

Abstract
Increased hyaluronan (HA) deposition is a common feature of inflamed tissues, including inflammatory bowel disease (IBD)‐involved intestines. However, whether HA accumulation promotes or is the result of intestinal inflammation is unknown. Using the mouse dextran sulfate sodium (DSS)‐induced experimental model of colitis, we investigated changes in HA deposition in the colon over time in conjunction with evolving pathological changes of tissue architecture. Profound changes in colon HA deposition occurred within 3–7 days of oral DSS administration and, more important, they preceded the inflammatory infiltrate. Interestingly, HA deposition within blood vessels of the colon is observed as early as 3 days during the course of colitis induction, well before any significant inflammatory infiltrate. HA deposition is also observed in blood vessels of inflamed human colon of IBD patients. We determined that human intestinal endothelial cells generate HA in response to proinflammatory stimuli by demonstrating a TNF‐α‐induced increase in hyaluronan synthase‐3 mRNA expression and the accumulation of HA cable‐like structures that are adhesive for leukocytes. Additionally, IBD mucosal endothelial cells produce higher levels of cell surface HA in response to TNF‐α than non‐IBD control cells. Therefore, HA deposition is an early event in inflamed gut tissue, preceding and likely promoting leukocyte infiltration.
Keywords: inflammatory bowel disease, intestine, inflammation, endothelium, colitis
Introduction
Hyaluronan (HA) is an abundant extracellular matrix component produced as a straight, chemically unmodified, polymer of repeating disaccharides of D‐glucuronic acid and N‐acetylglucosamine. 1 HA has no protein component, and this allows it to be produced at cell surfaces by one or more hyaluronan‐producing enzymes, the synthases 2 (HAS 1, HAS 2, and HAS 3), which are present in cell membranes. HA breakdown (i.e., depolymerization) is mediated principally by two hyaluronidases 3 (HYAL 1 and HYAL 2). A careful balance between synthesis and breakdown within tissues dictates the HA concentration and polymer size that are deposited in the extracellular matrix. Despite (or possibly because of) its very simple composition, HA communicates with many cell types. The data indicate that large polymers of HA (106–107 Da) favor homeostasis by inhibiting cell proliferation, migration, angiogenesis, and inflammation, while depolymerized HA fragments drive endothelial cell proliferation, migration, and angiogenic factor release, and promote macrophage chemotaxis and inflammatory cytokine production. 4 Most cells, including leukocytes, have receptors for HA, of note CD44, 5 the major receptor for HA, which was first identified as a leukocyte homing receptor. CD44 mediates monocyte and T‐cell adhesion to HA. 6 , 7
Increased HA deposition is a consistent feature of tissue after injury and during inflammation. 8 , 9 We have observed higher levels of HA staining in the inflamed intestine of patients with inflammatory bowel disease (IBD) as compared to samples from noninflamed IBD or control tissue. 10 Mechanistically, however, since the IBD patients typically have ongoing disease long before clinical diagnosis, it has yet to be determined whether HA deposition occurs before or after leukocyte influx. Since HA supports CD44‐mediated leukocyte adhesion, 6 , 7 the timing of deposition of this newly synthesized matrix component is likely to have a critical impact on leukocyte recruitment, retention, or activation within the intestine. To better understand this process, we employed a well‐established animal model of IBD to investigate the temporal relationship between HA deposition and inflammatory leukocyte influx.
Mice supplied with dextran sulfate sodium (DSS) in their drinking water reproducibly develop colitis in a concentration‐ and time‐dependent manner. 11 DSS compromises the epithelial barrier and the pathological effects are first observed in the distal colon in this model, where the bacterial burden is highest. This is important because of the well‐established notion that bacteria play a key role in initiating and perpetuating inflammation in human and experimental IBD. 12 , 13 , 14 , 15
HA has been implicated as a key player in leukocyte recruitment during inflammation both directly and indirectly through its association with the CD44 receptor. In vivo, CD44‐HA interactions have been shown to be crucial for inflammatory lymphocyte infiltration, and inflammation can be abrogated by either CD44 antibody or hyaluronidase treatment in a variety of experimental models. 16 , 17 , 18 In vitro, inflammatory cytokines and LPS have been shown to induce leukocyte‐adhesive HA production specifically by small vessel endothelium. 7 In this article we demonstrate that HA deposition is dramatically altered in gut inflammation and, more important, precedes the influx of inflammatory cells. Additionally, we identify HA in intestinal small vessels and demonstrate the ability of human mucosal endothelium to make leukocyte‐adhesive HA in response to TNF‐α, a key cytokine in the pathogenesis of IBD. We also show that IBD mucosal endothelium produces more leukocyte‐adhesive HA than control cells.
Materials and Methods
DSS murine model of colitis
Male C57/Bl6 mice were purchased from Jackson Lab and conventionally housed. All husbandry and treatments were conducted according to Institutional Animal Care and Use Committee (IACUC)‐approved protocols. To induce colitis, mice were allowed to drink sterile tap water with or without the addition of 2.5% dextran sodium sulfate (MP Biomedicals, Solon, OH, USA) ad libitum. Animals were euthanized on days 0, 3, 7, 10, and 13. The large intestine from the cecum to the rectum was dissected out and fixed whole in Histochoice (Sigma, St. Louis, MO, USA). Sample colon sections were taken directly adjacent to the cecum (proximal colon), and from the transverse and the rectal area (distal colon) from each large intestine sample and paraffin embedded.
Fluorescence histochemistry for confocal microscopy
Sections of mouse colon were deparaffinized and incubated in a solution of Hanks’ balanced salt solution (BSS) containing 2% fetal bovine serum (FBS) for 30 minutes. The slides were then incubated with a solution containing biotinylated hyaluronan binding protein (Cape Cod and Associates, MA, USA) (5 μg/mL) diluted in Hanks’ BSS containing 2% FBS for 16 hours at 4°C. The slides were subsequently washed three times with Hanks’ BSS, and then incubated with a solution containing Alexa‐488‐tagged streptavidin (1:500) in Hanks’ BSS containing 2% FBS. This secondary incubation was done for 1 hour at 25°C. The slides were washed three times in Hanks’ BSS and cover slips affixed to the slides in Vectashield mounting medium containing DAPI (Vector Labs, Inc., Burlingame, CA, USA). The slides were then sealed with nail polish and stored at −20°C.
Confocal images were obtained using a Leica TCS‐SP laser scanning confocal microscope (Leica, Heidelberg, Germany), which is equipped with three lasers and photodetectors that permit detection of three distinct fluorochromes. 10
Isolation and culture of human intestinal microvessel endothelial cells (HIMEC)
HIMECs were isolated from surgically resected human colon tissue and grown in culture as previously described. 18 Mucosal strips measuring 2–3 cm were dissected from the surgical specimens, rinsed in sterile Hanks’ BSS, and stirred for 30–45 minutes at room temperature in a solution of 1.5% dithiothreitol in Hanks’ BSS to remove mucus. Next, three sequential 45–60 minute washes using a 10% ethylenediaminetetraacetic acid (EDTA) solution were done to remove epithelial cells. Mucosal strips were then minced into 12‐mm pieces and digested in a 2 mg/mL type II collagenase solution (Worthington Biochemical Corp., Freehold, NJ, USA) at 37°C for 15 minutes. After the enzymatic digestion, the fragments were transferred into a sterile Petri dish containing endothelial MCDB 131 growth medium (Sigma Chemical Co., St. Louis, MO, USA), 20% heat‐inactivated fetal bovine serum (FBS), 2.5% penicillin‐streptomycin‐Fungizone solution, and 90 mg/mL heparin. Mechanical compression with the blunt edge of a No. 21 scalpel blade was applied to the center of each tissue fragment, squeezing in an outward direction to express clusters of microvascular endothelial cells. The culture medium containing expressed cells was filtered through a sterile 60‐μm‐pore nylon cell strainer (BD‐Falcon, Bedford, MA, USA) to remove tissue fragments. The filtered cells were suspended in 10–20 mL of endothelial growth medium. This cell suspension, containing clusters of endothelial cells, was then passed through a second nylon filter with a 15‐μm‐pore diameter (Nitex; Tetko Inc., Briarcliff Manor, NY, USA). Trapped cells, enriched for endothelial clusters, were flushed off the filter, collected, pelleted, and suspended in endothelial growth medium supplemented with 50 mg/mL of endothelial cell growth factor (ECGF; Boehringer Mannheim, Indianapolis, IN, USA). Endothelial cell clusters were plated onto 10 cm2tissue culture dishes precoated for 1 hour before plating with a 1 mg/mL (0.4 mg/cm2) solution of fibronectin (Boehringer Mannheim, Germany) in phosphate‐buffered saline (PBS). Plates were incubated in a 5% CO2 humidified chamber at 37°C and observed between 7 and 10 days. Clusters of endothelial cells were identified by their “cobblestone” appearance and positive von Willebrand factor staining (Dako). Endothelial cells were cultured from colon segments resected due to Crohn's disease (CD), ulcerative colitis (UC), or non‐IBD conditions (e.g., diverticular disease, benign polyps, and adenocarcinoma).
HIMEC mRNA analysis of HAS gene expression by RT‐PCR
RNA was extracted from HIMECs in lysis buffer, and captured on a silica‐based column contained in the RNeasy mini kit (Qiagen, Valencia, CA, USA). The RNA was eluted according to the manufacturer's protocol. Expression of mRNA was evaluated by reverse transcription of 1 μg of extracted RNA per reaction and the polymerase chain reaction (35 cycles, annealing temperature 55°C) using oligonucleotide primers for β‐actin, HAS 1, HAS 2, and HAS 3, as described previously. 19
Results
Intestinal HA deposition is altered and precedes inflammation during DSS colitis
C57/Bl6 mice consuming DSS consistently develop severe colitis over a 2‐week period. Symptoms typically include development of loose and bloody stools that coincides with tissue pathology. Figure 1 panel A and magnified panel C show the pathological changes that occur over time of DSS administration. The day 0 H&E image is typical of normal murine colon architecture. The mucosa provides the interface between the lumen contents and the sterile organism, and consists of compact, epithelial cell‐covered crypts that sit on a thin layer of smooth muscle cells. Beneath the smooth muscle cells is the thin region of the submucosa that contains vascularized loose connective tissue, and beyond this is the thick external muscle cell layer. Subtle changes are already evident on day 3 of DSS administration, including epithelial disruption, and mucosal smooth muscle thickening, but no leukocyte infiltration is visible. By day 7 there is a noticeable thickening of the intestinal wall (note all images are captured at the same magnification). While the epithelial barrier is highly eroded, the submucosal region is greatly expanded, and the external muscle layer appears nearly double in thickness. However, inflammatory cell infiltration is still not apparent. By day 10 the epithelial cells are essentially absent. There is now a dense inflammatory infiltrate in the mucosal and submucosal regions, and increased vascularity is apparent. The colon diameter is even greater. By day 13, the mucosal muscle cell layer is thickened and somewhat disorganized, and vascular density has further increased.
Figure 1.
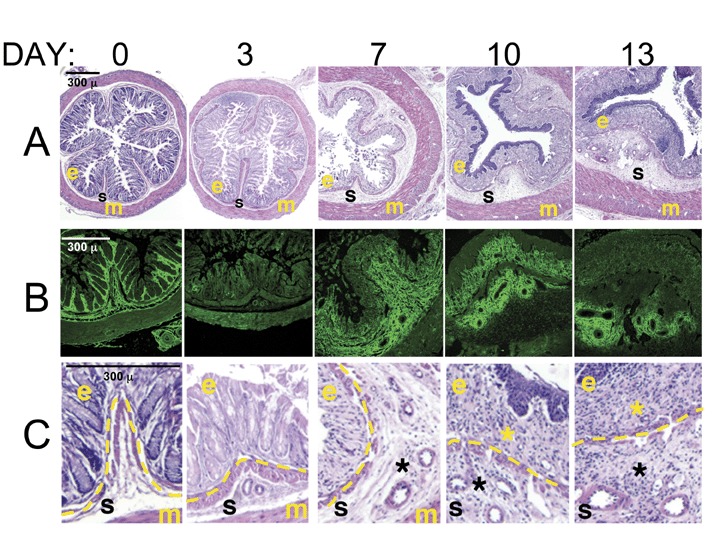
HA redistribution in distal colon precedes inflammation in the murine DSS model of colitis. Cross‐sections of distal colon samples collected from mice over time during a 2‐week course of colitis induction with 3% DSS were stained. (markers: e, epithelium; s, sub‐epithelial space; m, external muscle; *, leukocyte infiltrate; dashed line, smooth muscle layer that marks the transition from epithelial to subepithelial areas.) (A) Hematoxylin‐eosin staining highlights the cellular and structural changes in the intestine, including the course of inflammatory leukocyte infiltration. (B) HA staining (green) of serial sections demonstrates the changes in matrix deposition during colitis. (C) Enlarged areas from hematoxylin‐eosin stained sections to show cellular detail in the areas corresponding to the HA‐stained panels.
Figure 1B provides a view of serial sections stained for HA (green). At day 0, HA is present in a highly organized fashion surrounding the epithelial lined crypts and in the thin submucosal space below the mucosal muscle cells. Strikingly, however, by day 3, most of the structured HA has disappeared. By day 7, new HA deposition is observed in the submucosa, consistent with the expansion of that layer in the H&E image. HA staining is noticeably dense around the blood vessels of that region. Interestingly, on day 10, HA is present in the mucosal layer where the inflammatory infiltrate is highest. At day 13, HA staining is somewhat diminished, with the exception of the perivascular deposition. This model clearly demonstrates that HA redistribution in the colon during colitis precedes inflammation and vascular expansion.
HA deposition occurs in the small blood vessels of inflamed intestine
We have previously reported increased HA in human IBD colon tissue, 10 most abundantly around the smooth muscle cell layers and the connective tissues between intestinal crypts, a finding reproduced in the DSS model. More subtle HA deposits appear within the blood vessels of inflamed colon tissue. Using confocal microscopy and three‐dimensional (3D) reconstruction image software, we specifically investigated whether the small vessels of the intestine displayed specific HA staining. We observed HA within some of the vessels of inflamed tissue as early as day 3 in the DSS mouse colitis model (Figure 2A). That HA was truly within the vessel was confirmed in the 3‐D reconstruction analysis. In inflamed sections from IBD patient colons we also frequently observed delicate HA cable‐like structures, in small vessels (Figure 2B and inset 2C) in proximity to leukocytes. These structures are similar to those we previously reported as made by smooth muscle cells in vitro, 10 and are now shown as made by endothelium (Figure 3). We do not detect HA in the blood vessels of noninvolved intestine from mice and human sources using these methods (not shown).
Figure 2.
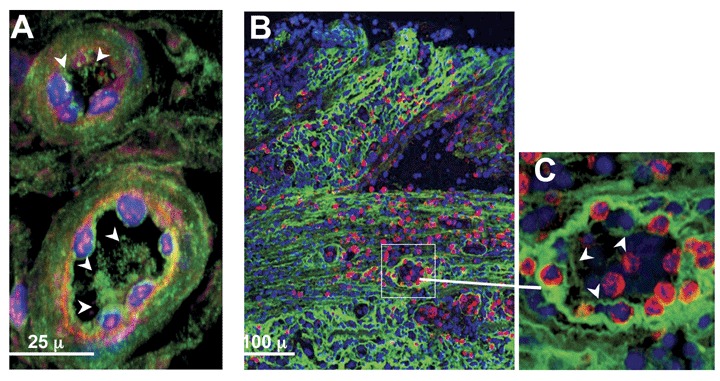
In situ detection of HA structures within the blood vessels of an inflamed colon of a mouse and a human IBD patient. (A) Mucosal blood vessels in a section of distal colon from a mouse treated 3 days with DSS have HA (green, with white arrowheads) within the vessel lumen. (Image derived from three‐dimensionally reconstructed confocal data.) (B) Colon sections from a CD patient show abundant HA staining (green) throughout the mucosa (top of image) and submucosa. (C) Enlargement of a blood vessel (inset) reveals relatively delicate HA structures (arrowheads) within the intestinal blood vessels, in proximity to leukocytes (red). Nuclei are blue.
Figure 3.
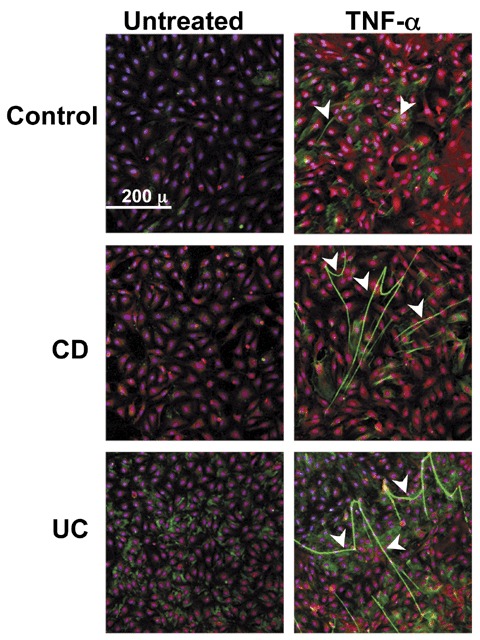
HA deposition and cable formation is increased in vitro on TNF‐α‐stimulated intestinal EC. Immunohistochemistry of three isolates of HIMEC that were treated without or with TNF‐α (10 ng/mL) for 18 hours and specifically stained for HA (green), von Willebrand Factor (red), and nuclei (blue). Arrowheads call attention to the cable‐like HA structures on the HIMEC surfaces that can span multiple cell lengths. The figure shown is representative of three or more different patient HIMEC isolates per patient category.
HIMECs generate HA in response to TNF‐αin vitro
Based on the observation that HA is generated in the lumen of small intestinal blood vessels, we investigated whether isolated mucosal endothelial cells produced HA under either unstimulated or stimulated conditions. Figure 3 shows representative confocal micrographs of three different HIMEC isolates of equivalent passage (non‐IBD control, CD, and UC) that were either untreated or treated with TNF‐α for 18 hours in the same experiment. Identity of the cells as endothelial is confirmed by Factor VIII staining (red) and delineates the cultured monolayer. HA is identified by green staining with a specific HA‐binding probe, and is more abundant in the TNF‐α‐treated cultures than on the unstimulated cells. The HA is arrayed in large cable‐like structures that span several cell lengths, similar to the structured HA we have previously shown to be important in the binding of nonstimulated leukocytes. 8 , Figure 3 also demonstrates that IBD HIMEC (i.e., both CD and UC) produce more HA as cable‐like structures in response to TNF‐α than the control HIMEC.
HA is not a protein and therefore production is not directly attributable to changes in gene expression. However, one of the factors that controls HA production is the expression of the enzymes that synthesize HA (i.e., HAS 1, HAS 2, and HAS 3). We sought to determine which HA synthase(s) was expressed and regulated in HIMEC by measuring changes in mRNA expression in replicate cultures that were untreated or treated with TNF‐α for 1, 3, 6, and 12 hours. Using reverse transcriptase PCR we found no detectable expression of HAS 1 and HAS 2, but found strong TNF‐α‐inducible expression of HAS 3 by HIMEC (Figure 4). Please note that the PCR reactions were run at different times and therefore we cannot assess whether the level of expression between HIMEC isolates differs. Poly I:C‐treated smooth muscle cell‐derived RNA served as positive control for HAS 2 expression (not shown).
Figure 4.
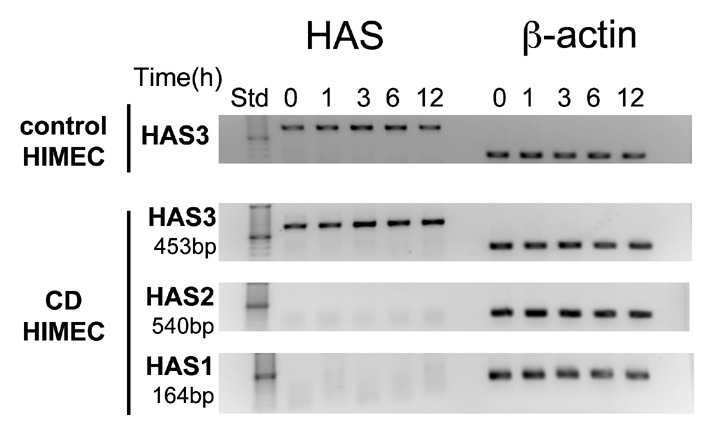
HAS3 is the predominant HA synthase expressed by HIMEC and regulated by TNF‐α. mRNA was isolated from cultures of HIMEC from two isolates (shown) that were treated with TNF‐α (10 ng/mL) for the times indicated and analyzed by RT‐PCR for the expression of HAS 3, HAS 2, HAS 1, and β‐actin as indicated.
HIMEC‐generated HA is adhesive for mononuclear leukocytes
TNF‐α regulation of endothelial cell‐mediated leukocyte binding via adhesion molecules including E‐selectin, VCAM‐1, and ICAM‐1 has been well‐documented. 20 We performed a series of experiments to determine whether TNF‐α‐induced HA also contributes to the binding of leukocytes to intestinal EC. Figure 5 A shows that HIMEC treated with TNF‐α for 18 hours binds a greater number of U937 monocytic cells than untreated HIMEC in a traditional leukocyte‐adhesion assay. 6 VCAM‐1 antibody blocked about half of the TNF‐α‐induced binding, confirming that this protein is an important leukocyte‐adhesion molecule under inflammatory conditions. Interestingly, however, almost one‐third of the bound U937 cells in these experiments were released by specific hyaluronidase treatment (Streptomyces hyaluronidase), demonstrating that HA also serves as one of the dominant HIMEC‐expressed adhesion molecules. Figure 5B provides confocal microscope images obtained from parallel cultures from this experiment and shows the increase in structured HA (green) by HIMEC after TNF‐α treatment, the increased binding of CD44 (HA receptor) positive U937 cells to the TNF‐α−treated cell layer, and the decrease in HA and leukocyte binding to TNF‐α−treated cultures after hyaluronidase treatment. Figure 5A demonstrates that the effects of the combination of VCAM‐1 blockade and hyaluronan digestion were additive and abrogated most (87%) of the induced leukocyte adhesion.
Figure 5.
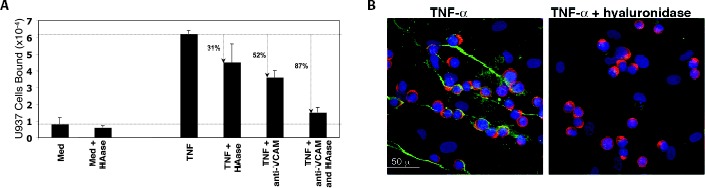
Monocytic cell adhesion to TNF‐α‐activated HIMEC is partially HA mediated. (A) Measurement of leukocyte adhesion: Confluent HIMEC were treated without or with TNF‐α (10 ng/mL) in MCDB 131 medium containing 5% FBS and incubated for 18 hours at 37°C. The adhesion of 51Cr‐labeled U937 was performed as described in Methods. To determine the number of VCAM‐1‐bound leukocytes, a monoclonal blocking antibody (10 μg/mL) was added to the wells during the 4°C adhesion step. To determine the proportion of HA‐bound leukocytes, cultures were treated without or with Streptomyces hyaluronidase (100 m Units/mL for 5 minutes at ambient temperature) after the leukocyte adhesion step.(B) Confocal images of monocytic cell adhesion to TNF‐α‐activated HIMEC and partial abrogation by HA removal. TNF‐α‐ treated HIMEC were incubated with U937 cells at 4°C. Unbound cells were washed away and the cultures were treated either with medium or Streptomyces hyaluronidase (100mUnits/mL for 5 minutes at ambient temperature). Cultures were fixed and stained for HA (HABP‐green), CD44 (A3D8 monoclonal antibody‐red), and nuclei (DAPI‐blue).
Discussion
The mouse DSS colitis model demonstrates that HA remodeling is a very early event during disease development and precedes inflammation. We can detect HA within small blood vessels at the early stages of pathological change in the distal colon and well before inflammatory cell infiltration. In vitro, human mucosal endothelial cells can make HA under inflammatory conditions, and that HA supports leukocyte adhesion. Together our results implicate HA as an important early mediator of intestinal inflammation. Additionally, the exacerbated inflammatory cytokine‐induced HA response by Crohn's disease and ulcerative colitis mucosal endothelium compared to control suggests this mechanism is important to the pathogenesis of IBD.
The natural triggers of increased HA deposition early in IBD flares, that is, before heavy leukocyte involvement, are obviously not known. Microbial challenge, permitted by a compromised epithelial border similar to the events in the DSS model, is one possibility. However, increased HA deposition also occurs during trauma‐associated edema and other forms of sterile injury. 8 Under these conditions, HA fragments apparently drive inflammation through CD44‐ and TLR4‐dependent mechanisms.
We did not expect to find an increase in TNF‐α−induced HA deposition on the surface of cultured HIMECs from IBD patients (both Crohn's and ulcerative colitis) compared to non‐IBD. HIMECs are maintained in culture many months after isolation and through multiple subcultures. The overproduction of HA by IBD HIMECs suggests a dysregulation intrinsic to the cell type, although it is unclear if this is due to genetic or epigenetic differences. HA overproduction could be the result of generalized hyperresponsiveness to TNF‐α, or either overexpression or overactivity of HA synthase enzymes (HAS 3). However, HIMEC from IBD patients have also previously been reported to bind greater numbers of leukocytes via traditional adhesion mechanisms in response to inflammatory cytokines. 18
The association of HA deposition and leukocyte recruitment is a more generalized phenomenon that extends past blood vessels of the intestinal mucosa. Smooth muscle cells also produce HA that mediates mononuclear leukocyte adhesion 6 and HA accumulates around areas rich in mucosal smooth muscle cells in IBD. 10 HA deposition in the mouse intestine during colitis development parallels the observation in human IBD, by showing heavy mucosal HA, which in the mouse precedes inflammatory cell recruitment. Interestingly, in the colitis model, we also see that after the major influx of leukocytes occurs (day 10), HA is somewhat diminished. This is consistent with the proposed role of leukocytes in hyaluronan breakdown. Teder et al. has demonstrated in a mouse lung injury model that the monocyte/macrophage mediates the breakdown of HA through a CD44‐dependent process, and that this event is crucial for resolution of inflammation. 21
We report that endothelial cells from intestinal small vessels produce HA cable‐like structures that can mediate monocytic cell adhesion in response to the prototypical IBD‐associated cytokine TNF‐α. This finding is in agreement with Mohamadzadeh et al., 7 who have shown that TNF‐α− and IL‐1‐stimulated endothelial cells derived from small blood vessels, but curiously not from large vessels, are capable of binding leukocytes via hyaluronan. Several reports from Siegelman and his colleagues have demonstrated that HA, via its interaction with CD44, is crucial for T‐cell infiltration into inflammatory sites. 16 DeGrendele showed abrogation of peritoneal leukocyte influx via CD44 blockade, 22 and Nandi intriguingly showed the cooperativity of CD44 and the VCAM‐1 ligand, VLA4, in T‐cell extravasation. 17
We suspect that HA's role as a leukocyte adhesion molecule during inflammation has been so far underappreciated for two reasons: first, most laboratories study the more easily isolated, larger vessel endothelium; and second most laboratories try to identify functional proteins while HA is purely a carbohydrate polymer. Simpson et al. 23 have shown HA‐mediated prostate cancer cell adhesion to bone marrow endothelial cells, but again not to larger vessel (umbilical vein) endothelium, and they correlate the adhesion with HAS 2 and HAS 3 overexpression. We demonstrate that only one of the three HA synthetic enzymes, HAS 3, is expressed and upregulated in HIMECs in response to TNF‐α. This result was unexpected since poly I:C‐induced generation of leukocyte‐adhesive HA structures by intestinal smooth muscle cells correlated with regulation of HAS 2, while HAS 3 was constitutively expressed (de la Motte and Strong‐unpublished). Currently it is not well understood whether the cell type or the specific stimulator dictates which HA synthases are expressed.
Endothelium in tissue sections of inflamed intestine of patients with IBD promotes greater homing of lymphocytes than noninflamed tissue. 24 HA may participate early in this leukocyte recruitment, via interaction with CD44 the homing receptor and HA‐binding molecule, thereby promoting inflammation at early stages. Later on, degradation of the intravascular as well as the mucosal deposits of HA likely results in small molecular weight HA breakdown fragments that are known to promote chemokine expression, induce chemotaxis, and promote angiogenesis 5 that could amplify inflammation. A less obvious but potentially important role for intravascular HA deposition is in promoting thrombotic events, an area gaining increasing attention in the pathophysiology of IBD. 25 HA is known to bind fibrin/fibrinogen. 26 Potentially the endothelium‐expressed HA may form foci for microthrombus generation in the small vessels of the colon during IBD. Therefore, HA, particularly that deposited in the mucosal vasculature, appears to be an early key player promoting intestinal inflammation.
Acknowledgments
The authors thank Dr. Vincent Hascall for advice and for many useful discussions. This work was supported by start‐up funds from the Department of Pathobiology at the Cleveland Clinic (CdlM) and by NIH grant DK069854 (CF).
References
- 1. Day AJ, Sheehan JK. Hyaluronan: polysaccharide chaos to protein organization. Curr Opin Struct Biol. 2001; 11: 617–622. [DOI] [PubMed] [Google Scholar]
- 2. Weigel PH, Hascall VC, Tammi M. Hyaluronan synthases. J Biol Chem. 1997; 272(22): 13997–14000. [DOI] [PubMed] [Google Scholar]
- 3. Stern R, Jedrzejas MJ. Hyaluronidases: their genomics, structures, and mechanisms of action. Chem Rev. 2006; 106: 818–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stern R, Asari AA, Sugahara KN. Hyaluronan fragments: an information‐rich system. Eur J Cell Biol. 2006; 85: 699–715. [DOI] [PubMed] [Google Scholar]
- 5. Aruffo A, Stamenkovic I, Melnick M, Underhill CB, Seed B. CD44 is the principal cell surface receptor for hyaluronate. Cell. 1990; 61: 1303–1313. [DOI] [PubMed] [Google Scholar]
- 6. De La Motte CA, Hascall VC, Calabro A, Yen‐Lieberman B, Strong SA. Mononuclear leukocytes preferentially bind via CD44 to hyaluronan on human intestinal mucosal smooth muscle cells after virus infection or treatment with poly I:C. J Biol Chem. 1999; 274: 30747–30755. [DOI] [PubMed] [Google Scholar]
- 7. Mohamadzadeh M, De Grendele H, Arizpe H, Hestess P, Siegelman M. Proinflammatory stimuli regulate endothelial hyaluronan expression and CD44/HA‐dependent primary adhesion. J Clin Invest. 1998; 101: 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Taylor KR, Yamasaki K, Radek KA, Di Nardo A, Goodarzi H, Golenbock D, Beutler B, Gallo RL. Recognition of hyaluronan released in sterile injury involves a unique receptor complex dependent on Toll‐like receptor 4, CD44, and MD‐2. J Biol Chem. 2007; 282(25): 18265–18275. [DOI] [PubMed] [Google Scholar]
- 9. Zaman A, Cui Z, Foley JP, Zhao H, Grimm PC, Delisser HM, Savani RC. Expression and role of the hyaluronan receptor RHAMM in inflammation after bleomycin injury. Am J Respir Cell Mol Biol. 2003; 33: 447–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. De La Motte CA, Hascall VC, Drazba, J , Bandyopadhyay S, Strong SA. Mononuclear leukocytes bind to specific hyaluronan structures on colon mucosal smooth muscle cells treated with polyI:C: inter‐a‐trypsin inhibitor is crucial to structure and function. Am J Path. 2003; 163: 121–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hibi T, Ogata H, Sakuraba A. Animal models of inflammatory bowel disease. J Gastroenterol. 2002; 37(6): 409–417. [DOI] [PubMed] [Google Scholar]
- 12. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, Almer S, Tysk C, O'Morain CA, Gassull M, Binder V, Finkel Y, Cortot A, Modigliani R, Laurent‐Puig P, Gower‐Rousseau R, Macry J, Colombel JF, Sahbatou M, Thomas G. Association of NOD2 leucine‐rich repeat variants with susceptibility to Crohn's disease. Nature. 2001; 411: 599–603. [DOI] [PubMed] [Google Scholar]
- 13. Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, Fukase K, Inamura S, Kusumoto S, Hashimoto M, Foster SJ, Moran AP, Fernandez‐Luna JL, Nunez G. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn's disease. J Biol Chem. 2003; 278: 5509–5512. [DOI] [PubMed] [Google Scholar]
- 14. Van Heel DA, Ghosh S, Butler M, Hunt KA, Lundberg AM, Ahmad T, McGovern DP, Onnie C, Negoro K, Goldthorpe S, Foxwell BM, Mathew CG, Forbes A, Jewell DP, Playford RJ. Muramyl dipeptide and toll‐like receptor sensitivity in NOD2‐associated Crohn's disease. Lancet. 2005; 365: 1794–1796. [DOI] [PubMed] [Google Scholar]
- 15. Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007; 448: 427–434. [DOI] [PubMed] [Google Scholar]
- 16. Siegelman MH, DeGrendele HC, Estess P. Activation and interaction of CD44 and hyaluronan in immunological systems. J Leukoc Biol. 1999; 66: 315–321. [DOI] [PubMed] [Google Scholar]
- 17. Nandi A, Estess P, Siegelman M. Bimolecular complex between rolling and firm adhesion receptors required for cell arrest; CD44 association with VLA‐4 in T cell extravasation. Immunity. 2004; 20: 455–465. [DOI] [PubMed] [Google Scholar]
- 18. Binion DG, West GA, Ina K, Ziats NP, Emancipator SN, Fiocchi C. Enhanced leukocyte binding by intestinal microvascular endothelial cells in inflammatory bowel disease. Gastroenterology. 1997; 112: 1895–1907. [DOI] [PubMed] [Google Scholar]
- 19. Selbi W, De La Motte C, Hascall V, Phillips A. BMP‐7 modulates hyaluronan‐mediated proximal tubular cell‐monocyte interaction. J Am Soc Nephrol. 2004; 15: 1199–1211. [DOI] [PubMed] [Google Scholar]
- 20. Chandrasekharan UM, Siemionow M, Unsal M, Yang L, Poptic E, Bohn J, Ozer K, Zhou Z, Howe PH, Penn M, DiCorleto PE. Tumor necrosis factor alpha (TNF‐alpha) receptor‐II is required for TNF‐alpha‐induced leukocyte‐endothelial interaction in vivo. Blood. 2007; 109: 1938–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Teder P, Vandivier RW, Jiang D, Liang J, Cohn L, Pure E, Henson PM, Noble PW. Resolution of lung inflammation by CD44. Science. 2002; 296: 155–158. [DOI] [PubMed] [Google Scholar]
- 22. DeGrendele HD, Estess P, Siegelman MH. Requirement for CD44 in activated T cell extravasation into an inflammatory site. Science. 1997; 278: 672–675. [DOI] [PubMed] [Google Scholar]
- 23. Simpson MA, Reiland J, Burger SR, Furcht LT, Spicer AP, Oegema TR, McCarthy JB. Hyaluronan synthase elevation in metastatic prostate carcinoma cells correlates with Hyaluronan surface retention, a prerequisite for rapid adhesion to bone marrow endothelial cells. J Biol Chem. 2001; 276: 17949–17957. [DOI] [PubMed] [Google Scholar]
- 24. Salmi M, Granfors K, MacDermott R, Jalkanen S. Aberrant binding of lamina propria lymphocytes to vascular endothelium in inflammatory bowel diseases. Gastroenterology. 1994; 106: 596–605. [DOI] [PubMed] [Google Scholar]
- 25. Danese S, Papa A, Saibeni S, Repici A, Malesci A, Vecchi M. Inflammation and coagulation in inflammatory bowel disease: the clot thickens. Am J Gastroenterol. 2007; 102: 174–186. [DOI] [PubMed] [Google Scholar]
- 26. LeBoeuf RD, Raja RH, Fuller GM, Weigel PH. Human fibrinogen specifically binds hyaluronic acid. J Biol Chem. 1986; 261: 12586–12592. [PubMed] [Google Scholar]