropathy. Nephron Clin Pract. 2012;121:c42–c53. doi: 10.1159/000342385. [DOI] [PubMed] [Google Scholar]
- 36.Ballarin J, Arce Y, Torra Balcells R, Diaz Encarnacion M, Manzarbeitia F, Ortiz A, et al. Acute renal failure associated to paroxysmal nocturnal haemoglobinuria leads to intratubular haemosiderin accumulation and CD163 expression. Nephrol Dial Transplant. 2011;26:3408–11. doi: 10.1093/ndt/gfr391. [DOI] [PubMed] [Google Scholar]
- 37.Tolosano E, Fagoonee S, Morello N, Vinchi F, Fiorito V. Heme scavenging and the other facets of hemopexin. Antioxid Redox Signal. 2010;12:305–20. doi: 10.1089/ars.2009.2787. [DOI] [PubMed] [Google Scholar]
- 38.Moestrup SK, Gliemann J, Pallesen G. Distribution of the alpha 2-macroglobulin receptor/low density lipoprotein receptor-related protein in human tissues. Cell Tissue Res. 1992;269:375–82. doi: 10.1007/BF00353892. [DOI] [PubMed] [Google Scholar]
- 39.Zarjou A, Bolisetty S, Joseph R, Traylor A, Apostolov EO, Arosio P, et al. Proximal tubule h-ferritin mediates iron trafficking in acute kidney injury. J Clin Invest. 2013;123:4423–34. doi: 10.1172/JCI67867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wolff NA, Liu W, Fenton RA, Lee WK, Thevenod F, Smith CP. Ferroportin 1 is expressed basolaterally in rat kidney proximal tubule cells and iron excess increases its membrane trafficking. J Cell Mol Med. 2011;15:209–19. doi: 10.1111/j.1582-4934.2009.00985.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang Z, Philips JD, Doty RT, Giraudi P, Ostrow JD, Tiribelli C, et al. Kinetics and specificity of feline leukemia virus subgroup c receptor (flvcr) export function and its dependence on hemopexin. J Biol Chem. 2010;285:28874–82. doi: 10.1074/jbc.M110.119131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol. 2005;16:3365–70. doi: 10.1681/ASN.2004090740. [DOI] [PubMed] [Google Scholar]
- 43.Uchino S, Kellum JA, Bellomo R, Doig GS, Morimatsu H, Morgera S, et al. Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA. 2005;294:813–8. doi: 10.1001/jama.294.7.813. [DOI] [PubMed] [Google Scholar]
- 44.American Society of Nephrology. American Society of Nephrology renal research report. J Am Soc Nephrol. 2005;16:1886–903. doi: 10.1681/ASN.2005030285. [DOI] [PubMed] [Google Scholar]
- 45.Baliga R, Ueda N, Shah SV. Increase in bleomycin-detectable iron in ischaemia/reperfusion injury to rat kidneys. Biochem J. 1993;291:901–5. doi: 10.1042/bj2910901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rosner MH, Okusa MD. Acute kidney injury associated with cardiac surgery. Clin J Am Soc Nephrol. 2006;1:19–32. doi: 10.2215/CJN.00240605. [DOI] [PubMed] [Google Scholar]
- 47.Baliga R, Zhang Z, Baliga M, Shah SV. Evidence for cytochrome p-450 as a source of catalytic iron in myoglobinuric acute renal failure. Kidney Int. 1996;49:362–9. doi: 10.1038/ki.1996.53. [DOI] [PubMed] [Google Scholar]
- 48.Baliga R, Zhang Z, Baliga M, Ueda N, Shah SV. In vitro and in vivo evidence suggesting a role for iron in cisplatin-induced nephrotoxicity. Kidney Int. 1998;53:394–401. doi: 10.1046/j.1523-1755.1998.00767.x. [DOI] [PubMed] [Google Scholar]
- 49.Baliga R, Zhang Z, Baliga M, Ueda N, Shah SV. Role of cytochrome p-450 as a source of catalytic iron in cisplatin-induced nephrotoxicity. Kidney Int. 1998;54:1562–9. doi: 10.1046/j.1523-1755.1998.00161.x. [DOI] [PubMed] [Google Scholar]
- 50.Paller MS, Jacob HS. Cytochrome p-450 mediates tissuedamaging hydroxyl radical formation during reoxygenation of the kidney. Proc Natl Acad Sci USA. 1994;91:7002–6. doi: 10.1073/pnas.91.15.7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baron P, Gomez-Marin O, Casas C, Heil J, Will N, Condie R, et al. Renal preservation after warm ischemia using oxygen free radical scavengers to prevent reperfusion injury. J Surg Res. 1991;51:60–5. doi: 10.1016/0022-4804(91)90070-3. [DOI] [PubMed] [Google Scholar]
- 52.Saad SY, Najjar TA, Al-Rikabi AC. The preventive role of deferoxamine against acute doxorubicin-induced cardiac, renal and hepatic toxicity in rats. Pharmacol Res. 2001;43:211–8. doi: 10.1006/phrs.2000.0769. [DOI] [PubMed] [Google Scholar]
- 53.Bando Y, Aki K. Superoxide-mediated release of iron from ferritin by some flavoenzymes. Biochem Biophys Res Commun. 1990;168:389–95. doi: 10.1016/0006-291x(90)92333-u. [DOI] [PubMed] [Google Scholar]
- 54.Biemond P, Swaak AJ, van Eijk HG, Koster JF. Superoxide dependent iron release from ferritin in inflammatory diseases. Free Radic Biol Med. 1988;4:185–98. doi: 10.1016/0891-5849(88)90026-3. [DOI] [PubMed] [Google Scholar]
- 55.Bolann BJ, Ulvik RJ. Release of iron from ferritin by xanthine oxidase. Role of the superoxide radical. Biochem J. 1987;243:55–9. doi: 10.1042/bj2430055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ueda N, Guidet B, Shah SV. Gentamicin-induced mobilization of iron from renal cortical mitochondria. Am J Physiol. 1993;265:F435–9. doi: 10.1152/ajprenal.1993.265.3.F435. [DOI] [PubMed] [Google Scholar]
- 57.Tangeras A, Flatmark T, Backstrom D, Ehrenberg A. Mitochondrial iron not bound in heme and iron-sulfur centers. Estimation, compartmentation and redox state. Biochim Biophys Acta. 1980;589:162–75. doi: 10.1016/0005-2728(80)90035-3. [DOI] [PubMed] [Google Scholar]
- 58.Shah SV. Role of reactive oxygen metabolites in experimental glomerular disease. Kidney Int. 1989;35:1093–106. doi: 10.1038/ki.1989.96. [DOI] [PubMed] [Google Scholar]
- 59.Paller MS, Hoidal JR, Ferris TF. Oxygen free radicals in ischemic acute renal failure in the rat. J Clin Invest. 1984;74:1156–64. doi: 10.1172/JCI111524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Paller MS. Hemoglobin- and myoglobin-induced acute renal failure in rats: role of iron in nephrotoxicity. Am J Physiol. 1988;255:F539–44. doi: 10.1152/ajprenal.1988.255.3.F539. [DOI] [PubMed] [Google Scholar]
- 61.Kirschner RE, Fantini GA. Role of iron and oxygen-derived free radicals in ischemia-reperfusion injury. J Am Coll Surg. 1994;179:103–17. [PubMed] [Google Scholar]
- 62.Green CJ, Gower JD, Healing G, Cotterill LA, Fuller BJ, Simpkin S. The importance of iron, calcium and free radicals in reperfusion injury: an overview of studies in ischaemic rabbit kidneys. Free Radic Res Commun. 1989;7:255–64. doi: 10.3109/10715768909087950. [DOI] [PubMed] [Google Scholar]
- 63.Shah SV, Walker PD. Evidence suggesting a role for hydroxyl radical in glycerol-induced acute renal failure. Am J Physiol. 1988;255:F438–43. doi: 10.1152/ajprenal.1988.255.3.F438. [DOI] [PubMed] [Google Scholar]
- 64.Walker PD, Shah SV. Hydrogen peroxide cytotoxicity in LLC-PK1 cells: a role for iron. Kidney Int. 1991;40:891–8. doi: 10.1038/ki.1991.290. [DOI] [PubMed] [Google Scholar]
- 65.Paller MS, Hedlund BE. Role of iron in postischemic renal injury in the rat. Kidney Int. 1988;34:474–80. doi: 10.1038/ki.1988.205. [DOI] [PubMed] [Google Scholar]
- 66.de Vries B, Walter SJ, von Bonsdorff L, Wolfs TG, van Heurn LW, Parkkinen J, et al. Reduction of circulating redox-active iron by apotransferrin protects against renal ischemia-reperfusion injury. Transplantation. 2004;77:669–75. doi: 10.1097/01.tp.0000115002.28575.e7. [DOI] [PubMed] [Google Scholar]
- 67.Ludwiczek S, Theurl I, Muckenthaler MU, Jakab M, Mair SM, Theurl M, et al. Ca2+ channel blockers reverse iron overload by a new mechanism via divalent metal transporter-1. Nat Med. 2007;13:448–54. doi: 10.1038/nm1542. [DOI] [PubMed] [Google Scholar]
- 68.Zager RA, Johnson AC, Becker K. Renal cortical hemopexin accumulation in response to acute kidney injury. Am J Physiol Renal Physiol. 2012;303:F1460–72. doi: 10.1152/ajprenal.00426.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Blachley JD, Hill JB. Renal and electrolyte disturbances associated with cisplatin. Ann Intern Med. 1981;95:628–32. doi: 10.7326/0003-4819-95-5-628. [DOI] [PubMed] [Google Scholar]
- 70.Jo SK, Rosner MH, Okusa MD. Pharmacologic treatment of acute kidney injury: why drugs haven’t worked and what is on the horizon. Clin J Am Soc Nephrol. 2007;2:356–65. doi: 10.2215/CJN.03280906. [DOI] [PubMed] [Google Scholar]
- 71.Schmidt-Ott KM, Mori K, Kalandadze A, Li JY, Paragas N, Nicholas T, et al. Neutrophil gelatinase-associated lipocalin-mediated iron traffic in kidney epithelia. Curr Opin Nephrol Hypertens. 2006;15:442–9. doi: 10.1097/01.mnh.0000232886.81142.58. [DOI] [PubMed] [Google Scholar]
- 72.Schrier RW, Wang W, Poole B, Mitra A. Acute renal failure: definitions, diagnosis, pathogenesis, and therapy. J Clin Invest. 2004;114:5–14. doi: 10.1172/JCI22353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Star RA. Treatment of acute renal failure. Kidney Int. 1998;54:1817–31. doi: 10.1046/j.1523-1755.1998.00210.x. [DOI] [PubMed] [Google Scholar]
- 74.Moriguchi J, Ezaki T, Tsukahara T, Furuki K, Fukui Y, Okamoto S, et al. Comparative evaluation of four urinary tubular dysfunction markers, with special references to the effects of aging and correction for creatinine concentration. Toxicol Lett. 2003;143:279–90. doi: 10.1016/s0378-4274(03)00181-4. [DOI] [PubMed] [Google Scholar]
- 75.Sato R, Suzuki Y, Takahashi G, Kojika M, Inoue Y, Endo S. A newly developed kit for the measurement of urinary liver-type fatty acid-binding protein as a biomarker for acute kidney injury in patients with critical care. J Infect Chemother. 2015;21:165–9. doi: 10.1016/j.jiac.2014.10.017. [DOI] [PubMed] [Google Scholar]
- 76.Kashani K, Al-Khafaji A, Ardiles T, Artigas A, Bagshaw SM, Bell M, et al. Discovery and validation of cell cycle arrest biomarkers in human acute kidney injury. Crit Care. 2013;17:R25. doi: 10.1186/cc12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Koyner JL, Vaidya VS, Bennett MR, Ma Q, Worcester E, Akhter SA, et al. Urinary biomarkers in the clinical prognosis and early detection of acute kidney injury. Clin J Am Soc Nephrol. 2010;5:2154–65. doi: 10.2215/CJN.00740110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Scindia Y, Dey P, Thirunagari A, Liping H, Rosin DL, Floris M, et al. Hepcidin mitigates renal ischemia-reperfusion injury by modulating systemic iron homeostasis. J Am Soc Nephrol. 2015;26:2800–14. doi: 10.1681/ASN.2014101037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Young GH, Huang TM, Wu CH, Lai CF, Hou CC, Peng KY, et al. Hemojuvelin modulates iron stress during acute kidney injury: improved by furin inhibitor. Antioxid Redox Signal. 2014;20:1181–94. doi: 10.1089/ars.2013.5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Niederkofler V, Salie R, Arber S. Hemojuvelin is essential for dietary iron sensing, and its mutation leads to severe iron overload. J Clin Invest. 2005;115:2180–6. doi: 10.1172/JCI25683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rodriguez Martinez A, Niemela O, Parkkila S. Hepatic and extrahepatic expression of the new iron regulatory protein hemojuvelin. Haematologica. 2004;89:1441–5. [PubMed] [Google Scholar]
- 82.Haase-Fielitz A, Mertens PR, Plass M, Kuppe H, Hetzer R, Westerman M, et al. Urine hepcidin has additive value in ruling out cardiopulmonary bypass-associated acute kidney injury: an observational cohort study. Crit Care. 2011;15:R186. doi: 10.1186/cc10339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pigeon C, Ilyin G, Courselaud B, Leroyer P, Turlin B, Brissot P, et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem. 2001;276:7811–9. doi: 10.1074/jbc.M008923200. [DOI] [PubMed] [Google Scholar]
- 84.Bolisetty S, Zarjou A, Hull TD, Traylor AM, Perianayagam A, Joseph R, et al. Macrophage and epithelial cell h-ferritin expression regulates renal inflammation. Kidney Int. 2015;88:95–108. doi: 10.1038/ki.2015.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mishra J, Dent C, Tarabishi R, Mitsnefes MM, Ma Q, Kelly C, et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet. 2005;365:1231–8. doi: 10.1016/S0140-6736(05)74811-X. [DOI] [PubMed] [Google Scholar]
- 86.Mishra J, Ma Q, Prada A, Mitsnefes M, Zahedi K, Yang J, et al. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J Am Soc Nephrol. 2003;14:2534–43. doi: 10.1097/01.asn.0000088027.54400.c6. [DOI] [PubMed] [Google Scholar]
- 87.Berger T, Togawa A, Duncan GS, Elia AJ, You-Ten A, Wakeham A, et al. Lipocalin 2-deficient mice exhibit increased sensitivity to Escherichia coli infection but not to ischemia-reperfusion injury. Proc Natl Acad Sci USA. 2006;103:1834–9. doi: 10.1073/pnas.0510847103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ferenbach DA, Kluth DC, Hughes J. Hemeoxygenase-1 and renal ischaemia-reperfusion injury. Nephron Exp Nephrol. 2010;115:e33–7. doi: 10.1159/000313828. [DOI] [PubMed] [Google Scholar]
- 89.Hull TD, Kamal AI, Boddu R, Bolisetty S, Guo L, Tisher CC, et al. Heme oxygenase-1 regulates myeloid cell trafficking in AKI. J Am Soc Nephrol. 2015;26:2139–51. doi: 10.1681/ASN.2014080770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nath KA. Heme oxygenase-1 and acute kidney injury. Curr Opin Nephrol Hypertens. 2014;23:17–24. doi: 10.1097/01.mnh.0000437613.88158.d3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Morimoto K, Ohta K, Yachie A, Yang Y, Shimizu M, Goto C, et al. Cytoprotective role of heme oxygenase (ho)-1 in human kidney with various renal diseases. Kidney Int. 2001;60:1858–66. doi: 10.1046/j.1523-1755.2001.01000.x. [DOI] [PubMed] [Google Scholar]
- 92.Zager RA, Johnson AC, Becker K. Plasma and urinary heme oxygenase-1 in AKI. J Am Soc Nephrol. 2012;23:1048–57. doi: 10.1681/ASN.2011121147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Askenazi DJ, Halloran B, Patil N, Keeling S, Saeidi B, Koralkar R, et al. Genetic polymorphisms of heme-oxygenase 1 (ho-1) may impact on acute kidney injury, bronchopulmonary dysplasia and mortality in premature infants. Pediatr Res. 2015;77:793–8. doi: 10.1038/pr.2015.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Baan C, Peeters A, Lemos F, Uitterlinden A, Doxiadis I, Claas F, et al. Fundamental role for HO-1 in the self-protection of renal allografts. Am J Transplant. 2004;4:811–8. doi: 10.1111/j.1600-6143.2004.00420.x. [DOI] [PubMed] [Google Scholar]
- 95.Katavetin P, Inagi R, Miyata T, Shao J, Sassa R, Adler S, et al. Erythropoietin induces heme oxygenase-1 expression and attenuates oxidative stress. Biochem Biophys Res Commun. 2007;359:928–34. doi: 10.1016/j.bbrc.2007.05.207. [DOI] [PubMed] [Google Scholar]
- 96.Lam CW, Getting SJ, Perretti M. In vitro and in vivo induction of heme oxygenase 1 in mouse macrophages following melanocortin receptor activation. J Immunol. 2005;174:2297–304. doi: 10.4049/jimmunol.174.4.2297. [DOI] [PubMed] [Google Scholar]
- 97.Lee TS, Chau LY. Heme oxygenase-1 mediates the antiinflammatory effect of interleukin-10 in mice. Nat Med. 2002;8:240–6. doi: 10.1038/nm0302-240. [DOI] [PubMed] [Google Scholar]
- 98.Mori K, Lee HT, Rapoport D, Drexler IR, Foster K, Yang J, et al. Endocytic delivery of lipocalin-siderophore-iron complex rescues the kidney from ischemia-reperfusion injury. J Clin Invest. 2005;115:610–21. doi: 10.1172/JCI23056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zou L, Sato N, Attuwaybi BO, Kone BC. Delayed administration of alpha-melanocyte-stimulating hormone or combined therapy with bay 11-7085 protects against gut ischemia-reperfusion injury. Shock. 2003;20:469–75. doi: 10.1097/01.shk.0000091205.08003.fd. [DOI] [PubMed] [Google Scholar]
- 100.Zarjou A, Balla J, Balla G, Agarwal A. Iron metabolism and oxidative stress. In: Miyata T, Eckardt K, Nanhaku M, editors. Studies on renal disorders. New York: Humana Press; 2011. [Google Scholar]