

Abstract
DIABETES MELLITUS IS A CHRONIC DISEASE that is growing in prevalence worldwide. Pharmacologic therapy is often necessary to achieve optimal glycemic control in the management of diabetes. Orally administered antihyperglycemic agents (OHAs) can be used either alone or in combination with other OHAs or insulin. The number of available OHAs has increased significantly in the last decade, which translates into more therapeutic options and complex decision-making for physicians. This review article is designed to help with these decisions. We review the mechanism of action, efficacy and side effects of the different classes of OHAs (α-glucosidase inhibitors, biguanides, insulin secretagogues, insulin sensitizers and intestinal lipase inhibitor) and discuss the current recommendations for their use.
Diabetes mellitus is a chronic disease that is growing in prevalence worldwide.1 Canadian data from the National Diabetes Surveillance Strategy demonstrate a prevalence of 4.8% among adults, with the vast majority having type 2 diabetes.2With the growing elderly Canadian population, the rising prevalence of obesity and the alarming increase in childhood and adolescent type 2 diabetes, the burden of this disease will continue to grow. Aggressive glycemic control has been demonstrated to decrease microvascular3,4,5 and perhaps macrovascular6,7 complications, although the latter claim remains controversial. The Canadian Diabetes Association 2003 Clinical Practice Guidelines for the Prevention and Management of Diabetes in Canada8 recommends a target hemoglobin A1c concentration of 7.0% or less for all patients with diabetes and, for those in whom it can be safely achieved, a target hemoglobin A1c concentration in the normal range (usually ≤ 6.0%).8 Although nonpharmacologic therapy (e.g., diet, exercise and weight loss) remains a critical component in the treatment of diabetes, pharmacologic therapy is often necessary to achieve optimal glycemic control. Orally administered antihyperglycemic agents (OHAs) can be used either alone or in combination with other OHAs or insulin. The number of available OHAs has increased significantly in the last decade, which translates into more therapeutic options and complex decision-making. This article reviews the mechanism of action, efficacy and side effects of each OHA drug class (α-glucosidase inhibitors, biguanides, insulin secretagogues, insulin sensitizers and intestinal lipase inhibitor) and the current recommendations for their use.
Pathogenesis of diabetes
In order to better understand the role of each drug class in the treatment of diabetes, it is important to have a basic understanding of the pathogenesis of diabetes (Fig. 1) and the interplay between insulin and glucose at different sites.
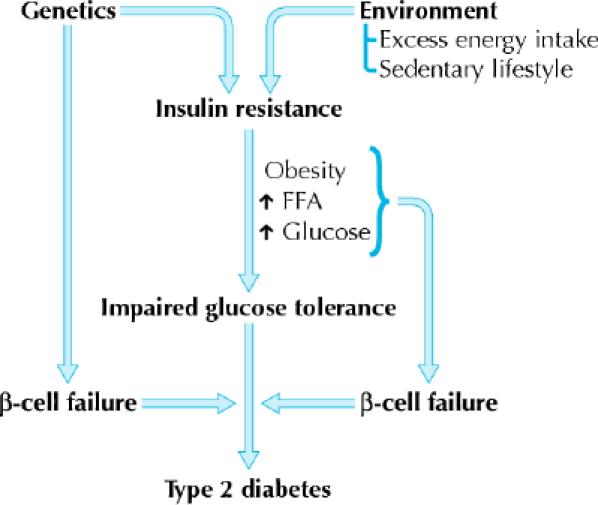
Fig. 1: Overview of the pathogenesis of type 2 diabetes mellitus. FFA = free fatty acids. Photo: Lianne Friesen and Nicholas Woolridge
Postprandial elevations in serum glucose levels stimulate insulin synthesis and release from pancreatic β cells. Insulin secreted into the systemic circulation binds to receptors in target organs (skeletal muscle, adipose tissue, liver). Insulin binding initiates a cascade of intracellular signal transduction pathways that inhibits glucose production in the liver, suppresses lipolysis in adipose tissue and stimulates glucose uptake into target cells (muscle and fat) by mechanisms such as the translocation of vesicles that contain glucose transporters to the plasma membrane.
Type 2 diabetes is a metabolic disorder that results from complex interactions of multiple factors and is characterized by 2 major defects: decreased secretion of insulin by the pancreas and resistance to the action of insulin in various tissues (muscle, liver and adipose), which results in impaired glucose uptake. The precise molecular mechanism of insulin resistance is not clearly understood, but deficits in the postinsulin receptor intracellular signalling pathways are believed to play a role.9,10 Insulin resistance, which is usually present before the onset of diabetes, is determined by a number of factors, including genetics, age, obesity and, later in the disease, hyperglycemia itself. Excess visceral adiposity, dyslipidemia and hypertension often accompany insulin resistance. Other findings may include impaired fibrinolysis, increased platelet aggregation, vascular inflammation, endothelial dysfunction and premature atherosclerosis.11 The inability to suppress hepatic glucose production is a major contributor to the fasting hyperglycemia seen in diabetes.12
The increase in lipolysis by adipose cells that are resistant to insulin and the subsequent increased levels of circulating free fatty acids also contribute to the pathogenesis of diabetes by impairing β-cell function, impairing glucose uptake in skeletal muscles and promoting glucose release from the liver. In addition to its role as a source of excess circulating free fatty acids, adipose tissue has emerged in the last decade as an endocrine organ. Adipose tissue is a source of a number of hormones (adipo-cytokines or “adipokines”) that appear to regulate insulin sensitivity (e.g., adiponectin, resistin), as well as appetite regulation (e.g., leptin), inflammation (e.g., tumour necrosis factor-α, interleukin-6) and coagulability (e.g., plasminogen activator inhibitor-1). Recent evidence suggests that the inflammatory cytokines are derived from infiltrating macrophages within adipose tissue beds rather than from the adipocytes themselves.13 A detailed discussion of this area is beyond the scope of this article, and the reader is referred to a recent review.14
The initial response of the pancreatic β cell to insulin resistance is to increase insulin secretion. Elevated insulin levels can be detected before the development of frank diabetes. As the disease progresses, pancreatic insulin production and secretion decreases, which leads to progressive hyperglycemia. Postprandial hyperglycemia can precede fasting hyperglycemia. Hyperglycemia itself exacerbates insulin resistance and impairs insulin secretion — so-called “glucotoxicity.” The cause of progressive pancreatic β-cell failure is not completely understood, but it appears to result from a number of factors, including genetic determinants, chronic inflammation, glucotoxicity and the deleterious effects of elevated levels of free fatty acids on β-cell function — so-called “lipotoxicity.”15,16
These interacting defects in multiple organs — muscle, liver, adipose tissue and pancreas — generate the pathogenic milieu that results in diabetes. Various classes of OHAs are now available that target the different pathophysiologic factors contributing to diabetes: α-glucosidase inhibitors to delay intestinal carbohydrate absorption, biguanides to target hepatic insulin resistance, insulin secretagogues to increase pancreatic insulin secretion, insulin sensitizers or thiazolidinediones to target adipocyte and muscle insulin resistance, and intestinal lipase inhibitor or orlistat to inhibit fat absorption and promote weight loss in obese patients (Fig. 2).
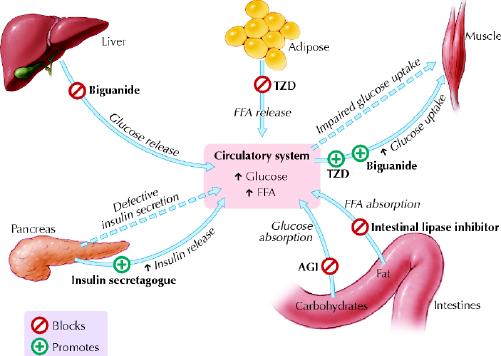
Fig. 2: Major target organs and actions of orally administered antihyperglycemic agents in type 2 diabetes mellitus. TZD = thiazolidinedione; FFA = free fatty acid; AGI = α-glucosidase inhibitor. Photo: Lianne Friesen and Nicholas Woolridge
α-Glucosidase inhibitors
Acarbose is the only α-glucosidase inhibitor available in Canada. Miglitol, another drug in this class, is available in the United States.
Mechanism of action
These drugs do not target a specific pathophysiologic aspect of diabetes. This class of OHA competitively inhibits enzymes in the small intestinal brush border that are responsible for the breakdown of oligosaccharides and disaccharides into monosaccharides suitable for absorption.17 It works primarily on α-glucosidase, which is found predominantly in the proximal half of the small intestine. The intestinal absorption of carbohydrates is therefore delayed and shifted to more distal parts of the small intestine and colon. This retards glucose entry into the systemic circulation and lowers postprandial glucose levels. α-Glucosidase inhibitors act locally at the intestinal brush border and are not absorbed. They are excreted in feces.
Efficacy and clinical use
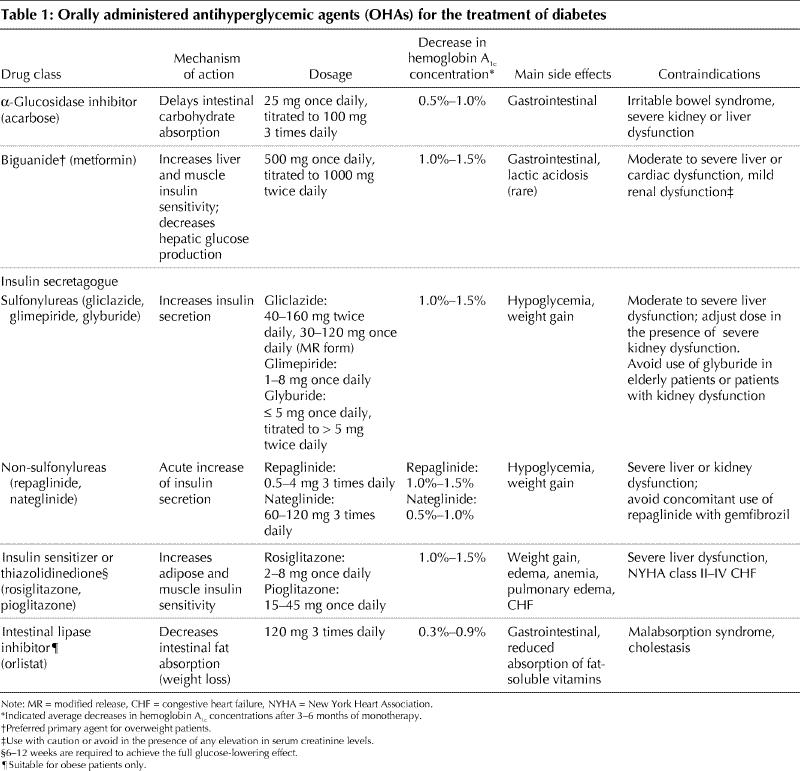
The blood glucose lowering effect of α-glucosidase inhibitors is less than that of most of the other classes of OHAs (Table 1). Compared with placebo, clinical trials have demonstrated an average hemoglobin A1c lowering effect of about 0.5%–1.0%.18 Not surprisingly, postprandial plasma glucose levels are improved more than fasting levels.17 A small reduction in triglyceride levels has also been demonstrated.19 Given the relatively poor efficacy compared with other OHAs, α-glucosidase inhibitors are rarely used alone and are not recommended as initial therapy for moderate to severe hyperglycemia (hemoglobin A1c concentration ≥ 9.0%).8 They are most useful in combination with other OHAs. Dosing should start low, at 25 mg once daily, and titrated upward as tolerated to 100 mg 3 times a day. Frequently, however, gastrointestinal side effects limit the tolerated dose to 50 mg.
Table 1
Side effects, cautions and contraindications
The main side effects of α-glucosidase inhibitors are gastrointestinal. Specifically, bloating, abdominal discomfort, diarrhea and flatulence occur in about 20% of patients.20 Initiation of therapy at a low dose with slow titration upward may minimize these side effects, and symptoms may diminish with continued use.17 Although hypoglycemia does not occur when a drug in this class is used alone, in patients who are using it in combination with another OHA or with insulin, hypoglycemia must be treated with glucose itself (e.g., dextrose tablets) instead of complex carbohydrates, since absorption of the latter is delayed. α-Glucosidase inhibitors are contraindicated in patients with irritable bowel syndrome or severe kidney or liver dysfunction. Inflammatory bowel disease is a relative contraindication.
Biguanides
Over 30 years ago various biguanides (e.g., metformin, phenformin, buformin) were used in different countries for the treatment of diabetes. All but metformin were removed from the international market in the 1970s because of the associated high risk of lactic acidosis.21 Since metformin had not been marketed in the United States at that time, it was only in 1995 that it was approved for use there, after safety concerns were satisfied by decades of experience in Canada, Europe and Asia.
Mechanisms of action
The mechanisms by which metformin exerts its antihyperglycemic effects are still not entirely clear. Its major action in patients with diabetes is to decrease hepatic glucose output, primarily by decreasing gluconeogenesis, but it may also, as a lesser effect, increase glucose uptake by skeletal muscles.22 In 2001, Zhou and colleagues discovered that metformin activates hepatic and muscle adenosine monophosphate-activated protein kinase (AMPK), an enzyme normally activated by adenosine monophosphate, the breakdown product of adenosine triphosphate and a cellular signal for increased energy requirements23 (Fig. 3). Activation of hepatic AMPK results in the phosphorylation and inhibition of acetyl-coenzyme A carboxylase, which catalyzes the rate-limiting step of lipogenesis. This block in fatty acid synthesis promotes fatty acid oxidation. In addition, activation of hepatic AMPK decreases expression of sterol-regulatory-element-binding-protein-1 (SREBP-1), a transcription factor implicated in the pathogenesis of insulin resistance, dyslipidemia and diabetes. Decreased SREBP-1 expression results in decreased gene expression of lipogenic enzymes, which further contributes to decreased triglyceride synthesis and hepatic steatosis. AMPK activation appears to be a critical step in the metformin-mediated reduction of hepatic glucose production and increase in skeletal muscle glucose uptake. Thus, AMPK is a major regulator of lipid and glucose metabolism and may be the key mediator of all the beneficial effects of metformin. Results of earlier studies, particularly those using other biguanide compounds (e.g., phenformin, buformin), suggest that another mechanism of action may be disruption of coupled oxidative phosphorylation in mitochondria.22 Whether this underlies the increase in AMPK activity because of a subtle increase in the ratio between adenosine monophosphate and adenosine triphosphate remains unclear. Other effects, such as increased expression of muscle hexokinase and the insulin-sensitive glucose transporter, may be secondary phenomena.24
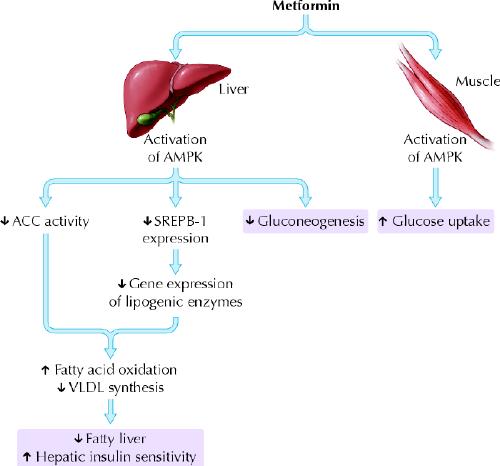
Fig. 3: Metformin activates AMPK in liver and muscle to improve glucose and lipid metabolism in type 2 diabetes. (A) shows the ion channel in a resting pancreatic β cell. (B) shows the action of insulin secretagogues on the cell. AMPK = adenosine monophosphate-activated protein kinase; ACC = acetyl-CoA carboxylase; SREPB-1 = sterol-regulatory-element-binding-protein-1; VLDL = very low density lipoprotein. Photo: Lianne Friesen and Nicholas Woolridge
Metformin is not protein bound and is widely distributed, with maximum accumulation in the small intestinal wall. It is excreted, unmodified, by the kidneys.22
Efficacy and clinical use
In placebo-controlled trials, metformin lowered hemoglobin A1c concentrations by about 1.0%–1.5%.25,26,27 The efficacy of metformin monotherapy is equivalent to that of sulfonylurea monotherapy.27,28 Metformin appears to have beneficial effects beyond glycemic control. It is associated with weight loss, or at least with no weight gain. Improvements in lipid profile have also been noted, with reductions in plasma levels of free fatty acids,29 triglycerides and very-low-density lipoproteins30 in patients whose baseline levels are elevated. Increased levels of plasminogen activator inhibitor-118 and C-reactive protein,31 both of which are associated with increased cardiovascular risk, were also reduced with metformin. The UK Prospective Diabetes Study examined the long-term effects of metformin compared with conventional diet therapy and intensive sulfonylurea or insulin therapy in a subgroup of overweight patients.27The metformin group experienced less hypoglycemia and weight gain than the intensive groups did. However, the most impressive findings were that the metformin group experienced a 36% relative risk reduction in all-cause mortality (p = 0.01), a 39% relative risk reduction in myocardial infarction (p = 0.01) and a 30% relative risk reduction in all macrovascular end points (p = 0.02) compared with the conventional group. Thus far, metformin is the only OHA to demonstrate significant cardiovascular benefit over and above its glucose lowering effect in diabetes.27
Because of its “insulin sensitizing” effect independent of insulin secretion, metformin has been used in type 1 diabetes to lower insulin requirements. Although no long-term clinical benefit has been demonstrated and this is not an approved indication for the use of metformin, there may be unique situations in which insulin resistance and obesity are seen in the context of type 1 diabetes in which metformin may be helpful.32,33
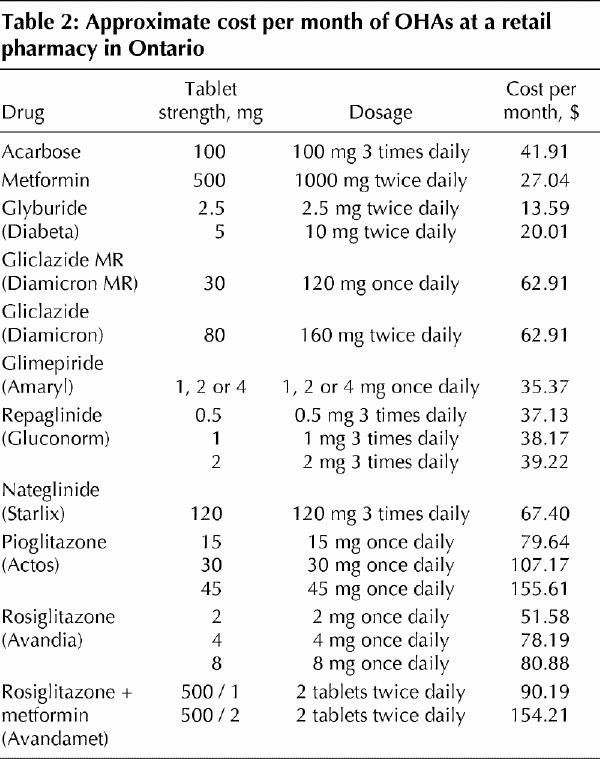
Metformin is approved for use in diabetes either as monotherapy or in combination with other OHAs, as well as with insulin (Box 1). It is recommended as first-line therapy for overweight patients with type 2 diabetes.8 It should be started at a low dose (500 mg once daily) and titrated upward at 1–2-week intervals to a maximum dose of 1000 mg twice daily. Metformin is covered by most provincial formularies and is relatively inexpensive (Table 2).
Box 1.

Table 2
Side effects, cautions and contraindications
Gastrointestinal side effects such as abdominal discomfort, anorexia, bloating and diarrhea are observed in 10%–15% of patients, depending on the dose. The reason for these effects is not known, but, like acarbose, metformin has been associated with decreased intestinal glucose absorption.34 These side effects usually improve with continued use and are minimal if started at a low dose (e.g., 250–500 mg/d) and slowly titrated upward. Discontinuation of therapy because of side effects occurs in less than 4% of patients.35 Since insulin secretion is not altered, hypoglycemia is not a side effect of metformin when used as monotherapy. Similarly, unlike some of the other OHAs, weight gain is not a side effect, and some patients experience weight loss.35
Although lactic acidosis was frequently seen with the earlier biguanide phenformin, its association with metformin has been rare. Monitoring of metformin safety over 56 000 patient-years of experience in Canada revealed a very low risk of lactic acidosis.36 In fact, a recent Cochrane Database systematic review of the incidence of fatal and nonfatal lactic acidosis with metformin compared with placebo and other glucose-lowering therapies in patients with type 2 diabetes demonstrated no increased association, with an incidence of lactic acidosis of 8.4 cases per 100 000 patient-years in the metformin group and 9 cases per 100 000 patient-years in the non-metformin group.37 The presence of another risk factor for lactic acidosis, such as acute renal or liver failure, cardiogenic or septic shock, or hypoxemia, and the inability to correlate lactate concentration or mortality with serum metformin concentrations in the metformin-associated cases make it difficult to discern the contribution of metformin.38 The main difference between metformin and phenformin is that metformin is rapidly excreted, unchanged, by the kidneys,36 whereas phenformin elimination requires conjugation and deactivation by the liver. Thus, in the absence of impaired renal function, metformin is less likely to accumulate. Metformin is contraindicated in patients with risk factors for lactic acidosis or drug accumulation, in other words in those with moderate to severe kidney, liver or cardiac dysfunction. Metformin may be used with extreme caution and in reduced doses in patients with mild renal dysfunction, bearing in mind that renal function may deteriorate rapidly in patients at risk for volume contraction. Insulin is thus a preferred agent in this setting.
Insulin secretagogues
Insulin secretagogues can be divided into 2 subclasses: sulfonylureas and non-sulfonylureas.
Sulfonylureas
Sulfonylureas that are currently available in Canada are gliclazide, glimepiride, glyburide, and the older agents chlorpropamide and tolbutamide. The last 2 are now rarely used.
Mechanism of action
Sulfonylureas bind to the sulfonylurea receptor on the surface of pancreatic β cells. The sulfonylurea receptor is intimately involved with subunits of an adenosine triphosphate-sensitive potassium channel (kir6.2). The binding of a sulfonylurea to the sulfonylurea receptor–kir6.2 complex results in closure of the potassium channels and inhibition of the efflux of potassium ions from the resting β cell. This results in depolarization of the cell membrane and, in turn, the opening of voltage-dependent calcium channels. The influx of calcium causes microtubules to contract and the exocytosis of insulin from vesicles (Fig. 4). Sulfonylureas do not directly affect insulin sensitivity. The increase in insulin sensitivity seen after treatment with these drugs is secondary to improved metabolic control. Sulfonylureas are predominantly metabolized by the liver and cleared by the kidneys. Several metabolites of glyburide are partially active, so that if clearance is impaired in the kidney, the accumulating metabolites can have a significant hypoglycemic effect. In contrast, gliclazide and glimepiride are metabolized by the liver to inactive metabolites.
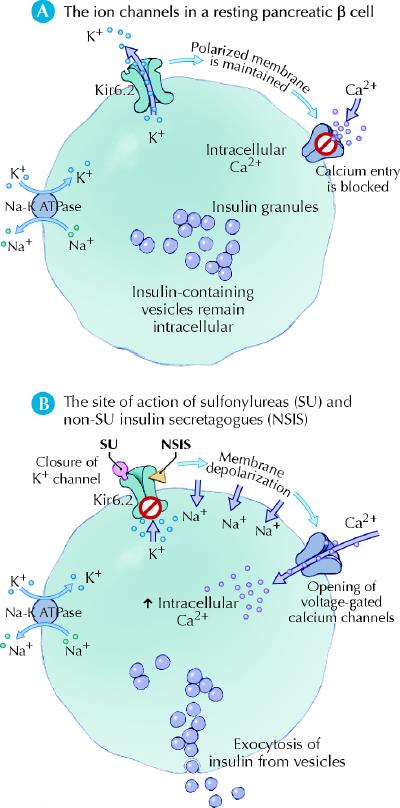
Fig. 4: Insulin secretagogues mimic glucose to close adenosine triphosphate-sensitive potassium channels (kir6.2) and stimulate insulin secretion. Photo: Lianne Friesen and Nicholas Woolridge
Efficacy and clinical use
When compared with placebo, sulfonylurea monotherapy produces an average reduction in hemoglobin A1c concentrations of about 1.0%–1.5%.4,39 Drugs in this class have similar efficacies.40,41The UK Prospective Diabetes Study demonstrated that intensive glycemic control with either sulfonylureas or insulin resulted in significant reductions in microvascular complications,4 and a subsequent epidemiologic analysis demonstrated a reduction in macrovascular complications associated with improved glycemic control.7 No increase in mortality was demonstrated, as was suggested by the findings of the older University Group Diabetes Program.42 The shortcomings of the latter study have been reviewed.43
In general, it is best to start with a low dose and titrate upward every 1–2 weeks to achieve the desired glycemic control and avoid hypoglycemia, particularly in elderly patients. Gliclazide is available in short- and long-acting formulations. The long-acting modified release formulation can be administered once daily. Glimepiride is also administered once daily. Glyburide may be administered once daily at 5 mg or less and twice daily at higher doses. Gliclazide and glimepiride, which are newer, are more expensive than glyburide. Glyburide is covered by provincial formularies, and gliclazide and glimepiride may be covered under special provisions by some provinces (e.g., Section 8 in Ontario for gliclazide).
Side effects, cautions and contraindications
The main side effects of sulfonylureas are hypoglycemia and weight gain.44 Given that these drugs directly stimulate insulin secretion from pancreatic β cells irrespective of plasma glucose levels, the risk of hypoglycemia is associated with all sulfonylureas. The results of several large clinical trials indicate an average incidence of hypoglycemia of 1%–2% per year.45 Most episodes are mild and easily treated with glucose in the form of fruit juice, sweetened beverages or glucose tablets. However, prolonged and severe hypoglycemia can occur, especially in the setting of renal or hepatic impairment or in frail, elderly patients. Gliclazide and glimepiride are less associated with hypoglycemia than is glyburide.18 Since these medications are metabolized in the liver, sulfonylureas are contraindicated in patients with moderate to severe liver dysfunction. The dose of glyburide should either be markedly reduced or avoided altogether in elderly patients and patients with moderate renal dysfunction. Dose adjustment is not required for gliclazide or glimepiride in patients with moderate kidney dysfunction. However, there are insufficient data to support their use in those with end-stage renal disease, in which case insulin is the preferred option. The weight gain seen with sulfonylureas, which is typically 2–5 kg, is likely related to the increase in plasma insulin levels.45 This may be discouraging in a population that is already prone to obesity and often struggling to lose weight. At the same time, metabolic control should not be compromised by withholding treatment in an attempt to avoid weight gain. Side effects of first-generation agents include skin rash, hyponatremia and alcohol-induced flushing.
Since sulfonylureas are sulfonamide derivatives, the question of whether patients with known allergies to sulfonamide antibiotics can take sulfonylureas is often raised. A recent retrospective cohort study in the United Kingdom demonstrated that patients with a previous allergic reaction to a sulfonamide antibiotic were at increased risk for an allergic reaction to a sulfonamide nonantibiotic (e.g., a sulfonylurea) compared with patients with no previous history (adjusted odds ratio 2.8; 95% confidence interval 2.1–3.7).46 However, the risk was lower than that associated with penicillin: patients with a previous sensitivity to penicillin were more likely than patients with a sensitivity to sulfonamide antibiotics to react to a sulfonamide nonantibiotic. The authors concluded that the association between previous sensitivity to sulfa and subsequent sensitivity to a sulfonamide nonantibiotic is due to a predisposition to allergic reactions rather than to cross-reactivity. However, as with all retrospective cohort studies, there were potential biases that may have influenced the results. Therefore, this issue remains controversial, and clinicians should recognize that there is the potential for cross-reactivity and perhaps use the severity of the initial reaction to sulfa to guide their decision whether to give a trial of sulfonylurea (or other sulfonamide nonantibiotic) to a patient with a previous hypersensitivity.
Non-sulfonylureas
This relatively new class of medications is currently represented by nateglinide and repaglinide. Repaglinide is a benzoic acid derivative, and nateglinide is a phenylalanine derivative.47,48
Mechanism of action
The mechanism of action of these drugs is similar to that of the sulfonylureas (closure of the potassium–adenosine triphosphate channel, leading to calcium-dependent insulin secretion). However, they bind to the sulfonylurea receptor at a different site and with different kinetics than the sulfonylureas (Fig. 4). Thus, the onset of action is faster and the half-life is shorter, which results in a brief stimulation of insulin release.47,48 These compounds are metabolized in the liver through the cytochrome p450 system into inactive biliary products.47,48
Efficacy and clinical use
The efficacy of repaglinide appears to be similar to that of sulfonylureas,49 and the efficacy of nateglinide appears to be somewhat less, with a reduction in hemoglobin A1c concentrations of 0.5%–1.0%.50 Given their rapid onset and short duration of action, non-sulfonylurea insulin secretagogues are best taken just before meals. They may be taken 3 or even 4 times daily. Postprandial hyperglycemia is well controlled.49,50 These medications are particularly useful for patients who require meal-time flexibility, elderly patients and patients with impaired renal function. For example, a dose may be omitted if a meal is skipped, and in the elderly patient with unpredictable food intake, the dose may be given immediately after the meal and titrated to the amount of food ingested. These medications can be used either as monotherapy or in combination with other OHAs (but not sulfonylureas) (Box 1). Because they have been introduced recently, there are no long-term outcome data for this class. They are fairly expensive and are not covered by most provincial formularies.
Side effects, cautions and contraindications
As with sulfonylureas, the main side effect of this class is hypoglycemia. However, the risk of hypoglycemia is lower than that with sulfonylureas.51 This difference is due in part to the shorter duration of action and in part to the glucose-dependent insulinotropic effects of nateglinide.48 Similarly, the amount of weight gain appears to be less than that seen with sulfonylureas,18 perhaps because of the limited duration of elevated insulin secretion. The non-sulfonylurea insulin secretagogues are contraindicated in patients with severe liver dysfunction, and the dose should be reduced in patients with severe kidney dysfunction.18 Given the metabolism of repaglinide through the cytochrome p450 isozyme CYP 3A4, glucose levels should be monitored carefully if the patient also receives a strong inhibitor or inducer of the CYP 3A4 system.52 The combination of gemfibrozil, a CYP 3A4 inhibitor, with repaglinide has been shown to dramatically increase the action of repaglinide and result in prolonged hypoglycemia.52 This combination should therefore be used cautiously or avoided. Nateglinide, on the other hand, is mostly metabolized via the CYP 2C9 isozyme and requires CYP 3A4 metabolism to a lesser extent. No interaction with gemfibrozil has been reported.48
Insulin sensitizers (thiazolidinediones)
The 2 thiazolidinediones currently available in Canada are rosiglitazone and pioglitazone. Troglitazone, an earlier thiazolidinedione introduced in 1997 in the United States, United Kingdom and Japan but not in Canada, was removed from the world market because of an unacceptable risk of fulminant hepatic failure.
Mechanism of action
Thiazolidinediones function as ligands for the peroxisome proliferator-activated receptor gamma (PPARγ), which is most highly expressed in adipocytes (Fig. 5). These nuclear receptors, which are ligand-activated transcription factors, play an integral part in the regulation of the expression of a variety of genes involved in carbohydrate and lipid metabolism. Thiazolidinediones improve insulin sensitivity, particularly in the peripheral tissues. Although unproven, this appears to be mainly mediated through an effect on adipocytes, since there are fewer PPARγ receptors in muscle tissue. In the adipocyte, differentiation is enhanced, lipolysis is reduced, and levels of circulating adipo-cytokines or “adipokines” are altered, namely a decrease in tumour necrosis factor-α and leptin and an increase in adiponectin.14 The recruitment of a greater number of smaller adipocytes, which is associated with improved lipogenesis and storage, results in a reduction in circulating free fatty acids. All these effects — decreased tumour necrosis factor-α and free fatty acid levels and increased adiponectin levels — are expected to enhance insulin sensitivity.14 Animal models have also demonstrated that thiazolidinediones improve and preserve pancreatic β-cell function.53,54 However, this finding has not been demonstrated definitively in humans.55
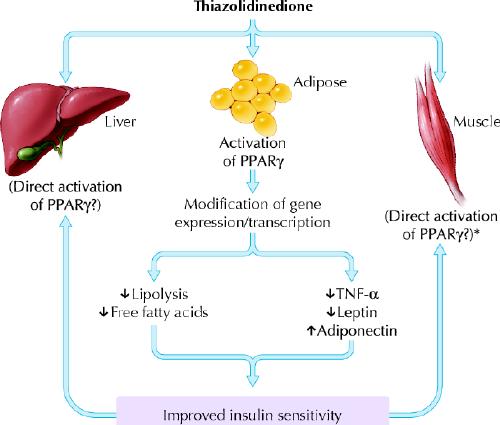
Fig. 5: Mechanism of action of thiazolidinediones. PPARγ = peroxisome proliferator-activated receptor-gamma; TNF-α = tumour necrosis factor-α. *There is recent evidence in rodents that skeletal muscle PPARγis an important mediator of the beneficial effects of TZDs on insulin sensitivity. Photo: Lianne Friesen and Nicholas Woolridge
Efficacy and clinical use
In placebo-controlled trials, thiazolidinediones lower hemoglobin A1c concentrations to the same extent as metformin and sulfonylureas.56,57 Direct comparisons of thiazolidinediones with metformin and sulfonylureas also demonstrate similar efficacy.58,59 Preliminary data suggest that thiazolidinediones may have beneficial effects beyond that of glycemic control. These include reduced urinary albumin excretion,60 increased levels of high-density lipoprotein cholesterol and reduced triglyceride levels,61 lower blood pressure18 and reduced levels of plasminogen activator inhibitor-1.18 Some studies have also demonstrated improvement in surrogate markers of atherosclerosis, such as intimal–medial thickness and neointimal proliferation after angioplasty.18 However, there are no long-term microvascular or macrovascular clinical outcome data available yet on the use of thiazolidinediones in patients with diabetes.
Thiazolidinediones are approved for use as monotherapy or in combination with metformin, sulfonylureas, non-sulfonylurea insulin secretagogues or α-glucosidase inhibitors (Box 1). Although some effect can be seen in 2–3 weeks, it may take 6–12 weeks to observe the full blood glucose lowering effect. Dose adjustments should be made accordingly. Patients should be appropriately screened by history, physical examination and laboratory investigations to rule out contraindications before therapy with thiazolidinediones is initiated.62 These drugs are expensive, but some provincial formularies cover them under special provisions (e.g., Section 8 in Ontario).
Side effects, cautions and contraindications
The major side effects of rosiglitazone and pioglitazone are weight gain, edema, anemia, pulmonary edema and congestive heart failure.62 The weight gain seen with thiazolidinediones is similar to that observed with sulfonylureas. However, the distribution of fat appears to be improved from a metabolic point of view — there is less visceral fat and more peripheral fat.63 Peripheral edema can occur in about 3%–5% of patients using thiazolidinedione as monotherapy62 and sometimes is severe enough that use of the medication is stopped. The incidence of peripheral edema is increased when use of the drug is combined with another glucose-lowering medication, particularly insulin.62 Thus, thiazolidinediones are not approved for use in combination with insulin in Canada.64,65 However, pioglitazone is approved for such combination therapy in the United States.66 Another adverse effect associated with thiazolidinedione use is anemia, which is considered to represent hemodilution from sodium and water retention.62 The more serious adverse events of pulmonary edema and congestive heart failure were infrequent in trials of monotherapy (about 1%) but increased in combination therapy with insulin (about 2%–3%).62 Subsequent epidemiological studies demonstrated a hazard ratio for congestive heart failure in patients receiving pioglitazone of 1.8.62 Although the absolute incidence is relatively small, the increased recognition of thiazolidinedione-induced congestive heart failure prompted the additional contraindication of use of this class of drugs in patients with New York Heart Association Class II, III or IV congestive heart failure.64,65 Also, postmarketing cases of thiazolidinedione-induced congestive heart failure have been reported in patients with normal systolic function. Patients with diabetes can have diastolic dysfunction even in the absence of hypertension or ischemic heart disease.62 The American Heart Association and the American Diabetes Association recently published a consensus statement on the issue of thiazolidinedione use, fluid retention and congestive heart failure that is worth reviewing.62 In addition to congestive heart failure, the use of thiazolidinediones is contraindicated in the presence of hepatic dysfunction.64,65 The fact that PPARγ receptors are present in other tissues, such as monocytes, macrophages, colonic epithelial cells and pituitary cells,67 raises the possibility of long-term adverse or beneficial actions that are yet to be determined.
Intestinal lipase inhibitor (orlistat)
Mechanism of action
Although not a “traditional” OHA, orlistat is an anti-obesity agent that acts as a selective inhibitor of gastric and pancreatic lipases and thereby inhibits the hydrolysis of dietary fat into absorbable free fatty acids and monoglycerides.68 The marked reduction in the absorption of fat results in decreased energy intake and weight loss. There is negligible systemic absorption of the drug. Metabolism occurs within the gastrointestinal wall, and fecal excretion is the main route of elimination.
Efficacy and clinical use
In the 2003 Canadian Diabetes Association clinical practice guidelines, orlistat was added to the list of OHAs for the treatment of diabetes.8 The addition of orlistat for 1 year in overweight or obese patients (body mass index 28–40 kg/m2) with diabetes treated with other OHAs or insulin has been shown to decrease body weight by about 4%–6.5% and improve hemoglobin A1c concentrations by 0.3%–0.9%.69,70,71 In some patients, the doses of OHAs or insulin could be lowered and metabolic control maintained when orlistat was added.
Orlistat's role in the treatment of diabetes is limited to obese patients. Since its blood glucose lowering effect is relatively small, it should be used in combination with other OHAs. Orlistat should be given at a dose of 120 mg with each meal. Appropriate dietary counselling must be provided to minimize the gastrointestinal side effects and improve long-term compliance.
Side effects, cautions and contraindications
The side effects of orlistat are gastrointestinal and typically occur in the first 3 months of treatment. These include flatulence with discharge, oily spotting, fecal urgency, steatorrhea, increased frequency of defecation and fecal incontinence. The incidence of these reactions is directly related to the fat content of the patient's diet. Patients who do not adhere to a diet containing 30% or less of energy intake from fat experience significant side effects, which results in either improved dietary compliance or discontinuation of the medication.68 Absorption of fat-soluble vitamins can be adversely affected, and patients on orlistat should take a daily multivitamin supplement that includes fat-soluble vitamins at least 2 hours before or following administration of the dose. Contraindications to orlistat are chronic malabsorption syndrome, cholestasis and known hypersensitivity.68
Summary of therapeutic recommendations
The 2003 Canadian Diabetes Association clinical practice guidelines recommend a treatment target hemoglobin A1c concentration of 7.0% or less and even lower (≤ 6.0%) in those who can safely achieve it, within 6–12 months of the initial diagnosis of diabetes.8 A multidisciplinary approach is critically important to achieve this goal, and the importance of education and lifestyle modifications cannot be overemphasized. These measures should continue alongside all other interventions. A therapeutic algorithm is depicted in Fig. 6. In patients with mild to moderate hyperglycemia (hemoglobin A1c concentration < 9.0%), lifestyle interventions should be implemented along with pharmacologic interventions as needed. If the hyperglycemia is very mild, one could consider instituting lifestyle interventions alone. If patients present with marked hyperglycemia (hemoglobin A1c concentrations ≥ 9.0%), pharmacologic measures should be started immediately, along with lifestyle modifications.8 Glycemic status should be reassessed frequently and necessary changes made to achieve target hemoglobin A1c concentrations within 6–12 months.8
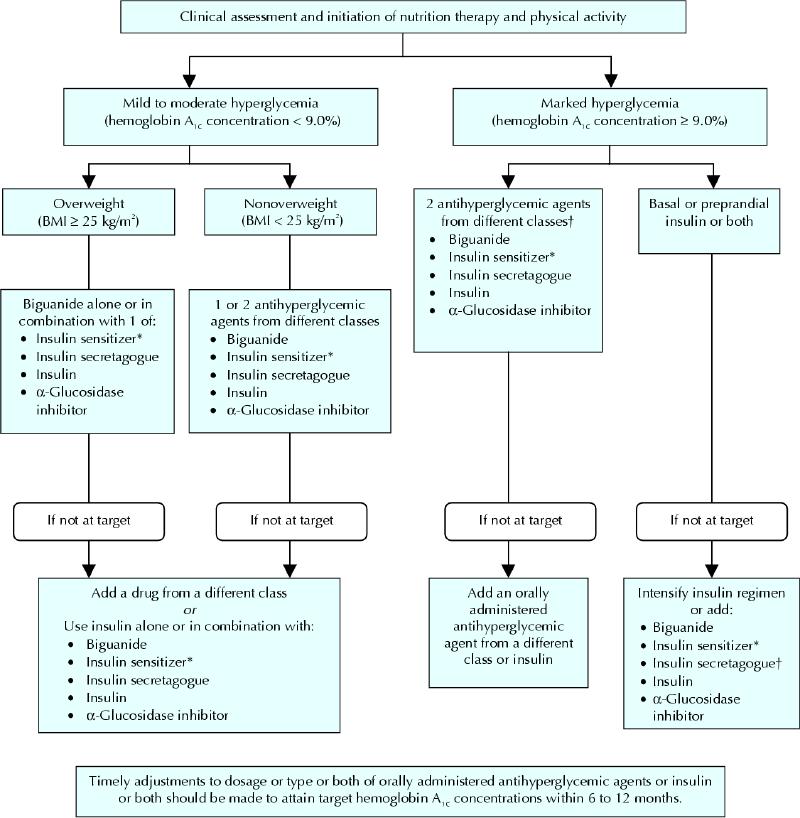
Fig. 6: Management of hyperglycemia in type 2 diabetes. BMI = body mass index. *When used in combination with insulin, insulin sensitizers may increase the risk of edema of congestive heart failure. The combination of an insulin sensitizer and insulin is currently not an approved indication in Canada. †If using preprandial insulin, do not add an insulin secretagogue. Reprinted with permission from Can J Diabetes 2003;27(Suppl 2):S39.
Metformin is recommended as the primary drug for overweight patients (body mass index > 25 kg/m2), unless contraindicated. For nonoverweight patients, other classes of OHAs (except orlistat) can be used as primary therapy. The current guidelines, in contrast to the 1998 guidelines, recommend that the addition of a different class of OHA be considered early to achieve glycemic targets. It has been shown that combinations of submaximal doses of OHAs produce greater reductions in hemoglobin A1c concentrations in a short period compared with maximum dose monotherapy.72,73,74,75 As one would expect, the incidence of side effects (particularly hypoglycemia) is higher with combination therapy than with monotherapy; however, the difference is not significant.72,73,74,75 Dose adjustments or addition of other classes of medications, or both, should occur in a timely fashion if targets are not achieved. A reasonable duration after which a response in hemoglobin A1c concentrations is expected is 3 months for α-glucosidase inhibitors, metformin and insulin secretagogues and 6 months for thiazolidinediones and orlistat. The choice of combinations requires knowledge of the mechanism of action of the different classes. Potential combinations and combinations to be avoided are listed in Box 1.
A common question in the management of diabetes is when and how to institute insulin therapy. Although the details of insulin use are beyond the scope of this review, the target hemoglobin A1c concentrations should remain the primary indicator. Thus, if maximally tolerated doses of combination OHA therapy does not achieve the desired glycemic targets, insulin should be started, either as monotherapy or in combination with OHAs. The combination of insulin with the following agents has been shown to have increased glucose-lowering effects: α-glucosidase inhibitor, metformin, sulfonylurea and thiazolidinedione.8 Sulfonylureas should not be combined with preprandial insulin because of an increased risk of hypoglycemia. However, they can be combined with basal insulin. Of note, the combination of thiazolidinedione and insulin is not approved in Canada because of an increased risk of peripheral edema and heart failure. The decision to use insulin alone or in combination with OHAs should be individualized and discussed with the patient. There are no long-term clinical outcome data to support or detract from using the combination approach. Regular follow-up and timely adjustments of medications in all patients are mandatory since worsening glycemic control may be expected, consistent with the natural history of diabetes.4 Therefore, choice and dose of OHAs need to be reassessed on an ongoing basis.
Prevention of diabetes
The worldwide epidemic of diabetes and the recognition that sedentary lifestyle and obesity are major epidemiologic determinants of the disease have led to a greater interest in prevention. Although this topic cannot be reviewed in detail here, it is important to note that the OHAs used in the treatment of diabetes have also been studied in the context of prevention. Multiple risk factors for diabetes have been identified.8 The greatest risk is impaired glucose tolerance, a precursor of diabetes. Thus, a number of type 2 diabetes prevention trials have included subjects with impaired glucose tolerance. These trials compared intensive lifestyle modifications (e.g., diet, exercise, weight loss), OHAs and placebo controls.76,77,78In brief, the greatest success thus far has been achieved with intensive lifestyle modification, with a 58% reduction in progression from impaired glucose tolerance to overt diabetes in 2 separate trials.76,77 In these same studies, the use of metformin resulted in a 33% relative reduction in the conversion to overt diabetes. However, a subsequent analysis performed after stopping the metformin demonstrated that a proportion of subjects were deriving benefit from treatment of diabetes rather than from its prevention, so that the preventive effect of metformin was actually lower, about 25%.79 In the STOP-NIDDM trial, acarbose was associated with a 25% relative reduction in the conversion of impaired glucose tolerance to diabetes after 3.3 years.20 In the TRIPOD study, troglitazone, a thiazolidinedione that is no longer available, decreased the incidence of diabetes compared with placebo in a group of high-risk Hispanic women with a history of gestational diabetes.80 Finally, orlistat combined with lifestyle changes also decreased progression from impaired glucose tolerance to diabetes over 4 years compared with lifestyle modification plus placebo.81 Currently, diabetes prevention trials using thiazolidinedione and non-sulfonylurea insulin secretagogues, alone and in combination with other agents such as angiotensin-converting-enzyme inhibitors, are underway. One encouraging observation is that, in the acarbose prevention trial, the treated group also had a 49% relative risk reduction in cardiovascular events and a 34% relative risk reduction in new cases of hypertension.82 These data support the concept that prevention of diabetes will be associated with lower morbidity and mortality. However, OHAs are currently approved for only the treatment and not the prevention of diabetes. This may change as more data become available demonstrating efficacy, safety and cost-effectiveness. However, it is imperative for clinicians to recognize that intensive and structured lifestyle modifications, including diet, physical activity and weight loss, have the greatest impact on diabetes prevention and remain the intervention of choice.8
Footnotes
This article has been peer reviewed.
Contributors: Dr. Cheng drafted the first version of the article, searched the literature and designed the figures. Dr. Fantus reviewed and revised the manuscript and the final figures and provided additional information. Both authors approved the final version.
Competing interests: None declared.
Correspondence to: Dr. I. George Fantus, Mount Sinai Hospital, 600 University Ave., Lebovic Bldg., Room 5–028, Toronto ON M5G 1X5; fax 416 361-2657; fantus@mshri.on.ca
References
- 1.Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature 2001;414:782-7. [DOI] [PubMed]
- 2.Health Canada. Diabetes in Canada. 2nd ed. Ottawa: Centre for Chronic Disease Prevention and Control, Population and Public Health Branch; 2002. Available at: www.hc-sc.gc.ca/pphb-dgspsp/publicat/dic-dac2/english/01cover_e.html (accessed 2004 Nov 22).
- 3.The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 1993;329:977-86. [DOI] [PubMed]
- 4.UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998;352:837-53. [PubMed]
- 5.Ohkubo Y, Kishikawa H, Araki E, Miyata T, Isami S, Motoyoshi S, et al. Intensive insulin therapy prevents the progression of diabetic microvascular complications in Japanese patients with non-insulin dependent diabetes mellitus: a randomized prospective 6-year study. Diabetes Res Clin Pract 1995; 28 (2): 103 -17. [DOI] [PubMed]
- 6.Lawson ML, Gerstein HC, Tsui E, Zinman B. Effect of intensive therapy on early macrovascular disease in young individuals with type 1 diabetes: a systematic review and meta-analysis. Diabetes Care 1999;22(S2):B35-9. [PubMed]
- 7.Stratton IM, Adler AI, Neil HA, Matthews DR, Manley SE, Cull CA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ 2000;321(7258):405-12. [DOI] [PMC free article] [PubMed]
- 8.Canadian Diabetes Association Clinical Practice Guidelines Expert Committee. Canadian Diabetes Association 2003 Clinical Practice Guidelines for the Prevention and Management of Diabetes in Canada. Can J Diabetes 2003; 27 (suppl 2): S1-S152. [DOI] [PubMed]
- 9.Virkamaki A, Ueki K, Kahn CR. Protein–protein interaction in insulin signaling and the molecular mechanisms of insulin resistance. J Clin Invest 1999;103;931-43. [DOI] [PMC free article] [PubMed]
- 10.Saltiel AR, Kahn CR. Insulin signaling and the regulation of glucose and lipid metabolism. Nature 2001;414;799-806. [DOI] [PubMed]
- 11.Fonseca VA. Management of diabetes mellitus and insulin resistance in patients with cardiovascular disease. Am J Cardiol 2003;92(suppl):50J-60J. [DOI] [PubMed]
- 12.Firth R, Bell P, Pizza R. Insulin action in non-insulin-dependent-diabetes mellitus: the relationship between hepatic and extrahepatic insulin resistance and obesity. Metabolism 1987;36:1091-5. [DOI] [PubMed]
- 13.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr, et al. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 2003;112(12):1796-808. [DOI] [PMC free article] [PubMed]
- 14.Aldhahi W, Hamdy O. Adipokines, inflammation and the endothelium in diabetes. Curr Diab Rep 2003;3:293-8. [DOI] [PubMed]
- 15.Buchanan TA. Pancreatic beta-cell loss and preservation in type 2 diabetes. Clin Ther 2003;25(Suppl B):B32-46. [DOI] [PubMed]
- 16.Kashyap S, Belfort R, Gastaldelli A, Pratipanawatr T, Berria R, Pratipanawatr W, et al. A sustained increase in plasma free fatty acids impairs insulin secretion in nondiabetic subjects genetically predisposed to develop type 2 diabetes. Diabetes 2003;25(10):2461-74. [DOI] [PubMed]
- 17.Lebovitz HE. Alpha-glucosidase inhibitors. Endocrinol Metab Clin North Am 1997;26:539-51. [DOI] [PubMed]
- 18.Inzucchi SE. Oral antihyperglycemic therapy for type 2 diabetes. JAMA 2002;287:360-72. [DOI] [PubMed]
- 19.Bayraktar M, Van Thiel DH, Adalar N. A comparison of acarbose versus metformin as an adjuvant therapy in sulfonylurea-treated NIDDM patients. Diabetes Care 1996;19:252-4. [DOI] [PubMed]
- 20.Chiasson JL, Josse RG, Gomis R, Hanefeld M, Karasik A, Laakso M; STOP-NIDDM Trail Research Group. Acarbose for prevention of type 2 diabetes mellitus: the STOP-NIDDM randomized trial. Lancet 2002;359(9323):2072-7. [DOI] [PubMed]
- 21.Bailey CJ, Turner RC. Metformin. N Engl J Med 1996;334:574-9. [DOI] [PubMed]
- 22.Kirpichnikov D, McFarlane SI, Sowers JR. Metformin: an update. Ann Intern Med 2002;137:25-33. [DOI] [PubMed]
- 23.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-acitvated protein kinase in mechanism of metformin action. J Clin Invest 2001;108(8):1167-74. [DOI] [PMC free article] [PubMed]
- 24.Holmes BF, Kurth-Kraczek EJ, Winder WW. Chronic activation of 5'-AMP-activated protein kinase increases GLUT-4, hexokinase, and glycogen in muscle. J Appl Physiol 1999;87:1990-5. [DOI] [PubMed]
- 25.Garber AJ, Duncan TG, Goodman AM, Mills DJ, Rohlf JL. Efficacy of metformin in type II diabetes: results of a double-blind, placebo-controlled, dose-response trial. Am J Med 1997;103(6):491-7. [DOI] [PubMed]
- 26.Hoffmann J, Spengler M. Efficacy of 24-week monotherapy with acarbose, metformin, or placebo in dietary-treated NIDDM patients: the Essen-II Study. Am J Med 1997;103:483-90. [DOI] [PubMed]
- 27.UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 1998;352:854-865. [PubMed]
- 28.Tessier D, Maheux P, Khalil A, Fulop T. Effects of gliclazide versus metformin on the clinical profile and lipid peroxidation markers in type 2 diabetes. Metab Clin Exp 1999;48:897-903. [DOI] [PubMed]
- 29.Abbasi F, Kamath V, Rizvi AA, Carantoni M, Chen YD, Reaven GM. Results of a placebo-controlled study of the metabolic effects of the addition of metformin to sulfonylurea-treated patients. Evidence for a central role of adipose tissue. Diabetes Care 1997;20(12):1863-9. [DOI] [PubMed]
- 30.Landin K, Tengborn L, Smith U. Treating insulin resistance in hypertension with metformin reduces both blood pressure and metabolic risk factors. J Intern Med 1991;229:181-7. [DOI] [PubMed]
- 31.Morin-Papunen L, Rautio K, Ruokonen A, Hedberg P, Puukka M, Tapanainen JS. Metformin reduces servum C-reactive protein levels in women with polycystic ovary syndrome. J ClinEndocrinol Metab 2003; 88 (10): 4649-54. [DOI] [PubMed]
- 32.Sarnblad S, Kroon M, Aman J. Metformin as additional therapy in adolescents with poorly controlled type 1 diabetes: randomized placebo-controlled trial with aspects on insulin sensitivity. Eur J Endocrinol 2003;149:323-9. [DOI] [PubMed]
- 33.Meyer L, Bohme P, Delbachian I, Lehert P, Cugnardey N, Drouin, et al. The benefits of metformin therapy during continuous subcutaneous insulin infusion treatment of type 1 diabetic patients. Diabetes Care 2002;25:2153-8. [DOI] [PubMed]
- 34.Ikeda T, Iwata K, Murakami H. Inhibitory effect of metformin on intestinal glucose absorption in perfused rat intestine. Biochem Pharmacol 2000;59:887-90. [DOI] [PubMed]
- 35.DeFronzo RA, Barzilai N, Simonson DC. Mechanism of metformin action in obese and lean noninsulin-dependent diabetic mellitus. N Engl J Med 1995; 333: 550-4. [DOI] [PubMed]
- 36.Lucis OJ. The status of metformin in Canada. CMAJ 1983;128:24-6. [PMC free article] [PubMed]
- 37.Salpeter S, Greyber E, Pasternak G, Salpeter E. Risk of fatal and nonfatal lactic acidosis with metformin use in type 2 diabetes mellitus. [Cochrane review]. In: The Cochrane Library; Issue 4, 2004. Oxford: Update Software. [DOI] [PMC free article] [PubMed]
- 38.Stades AM, Heikens JT, Erkelens DW, Holleman F, Hoekstra JB. Metformin and lactic acidosis: cause or coincidence? A review of case reports. J Intern Med 2004;255(2):179-87. [DOI] [PubMed]
- 39.Klepzig H, Kober G, Matter C, Luus H, Schneider H, Boedeker KH, et al. Sulfonylureas and ischaemic preconditioning: a double-blind, placebo-controlled evaluation of glimepiride and glibenclamide. Eur Heart J 1999; 20 (6): 403-5. [DOI] [PubMed]
- 40.Tessier D, Dawson K, Tetrault JP, Bravo G, Meneilly GS. Glibenclamide vs gliclazide in type 2 diabetes of the elderly. Diabet Med 1994;11(10):974-80. [DOI] [PubMed]
- 41.Dills DG, Schneider J. Clinical evaluation of glimepiride versus glyburide in NIDDM in a double-blind comparative study. Horm Metab Res 1996;28:426-9. [DOI] [PubMed]
- 42.Goldner MG, Knatterud GL, Prout TE. Effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes, III: clinical implication of UGDP results. JAMA 1971;218:1400-10. [PubMed]
- 43.Kilo C, Miller JP, Williamson JR. The crux of the UGDP. Spurious results and biologically inappropriate data analysis. Diabetologiai 1980;18:179-85. [DOI] [PubMed]
- 44.Zimmerman BR. Sulfonylureas. Endocrinol Metab Clin North Am 1997;26:511-21. [DOI] [PubMed]
- 45.Lebovitz HE. Oral therapies for diabetic hyperglycemia. Endocrinol Metab Clin North Am 2001;30;909-33. [DOI] [PubMed]
- 46.Strom BL, Schinnar R, Apter AJ, Margolis DJ, Lautenbach E, Hennessy S, et al. Absence of cross-reactivity between sulfonamide antibiotics and sulfonamide nonantibiotics. N Engl J Med 2003;349(17):1628-35. [DOI] [PubMed]
- 47.Hatorpe V. Clinical pharmacokinetics and pharmacodynamics of repaglinide. Clin Pharmacokinet 2002;41:471-83. [DOI] [PubMed]
- 48.McLeod JF. Clinical pharmacokinetics of nateglinide. Clin Pharmacokinet 2004; 43:97-120. [DOI] [PubMed]
- 49.Marbury T, Huang WC, Strange P, Lebovitz H. Repaglinide versus glyburide: a one-year comparison trial. Diabetes Res Clin Pract 1999;43(3):155-66. [DOI] [PubMed]
- 50.Hanefeld M, Bouter KP, Dickinson S, Guitard C. Rapid and short-acting mealtime insulin secretion with nateglinide controls both prandial and mean glycemia. Diabetes Care 2000;23(2):202-7. [DOI] [PubMed]
- 51.Nattrass M, Lauritzen T. Review of prandial glucose regulation with repaglinide: a solution to the problem of hypoglycaemia in the treatment of type 2 diabetes? Int J Obes Relat Metab Disord 2000;24(suppl 3):S21-S31. [DOI] [PubMed]
- 52.Hatorp V, Hansen KT, Thomsen MS. Influence of drugs interacting with CYP3A4 on the pharmacokinetics, pharmacodynamics, and safety of the prandial glucose regulator repaglinide. J Clin Pharmacol 2003;43:649-60. [PubMed]
- 53.Lister CA, Moore GBT, Piercy V, et al. Rosiglitazone, but not metformin or glibenclamide, improves glycaemic control and increases islet insulin content. Diabetologia 1999;42(suppl 1):A150 (abstract).
- 54.Finegood DT, McArthur MD, Kojwang D, Thomas MJ, Topp BG, Leonard T, et al. Beta-cell mass dynamics in Zucker diabetic fatty rats: rosiglitazone prevents the rise in net cell death. Diabetes 2001;50(5):1021-9. [DOI] [PubMed]
- 55.Bell DSH. Beta-cell rejuvenation with thiazolidinediones. Am J Med 2003; 115: 20S-3S. [DOI] [PubMed]
- 56.Aronoff S, Rosenblatt S, Braithwaite S, Egan JW, Mathisen AL, Schneider RL. Pioglitazone hydrochloride monotherapy improves glycemic control in the treatment of patients with type 2 diabetes: a 6-month randomized placebo-controlled dose-response study. The Pioglitazone 001 Study Group. Diabetes Care 2000;23:1605-11. [DOI] [PubMed]
- 57.Lebovitz HE, Dole JF, Patwardhan R, Rappaport EB, Freed MI; Rosiglitazone Clinical Trials Study Group. The rosiglitazone clinical trials study group: rosiglitazone monotherapy is effective in patients with type 2 diabetes. J Clin Endocrinol Metab 2001;86:280-8. [DOI] [PubMed]
- 58.Inzucchi SE, Maggs DG, Spollett GR, Page SL, Rife FS, Walton V, et al. Efficacy and metabolic effects of metformin and troglitazone in type II diabetes mellitus. N Engl J Med 1998;338(13):867-72. [DOI] [PubMed]
- 59.Horton ES, Whitehouse F, Ghazzi MN, Venable TC, Whitcomb RW. Troglitazone in combination with sulfonylureas restores glycemic control in patients with type 2 diabetes. Diabetes Care 1998;21(9):1462-9. [DOI] [PubMed]
- 60.Bakris G, Viberti G, Weston WM, Heise M, Porter LE, Freed MI. Rosiglitazone reduces urinary albumin excretion in type II diabetes. J Hum Hypertens 2003; 17(1):7-12. [DOI] [PubMed]
- 61.Herz M, Johns D, Reviriego J, Grossman LD, Godin C, Duran S, et al. A randomized, double-blind, placebo-controlled, clinical trial of the effects of pioglitazone on glycemic control and dyslipidemia in oral antihyperglycemic medication-naive patients with type 2 diabetes mellitus. Clin Ther 2003; 25 (4): 1074-95. [DOI] [PubMed]
- 62.Nesto RW, Bell D, Bonow RO, Fonseca V, Grundy SM, Horton ES, et al; American Heart Association; American Diabetes Association. Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the American Heart Association and American Diabetes Association. October 7, 2003. Circulation 2003;108(23):2941-8. [DOI] [PubMed]
- 63.Kelly IE, Hans TS, Walsh K, Lean ME. Effects of a thiazolidinediones compound on body fat and fat distribution of patients with type 2 diabets. Diabetes Care 1999;22:288-93. [DOI] [PubMed]
- 64.Avandia®, rosiglitazone maleate tablets [product monograph]. Mississauga: GlaxoSmith-Kline; 2001.
- 65.Actos®, (pioglitazone hydrochloride) tablets [product monograph]. Toronto: Eli Lilly Canada Inc.; 2001.
- 66.Takeda Pharmaceuticals North America Inc, Eli Lilly and Company. Dear Colleague letter about Actos. 2002. Available: www.fda.gov/medwatch/SAFETY/2002/summary-actos-avandia.PDF (accessed 2004 Nov 22).
- 67.Lee CH, Olson P, Evans RM. Minireview: lipid metabolism, metabolic diseases, and peroxisome proliferators-activated receptors. Endocrinology 2003; 144: 2201-7. [DOI] [PubMed]
- 68.Guerciolini R. Mode of action of orlistat. Int J Obes 1997;21(suppl 3):S12-S23. [PubMed]
- 69.Hollander PA, Elbein SC, Hirsch IB, Kelley D, McGill J, Taylor T, et al. Role of orlistat in the treatment of obese patients with type 2 daibetes. Diabetes Care 1998;21(8):1288-94. [DOI] [PubMed]
- 70.Hanefeld M, Sachse G. The effects of orlistat on body weight and glycaemic control in overweight patients with type 2 diabetes. Diabetes Obes Metab 2002; 4: 415-23. [DOI] [PubMed]
- 71.Kelley DE, Bray GA, Pi-Sunyer FX, Klein S, Hill J, Miles J, et al. Clinical efficacy of orlistat therapy in overweight and obese patients with insulin-treated type 2 diabetes: a 1-year randomized controlled trial. Diabetes Care 2002; 25 (6): 1033-41. [DOI] [PubMed]
- 72.Hermann LS, Schersten B, Melander A. Antihyperglycaemic efficacy, response prediction and dose-response relations of treatment with metformin and sulphonylurea, alone and in primary combination. Diabet Med 1994; 11: 953- 60. [DOI] [PubMed]
- 73.Rosenstock J, Shen SG, Gatlin MR, Foley JE. Combination therapy with nateglinide and thiazolidinedione improves glycemic control in type 2 diabetes. Diabetes Care 2002;25:1529-33. [DOI] [PubMed]
- 74.Garber AJ, Larsen J, Schneider SH, Piper BA, Henry D; Glyburide/Metformin Initial Therapy Study Group. Simultaneous glyburide/metformin therapy is superior to component monotherapy as an initial pharmacological treatment for type 2 diabetes. Diabetes Obes Metab 2002;4(3):201-8. [DOI] [PubMed]
- 75.Horton ES, Foley JE, Shen SG, Baron MA. Efficacy and tolerability of initial combination therapy with nateglinide and metformin in treatment-naïve patients with type 2 diabetes. Curr Med Res Opin 2004;20:883-9. [DOI] [PubMed]
- 76.Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, et al; Diabetes Prevention Program Research Grou9. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002;346(6):393-403. [DOI] [PMC free article] [PubMed]
- 77.Tuomilehto J, Lindstrom J, Eriksson JG, Valle TT, Hamalainen H, Ilanne-Parikka P, et al; Finnish Diabetes Prevention Study Group. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med 2001;344(18):1343-50. [DOI] [PubMed]
- 78.Pan XR, Li GW, Hu YH, Wang JX, Yang WY, An ZX, et al. Effects of diet and exercise in preventing NIDDM in people with impaired glucose tolerance. The Da Qing IGT and Diabetes Study. Diabetes Care 1997;20:537-44. [DOI] [PubMed]
- 79.Diabetes Prevention Program Research Group. Effects of withdrawal from metformin on the development of diabetes in the diabetes prevention program. Diabetes Care 2003;26:977-80. [DOI] [PMC free article] [PubMed]
- 80.Buchanan TA, Xiang AH, Peters RK, Kjos SL, Marroquin A, Goico J, et al. Preservation of pancreatic beta-cell function and prevention of type 2 diabetes by pharmacological treatment of insulin resistance in high-risk Hispanic women. Diabetes 2002;51(9):2796-803. [DOI] [PubMed]
- 81.Torgerson JS, Hauptman J, Boldrin MN, Sjostrom L. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care 2004;27(1):155-61. [DOI] [PubMed]
- 82.Chiasson JL, Josse RG, Gomis R, Hanefeld M, Karasik A, Laakso M; STOP-NIDDM Trial Research Group. Acarbose treatment and the risk of cardiovascular disease and hypertension in patients with impaired glucose tolerance (the STOP-NIDDM trial). JAMA 2003290(4):486-94. [DOI] [PubMed]