

Abstract
The dogma that the heart is a static organ which contains an irreplaceable population of cardiomyocytes prevailed in the cardiovascular field for the last several decades. However, the recent identification of progenitor cells that give rise to differentiated myocytes has prompted a re‐interpretation of cardiac biology. The heart cannot be viewed any longer as a postmitotic organ characterized by a predetermined number of myocytes that is defined at birth and is preserved throughout life. The myocardium constitutes a dynamic entity in which new young parenchymal cells are formed to substitute old damaged dying myocytes. The regenerative ability of the heart was initially documented with a classic morphometric approach and more recently with the demonstration that DNA synthesis, mitosis, and cytokinesis take place in the newly formed myocytes of the normal and pathologic heart. Importantly, replicating myocytes correspond to the differentiated progeny of cardiac stem cells. These findings point to the possibility of novel therapeutic strategies for the diseased heart.
Keywords: heart failure, myocyte proliferation, cardiac progenitor cells
Introduction
Historically, the adult human heart was considered an organ capable of increasing its muscle mass by hyperplasia and hypertrophy of existing myocytes. 1 This view was based on the assumption that cardiomyocytes retain the ability to reenter the cell cycle and divide throughout the lifespan of the organ and organism. However, no evidence in favor of this possibility was provided. In the early 1920s, more rigorous studies challenged this conviction and claimed that myocyte hypertrophy was the only growth reserve mechanism available to accommodate increases in pressure and/or volume loads on the adult heart. 2 This conclusion was dictated by the lack of observations of mitotic figures in myocytes, which prompted the claim that myocyte regeneration does not occur in the diseased human heart. The concept that cardiomyocytes are permanently withdrawn from the cell cycle gained support in the late 1960s from autoradiographic results of tritiated thymidine incorporation obtained experimentally in the myocardium during post‐natal development and pathological overloads. 3 , 4 , 5 , 6 DNA synthesis in myocyte nuclei was shown to be negligible, strengthening the argument that myocytes can increase in volume but not in number. 7
These important but qualitative data were contrasted by quantitative measurements collected by Linzbach and collaborators in the 1940s and early 1950s documenting that myocyte proliferation constituted the major adaptive cellular response of the hypertrophied heart when cardiac weight reached a value equal to or greater than 500 g. 8 , 9 From the 1970s to the 1990s, the debate concerning the regenerative potential of the heart was silent and the field was dominated by the search for the mechanical, biochemical, and coronary blood flow abnormalities present in the hypertrophied heart. Similarly, the explosion of molecular cardiology and cell signaling considered heart failure as the consequence of a defective myocyte hypertrophy in which alterations in the effector pathways regulating myocyte growth and contractility were responsible for the depression in organ, tissue, and cell function. 10 , 11 The dogma that the heart is a static postmitotic organ in which cardiomyocytes cannot be replaced by division of a subpopulation of nonterminally differentiated myocytes or by activation and commitment of a pool of primitive cells, has profoundly conditioned basic and clinical research in cardiology for the last three decades. 12 , 13
The recognition that exogenous and endogenous progenitor cells have the ability to promote myocardial regeneration is having an unprecedented impact on cardiovascular science and cardiology. A paradigm shift concerning the biology of the heart is in progress and this may change dramatically our understanding of cardiac homeostasis and pathology and may have extraordinary consequences on the treatment of the human disease.
Human Pathology
The possibility that myocyte hypertrophy is not the only cellular mechanism involved in the increase in myocardial mass of the human heart proposed in the 1940s and 1950s was reinforced by sporadic quantitative studies performed in the following 40 years. 14 , 15 , 16 The number of ventricular myocytes was found to nearly double in the decompensated human heart. Additionally, several reports have provided convincing evidence that DNA synthesis in myocyte nuclei occurs throughout life with the formation of ploidy, 17 suggesting that the machinery for DNA replication is not irreversibly repressed in cardiomyocytes. A similar phenomenon was also documented experimentally in the overloaded heart. 5 Two major daily observations of human cardiac pathology have not been properly appreciated and have failed to attract the attention of most experienced pathologists. At autopsy, human hearts weighing nearly two pounds or more are not uncommon and routine histological examination of myocyte dimension shows a striking lack of correspondence between myocyte size and the remarkable increase in ventricular muscle mass. 8 , 14 , 18 , 19 The majority of myocytes is slightly hypertrophied or of normal volume, which is in sharp contrast with the unusual magnitude of organ hypertrophy (Figure 1). This simple finding points to myocyte formation as the prevailing cellular growth mechanism capable of accounting for the massive increase in cardiac weight. Significantly enlarged myocytes with bizarre nuclei are also present but they constitute the exception rather than the predominant finding.
Figure 1.
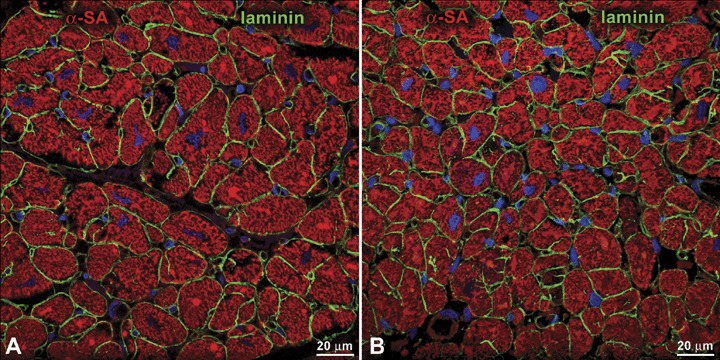
Cardiac hypertrophy and myocyte size. Section of a normal (A, heart weight = 220 g) and hypertrophied (B, heart weight = 700 g) human myocardium. The boundary of myocytes is defined by laminin (green). Note that the myocyte cross‐sectional area in both preparations is comparable, suggesting the presence of a higher number of myocytes in the heavier heart α‐SA, α‐sarcomeric actin (red).
The second important variable that has not been considered properly is the extent and characteristics of myocardial damage present in the diseased human heart, whether it is affected by chronic coronary artery disease or idiopathic dilated cardiomyopathy. 20 , 21 Scar formation, foci of replacement fibrosis scattered throughout the ventricular wall, diffuse interstitial fibrosis, and ongoing myocyte death by apoptosis and necrosis (Figure 2) are typically present in the hypertrophied failing heart. 21 , 22 Importantly, the deposition of 1 mm3 of collagen reflects the loss of 50 × 103 myocytes. 23 Even if we exclude the extensive magnitude of established myocardial pathology, the degree of constant myocyte death in the absence of cell regeneration would rapidly lead to a dramatic reduction in myocyte number which would be incompatible with life in humans.
Figure 2.

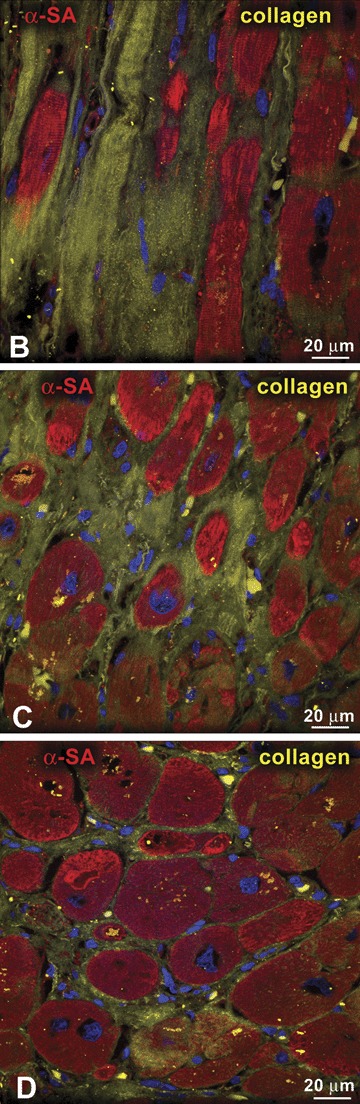
Myocardial damage. Panels A–D represent various aspects of myocardial fibrosis. Replacement fibrosis is recognized by foci of accumulation of collagen type I and type III (yellow) in hearts affected by chronic ischemia (A and B). Interstitial fibrosis is documented by the presence of collagen in the interstitial space between neighboring myocytes (C and D). Panel E illustrates a myocyte undergoing necrosis. Blunt‐end DNA strand breaks, typical of this type of cell death, are shown by in situ ligation of a Pfu oligonucleotide probe (yellow, arrow). Additionally, loss of plasmamembrane integrity (arrowheads) is visualized by a discontinuous vinculin staining (green). Panel E: modified from Ref. 26.
In a man 45 years of age, there are 5.8 × 109 myocytes in the left ventricle. 24 The rate of myocyte apoptosis and necrosis in the failing heart is 0.1% to 0.2% and 0.6% to 1.2%, respectively. 25 , 26 Since apoptosis is completed in approximately 2 hours and necrosis in approximately 48 hours, 27 the diseased heart would lose 0.1 × 109 myocytes per day. Irreversible cardiogenic shock occurs with a loss of 46% of myocytes, 28 which would develop in 26 days. The entire left ventricle would disappear in 2 months. The levels of apoptosis and necrosis utilized in this computation are conservative and much higher values have been reported by several laboratories. 29 , 30 Therefore, it is impossible to reconcile these pathological variables with the contention that the postnatal heart is composed of a fixed number of myocytes which, if they die, are permanently lost and the myocardium has to sustain its function with a reduced number of cells. Based on this belief, organism, organ, and myocyte aging should coincide, suggesting that the lifespan of the heart, approximately 80 years, is identical to the lifespan of ventricular myocytes. Occasionally, human beings live 100 years and longer and myocytes 100 years of age and older should be present. These myocytes would have contracted 3.7 billion times and still be functional in the senescent heart. 31 The discussion below tends to project a more realistic perspective of the biology of the heart and its compensatory mechanisms.
Myocyte Regeneration
The majority of organs contain dividing and nondividing cells. Newly formed replicating cells are expected to replace the dying population as a result of the programmed turnover of cells that accompanies the progression of life and regulates tissue homeostasis. Cell regeneration would be anticipated to be enhanced in the presence of injury in an attempt to attenuate organ damage and restore its physiological function. These cellular growth processes have not been considered feasible in the myocardium, and the theory has been formulated that ventricular myocytes cannot be replaced once cell division ceases immediately after birth in the mammalian heart. 32 , 33 , 34 , 35
Surprisingly, the promoters of myocyte proliferations also felt that the regeneration potential of the adult human heart could not be mediated by undifferentiated myocytes traversing the cell cycle and undergoing karyokinesis and cytokinesis. 36 The dramatic increases in myocyte number documented morphometrically in the failing heart were not supported by images of myocytes in metaphase, early and late anaphase, or in the process of cell division. The general consensus was that matured hypertrophied myocytes could divide in half through longitudinal splitting of the cell cytoplasm, resulting in the generation of two cells with similar structural characteristics. 14 , 18 , 19 How the nuclei would separate in the two forming cells, however, remained unresolved. For decades, several generations of pathologists and cardiovascular scientists have believed that mitotic myocytes are not to be found in the adult myocardium. Sadly, this view is still strongly engrained in part of the scientific community that vigorously opposes current knowledge and rejects published results. 32 , 33 , 34 , 35
In the last decade, seminal observations were made in favor of the notion that the human heart is not a postmitotic organ and myocyte regeneration may contribute to the increase in muscle mass of the adult myocardium. Mitotic images in myocytes have been identified unequivocally in the heart of patients affected by end‐stage ischemic cardiomyopathy, idiopathic dilated cardiomyopathy, and chronic aortic stenosis 36 , 37 , 38 , 39 (Figure 3). Additionally, the cell cycle protein Ki67 has been detected in a large fraction of myocytes located in proximity of the border zone of acute infarcts and in the distant myocardium. 37 And similar results have been obtained in the pressure overloaded heart. 38 Ki67 is a nuclear antigen expressed in all phases of the cell cycle except in G0. 40 , 41 Ki67 is apparent mainly in the late S phase, increases further in G2, persists during prophase and metaphase, and decreases in anaphase and telophase. 42 , 43 Ki67 is preferable to thymidine, bromodeoxyuridine, and proliferating cell nuclear antigen for labeling of dividing cells, because it is not involved in DNA repair. 44 Further evidence of myocyte division was also found. During mitosis, microtubules form the mitotic spindle, allowing each chromatid to be pulled toward the spindle pole by the kinetochore microtubules. 45 This process occurs in anaphase and lasts only a few minutes. 46 The arrangement of microtubules in the mitotic spindle of dividing myocytes was recognized by immunolabeling and confocal microscopy. 37 In addition, the accumulation of actin and its assembly in the contractile ring were identified. 37 Importantly, mitotic myocytes were detected in control human myocardium although their frequency was significantly lower than in the decompensated heart. 36 , 37 Importantly, similar data were obtained experimentally in small and large animal models, 47 , 48 , 49 , 50 , 51 , 52 , 53 , 54 strengthening the human results. In a manner comparable to humans, acute and chronic heart failure induced by myocardial infarction 48 , 49 or as a consequence of aging alone 47 , 54 was associated with a remarkable increase in the number of dividing myocytes.
Figure 3.
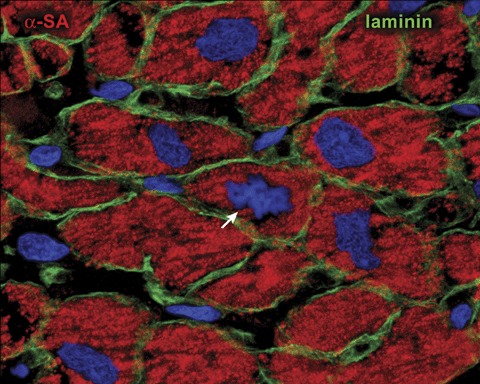
Myocyte proliferation. Myocyte generation in the diseased human heart. Metaphase chromosomes (arrow) are apparent in the dividing cardiomyocyte. Laminin (green) marks the boundaries between cells. Note the small size of the mitotic myocyte. Reprinted from Ref. 31.
Together, these findings began to challenge the perennial view of the heart as a terminally differentiated organ unable to replace parenchymal cells and raised the possibility that myocyte proliferation may have to be regarded as a component of the growth reserve of the myocardium. The presence of cell division in the nonpathological adult heart also suggested that a continuous turnover of myocytes occurs during the lifespan of the organism. However, these results left unanswered the question concerning the origin of replicating myocytes in the normal and diseased human heart.
Origin of Replicating Myocytes
The documentation that a class of myocytes can proliferate and expand significantly the muscle compartment of the overloaded myocardium imposed the inevitable question regarding the source of this pool of nonterminally differentiated cells. These replicating myocytes may reside in the heart or represent the committed progeny of circulating primitive cells that homed to the myocardium. 55 Studies of cardiac chimerism following sex‐mismatched heart transplantation have provided consistent results concerning the migration of progenitor cells from the host to the graft. 56 , 57 , 58 , 59 , 60 , 61 , 62 , 63 , 64 , 65 , 66 , 67 , 68 , 69 , 70 , 71 , 73 , 74 Following engraftment, host progenitor cells undergo replication and differentiation generating cardiomyocytes and vascular structures. 58 , 60 , 62 , 67 , 68 Although there is controversy on the magnitude of myocyte formation in the transplanted donor heart, 75 , 76 there is no disagreement on the occurrence of this phenomenon. For cardiomyocytes, however, the published values vary from 18% to 0.02%, 58 , 59 , 62 with numerous intermediate results between these two extremes.
During cardiac transplantation, portions of the atria of the recipient are sutured to the partially dissected atria of the donor. The presence of hybrid atria raises the problem whether undifferentiated cells migrate from the host to the graft through the systemic circulation or homed to the ventricle from the native atrial myocardium. 55 These two possibilities are not mutually exclusive but question the actual origin of the repopulating progenitor cell pool. Although this critical issue remains uncertain, the high degree of cardiac chimerism reported by us and others 58 , 60 , 62 , 67 , 68 is consistent with the needs of the donor heart to reverse the increased hemodynamic load and the clinical manifestations of heart failure in the recipient. 77 , 78 These mechanical factors in combination with the synthesis and secretion of multiple growth factors may trigger the translocation of progenitor cells nested in the native atria and concurrently activate resident cells in the donor heart. Locally distributed primitive cells together with those migrated from the bone marrow via the systemic circulation to the host myocardium may contribute to optimizing the cardiac mass and restoring ventricular function in terminally ill patients.
Cardiac Stem Cells
Nondividing cells can rest in G0 and reenter the cell cycle following growth activation or become terminally differentiated and die without undergoing further division. 79 Because the majority of cardiomyocytes in the adult human heart are permanently withdrawn from the cell cycle and cannot divide, replicating myocytes may originate from transdifferentiation of bone marrow progenitor cells homed to the heart, 62 , 70 from the residual growth of a subgroup of nonterminally differentiated myocytes, 1 , 27 or activation of cardiac progenitor cells resident within the myocardium. 38 , 39
In the first case, the inevitable implication would be the recognition that hematopoietic stem cells retain a remarkable degree of developmental plasticity and differentiate into cell lineages distinct from the organ in which they reside. This possibility views the bone marrow as the critical determinant of tissue homeostasis and repair of “postmitotic” organs such as the heart and the brain. 80 , 81 , 82 Experimentally, under physiological and pathological conditions, the spontaneous contribution of bone marrow cells to the turnover and replacement of cardiomyocytes has been found to be modest. 83 , 84 Conversely, the local injection of bone marrow cells to the infarcted mouse heart results in significant reconstitution of the necrotic myocardium and remarkable improvement in ventricular function. 85 , 86 , 87 Similarly, the delivery of bone marrow cells to the acutely and chronically infarcted human heart has significant health benefits 88 , 89 , 90 but whether these positive effects are mediated by myocardial regeneration is open to question. 91
In the second case, myocyte regeneration should be self‐limiting and restricted to the acute phases of growth adaptation. In the presence of an increased pressure and/or volume load on the myocardium, these quiescent but potentially proliferating myocytes would be expected to respond to the mechanical stress by reactivation of the cell cycle machinery, karyokinesis, and cytokinesis. Although the presence of dividing myocytes in animals and humans has been documented repeatedly, 1 , 12 , 13 , 27 , 36 , 37 , 38 , 47 , 54 evidence in favor of the ability of adult fully differentiated myocytes to replicate is lacking. Dividing myocytes are small, commonly one‐tenth of adult cells, 1 , 27 , 38 , 53 challenging the likelihood that a pool of matured myocytes can divide and increase the myocyte compartment of the heart. If this were possible, cardiomyocytes approximately 25,000 to 30,000 mm3 in volume should be able to traverse the S phase and enter mitosis. Before cell division, the replicating myocytes would have to acquire a volume of approximately 50,000 to 60,000 mm3 in order to generate two daughter cells, each approximately 25,000 to 30,000 mm3 in volume. So far, there is not a single demonstration of dividing myocytes of this size. In this regard, the overexpression of cyclins promoting S phase entry has been claimed to reestablish the proliferative potential of terminally differentiated myocytes. 92 , 93 Unfortunately, this study has failed to provide any form of documentation that this phenomenon actually occurs. Newly formed myocytes do not appear to represent the progeny of a class of differentiated proliferating myocytes.
In the third case, the human heart would be expected to contain primitive and early committed cells resulting from lineage differentiation of the stem cell compartment. 12 , 13 Resident progenitor cells should be nested in niches, express typical stem cell antigens 94 , 95 , 96 and telomerase, 97 , 98 be negative for hematopoietic markers, 94 , 95 , 96 divide rapidly upon stimulation 98 , 99 and give rise to myocytes at various stages of maturation. 12 , 13 , 38 , 39 Recently, a class of human c‐kit‐positive cardiac cells which possess all these fundamental properties of stem cells has been identified. 96 Additionally, these cells are self‐renewing, clonogenic, and multipotent in vitro and in vivo (Figure 4). In vitro, human cardiac stem cells differentiate predominantly into cardiomyocytes and to a lesser extent into vascular smooth muscle cells and endothelial cells. Importantly, other laboratories have been able to isolate and expand progenitor cells from the human heart and show that they have a remarkable ability to grow and differentiate. 100 , 101 , 102
Figure 4.
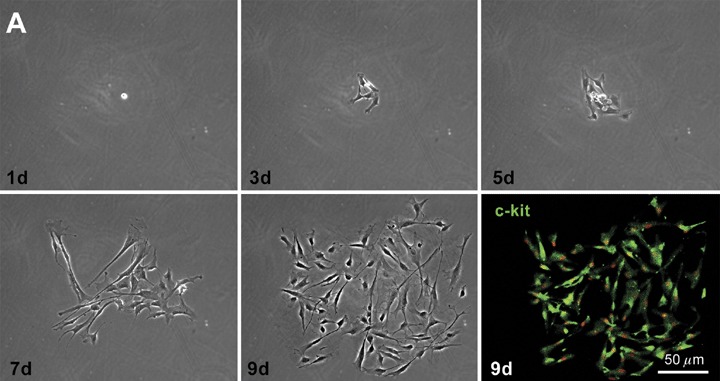
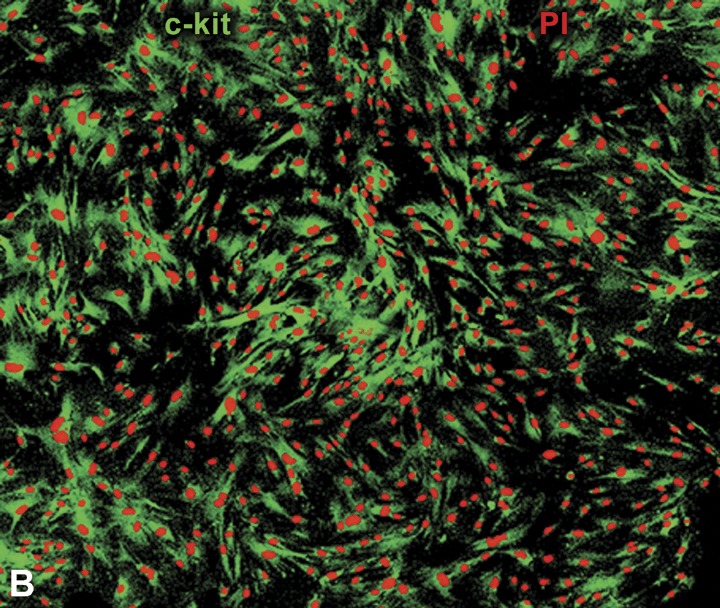
Clonogenicity of human cardiac stem cells. (A) Growth of a clone from a single c‐kit positive cell over a period of 9 days (d). The lower right panel documents that cells in the clone retain the c‐kit antigen (green). (B) Higher magnification of a larger clone of human cardiac stem cells derived from a single c‐kit positive cell. Panel A: reprinted from Ref. 96.
When locally injected in the infarcted myocardium of immunodeficient mice and immunosuppressed rats, human cardiac stem cells generate a chimeric heart, which contains human myocardium composed of myocytes, coronary resistance arterioles, and capillary profiles. Importantly, the differentiated human cardiac cells possess only one set of human sex chromosomes excluding cell fusion. Although the human myocardium shows an immature phenotype, it is functionally integrated with the rodent myocardium; it contracts regionally and contributes to the improvement in the hemodynamic performance of the infarcted heart. Thus, human cardiac stem cells can be isolated and expanded in vitro from samples of myocardium, which is a prerequisite for autologous stem cell therapy in humans.
Conclusions
Collectively, the discussion above suggests that the human heart cannot be considered any longer a terminally differentiated postmitotic organ incapable of significant myocardial regeneration. Intense myocyte formation can occur through the commitment and differentiation of cardiac stem cells to the myocyte lineage. The recognition that the heart belongs to organs with self‐renewing properties imposes that the theme of the controversy on myocyte regeneration be reexamined. Years ago, we proposed that a subpopulation of small developing myocytes in the adult heart is capable of reentering the cell cycle and undergoing cytokinesis. 1 , 36 , 37 This notion was opposed by negative results obtained after the in vivo injection of proteins interfering with cell cycle inhibitors. 103 Myocyte division was not detected and the attempts of the cells to replicate resulted in apoptotic death. Either position had valid points but could not provide a logical understanding of myocyte growth or its developmental block. At present, the controversy no longer exists. Dividing myocytes are amplifying cells that can experience a finite number of divisions before reaching terminal differentiation and growth arrest. Similarly, forced recall of differentiated myocytes into the cell cycle fails to trigger DNA synthesis and activate the endogenous cell death program.
Acknowledgment
This study was supported by NIH grants.
References
- 1. Anversa P, Kajstura J. Ventricular myocytes are not terminally differentiated in the adult mammalian heart. Circ Res. 1998; 83: 1–14. [DOI] [PubMed] [Google Scholar]
- 2. Karsner HT, Saphir O, Todd TW. The state of the cardiac muscle in hypertrophy and atrophy. Am J Pathol. 1925; 1: 351–371. [PMC free article] [PubMed] [Google Scholar]
- 3. Petersen RO, Baserga R. Nucleic acid and protein synthesis in cardiac muscle of growing and adult mice. Exp Cell Res. 1965; 40: 340–352. [DOI] [PubMed] [Google Scholar]
- 4. Morkin E, Ashford TP. Myocardial DNA synthesis in experimental cardiac hypertrophy. Am J Physiol. 1968; 215: 1409–1413. [DOI] [PubMed] [Google Scholar]
- 5. Grove D, Nair KG, Zak R. Biochemical correlates of cardiac hypertrophy, III: changes in DNA content: the relative contributions of polyploidy and mitotic activity. Circ Res. 1969; 25: 463–471. [DOI] [PubMed] [Google Scholar]
- 6. Grove D, Zak R, Nair KG, Aschenbrenner V. Biochemical correlates of cardiac hypertrophy, IV: observation on the cellular organization of growth during myocardial hypertrophy in the rat. Circ Res. 1969; 25: 473–485. [DOI] [PubMed] [Google Scholar]
- 7. Zak R. Development and proliferative capacity of cardiac muscle cells. Circ Res. 1974; 35: 17–26. [PubMed] [Google Scholar]
- 8. Linzbach AJ. Mikrometrische und histologische Analyse hypertropher menschlicher Herzen. Virchow Arch Pathol Anat Physiol. 1947; 314: 534–594. [PubMed] [Google Scholar]
- 9. Linzbach AJ. Muskelfaserkonstante und das Wachstumsgesetz der menschlichen Herzkammern. Virchows Arch. 1950; 318: 575–618. [DOI] [PubMed] [Google Scholar]
- 10. Pasumarthi KB, Field LJ. Cardiomyocyte cell regulation. Circ Res. 2002; 90: 1044–1054. [DOI] [PubMed] [Google Scholar]
- 11. Field LJ. Modulation of the cardiomyocyte cell cycle in genetically altered animals. Ann NY Acad Sci. 2004; 1015: 160–170. [DOI] [PubMed] [Google Scholar]
- 12. Leri A, Kajstura J, Anversa P. Cardiac stem cells and mechanisms of myocardial regeneration. Physiol Rev. 2005; 85: 1373–1416. [DOI] [PubMed] [Google Scholar]
- 13. Anversa P, Kajstura J, Leri A, Bolli R. Life and death of cardiac stem cells. A paradigm shift in cardiac biology. Circulation. 2006; 113: 1451–1463. [DOI] [PubMed] [Google Scholar]
- 14. Astorri E, Bolognesi R, Colla B, Chizzola A, Vilioli O. Left ventricular hypertrophy: a cytometric study on 42 human hearts. J Mol Cell Cardiol. 1977; 9: 763–775. [DOI] [PubMed] [Google Scholar]
- 15. Grajek S, Lesiak M, Pyda M, Zajac M, Paradowski S, Kaczmarek E. Hypertrophy or hyperplasia in cardiac muscle. Post‐mortem human morphometric study. Eur Heart J. 1993; 14: 40–47. [DOI] [PubMed] [Google Scholar]
- 16. Olivetti G, Melissari M, Balbi T, Quaini F, Sonnenblick EH, Anversa P. Myocyte nuclear and possible cellular hyperplasia contribute to ventricular remodeling in the hypertrophic senescent heart in humans. J Am Coll Cardiol. 1994; 24: 140–149. [DOI] [PubMed] [Google Scholar]
- 17. Adler CP, Friedburg H. Myocardial DNA content, ploidy level and cell number in geriatric hearts: post‐mortem examination of human myocardium in old age. J Mol Cell Cardiol. 1986; 18: 39–53. [DOI] [PubMed] [Google Scholar]
- 18. Linzbach AJ. Heart failure from the point of view of quantitative anatomy. Am J Cardiol. 1960; 5: 370–382. [DOI] [PubMed] [Google Scholar]
- 19. Astorri E, Chizzola A, Visioli O, Anversa P, Olivetti G, Vitali‐Mazza L. Right ventricular hypertrophy: a cytometric study on 55 human hearts. J Mol Cell Cardiol. 1971; 2: 99–110. [DOI] [PubMed] [Google Scholar]
- 20. Beltrami CA, Finato N, Rocco M, Feruglio GA, Puricelli C, Cigola E, Quaini F, Sonnenblick EH, Olivetti G, Anversa P. Structural basis of end‐stage failure in ischemic cardiomyopathy in humans. Circulation. 1994; 89: 151–163. [DOI] [PubMed] [Google Scholar]
- 21. Beltrami CA, Finato N, Rocco M, Feruglio GA, Puricelli C, Cigola E, Sonnenblick EH, Olivetti G, Anversa P. The cellular basis of dilated cardiomyopathy in humans. J Mol Cell Cardiol. 1995; 27: 291–305. [DOI] [PubMed] [Google Scholar]
- 22. Anversa P, Olivetti G. Cellular basis of physiological and pathological myocardial growth In: Fozzard HA, Solaro RJ, eds. Handbook of Physiology: the Cardiovascular System. The Heart. New York : Oxford University Press; 2002: 75–144. [Google Scholar]
- 23. Anversa P, Beghi C, Kikkawa Y, Olivetti G. Myocardial infarction in rats. Infarct size, myocyte hypertrophy, and capillary growth. Circ Res. 1986; 58: 26–37. [DOI] [PubMed] [Google Scholar]
- 24. Olivetti G, Melissari M, Capasso JM, Anversa P. Cardiomyopathy of the aging human heart. Myocyte loss and reactive cellular hypertrophy. Circ Res. 1991; 68: 1560–1568. [DOI] [PubMed] [Google Scholar]
- 25. Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, Quaini E, Di Loreto C, Beltrami CA, Krajewski S, Reed JC, Anversa P. Apoptosis in the failing human heart. N Engl J Med. 1997; 336: 1131–1141. [DOI] [PubMed] [Google Scholar]
- 26. Guerra S, Leri A, Wang X, Finato N, Di Loreto C, Beltrami CA, Kajstura J, Anversa P. Myocyte death in the failing human heart is gender dependent. Circ Res. 1999; 85: 856–866. [DOI] [PubMed] [Google Scholar]
- 27. Anversa P, Leri A, Beltrami CA, Guerra S, Kajstura J. Myocyte death and growth in the failing heart. Lab Invest. 1998; 78: 767–786. [PubMed] [Google Scholar]
- 28. Page DL, Caulfield JB, Kastor JA, DeSanctis RW, Sanders CA. Myocardial changes associated with cardiogenic shock. N Engl J Med. 1971; 285: 133–137. [DOI] [PubMed] [Google Scholar]
- 29. Mallat Z, Tedgui A, Fontaliran F, Frank R, Durigon M, Fontaine G. Evidence of apoptosis in arrhythmogenic right ventricular dysplasia. N Engl J Med. 1996; 335: 1190–1196. [DOI] [PubMed] [Google Scholar]
- 30. Adams JW, Sakata Y, Davis MG, Sah VP, Wang Y, Liggett SB, Chien KR, Brown JH, Dorn GW 2nd. Enhanced Galphaq signaling: a common pathway mediates cardiac hypertrophy and apoptotic heart failure. Proc Natl Acad Sci USA. 1998; 95: 10140–10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Anversa P, Leri A, Rota M, Hosoda T, Bearzi C, Urbanek K, Kajstura J, Bolli R. Stem cells, myocardial regeneration, and methodological artifacts. Stem Cells. 2007; 25: 589–601. [DOI] [PubMed] [Google Scholar]
- 32. Soonpaa MH, Field LJ. Survey of studies examining mammalian cardiomyocyte DNA synthesis. Circ Res. 1998; 83: 15–26. [DOI] [PubMed] [Google Scholar]
- 33. Rubart M, Field LJ. Cardiac regeneration: repopulating the heart. Annu Rev Physiol. 2006; 68: 29–49. [DOI] [PubMed] [Google Scholar]
- 34. Chien KR. Stem cells: lost in translation. Nature. 2004; 428: 607–608. [DOI] [PubMed] [Google Scholar]
- 35. Laflamme MA, Murry CE. Regenerating the heart. Nature Biotech. 2005; 23: 845–856. [DOI] [PubMed] [Google Scholar]
- 36. Kajstura J, Leri A, Finato N, Di Loreto C, Beltrami CA, Anversa P. Myocyte proliferation in end‐stage cardiac failure in humans. Proc Natl Acad Sci USA. 1998; 95: 8801–8805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Beltrami AP, Urbanek K, Kajstura J, Yan SM, Finato N, Bussani R, Nadal‐Ginard B, Silvestri F, Leri A, Beltrami CA, Anversa P. Evidence that human cardiac myocytes divide after myocardial infarction. N Engl J Med. 2001; 344:1750–1757. [DOI] [PubMed] [Google Scholar]
- 38. Urbanek K, Quaini F, Tasca G, Torella D, Castaldo C, Nadal‐Ginard B, Leri A, Kajstura J, Quaini E, Anversa P. Intense myocyte formation from cardiac stem cells in human cardiac hypertrophy. Proc Natl Acad Sci USA. 2003; 100: 10440–10445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Urbanek K, Torella D, Sheikh F, De Angelis A, Nurzynska D, Silvestri F, Beltrami CA, Bussani R, Beltrami AP, Quaini F, Bolli R, Leri A, Kajstura J, Anversa P. Myocardial regeneration by activation of multipotent cardiac stem cells in ischemic heart failure. Proc Natl Acad Sci USA. 2005; 102: 8692–8697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Endl E, Gerdes J. The Ki‐67 protein: fascinating forms and an unknown function. Exp Cell Res. 2000; 257: 231–237. [DOI] [PubMed] [Google Scholar]
- 41. Scholzen T, Gerdes J. The Ki‐67 protein: from the known and the unknown. J Cell Physiol. 2000; 182: 311–322. [DOI] [PubMed] [Google Scholar]
- 42. Bullwinkel J, Baron‐Lühr B, Lüdemann A, Wohlenberg C, Gerdes J, Scholzen T. Ki‐67 protein is associated with ribosomal RNA transcription in quiescent and proliferating cells. J Cell Physiol. 2006; 206: 624–635. [DOI] [PubMed] [Google Scholar]
- 43. Endl E, Kausch I, Baack M, Knippers R, Gerdes J, Scholzen T. The expression of Ki‐67, MCM3, and p27 defines distinct subsets of proliferating, resting, and differentiated cells. J Pathol. 2001; 195: 457–462. [DOI] [PubMed] [Google Scholar]
- 44. Birner P, Ritzi M, Musahl C, Knippers R, Gerdes J, Voigtländer T, Budka H, Hainfellner JA. Immunohistochemical detection of cell growth fraction in formalin‐fixed and paraffin‐embedded murine tissue. Am J Pathol. 2001; 158: 1991–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kline‐Smith SL, Sandall S, Desai A. Kinetochore‐spindle microtubule interactions during mitosis. Curr Opin Cell Biol. 2005; 17: 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ganem NJ, Compton DA. Functional roles of poleward microtubule flux during mitosis. Cell Cycle. 2006; 5: 481–485. [DOI] [PubMed] [Google Scholar]
- 47. Anversa P, Fitzpatrick D, Argani S, Capasso JM. Myocyte mitotic division in the aging mammalian rat heart. Circ Res. 1991; 69: 1159–1164. [DOI] [PubMed] [Google Scholar]
- 48. Kajstura J, Zhang X, Reiss K, Szoke E, Li P, Lagrasta C, Cheng W, Darzynkiewicz Z, Olivetti G, Anversa P. Myocyte cellular hyperplasia and myocyte cellular hypertrophy contribute to chronic ventricular remodeling in coronary artery narrowing‐induced cardiomyopathy in rats. Circ Res. 1994; 74: 383–400. [DOI] [PubMed] [Google Scholar]
- 49. Reiss K, Kajstura J, Zhang X, Li P, Szoke E, Olivetti G, Anversa P. Acute myocardial infarction leads to upregulation of the IGF‐1 autocrine system, DNA replication, and nuclear mitotic division in the remaining viable cardiac myocytes. Exp Cell Res. 1994; 213: 463–472. [DOI] [PubMed] [Google Scholar]
- 50. Liu Y, Cigola E, Cheng W, Kajstura J, Olivetti G, Hintze TH, Anversa P. Myocyte nuclear mitotic division and programmed myocyte cell death characterize the cardiac myopathy induced by rapid ventricular pacing in dogs. Lab Invest. 1995; 73: 771–787. [PubMed] [Google Scholar]
- 51. Setoguchi M, Leri A, Wang S, Liu Y, De Luca A, Giordano A, Hintze TH, Kajstura J, Anversa P. Activation of cyclins and cyclin‐dependent kinases, DNA synthesis, and myocyte mitotic division in pacing‐induced heart failure in dogs. Lab Invest. 1999; 79: 1545–1558. [PubMed] [Google Scholar]
- 52. Leri A, Barlucchi L, Limana F, Deptala A, Darzynkiewicz Z, Hintze TH, Kajstura J, Nadal‐Ginard B, Anversa P. Telomerase expression and activity are coupled with myocyte proliferation and preservation of telomeric length in the failing heart. Proc Natl Acad Sci USA. 2001; 98: 8626–8631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Limana F, Urbanek K, Chimenti S, Quaini F, Leri A, Kajstura J, Nadal‐Ginard B, Izumo S, Anversa P. bcl‐2 overexpression promotes myocyte proliferation. Proc Natl Acad Sci USA. 2002; 99: 6257–6262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Torella D, Rota M, Nurzynska D, Musso E, Monsen A, Shiraishi I, Zias E, Walsh K, Rosenzweig A, Sussman MA, Urbanek K, Nadal‐Ginard B, Kajstura J, Anversa P, Leri A. Cardiac stem cell and myocyte aging, heart failure, and insulin‐like growth factor‐1 overexpression. Circ Res. 2004; 94: 514–524. [DOI] [PubMed] [Google Scholar]
- 55. Schwartz RS, Curfman GD. Can the heart repair itself? N Engl J Med. 2002; 346: 2–4. [DOI] [PubMed] [Google Scholar]
- 56. Laflamme MA, Myerson D, Saffitz JE, Murry CE. Evidence for cardiomyocyte repopulation by extracardiac progenitors in transplanted human hearts. Circ Res. 2002; 90: 634–640. [DOI] [PubMed] [Google Scholar]
- 57. Minami E, Laflamme A, Saffitz JE, Murry CE. Extracardiac progenitor cells repopulate most major cell types in the transplanted human heart. Circulation. 2005; 112: 2951–2958. [DOI] [PubMed] [Google Scholar]
- 58. Quaini F, Urbanek K, Beltrami AP, Finato N, Beltrami CA, Nadal‐Ginard B, Kajstura J, Leri A, Anversa P. Chimerism of the transplanted heart. N Engl J Med. 2002; 346: 5–15. [DOI] [PubMed] [Google Scholar]
- 59. Hruban RH, Long PP, Perlman EJ, Hutchins GM, Baumgartner WA, Baughman KL, Griffin CA. Fluorescence in situ hybridization for the Y‐chromosom e can be used to detect cells of recipient origin in allografted hearts following cardiac transplantation. Am J Pathol. 1993; 142: 975–980. [PMC free article] [PubMed] [Google Scholar]
- 60. Müller P, Pfeidder P, Koglin J, Schäfers HJ, Seeland U, Janzen I, Urbschat S, Böhm M. Cardiomyocytes of noncardiac origin in myocardial biopsies of human transplanted hearts. Circulation. 2002; 106: 31–35. [DOI] [PubMed] [Google Scholar]
- 61. Bayes‐Genis A, Salido M, Solé Ristol F, Puig M, Brossa V, Campreciós M, Corominas JM, Mariñoso ML, Baró T, Vela MC, Serrano S, Padró JM, Bayes de Luna A, Cinca J. Host cell‐derived cardiomyocytes in sex‐mismatched cardiac allografts. Cardiovasc Res. 2002;56: 404–410. [DOI] [PubMed] [Google Scholar]
- 62. Thiele J, Varus E, Wickenhauser C, Kvasnicka HM, Metz K, Schaefer UW, Beelen DW. Chimerism of cardiomyocytes and endothelial cells after allogeneic bone marrow transplantation in chronic myeloid leukemia. An autopsy study. Pathologe. 2002; 23: 405–410. [DOI] [PubMed] [Google Scholar]
- 63. Glaser R, Lu MM, Narula N, Epstein JA. Smooth muscle cells, but not myocytes, of host origin in transplanted human hearts. Circulation. 2002; 106: 17–19. [DOI] [PubMed] [Google Scholar]
- 64. Körbling M, Katz RL, Khanna A, Ruifrok AC, Rondon G, Albitar M, Champlin RE, Estrov Z. Hepatocytes and epithelial cells of donor origin in recipients of peripheral‐blood stem cells. N Engl J Med. 2002; 346: 738–746. [DOI] [PubMed] [Google Scholar]
- 65. Caplice NM, Bunch TJ, Stalboerger PG, Wang S, Simper D, Miller DV, Russell SJ, Litzow MR, Edwards WD. Smooth muscle cells in human coronary atherosclerosis can originate from cells administered at marrow transplantation. Proc Natl Acad Sci USA. 2003; 100: 4754–4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kleeberger W, Versmold A, Rothämel T, Glöckner S, Bredt M, Haverich A, Lehmann U, Kreipe H. Increased chimerism of bronchial and alveolar epithelium in human lung allografts undergoing chronic injury. Am J Pathol. 2003; 162: 1487–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Deb A, Wang S, Skelding KA, Miller D, Simper D, Caplice NM. Bone marrow‐derived cardiomyocytes are present in adult human heart. A study of gender‐mismatched bone marrow transplantation patients. Circulation. 2003; 107: 1245–1247. [DOI] [PubMed] [Google Scholar]
- 68. Thiele J, Varus E, Wickenhauser C, Kvasnicka HM, Lorenzen J, Gramley F, Metz KA, Rivero F, Beelen DW. Mixed chimerism of cardiomyocytes and vessels after allogeneic bone marrow and stem‐cell transplantation in comparison with cardiac allografts. Transplantation. 2004; 77: 1902–1905. [DOI] [PubMed] [Google Scholar]
- 69. Höcht‐Zeisberg E, Kahnert H, Guan K, Wulf G, Hemmerlein B, Schlott T, Tenderich G, Körfer R, Raute‐Kreinsen U, Hasenfuss G. Cellular repopulation of myocardial infarction in patients with sex‐mismatched heart transplantation. Eur Heart J. 2004; 25: 749–758. [DOI] [PubMed] [Google Scholar]
- 70. Mathur A, Martin JF. Stem cells and repair of the heart. Lancet. 2004; 364: 183–192. [DOI] [PubMed] [Google Scholar]
- 71. Von Harsdorf R, Poole‐Wilson PA, Dietz R. Regenerative capacity of the myocardium: implications for treatment of heart failure. Lancet. 2004; 363: 1306–1313. [DOI] [PubMed] [Google Scholar]
- 73. Cogle CR, Yachnis AT, Laywell ED, Zander DS, Wingard JR, Steindler DA. Bone marrow transdifferentiation in brain after transplantation: a retrospective study. Lancet. 2004; 363: 1432–1437. [DOI] [PubMed] [Google Scholar]
- 74. Angelini P, Markwald RR. Stem cell treatment of the heart. Tex Heart Inst J. 2005; 32: 479–488. [PMC free article] [PubMed] [Google Scholar]
- 75. Taylor DA, Hruban R, Rodriguez ER et al Cardiac chimerism as a mechanism for self‐repair. Does it happen and if so to what degree? Circulation. 2002; 106: 2–4. [DOI] [PubMed] [Google Scholar]
- 76. Anversa P, Nadal‐Ginard B. Cardiac chimerism: methods matter. Circulation. 2002;106: e129–131. [DOI] [PubMed] [Google Scholar]
- 77. Fyfe B, Loh E, Winters GL, Couper GS, Kartashov AI, Schoen FJ. Heart transplantation‐associated perioperative ischemic myocardial injury. Morphological features and clinical significance. Circulation. 1996; 93: 1133–1140. [DOI] [PubMed] [Google Scholar]
- 78. Beltrami CA, Di Loreto C, Finato N, Rocco M, Artico D, Cigola E, Gambert SR, Olivetti G, Kajstura J, Anversa P. Proliferating cell nuclear antigen (PCNA), DNA synthesis and mitosis in myocytes following cardiac transplantation in man. J Mol Cell Cardiol. 1997; 29: 2789–2802. [DOI] [PubMed] [Google Scholar]
- 79. Zhu O, Skoultchi AI. Coordinating cell proliferation and differentiation. Curr Opin Genet Dev. 2001; 1: 91–97. [DOI] [PubMed] [Google Scholar]
- 80. Brazelton TR, Rossi FM, Keshet GI, Blau HM. From marrow to brain: expression of neuronal phenotypes in adult mice. Science. 2000; 290: 1775–1779. [DOI] [PubMed] [Google Scholar]
- 81. Mezey E, Chandross KJ, Harta G, Maki RA, McKercher SR. Turning blood into brain: cells bearing neuronal antigens generated in vivo from bone marrow. Science. 2000; 290: 1779–1782. [DOI] [PubMed] [Google Scholar]
- 82. Dawn B, Bolli R. Adult bone‐marrow‐derived cells: regenerative potential, plasticity, and tissue commitment. Basic Res Cardiol. 2005; 100: 494–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Jackson KA, Majka SM, Wang H, Pocius J, Hartley CJ, Majesky MW, Entman ML, Michael LH, Hirschi KK, Goodell MA. Regeneration of ischemic cardiac muscle and vascular endothelium by adult stem cells. J Clin Invest. 2001; 107: 1395–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Dawn B, Guo Y, Rezazadeh A, Huang Y, Stein AB, Hunt G, Tiwari S, Varma J, Gu Y, Prabhu SD, Kajstura J, Anversa P, Ildstad ST, Bolli R. Postinfarct cytokine therapy regenerates cardiac tissue and improves left ventricular function. Circ Res. 2006; 98: 1098–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B, Pickel J, McKay R, Nadal‐Ginard B, Bodine DM, Leri A, Anversa P. Bone marrow cells regenerate infarcted myocardium. Nature. 2001; 410: 701–705. [DOI] [PubMed] [Google Scholar]
- 86. Kajstura J, Rota M, Whang B, Cascapera S, Hosoda T, Bearzi C, Nurzynska D, Kasahara H, Zias E, Bonafé M, Nadal‐Ginard B, Torella D, Nascimbene A, Quaini F, Urbanek K, Leri A, Anversa P. Bone marrow cells differentiate in cardiac cell lineages after infarction independently of cell fusion. Circ Res. 2005; 96: 127–137. [DOI] [PubMed] [Google Scholar]
- 87. Rota M, Kajstura J, Hosoda T, Bearzi C, Vitale S, Esposito G, Iaffaldano G, Padin‐Iruegas ME, Gonzalez A, Rizzi R, Small N, Muraski J, Alvarez R, Chen X, Urbanek K, Bolli R, Houser SR, Leri A, Sussman MA, Anversa P. Bone marrow cells adopt the cardiomyogenic fate in vivo. Proc Natl Acad Sci USA. 2007; 104: 17783–17788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Schächinger V, Erbs S, Elsässer A, Haberbosch W, Hambrecht R, Hölschermann H, Yu J, Corti R, Mathey DG, Hamm CW, Süselbeck T, Werner N, Haase J, Neuzner J, Germing A, Mark B, Assmus B, Tonn T, Dimmeler S, Zeiher AM, Investigators REPAIR‐AMI. Improved clinical outcome after intracoronary administration of bone‐marrow‐derived progenitor cells in acute myocardial infarction: final 1‐year results of the REPAIR‐AMI trial. Eur Heart J. 2006; 27: 2775–2783. [DOI] [PubMed] [Google Scholar]
- 89. Schächinger V, Erbs S, Elsässer A, Haberbosch W, Hambrecht R, Hölschermann H, Yu J, Corti R, Mathey DG, Hamm CW, Süselbeck T, Assmus B, Tonn T, Dimmeler S, Zeiher AM, Investigators REPAIR‐AMI. Intracoronary bone marrow‐derived progenitor cells in acute myocardial infarction. N Engl J Med. 2006; 355: 1210–1221. [DOI] [PubMed] [Google Scholar]
- 90. Assmus B, Honold J, Schächinger V, Britten MB, Fischer‐Rasokat U, Lehmann R, Teupe C, Pistorius K, Martin H, Abolmaali ND, Tonn T, Dimmeler S, Zeiher AM. Transcoronary transplantation of progenitor cells after myocardial infarction. N Engl J Med. 2006; 355: 1222–1232. [DOI] [PubMed] [Google Scholar]
- 91. Dimmeler S, Zeiher AM, Schneider MD. Unchain my heart: the scientific foundations of cardiac repair. J Clin Invest. 2005; 115: 572–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Pasumarthi KB, Nakajima H, Nakajima HO, Soonpaa MH, Field LJ. Targeted expression of cyclin D2 results in cardiomyocyte DNA synthesis and infarct regression in transgenic mice. Circ Res. 2005; 96: 110–118. [DOI] [PubMed] [Google Scholar]
- 93. Soonpaa MH, Koh GY, Pajak L, Jing S, Wang H, Franklin MT, Kim KK, Field LJ. Cyclin D1 overexpression promotes cardiomyocyte DNA synthesis and multinucleation in transgenic mice. J Clin Invest. 1997; 99: 2644–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Kajstura J, Nadal‐Ginard B, Anversa P. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003; 114: 763–766. [DOI] [PubMed] [Google Scholar]
- 95. Urbanek K, Cesselli D, Rota M, Nascimbene A, De Angelis A, Hosoda T, Bearzi C, Boni A, Bolli R, Kajstura J, Anversa P, Leri A. Stem cell niches in the adult mouse heart. Proc Natl Acad Sci USA. 2006; 103: 9226–9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Bearzi C, Rota M, Hosoda T, Tillmanns J, Nascimbene A, De Angelis A, Yasuzawa‐Amano S, Trofimova I, Siggins RW, Lecapitaine N, Cascapera S, Beltrami AP, D'Alessandro DA, Zias E, Quaini F, Urbanek K, Michler RE, Bolli R, Kajstura J, Leri A, Anversa P. Human cardiac stem cells. Proc Natl Acad Sci USA. 2007; 104: 14068–14073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Greider CW. Telomerase activity, cell proliferation, and cancer. Proc Natl Acad Sci USA. 1998; 95: 90–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Leri A, Barlucchi L, Limana F, Deptala A, Darzynkiewicz Z, Hintze TH, Kajstura J, Nada‐Ginard B, Anversa P. Telomerase expression and activity are coupled with myocyte proliferation and preservation of telomeric length in the failing heart. Proc Natl Acad Sci USA. 2001; 98: 8626–8631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Yui J, Chiu CP, Lansdorp P M. Telomerase activity in candidate stem cells from fetal liver and adult bone marrow. Blood. 1998; 91: 3255–3262. [PubMed] [Google Scholar]
- 100. Messina E, De Angelis L, Frati G, Morrone S, Chimenti S, Fiordaliso F, Salio M, Battaglia M, Latronico MV, Coletta M, Vivarelli E, Frati L, Cossu G, Giacomello A. Isolation and expansion of adult cardiac stem cells from human and murine heart. Circ Res. 2004; 95: 911–921. [DOI] [PubMed] [Google Scholar]
- 101. Smith RR, Barile L, Cho HC, Leppo MK, Hare JM, Messina E, Giacomello A, Abraham MR, Marbán E. Regenerative potential of cardiosphere‐derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation. 2007; 115: 896–908. [DOI] [PubMed] [Google Scholar]
- 102. Pfister O, Mouquet F, Jain M, Summer R, Helmes M, Fine A, Colucci WS, Liao R. CD31‐ but Not CD31+ cardiac side population cells exhibit functional cardiomyogenic differentiation. Circ Res. 2005; 97: 52–61. [DOI] [PubMed] [Google Scholar]
- 103. Agah R, Kirshenbaum LA, Abdellatif M, Truong LD, Chakraborty S, Michael LH, Schneider MD. Adenoviral delivery of E2F‐1 directs cell cycle reentry and p53‐independent apoptosis in postmitotic adult myocardium in vivo. J Clin Invest. 1997; 100: 2722–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]