

Abstract
Circulating inflammatory mediators including complement activation products participate in the pathogenesis of cardiovascular diseases. As such, previous reports demonstrating the presence of complement proteins within atherosclerotic plaque and on the luminal surface would be anticipated. In contrast, we have recently made the unexpected observation that complement proteins also deposit along the external elastic lamina of mouse aortas in the absence of luminal deposition or plaque development. This suggests that complement activation may play a critical role in the pathogenesis of vascular stiffness and atherosclerosis through a mechanism initiated within the adventitia rather than on the endothelial surface. This hypothesis was tested in the current study by ultrastructural identification of the C3‐ and C4‐binding targets within the adventitia of the mouse aorta. The results demonstrate extensive binding of C3 and C4 to both collagen and elastin fibers within the adventitia in both ApoE(−/−) and C57Bl/6J control mice, as well as the presence of C3 and C4 within perivascular adipose tissue. These observations suggest a potential “outside‐in” mechanism of vascular stiffness during which perivascular adipose may produce C3 and C4 that bind to collagen and elastin fibers within the adventitia through covalent thiolester bonds, leading to increased vascular stiffness. Clin Trans Sci 2011; Volume 4: 146–152
Keywords: atherosclerosis, aorta, immunology, immunohistochemistry, inflammation, vasculature
Introduction
Traditionally, perivascular adipose tissue (PVAT) has been regarded as an inert fat storage organ and structrual support for the vasculature. More recently, visceral adipose has been increasingly recognized as the largest endocrine organ in the body and a source of numerous anti‐and proinflammatory mediators. 1 , 2 The proximity of PVAT adipocytes to the adventitia and access to the vasa vasorum suggests that secretion of adipokines, cytokines, and other mediators may directly influence vascular inflammation and participate in the pathogenesis of vascular stiffness and atherosclerosis. 3 , 4 , 5 Despite increasing recognition of a potential role for depot‐specific visceral adipose in the pathogenesis of cardiovascular disease, 1 links among PVAT, complement activation, vascular stiffening, and atherosclerosis have not been explored.
We have recently demonstrated specific deposition of complement proteins C3 and C4 along the peripheral aspect of the external elastic lamina (EEL) in murine aortas of both control and ApoE(−/−) models prior to endothelial plaque development. 6 We have also demonstrated that increasing amounts of complement deposition are associated with age‐related and disease‐related vascular stiffness in these mice in the absence of plaque or other evidence of atherosclerosis. 6 The current investigation was designed to determine the three‐dimensional morphology and ultrastructural features of the mouse aortic wall, to identify the targets of C3 and C4 binding within vascular adventitia, and to determine if PVAT may be a source of complement proteins that participate in this pathogenic process.
Methods
Animals
Atherosclerotic prone apolipoprotein deficient (ApoE[−/−]) mice with a B6 background and control mice (C57BL/6J) were purchased from Jackson Laboratories, Bar Harbor, ME, USA. We distinguished between young (#15 weeks) and old (>15 weeks) time points based on development of fatty streaks within the aorta and luminal lesion development along with increased vascular stiffness. Mice were housed at the University of Pittsburgh under standard conditions and fed a normal chow diet ad libitum. All experimental protocols were carried out in a blinded fashion and approved by the Institutional Animal Care and Use Committee.
Complement C3 and C4: immunofluorescence (IF)
Thoracic aortas were harvested after euthanasia by carbon dioxide inhalation. The descending aortic arch and celiac trunk were used as landmarks to define the beginning and end of the thoracic aorta. Immediately following excision, the aorta was placed in PBS (pH 7.3; BP665–1, Fisher Scientific, Pittsburgh, PA, USA). Vessels were cryopreserved by filling the lumen with OCT freezing medium (Sakura Finetek, 4583, Torrance, CA, USA), embedded vertically in a 5 mL beaker of OCT, and snap frozen with liquid nitrogen. Using a cryostat (Shandon Lipshaw, 620E, Pittsburgh, PA, USA), serial sections were cut at −17°C and mounted on Superfrost Plus glass slides (VWR, 48311–703, West Chester, PA, USA) and stored at −20°C until use.
Monoclonal C3 and C4 rat anti‐mouse primary antibodies (Cedarlane Laboratories, Burlington, NC, USA) and an isotype control IgG2a (BD Pharmingen, San Jose, CA, USA) were used with a fluorochrome‐conjugated goat anti‐rat secondary antibody Alex Fluor 555 (Invitrogen, Carlsbad, CA, USA) for IF analysis.
IF analysis
Slides were placed in a humidifying chamber, thawed, rehydrated with PBS, and washed in 0.5% BSA. A 2% BSA block was applied with subsequent 0.5% BSA washes and application of the C3 or C4 primary antibody or isotype at 5 μg/mL for 60 minutes. The slides were washed in 0.5% BSA and the secondary antibody was applied to all slides for 60 minutes. Selected samples were stained with DAPI to label nuclei. The slides were washed with 0.5% BSA followed by a PBS wash and coverslipped with Permafluor (Thermo Scientific, Brookfield, WI, USA). Slides were refrigerated and imaged under a confocal microscope (Olympus, Fluoview 500, Center Valley, PA, USA) within 24 hours.
Scanning electron microscopy (SEM)
Excised aortas were perfused and placed within a 2.5% glutaraldehyde bath and refrigerated for 60 minutes for morphological preservation. The aortas were sectioned longitudinally and cross‐sectionally to produce en face and transverse sections, respectively, washed in PBS, and submerged in 1% osmium oxide for 60 minutes. The specimens were dehydrated in a series of ethanol washes and rinsed in hexamethyldisilazane (HMDS). The specimens were placed onto electron microscopy (EM) stubs with conductive copper biadhesive tape, sputter coated with gold, and placed in the field emission scanning electron microscope (JEOL, Tokyo, Japan) at 3 kV.
Complement C3 and C4: immuno‐scanning electron microscopy (iSEM)
Monoclonal C3 and C4 rat anti‐mouse primary antibodies (Cedarlane Laboratories) and an isotype control IgG2a (BD Pharmingen) were used with a goat anti‐rat 18‐nm colloidal gold particle secondary antibody (Jackson ImmunoResearch, West Grove, PA, USA). Slides were processed in the same manner as the IF slides except that normal goat serum was used as the blocking agent and the secondary antibody was applied and refrigerated overnight. Upon completion, the slides were placed in 2.5% glutaraldehyde for 60 minutes at 4°C. The slides were dehydrated through a series of ethanol washes and processed with HMDS. The slides were placed on EM stubs with copper biadhesive tape, carbon coated, and placed in the field emission scanning electron microscope at 10 kV for back scatter imaging (BSI) and secondary electron imaging (SEI).
For iSEM analysis, two images were jointly obtained to capture both the nano‐scale gold primary at an extreme magnification (BSI approximately ×27,000) and a reduced macro‐scale magnification of the specimen (SEI approximately ×1,000) orienting the viewer to the labeling location on the specimen. For ease of identification, the 15‐nm and 18‐nm gold label secondary antibody attachment of the BSI were colored in red using Adobe Photoshop Elements 8.0 (Seattle, WA, USA).
Results
Nonuniform PVAT distribution surrounding the murine aorta
Initial studies were performed to determine the gross and ultrastructural morphology of the PVAT and its geometry with respect to the descending aorta. Histologic and SEM images demonstrated extensive PVAT surrounding the descending aorta, in both ApoE(−/−) and control mice. The aorta was completely surrounded by PVAT beginning at the aortic arch. However, progressing distally, the PVAT along the descending aorta developed into a trileaflet pyramidal distribution not previously reported in mice but observed in computed tomography anatomy studies of humans 7 ( Figure 1 ). The PVAT appeared to be tethered to the wall of the aorta at three specific sites on the vascular circumference.
Figure 1.
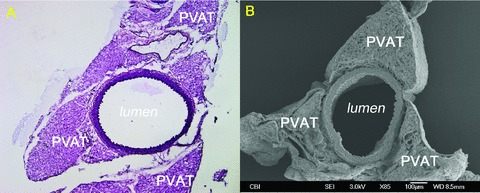
Trileaflet PVAT configuration. A 13‐week‐old control: (A) H&E (×10) and (B) SEM (×85).
Age and strain‐dependent complement accumulation in the vascular wall and surrounding PVAT
In previous studies, demonstrating C3 and C4 deposition along the EEL, it was not possible to determine if PVAT might be a source of the complement proteins because it had been stripped from the vessel wall during harvesting. Therefore, in these studies we confirmed the linear deposition of complement along the EEL and also demonstrated the presence of C3 and C4 throughout the surrounding PVAT in both control and ApoE(−/−) mouse strains at all ages. The high concentration of complement proteins at the EEL was maintained at young ages without deposition in the vascular wall or on the luminal surface prior to atherosclerotic lesion development within the lumen ( Figure 2 ). No substantial differences between mouse strains were noted. We also detected linear deposition of C3 and C4 surrounding the PVAT in both mouse strains at all ages ( Figure 3C ). At older ages, the ApoE(−/−) strain maintained complement deposition throughout the PVAT. With the development of atherosclerotic lesions in the lumen, the complement accumulation in the ApoE(−/−) mice then extended from the PVAT through the arterial wall and into the plaque ( Figure 3D–F ). Control mice at 36 weeks had the same accumulation of complement throughout the PVAT, but the deposition was interrupted at the EEL with limited labeling of the media ( Figure 3A–C ). We also noted increased cellular infiltrates in ApoE(−/−) specimens ( Figure 3D–F ).
Figure 2.
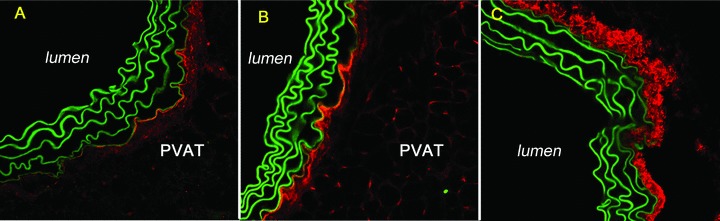
Linear deposition of complement proteins along the external elastic lamina (EEL) and throughout the surrounding PVAT. A 13‐week‐old control: (A) C3 40×, red; (B) C4 40×, red. Collagen type I pattern for control. (C) Collagen type I 40×, red.
Figure 3.
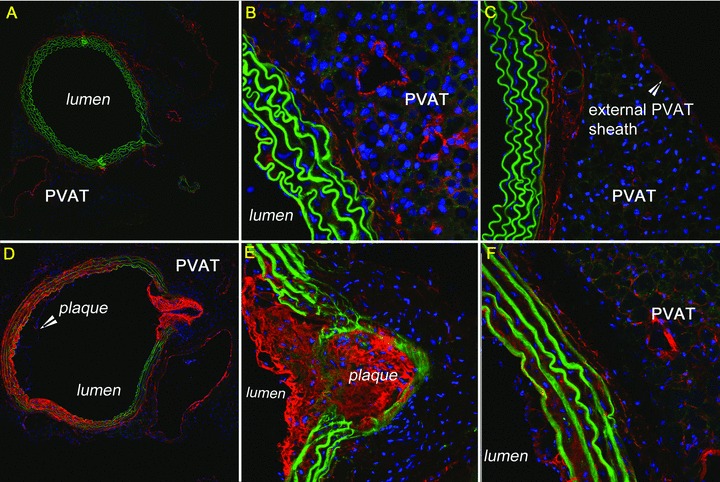
Deposition of complement proteins throughout surrounding PVAT. A 36‐week‐old control: (A) C3 10×, red; (B) C3 40×, red; (C) C4 40×, red. A 36‐week‐old ApoE(−/−) with plaque and lumen disruption: (D) C3 10×, red; (E) C3 40×, red; (F) C4 40×, red. Green = auto‐fluorescing elastin. Blue = nuclear stain.
Collagen Types I and III are present in the vascular wall and surrounding PVAT independent of age or strain
The distinct complement patterns detected along the EEL, throughout the adventitia, and surrounding the PVAT suggested structural proteins including collagen and elastin as possible complement‐binding sites. As seen in Figures 2 and 3 , the large bands of elastin fibers autofluoresce. Consequently, we evaluated the collagen distribution in the vascular wall and surrounding PVAT through confocal IF ( Figure 2C ). In both mouse strains, a heterogeneous mix of collagen types I and III (not shown) was identified throughout the media and adventitia extending into the extracellular matrix of the surrounding PVAT. In addition, a collagen type I sheath was detected surrounding the PVAT in both mouse strains at all ages ( Figure 3C ).
Morphological identification of collagen and elastin through SEM analysis
Prior to explicitly identifying the binding site(s) of complement proteins C3 and C4, we performed a rigorous characterization of the vascular wall and surrounding PVAT using SEM. In both mouse strains, regardless of age, the ECM components of the arterial wall including the thick, rolling elastin layers and the characteristic 67‐nm banding pattern of collagen were well preserved and were morphologically identifiable without the need for immunohistochemistry ( Figure 4A and B ). The luminal surface appeared undamaged and relatively smooth for both mouse strains in the absence of plaque, with the elastic lamina producing a rippled effect due to the preservation of aortic tissue in an unstressed or nonpressurized state ( Figure 4C ).
Figure 4.
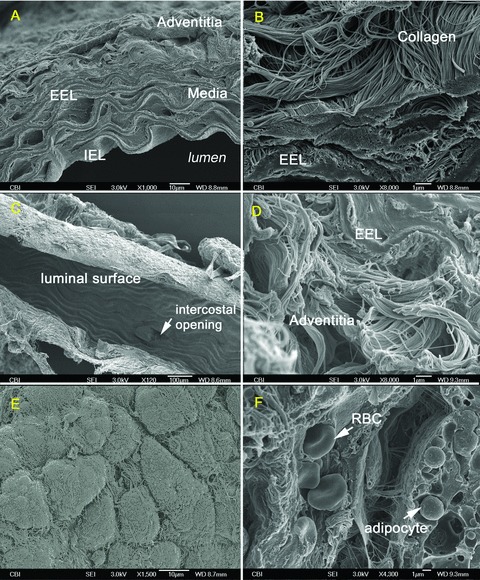
Three‐dimensional morphology and ultrastructure of aortic vascular wall through SEM. A 7‐week‐old control: (A) Aortic vascular wall, (B) EEL and adventitial collagen, (C) Luminal surface and intercostal opening. A 15‐week‐old ApoE(−/−): (D) EEL and adventitial collagen, (E) PVAT covered with collagen type I sheath, (F) RBC pressing through capillary within surrounding PVAT.
The surrounding PVAT was also readily identifiable surrounding the aorta in both strains regardless of age ( Figure 4E and F ) along with the external collagen type I sheath previously detected through IF ( Figure 4E ). An interesting observation made possible through SEM characterization was the apparent vascularization of the murine PVAT. This suggests an underlying vasa vasorum not previously described or thought to be necessary due to the small diameter of the murine vasculature ( Figure 4F ).
Complement C3 and C4 proteins bind to collagen and elastin
The distinct complement‐binding patterns observed through confocal IF suggested that the extracellular elastin and collagen fibers might be specific targets. Given the detail inherent in the SEM images and ability to identify both collagen and elastin, we utilized iSEM to determine specifically the structures that complement proteins were binding.
The iSEM studies confirmed the confocal IF results, demonstrating predominant binding of anti‐C3 ( Figure 5 ) and anti‐C4 ( Figure 6 ) monoclonal antibodies to the peripheral aspect of the EEL, throughout the adventitia, and to the surrounding PVAT at all ages regardless of strain. This is of note considering there is no evidence of luminal lesion development in either strain, yet there is a clear demarcation of complement at the EEL, adventitia, and PVAT at young ages.
Figure 5.
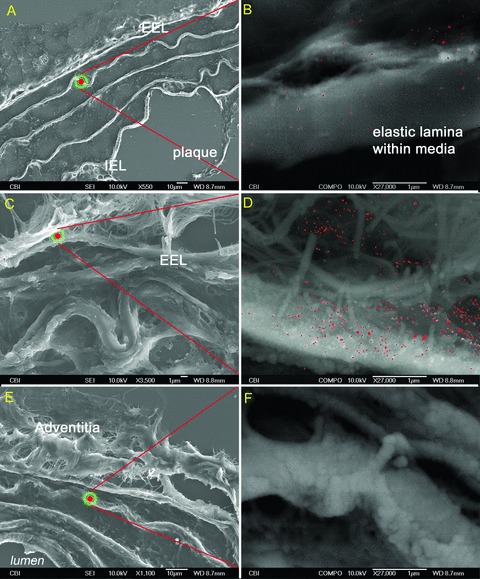
Complement protein C3 primary antibodies highlighted in red on 5‐μm sections of murine aortas. A 36‐week‐old ApoE(−/−): (A and B) SEI‐BSI, elastic lamina within media, 36‐week‐old control: (C and D) SEI‐BSI, isotype ApoE(−/−): (E and F) SEI‐BSI, elastic lamina within media, no complement label on specimen.
Figure 6.
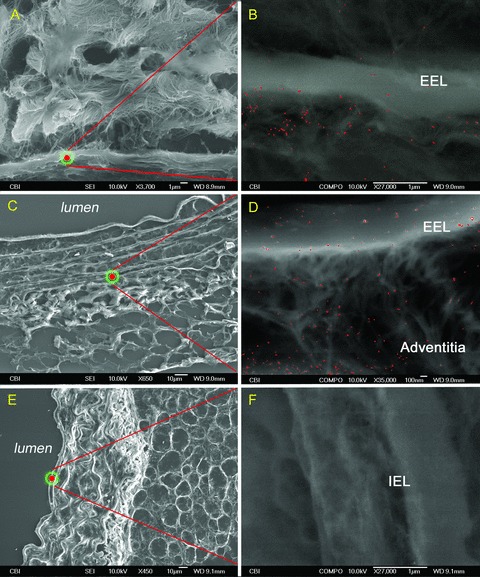
Complement protein C4 antibodies highlighted in red on 5‐μm sections of murine aortas. A 36‐week‐old ApoE(−/−): (A and B) SEI‐BSI, EEL into adventitia. A 36‐week‐old control: (C and D) SEI‐BSI, EEL extending through adventitia and PVAT. Isotype ApoE(−/−): (E and F) SEI‐BSI, internal elastic lamina, no complement label on specimen.
At older ages, this same pattern was present in both strains (C3: Figure 5A–D and C4: Figure 6A–D ); however, with the onset of luminal lesion formation in the ApoE(−/−) strain, the EEL boundary was no longer a line of demarcation and the accumulation of complement extended into the media, the internal elastic lamina, and atherosclerotic lesions (C3: Figure 5A and B and C4: Figure 6A and B ). The presence of C3 and C4 within the aorta of the control strain was consistently maintained within the EEL boundary (C3: Figure 5C and D and C4: Figure 6C and D ).
Discussion
This is the first study to rigorously characterize the three‐dimensional morphology and ultrastructure of the mouse aorta. The most important advance of these efforts is our capacity to demonstrate the binding of C3‐ and C4‐derived ligands to collagen and elastin fibers within the adventitia of the vascular wall. We also present evidence suggesting that perivascular adipose serves as the source of these complement proteins. Together, these observations support an “outside‐in” model of vascular stiffness and atherosclerosis. 8
High circulating levels of C3 and C4 have been correlated with increased rates of myocardial infarction in both healthy women and those with established coronary artery disease. 9 , 10 Traditional models have suggested this systemic source of complement as a participant in the inflammatory responses that occur during development of luminal atherosclerotic lesions. 11 , 12 , 13 , 14 This model can be referred to as the “inside‐out” theory of vascular pathogenesis. However, our previous studies had suggested an alternative role for complement proteins during progression of vascular stiffness and atherosclerosis. 6 In those studies, we had identified colocalization of C3 and C4 with collagen and elastin fibers in the adventitia not only during atherosclerosis but also during normal aging in the absence of plaque or other evidence of inflammation. In the current studies, we expanded these observations to examine the ultrastructural proximity of C3 and C4 to elastin and collagen and to identify a potential source of these proteins that were apparently targeted to the adventitial structures. The results demonstrate specific binding of C3‐ and C4‐derived ligands to elastin and collagen fibers and not to other components of the adventitia.
There are several plausible mechanisms that may be responsible for binding of complement proteins to collagen and elastin. Complement receptors are cell specific, ruling out a receptor–ligand interaction. Therefore, one particularly attractive mechanism would be covalent attachment of C3‐ and C4‐derived ligands via the unique thiolester bonds present within these molecules that are capable of forming ester or amide linkages after recognizing free hydroxyl or amino groups, respectively. 15 In the absence of complement regulatory proteins on elastin or collagen fibers, hydrolysis of C3 and/or C4 within the adventitia could readily lead to the patterns of deposition observed in the current study. Such direct binding of C3‐ and C4‐derived ligands to collagen and elastin might contribute to, or be a primary determinant of, increased vascular stiffness. This complement deposition may also stimulate or contribute to atherosclerosis.
In addition to, or as an alternative to, direct covalent attachment, C3‐ and C4‐derived molecules may bind indirectly to elastin and collagen. For example, fibromodulin, lumican, and decorin are members of the leucine‐rich repeat glycoprotein/proteoglycan family, which bind to the types of collagen found in the vascular wall. 16 , 17 , 18 Fibromodulin been found to bind C1q and activate the classical pathway, and has been identified throughout the aortic wall and atherosclerotic lesions. 19 Another participant in this mechanistic process might be adiponectin, which is produced by adipocytes, binds to the collagens of the vascular wall, and initiates complement activation. 20 , 21
In our previous studies, we had identified colocalization of C3 and C4 within elastin and collagen in the adventitia 6 , however the potential source of these proteins was not apparent, and the possible role of PVAT could not be determined, because it had been stripped from the aorta during harvest of the specimen. In these studies, the PVAT was preserved, allowing us to demonstrate the presence of C3 and C4 in a cytoplasmic pattern within adipose, suggesting the capacity of adipocytes to produce C3 and C4, which may then be targeted to the elastin and collagen within the adventitia under appropriate conditions. PVAT is in unique proximity to the external layers of the vascular wall. Previous studies have suggested a potential link between adipose and complement activation. 22 Several studies demonstrate that adipose inflammation is directly linked to the alternative pathway through the adipocyte production of adipsin (adipocyte‐derived human factor D), 23 , 24 factor B, and C3, 25 and in vivo data suggest adipose tissue activates complement in normal mice. 24 Dysfunction of the classical pathway has also been associated with murine models of obesity and insulin resistance through abnormal in vivo adipocyte and immune cell production of C1 complex proteins (C1q, C1r, C1s). 26 Most recently, a direct influence of adipose‐derived complement on the vasculature was demonstrated by a report showing that PVAT‐dervied C3 stimulated adventitial fibroblast migration and differentiation via the c‐Jun N‐terminal kinase pathway in a rat model of hypertension. 27
One unanswered question is the mechanism by which C3 and C4 and their derived ligands penetrate the EEL. During repair of vascular injury (i.e., atheroma) or remodeling of the vascular wall due to nonnormal stresses (i.e., hypertension), various mechanisms influence the integrity of the vascular wall including pro‐ and antiinflammatory production of chemokines, cytokines, and more recently noted adipokines that facilitate cellular migration through the vascular wall and increase vascular permeability. In addition, sustained vascular dysfunction can physically tear and degrade the vessel wall structural components including elastin and collagen, which triggers an additional inflammatory response to repair the injury. 28 , 29 , 30 This degradation of elastin and increased permeability of the vascular wall may facilitate complement deposition from both the PVAT as well as from the circulation, thus producing an inflammatory feedback loop.
Conclusion
Although complement activation is known to occur within atherosclerotic plaque and luminal lesions, we have demonstrated that the complement system may also independently target the adventitia during progression of vascular stiffness and atherosclerosis. C3‐ and C4‐derived ligands, generated from native proteins produced in the PVAT, may bind directly or indirectly to elastin and collagen fibers and contribute to an “outside‐in” mechanism of vascular pathogenesis.
Acknowledgments
The authors wish to thank Jonathan Franks for his instruction with use of the SEM and Dr. Kim Sutton‐Tyrrell and Dr. Janice Sabatine for their editorial assistance.
References
- 1. Thalmann S, Meier CA. Local adipose tissue depots as cardiovascular risk factors. Cardiovasc Res. 2007; 75(4): 690–701. [DOI] [PubMed] [Google Scholar]
- 2. Wang P, Xu TY, Guan YF, Su DF, Fan GR, Miao CY. Perivascular adipose tissue‐derived visfatin is a vascular smooth muscle cell growth factor: role of nicotinamide mononucleotide. Cardiovasc Res. 2009; 81(2): 370–380. [DOI] [PubMed] [Google Scholar]
- 3. Rajsheker S, Manka D, Blomkalns AL, Chatterjee TK, Stoll LL, Weintraub NL. Crosstalk between perivascular adipose tissue and blood vessels. Curr Opin Pharmacol. 10(2):191–196. [Epub Jan 7 2010]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chatterjee TK, Stoll LL, Denning GM, Harrelson A, Blomkalns AL, Idelman G, Rothenberg FG, Neltner B, Romig‐Martin SA, Dickson EW, et al Proinflammatory phenotype of perivascular adipocytes: influence of high‐fat feeding. Circ Res. 2009; 104(4): 541–549. [Epub Jan 2 2009]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. 1999; 340(2): 115–126. [DOI] [PubMed] [Google Scholar]
- 6. Santelices LC, Calano SJ, Erhart JC, Prantil RL, Haney JL, Vorp DA, Ahearn JM. Experimental system for ex vivo measurement of murine aortic stiffness. Physiol Meas. 2007; 28(8): N39–N49. [DOI] [PubMed] [Google Scholar]
- 7. Dean D, Herbener TE. Cross sectional human anatomy. Philadelphia: Lippincott Williams and Wilkins2000.
- 8. Maiellaro K, Taylor WR. The role of the adventitia in vascular inflammation. Cardiovasc Res. 2007; 75(4): 640–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Szeplaki G, Prohaszka Z, Duba J, Rugonfalvi‐Kiss S, Karadi I, Kokai M, Kramer J, Fust G, Kleiber M, Romics L, et al Association of high serum concentration of the third component of complement (C3) with pre‐existing severe coronary artery disease and new vascular events in women. Atherosclerosis. 2004; 177: 383–389. [DOI] [PubMed] [Google Scholar]
- 10. Muscari A, Bozzoli C, Puddu GM, Sangiorgi Z, Dormi A, Rovinetti C, Descovich GC, Puddu P. Association of serum C3 levels with the risk of myocardial infarction. Am J Med. 1995; 98: 357–364. [DOI] [PubMed] [Google Scholar]
- 11. Hansson GK, Jonasson L, Seifert PS, Stemme S. Immune mechanisms in atherosclerosis. Arteriosclerosis. 1989; 9(5): 567–578. [DOI] [PubMed] [Google Scholar]
- 12. Schwartz CJ, Kelley JL, Nerem RM, Sprague EA, Rozek MM, Valente AJ, Edwards EH, Prasad AR, Kerbacher JJ, Logan SA. Pathophysiology of the atherogenic process. Am J Cardiol. 1989; 64(13): 23G–30G. [DOI] [PubMed] [Google Scholar]
- 13. van der Wal AC, Das PK, Bentz van de Berg D, van der Loos CM, Becker AE. Laboratory Investigation. Atherosclerotic lesions in humans. In situ immunophenotypic analysis suggesting an immune mediated response. 1989; 61(2): 166–170. [PubMed] [Google Scholar]
- 14. Laine P, Pentikainen MO, Wurzner R, Penttila A, Paavonen T, Meri S, Kovanen PT. Evidence for complement activation in ruptured coronary plaques in acute myocardial infarction. Am J Cardiol. 2002; 90: 404–408. [DOI] [PubMed] [Google Scholar]
- 15. Law SK, Dodds AW. The internal thioester and the covalent binding properties of the complement proteins C3 and C4. Protein Sci. 1997; 6(2): 263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Svensson L, Närlid I, Oldberg A. Fibromodulin and lumican bind to the same region on collagen type I fibrils. FEBS Lett. 2000; 470(2): 178–182. [DOI] [PubMed] [Google Scholar]
- 17. Hedbom E, Heinegård D. Interaction of a 59‐kDa connective tissue matrix protein with collagen I and collagen II. J Biol Chem. 1989; 264(12): 6898–6905. [PubMed] [Google Scholar]
- 18. Scott JE. Proteodermatan and proteokeratan sulfate (decorin, lumican/fibromodulin) proteins are horseshoe shaped. Implications for their interactions with collagen. Biochemistry. 1996; 35(27): 8795–8799. [DOI] [PubMed] [Google Scholar]
- 19. Ström A, Ahlqvist E, Franzén A, Heinegård D, Hultgårdh‐Nilsson A. Extracellular matrix components in atherosclerotic arteries of Apo E/LDL receptor deficient mice: an immunohistochemical study. Histol Histopathol. 2004; 19(2): 337–347. [DOI] [PubMed] [Google Scholar]
- 20. Peake PW, Shen Y, Walther A, Charlesworth JA. Adiponectin binds C1q and activates the classical pathway of complement. Biochem Biophys Res Commun. 2008; 367(3): 560–565. [DOI] [PubMed] [Google Scholar]
- 21. Peake P, Shen Y. Factor H binds to the N‐terminus of adiponectin and modulates complement activation. Biochem Biophys Res Commun. 2010; 397(2): 361–366. [DOI] [PubMed] [Google Scholar]
- 22. van Greevenbroek MM. The expanding role of complement in adipose tissue metabolism and lipoprotein function. Curr Opin Lipidol. 2009; 20(4): 353–354. [DOI] [PubMed] [Google Scholar]
- 23. White RT, Damm D, Hancock N, Rosen BS, Lowell BB, Usher P, Flier JS, Spiegelman BM. Human adipsin is identical to complement factor D and is expressed at high levels in adipose tissue. J Biol Chem. 1992; 267(13): 9210–9213. [PubMed] [Google Scholar]
- 24. Choy LN, Rosen BS, Spiegelman BM. Adipsin and an endogenous pathway of complement from adipose cells. J Biol Chem. 1992; 267(18): 12736–12741. [PubMed] [Google Scholar]
- 25. Choy LN, Spiegelman BM. Regulation of alternative pathway activation and C3a production by adipose cells. Obes Res. 1996; 4(6): 521–532. [DOI] [PubMed] [Google Scholar]
- 26. Zhang J, Wright W, Bernlohr DA, Cushman SW, Chen X. Alterations of the classic pathway of complement in adipose tissue of obesity and insulin resistance. Am J Physiol Endocrinol Metab. 2007; 292: E1433–E1440. [DOI] [PubMed] [Google Scholar]
- 27. Ruan C‐C, Zhu D‐L, Chen Q‐Z, Chen J, Guo S‐J, Li X‐D, Gao P‐J. Perivascular adipose tissue‐derived complement 3 is required for adventitial fibroblast functions and adventitial remodeling in deoxycorticosterone acetate‐salt hypertensive rats. Arterioscler Thromb Vasc Biol. 2010; 30: 2568–2574. [DOI] [PubMed] [Google Scholar]
- 28. Sprague AH, Khalil RA. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol. 2009; 78(6): 539–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Onat A, Can G, Rezvani R, Cianflone K. Complement C3 and cleavage products in cardiometabolic risk. Clinica Chimica Acta. 2011; 412: 1171–1179. [DOI] [PubMed] [Google Scholar]
- 30. Prévost G, Bulckaen H, Gaxatte C, Boulanger E, Béraud G, Creusy C, Puisieux F, Fontaine P. Structural modifications in the arterial wall during physiological aging and as a result of diabetes mellitus in a mouse model: are the changes comparable Diabetes & Metabolism. 2011; 37: 106–111. [DOI] [PubMed] [Google Scholar]