

Abstract
Purpose: Dominant‐negative growth hormone gene (GH1) mutations cause familial isolated growth hormone deficiency type II (IGHD II), which is characterized by GH deficiency, occasional multiple anterior pituitary hormone deficiencies, and anterior pituitary hypoplasia. We have previously shown that 17.5‐/22‐kDa GH1 transcript ratios correlate with the severity of the IGHD II phenotype. We hypothesized that different pharmaceutical agents could affect the GH1 transcript ratio by modulating alternative splicing.
Methods: We exposed peripheral blood mononuclear cells from IGHD II patients and unaffected family members to different pharmacologic agents and then determined the 17.5‐/22‐kDa transcript ratios by real‐time PCR.
Results: Dexamethasone and digoxin significantly increased the 17.5‐/22‐kDa transcript ratio, while sodium butyrate and 5‐iodotubericidin significantly decreased the ratio.
Conclusion: Since we have previously shown that the ratio of the 17.5‐/22‐kDa GH1 transcripts correlates with severity of the IGHD II phenotype, our findings here suggest that selected previously unconsidered agents could possibly reduce the severity of IGHD II, while other agents could possibly exacerbate the disease phenotype. Clin Trans Sci 2011; Volume 4: 175–179
Keywords: IGHD II, variable expressivity, dominant‐negative, splicing modulation
Introduction
The human growth hormone gene (GH1) is a member of the somatotropin/prolactin family of hormones and plays a key role in growth. The human GH1 transcript consists of five exons that are alternatively spliced to produce five major transcripts and proteins. The two major transcripts and their associated proteins are a 22‐kDa protein, which forms the majority of circulating GH and a 17.5‐kDa protein, which is missing exon 3 of GH1 (in GH1 literature the GH1 splice isoforms have traditionally referred by their protein identification rather than their National Center for Biotechnology Information reference numbers that is NM_000515.3 for the full length 22‐kDa protein and NM_022559.2 for the 17.5‐kDa protein). 1 , 2 Interestingly, the splice sites surrounding exon 3 of GH1 are relatively weak and three splicing enhancers are required to ensure exon 3 inclusion. 3 One of these enhancers (exon splice enhancer 1 [ESE1]) comprises the first seven bases of exon 3 and it augments use of the upstream 3% splice site and suppresses a downstream cryptic splice site. 4 Normal individuals produce small amounts of the 17.5‐kDa transcript/protein as part of the normal alternative splicing of GH1, however, the amounts are typically too low to disrupt GH1 function. However, disruption of ESE1 by mutations can cause increased exon 3 skipping, which can lead to significantly higher amounts of the smaller 17.5‐kDa isoform. This can cause autosomal dominant familial isolated growth hormone deficiency type‐II (IGHD‐II), due to a dominant‐negative effect of the 17.5‐kDa isoform that prevents secretion of normal GH in tissue culture cells and transgenic mice. 1 , 2 , 5 , 6 , 7
IGHD II is a disease characterized by GH deficiency, deficient levels of other hormones, and anterior pituitary hypoplasia; these features show variable onset and progression in family members heterozygous for the same GH1 mutation. We have recently shown that GH1 transcript levels play an important phenotype‐modifying role in IGHD II patients. In a kindred with a guanine to adenine transition in the first base of exon 3 (E3+1 G/A) that disrupted ESE1 sufficiently to cause exon 3 skipping and resultant IGHD II, 4 we found that the severity of IGHD II phenotype was directly proportional to the 17.5‐/22‐kDa transcript ratios. 8
Individuals with IGHD II can be treated with recombinant human GH. However, since these individuals continue to make the 17.5‐kDa product, which is toxic to somatotrophs, GH1 replacement does not prevent eventual pituitary failure and the resultant multiple endocrine abnormalities typically seen in severe cases of IGHD II. 9 Thus, a way to alter the splicing of GH1 mRNA in vivo would be an attractive therapeutic avenue in this dominant‐negative condition.
A recent study showed that using shRNA to knockdown the 17.5‐kDa isoform improved wild‐type GH secretion in rat pituitary tumor growth cells, but this type of approach is not yet feasible in human patients. 10 Since we recently showed the 17.5‐/22‐kDa transcript ratios were a key determinant of disease phenotype, 8 we investigated the possibility that pharmacologic agents could be used to change this ratio.
Recent studies have shown that many pharmacologic agents commonly used in clinical settings can modulate alternative splicing. 10 Chang et al. identified sodium butyrate (SB) as a splicing modulator in peripheral blood mononuclear cells (PBMNCs) from individuals with spinal muscular atrophy (SMA) where it was shown to increase alternative splicing. SMA, like IGHD II, also results from aberrant splicing and several studies have shown that pharmacologic agents affect splicing in this disorder. 11 In addition, a high‐throughput screen identified digoxin as an alternative splicing modulator. 12 The exact mechanism(s) by which these and other pharmacologic compounds modulate alternative splicing are poorly understood, but possibilities include modification of chromatin structure or effects on the expression or phosphorylation of serine/arginine rich (SR) proteins. 10
Here, we present the results of a focused drug screen conducted to identify pharmacologic agents that may alter the 17.5‐/22‐kDa transcript ratios in patient‐derived cells. These cells were isolated from affected individuals in a kindred that contained the E3+1 G/A transition that disrupted ESE1 sufficiently to cause exon 3 skipping and produce IGHD II. Our data show that four agents could significantly alter this ratio. Our findings have implications for the use of pharmacologic agents in the modification of genetic phenotypes and clinical management of dominant‐negative conditions such as IGHD II and other disorders characterized by aberrant alternative splicing.
Materials and Methods
Establishment of PBMNC lines
Study subjects were members of a previously described Caucasian family with IGHD II (III‐1 referred to as control or unaffected family member with height standard deviation (Ht SDS) score of 0.46 and III‐6 referred to as patient or affected family member with Ht SDS score of −4.13). 8 Cell lines were established and PBMNCs were immortalized with Epstein‐Barr Virus as previously described. 13 Samples were collected under a Vanderbilt University institutional review board protocol.
Cell culture and drug screen
PBMNCs were grown in 15% fetal bovine serum in Roswell park memorial institute medium 1640 with 2 mm L‐glutamine. Exponentially growing cultures were exposed to agents listed in Table 1 . Cultures were done in triplicate for each drug, and medium and drug were replaced at 24 hours. All experiments were repeated at least three times.
Table 1.
List of pharmacological agents known to affect splicing evaluated in this report.
| No | Pharmacological agent name | Mechanism | Reference |
|---|---|---|---|
| 1 | Aclarubicin | Topo 1 | 21 |
| 2 | Camphothecin | Topo 1 | 22 |
| 3 | Cantharidin | PP1 inhibition | 23 |
| 4 | Dexamethasone | Coupling of transcription and splicing | 24 |
| 5 | Digoxin | Change in ion gradient | 12 |
| 6 | Etoposide | Topo II inhibition | 22 |
| 7 | 5‐(N‐ethyl‐N‐isopropyl) amiloride (EIPA) | Change in ion gradient | 25 |
| 8 | Sodium butyrate | HDAC inhibitor | 11 |
| 9 | Valproic Acid | HDAC inhibitor | 26 |
| 10 | 5‐iodotubercidin | Adenosine kinase inhibitor | 12 |
| 11 | Tyrphostin‐9 | Kinase inhibitor | 12 |
Analysis of GH transcripts by real‐time
cDNA was synthesized from 1 μg of cellular RNA from each culture using the SuperScript VILO cDNA Synthesis Kit (Invitrogen Life Technologies, Carlsbad, CA, USA). We used pre designed ABI Taqman assays for both 17.5‐ (Hs 00737954_gL that overlaps junction of exon 2 and 4) and 22‐kDa (Hs 00792200_gL that overlaps the junction of exon 2 and 3) GH1 transcript detection. Real‐time PCR analysis was done using Taqman Universal Master Mix and a 7500 real‐time PCR system according to the manufacturer’s instructions (Applied Biosystems, Foster City, CA, USA). We used TaqMan human endogenous control plate (catalog number 4309199) to analyze 13 genes as potential housekeeping genes (18S rRNA, acidic ribosomal protein, beta‐actin, cyclophilin, glyceraldehyde‐3‐phosphate dehydrogenase, phosphoglycerokinase, β2‐microglobulin, β‐glucronidase, hypoxanthine ribosyl transferase (HPRT), transcription factor IID, TATA binding protein, transferrin receptor, and Abelson) for data normalization. HPRT was chosen because it showed the least variability between tissues and was similar in abundance to GH1 isoforms. 14 An initial denaturation at 95°C for 10 minutes was followed by 40 cycles of denaturation at 95°C for 15 seconds and annealing and extension at 60°C for 1 minute. Relative expression levels were calculated using the comparative threshold method. 6 , 15 , 16
Statistics
Data are expressed as mean ± standard error of means (SEM). Comparisons between samples were performed with student’s t‐test and all tests were two‐tailed. Statistical analyses were performed with the statistical package GraphPad Prism for Mac (GraphPad Software Inc., La Jolla, CA, USA).
Results
Selection of drugs that affect GH 1 splicing and determination of optimal dosage and exposure time
We first conducted a thorough literature search to identify drugs that might regulate alternative splicing. 10 , 12 , 17 , 18 Examples of drugs and the putative mechanisms by which they affect cell function are shown in our Table 1 . 11 , 12 , 21 , 22 , 23 , 24 , 25 , 26 To evaluate the possible effects of drugs, we selected and screened an agent from each class in Table 1 , when practical in short‐term cultures. We used real‐time PCR analysis to initially test 11 drugs (see Table 1 ) for their effects on 17.5‐kDa and 22‐kDa mRNA abundance in exponentially growing PBMNCs cultures. PBMNCs are one of the few tissues of the body other than somatotrophs that express GH1 and as such have been used extensively as model to study growth hormone expression. 8 , 19 The four agents studied in more detail in this report were selected because the ratios of 17.5‐/22‐kDa transcripts of cultured PBMNCs derived from IGHD II and controls, exhibited significant increases (dexamethasone and digoxin) or decreases (SB and 5‐iodotubericidin [5‐IT]). For each of these drugs, we then established optimal exposure time and dose by time course and dose‐range exposure experiments (see Supplementary Figure 1 for an example of data from one such experiment). Drug concentrations and exposure times were selected for further studies by choosing conditions that did not result in extensive cell death based on trypan blue exclusion and manual counting. The optimal dose and time for the four drugs were as follows; digoxin 100 nm for 48 hours, dexamethasone 1 μm for 48 hours, SB 5 mm for 48 hours, and 5‐IT 1 μm for 48 hours.
Dexamethasone and digoxin increase the 17.5‐/22‐kDa transcript ratio
We hypothesized that we can identify pharmacological agents that affect alternative splicing and thereby increase the 17.5‐/22‐kDa ratio in PBMNCs from affected and control individuals. Based on our initial screen, we exposed exponentially growing patient and control PBMNCs to dexamethasone or digoxin. After 48 hours, the abundance of 17.5‐ and 22‐kDa transcripts was determined by real‐time PCR and the relative 17.5‐/22‐kDa ratios calculated. We found that both dexamethasone and digoxin increased this relative ratio. Dexamethasone exposure of PBMNCs from the affected and the control individual resulted in an increase in their 17.5‐/22‐kDa transcript ratios from 0.63 to 1.23 (p < 0.003) and 0.05 to 1.03 (p < 0.0001), respectively ( Figure 1 ). Similarly, digoxin also increased the 17.5‐/22‐kDa transcript ratios in both the affected and the control individual PBMNCs (Supplementary Figure 2). The increased ratios following dexamethasone or digoxin exposure were due to an increase in the 17.5‐kDa transcripts in both the affected and the control individual, though in the control individual, decreased amount of 22‐kDa GH1 transcript also contributed to the observed increased ratio ( Figure 1 ). Importantly, dexamethasone caused the 17.5‐/22‐kDa ratios of the control PBMNCs to resemble the relative ratio seen in the affected individual ( Figure 1 ).
Figure 1.
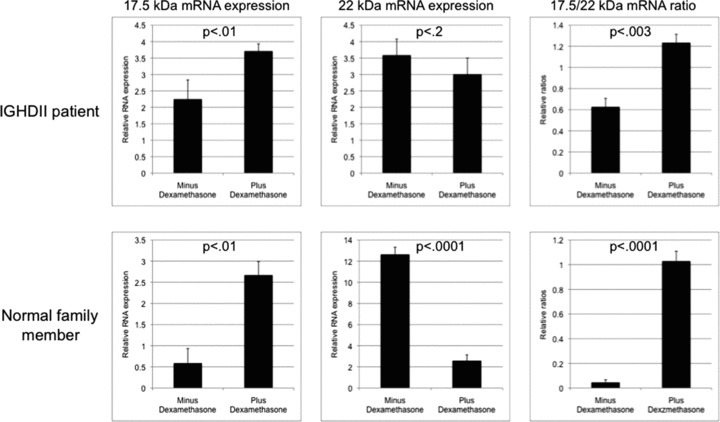
Effect of 48‐hour exposure of 1 μM dexamethasone on GH1 17.5‐kDa and 22‐kDa transcript levels and the 17.5‐/22‐kDa transcript ratios. Analysis was done with relative real‐time PCR in PBMNCs derived from an IGHD II patient and a first‐degree unaffected family member. Each sample was done in triplicate and analysis was repeated three times. Error bars represent SEM; p‐values were calculated by using student’s t‐test. Please note that scales of the graphs are slightly different for better visualization of differences between minus and plus samples.
Sodium butyrate and 5‐IT decrease the 17.5‐/22‐kDa transcript ratio
We then proceeded to further analyze pharmacological agents that were identified in our initial PBMNC‐based screen as decreasing the 17.5‐/22‐kDa transcript ratios. Exponentially growing PBMNCs from affected and control individual were treated with SB or 5‐IT. After 48 hours, the abundance of 17.5‐ and 22‐kDa transcripts was determined by real‐time PCR and the relative 17.5‐/22‐kDa ratios calculated. Treatment with SB decreased the ratio from 0.24 to 0.13 (p < 0.02) and from 0.013 to 0.0081 (p < 0.004) in PBMNCs from affected and control individual, respectively ( Figure 2 ). Exposure to 5‐IT had similar effect (Supplementary Figure 3). The decrease in the ratio was due to an increase in the 22‐kDa transcript in both the affected as well as the control family member PBMNCs ( Figure 2 ). However, treatment with SB did not decrease the ratio in affected patient derived PBMNCs to control levels ( Figure 2 ).
Figure 2.
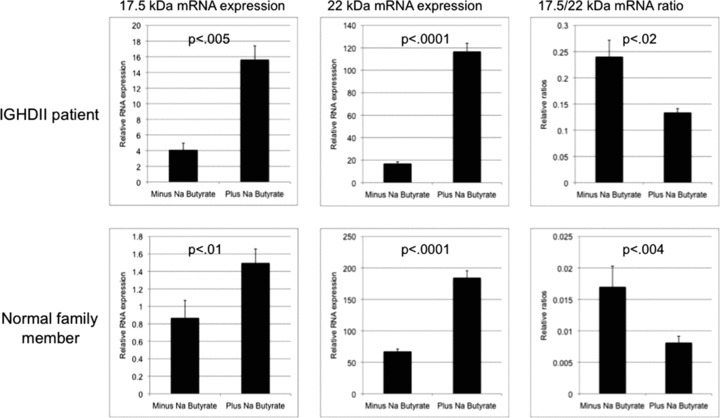
Effect of 48‐hour exposure of 5 mm sodium butyrate on GH1 17.5‐kDa and 22‐kDa transcript levels and the 17.5‐/22‐kDa transcript ratios. Analysis was done with relative real‐time PCR in PBMNCs derived from an IGHD II patient and a first‐degree unaffected family member. Each sample was done in triplicate and analysis was repeated three times. Error bars represent SEM; p‐values were calculated by using student’s t‐test. Please note that scales of the graphs are slightly different for better visualization of differences between minus and plus samples.
Discussion
While growth hormone replacement therapy in IGHD II patients helps growth, it does not prevent the development of other pituitary hormone deficiencies that eventually complicate the clinical picture in many of these patients. Thus, there remains a need for alternative approaches to IGHD II therapy. IGHD II is primarily a splicing disorder where mutations disrupting splicing enhancers result in exon exclusion and resultant overexpression of a detrimental dominant‐negative 17.5‐kDa GH1 product. In this study, we demonstrate that focused screen of pharmaceutical agents resulted in identification of potential modifiers of alternative splicing of GH1. We identified agents that can significantly alter the 17.5‐/22‐kDa GH1 transcript ratios, which has been established as a modifier of IGHD II severity. 8 , 20
We found that, depending on the drug used, the ratio can either increase (predicted to be detrimental for the patient) or decrease (predicted to be beneficial for the patient). Among the drugs screened, dexamethasone and digoxin increased the ratio, which in theory would be predicted to possibly exacerbate the IGHD II phenotype. Our finding that dexamethasone increases the ratio is interesting since related agents are sometimes used to treat the endocrine abnormalities seen in IGHD II patients. Furthermore, it supports the findings of Salemi and colleagues who showed that dexamethasone can increase the 17.5‐kDa isoform in mouse pituitary cells. 20 Dexamethasone has also been shown to affect splicing of the insulin receptor mRNA, further showing that its effects on splicing might be widespread and potentially detrimental. 17
We also found two agents that had the desired effect of decreasing the ratio. One of these agents was 5‐IT, an adenosine derivative that initiates glycogen synthesis in hepatocytes. Because this agent inhibits a range of protein kinases, its possible therapeutic potential may be limited because in vivo dosing for splicing modulation versus kinase inhibition would ultimately need to be determined. SB also decreased the GH1 transcript ratio, and importantly, has been found to change the splicing pattern of the SMN2 gene and ameliorate symptoms in a mouse model of SMA. 11 Its ability to alter the splicing ratio in two distinct genetic diseases that result from aberrant alternative splicing makes SB an intriguing therapeutic possibility.
Another key conclusion of our results is that the effect of a specific drug on alternative splicing of a particular gene maybe gene specific. For example, our data show that Na Butyrate decreased the mutant/normal GH1 transcript ratio in IGHD II and control derived PBMNCs while it increased SMN2/SMN transcript ratio in SMA derived cells. 11 This suggests that the identification of agents that could ameliorate abnormal splicing that causes a disease may need to be done experimentally on a disease‐by‐disease basis (rather then what is reported in the literature).
Further studies will now be needed to confirm in vivo the potency of these agents in animal models of GH1 deficiency and to determine what kinds of proteins in somatotrophs are being targeted by these drugs. Studies using these agents in other systems indicate that these drugs may have direct interactions (such as alteration of phosphorylation status) with the RNA‐binding SR proteins. Additionally, it will be important to determine any in vivo deleterious side effects of these drugs. In this context, it is encouraging that several of the agents identified here, including dexamethasone, digoxin, and SB, have been extensively used clinically with known and or limited toxic side effects. Further, in vivo validation of our findings may thus identify additional therapies for IGHD II.
In summary, our data show that pharmacological agents can alter the 17.5‐/22‐kDa GH1 transcript ratios, which have been shown to affect the phenotype/severity of IGHD II, opening the door for more widespread investigation into the treatment of dominant‐negative and alternative splicing disorders.
Conflict of Interest
The authors have no commercial association that creates a conflict of interest with the information presented herein. The work was entirely supported by NIH grant R01 DK35592 (John A. Phillips).
Supporting information
Figure S1. Effect of 100‐nm digoxin exposure over a 48‐hour interval on GH1 17.5‐kDa and 22‐kDa transcript levels and the 17.5‐/22‐kDa transcript ratios.
Figure S2. Effect of 48‐hour exposure of 100‐nm digoxin on the 17.5‐/22‐kDa transcript ratios.
Figure S3. Effect of 48‐hour exposure of 5‐IT 1 μM on the 17.5‐/22‐kDa transcript ratios.
Supporting info item
Supporting info item
Supporting info item
Acknowledgments
We would like to thank Cindy Holladay for providing technical support for some of the preliminary experiments.
Grants supporting paper: R01 DK35592 (John A. Phillips)
References
- 1. Cogan JD, Phillips JA 3rd, Schenkman SS, Milner RD, Sakati N. Familial growth hormone deficiency: a model of dominant and recessive mutations affecting a monomeric protein. J Clin Endocrinol Metab. 1994; 79(5): 1261–1265. [DOI] [PubMed] [Google Scholar]
- 2. Procter AM, Phillips JA 3rd, Cooper DN. The molecular genetics of growth hormone deficiency. Hum Genet. 1998; 103(3): 255–272. [DOI] [PubMed] [Google Scholar]
- 3. Ryther RC, Flynt AS, Harris BD, Phillips JA 3rd, Patton JG. GH1 splicing is regulated by multiple enhancers whose mutation produces a dominant‐negative GH isoform that can be degraded by allele‐specific small interfering RNA (siRNA). Endocrinology. 2004; 145(6): 2988–2996. [DOI] [PubMed] [Google Scholar]
- 4. Ryther RC, McGuinness LM, Phillips JA 3rd, Moseley CT, Magoulas CB, Robinson IC, Patton JG. Disruption of exon definition produces a dominant‐negative growth hormone isoform that causes somatotroph death and IGHD II. Hum Genet. 2003; 113(2): 140–148. [DOI] [PubMed] [Google Scholar]
- 5. McGuinness L, Magoulas C, Sesay AK, Mathers K, Carmignac D, Manneville JB, Christian H, Phillips JA 3rd, Robinson IC. Autosomal dominant growth hormone eficiency disrupts secretory vesicles in vitro and in vivo in transgenic mice. Endocrinology. 2003; 144(2): 720–731. [DOI] [PubMed] [Google Scholar]
- 6. Shariat N, Holladay CD, Cleary RK, Phillips JA 3rd, Patton JG. Isolated growth hormone deficiency type II caused by a point mutation that alters both splice site strength and splicing enhancer function. Clin Genet. 2008; 74(6): 539–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shariat N, Ryther RC, Phillips JA 3rd, Robinson IC, Patton JG. Rescue of pituitary function in a mouse model of isolated growth hormone deficiency type II by RNA interference. Endocrinology. 2008; 149(2): 580–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hamid R, Phillips JA 3rd, Holladay C, Cogan JD, Austin ED, Backeljauw PF, Travers SH, Patton JG. A molecular basis for variation in clinical severity of isolated growth hormone deficiency type II. J Clin Endocrinol Metab. 2009; 94(12): 4728–4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mullis PE, Robinson IC, Salemi S, Eblé A, Besson A, Vuissoz JM, Deladoey J, Simon D, Czernichow P, Binder G. Isolated autosomal dominant growth hormone deficiency: an evolving pituitary deficit? A multicenter follow‐up study. J Clin Endocrinol Metab. 2005; 90(4): 2089–2096. [DOI] [PubMed] [Google Scholar]
- 10. Sumanasekera C, Watt DS, Stamm S. Substances that can change alternative splice‐site selection. Biochem Soc Trans. 2008; 36(Pt 3): 483–490. [DOI] [PubMed] [Google Scholar]
- 11. Chang JG, Hsieh‐Li HM, Jong YJ, Wang NM, Tsai CH, Li H. Treatment of spinal muscular atrophy by sodium butyrate. Proc Natl Acad Sci U S A. 2001; 98(17): 9808–9813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stoilov P, Lin CH, Damoiseaux R, Nikolic J, Black DL. A high‐throughput screening strategy identifies cardiotonic steroids as alternative splicing modulators. Proc Natl Acad Sci U S A. 2008; 105(32): 11218–11223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Oh HM, Oh JM, Choi SC, Kim SW, Han WC, Kim TH, Park DS, Jun CD. An efficient method for the rapid establishment of Epstein‐Barr virus immortalization of human B lymphocytes. Cell Prolif. 2003; 36(4): 191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hamid R, Patterson J, Brandt SJ. Genomic structure, alternative splicing and expression of TG‐interacting factor, in human myeloid leukemia blasts and cell lines. Biochim Biophys Acta. 2008; 1779(5): 347–355. [DOI] [PubMed] [Google Scholar]
- 15. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods. 2001; 25(4): 402–408. [DOI] [PubMed] [Google Scholar]
- 16. Ogunkolade BW, Boucher BJ, Prahl JM, Bustin SA, Burrin JM, Noonan K, North BV, Mannan N, McDermott MF, DeLuca HF, Hitman GA. Vitamin D receptor (VDR) mRNA and VDR protein levels in relation to vitamin D status, insulin secretory capacity, and VDR genotype in Bangladeshi Asians. Diabetes. 2002; 51(7): 2294–2300. [DOI] [PubMed] [Google Scholar]
- 17. Wu S, Romfo CM, Nilsen TW, Green MR. Functional recognition of the 3′ splice site AG by the splicing factor U2AF35. Nature. 1999; 402(6763): 832–835. [DOI] [PubMed] [Google Scholar]
- 18. Zorio DA, Blumenthal T. Both subunits of U2AF recognize the 3′ splice site in Caenorhabditis elegans. Nature. 1999; 402(6763): 835–838. [DOI] [PubMed] [Google Scholar]
- 19. Hattori N. Expression, regulation and biological actions of growth hormone (GH) and ghrelin in the immune system. Growth Horm IGF Res. 2009; 19(3): 187–197. [DOI] [PubMed] [Google Scholar]
- 20. Salemi S, Yousefi S, Lochmatter D, Eblé A, Deladoëy J, Robinson IC, Simon HU, Mullis PE. Isolated autosomal dominant growth hormone deficiency: stimulating mutant GH‐1 gene expression drives GH‐1 splice‐site selection, cell proliferation, and apoptosis. Endocrinology. 2007; 148(1): 45–53. [DOI] [PubMed] [Google Scholar]
- 21. Andreassi C, Jarecki J, Zhou J, Coovert DD, Monani UR, Chen X, Whitney M, Pollok B, Zhang M, Androphy E, Burghes AH. Aclarubicin treatment restores SMN levels to cells derived from type I spinal muscular atrophy patients. Hum Mol Genet. 2001; 10: 2841–2849. [DOI] [PubMed] [Google Scholar]
- 22. Solier S, Lansiaux A, Logette E, Wu J, Soret J, Tazi J, Bailly C, Desoche L, Solary E, Corcos L. Topoisomerase I and II inhibitors control caspase‐2 pre‐messenger RNA splicing in human cells. Mol Cancer Res. 2004; 2: 53–61. [PubMed] [Google Scholar]
- 23. Novoyatleva T, Heinrich B, Tang Y, Benderska N, Butchbach ME, Lorson CL, Lorson MA, Ben‐Dov C, Fehlbaum P, Bracco L, Burghes AH, Bollen M, Stamm S. Protein phosphatase 1 binds to the RNA recognition motif of several splicing factors and regulates alternative pre‐mRNA processing. Hum Mol Genet. 2008; 17: 52–70. [DOI] [PubMed] [Google Scholar]
- 24. Kosaki A, Webster NJ. Effect of dexamethasone on the alternative splicing of the insulin receptor mRNA and insulin action in HepG2 hepatoma cells. J Biol Chem. 1993; 268: 21990–21996. [PubMed] [Google Scholar]
- 25. Yuo CY, Lin HH, Chang YS, Yang WK, Chang JG. 5‐(N‐ethyl‐N‐isopropyl) amiloride enhances SMN2 exon 7 inclusion and protein expression in spinal muscular atrophy cells. Ann Neurol. 2008; 63: 26–34. [DOI] [PubMed] [Google Scholar]
- 26. Brichta L, Hofmann Y, Hahnen E, Siebzehnrubl FA, Raschke H, Blumcke I, Eyupoglu IY, Wirth B. Valproic acid increases the SMN2 protein level: a well‐known drug as a potential therapy for spinal muscular atrophy. Hum Mol Genet. 2003; 12: 2481–2489. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Effect of 100‐nm digoxin exposure over a 48‐hour interval on GH1 17.5‐kDa and 22‐kDa transcript levels and the 17.5‐/22‐kDa transcript ratios.
Figure S2. Effect of 48‐hour exposure of 100‐nm digoxin on the 17.5‐/22‐kDa transcript ratios.
Figure S3. Effect of 48‐hour exposure of 5‐IT 1 μM on the 17.5‐/22‐kDa transcript ratios.
Supporting info item
Supporting info item
Supporting info item