

Abstract
(‐)‐Epicatechin (Epi), a flavanol in cacao stimulates mitochondrial volume and cristae density and protein markers of skeletal muscle (SkM) mitochondrial biogenesis in mice. Type 2 diabetes mellitus (DM2) and heart failure (HF) are diseases associated with defects in SkM mitochondrial structure/function. A study was implemented to assess perturbations and to determine the effects of Epi‐rich cocoa in SkM mitochondrial structure and mediators of biogenesis. Five patients with DM2 and stage II/III HF consumed dark chocolate and a beverage containing approximately 100 mg of Epi per day for 3 months. We assessed changes in protein and/or activity levels of oxidative phosphorylation proteins, porin, mitofilin, nNOS, nitric oxide, cGMP, SIRT1, PGC1α, Tfam, and mitochondria volume and cristae abundance by electron microscopy from SkM. Apparent major losses in normal mitochondria structure were observed before treatment. Epi‐rich cocoa increased protein and/or activity of mediators of biogenesis and cristae abundance while not changing mitochondrial volume density. Epi‐rich cocoa treatment improves SkM mitochondrial structure and in an orchestrated manner, increases molecular markers of mitochondrial biogenesis resulting in enhanced cristae density. Future controlled studies are warranted using Epi‐rich cocoa (or pure Epi) to translate improved mitochondrial structure into enhanced cardiac and/or SkM muscle function. Clin Trans Sci 2012; Volume 5: 43–47
Keywords: heart failure, type 2 diabetes, epicatechin, mitochondrial biogenesis, skeletal muscle
Introduction
Mitochondrial dysfunction is thought to be fundamental in the pathophysiology of both type 2 diabetes mellitus (DM2) and heart failure (HF). Abnormalities in skeletal muscle (SkM) of DM2 and HF patients include reduced mitochondrial numbers and/or cristae abundance which can lead to decreased oxidative capacity thereby compromising tissue bioenergetics and facilitating the development of muscle fatigue. 1 , 2 In animal models of DM2 and HF, signaling pathways involved in mitochondrial biogenesis including nitric oxide synthase (NOS) dependent nitric oxide (NO) production, cyclic guanosine monophosphate (cGMP), silent mating type information regulation 2 homolog (SIRTl), peroxisome activated receptor‐γ coactivator 1α (PGC1α) and transcription factor A, mitochondrial (Tfam) seem compromised and may explain mitochondrial abnormalities. 3 However, the involvement of these pathways has not been examined in SkM of DM2 and HF patients. A thorough understanding of possible perturbations in these pathways is necessary for the development of safe and effective therapies aimed at stimulating mitochondria biogenesis for the prevention and/or treatment of these and other diseases. Interestingly, only exercise‐based strategies have so far been demonstrated to stimulate mitochondria biogenesis in normal subjects or in patients with DM2 and/or HF.
We recently demonstrated that (‐)‐epicatechin (Epi), a flavanol present in cacao stimulates mitochondrial biogenesis in heart and SkM of mice leading to improved exercise performance. 4 Of relevance is that the consumption of dark chocolate has been associated with improved insulin sensitivity and reductions in the incidence cardiovascular diseases including HF. 5 , 6 A small preliminary proof of concept study was implemented to compare perturbations in mitochondrial structure and in molecular signatures of biogenesis in SkM of DM2 and HF patients before and after treatment with Epi rich cocoa.
Materials and Methods
Five patients with stable New York Heart Association (NYHA) stage II and III HF and DM2 were recruited from the San Diego Veterans Administration Medical Center. Patient exclusion criteria were age >75, creatinine clearance <30, or thiazolidinedione treatment. All patients had standard therapy for HF and DM2 and no changes were made to treatment during the course of the trial. This open label protocol involved patients eating four small chocolate squares and two cocoa research beverages per day for 3 months. Total Epi content per day was 100 mg with 390 calories and 18 g of fat. Patients had blood drawn for the measurement of hemoglobin A1c (HbA1c), lipid profile (cholesterol, HDL, LDL, triglycérides) and brain natriuretic peptide (BNP) levels and muscle biopsies from quadriceps femoris before and after cocoa consumption. This protocol was approved by the Institutional Review Board at University of California, San Diego and all subjects gave informed consent. Details of our methods are elaborated upon in the appendix.
Data analysis
Paired t‐test was used to analyze Western blots densitometric values, biochemical and electron microscopy results. BNP was analyzed by Wilcoxon nonparametric test, p < 0.05 was considered significant.
Results
All patients (age range 47–71 years) reported no adverse effects with treatment. Modest improvements (mean ± SD) were noted in HDL (38.2 ± 7.2 mg/dL vs. 44 ± 7.4 mg/dL, p < 0.05) and BNP levels (218.8 ± 206.3 pg/mL vs. 108.4 ± 94 pg/mL, p= 0.06) whereas no differences were noted in total cholesterol, LDL, triglycerides, HbAlc, blood pressure or body weight (data not shown). The average increase in plasma Epi concentration after cocoa administration was 1.06 ± 0.3 μM (SD). Images obtained from Western blots illustrate the stimulatory effects observed in SkM nNOS, p‐nNOS ( Figure 1A ), SIRT1, PGC1α (total and acetylated), Tfam ( Figure 2A‐C ), porin, mitofilin, Complex I (subunit NDUFB8), Complex V (α subunit) after treatment ( Figure 3A and B ). The analysis of densitometric values (before vs. after) shows statistically significant differences for all of the measured variables (p < 0.05). We were unable to detect eNOS in SkM. Cocoa also significantly increased production of NO in SkM (indirectly measured as nitrates/nitrites) ( Figure 1B ) and cGMP levels ( Figure 1C ). A representative example of changes in SkM mitochondrial ultrastructure before and after treatment is shown in Figure 4 . Cristae abundance increased significantly, whereas no differences were observed in mitochondrial volume density ( Figure 4 ).
Figure 1.
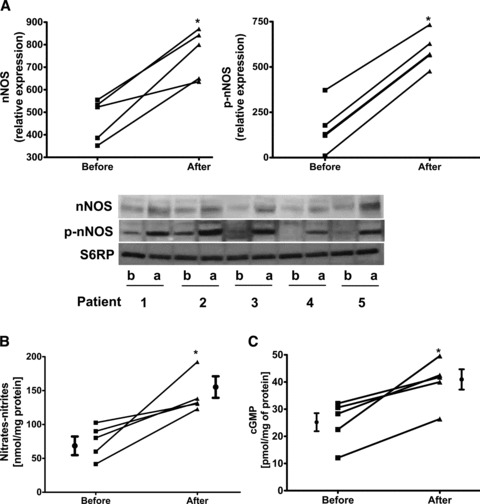
Comparison of changes observed in protein or enzyme activity levels before (b) and after (a) treatment with Epi‐rich cocoa. (A) neuronal‐NOS and phosphorylated‐NOS. Western blots were normalized against S6RP. (B) Changes in skeletal muscle NO production (measured as nitrates/nitrites) and (C) changes in cGMP levels. Significant differences (*p < 0.05) were observed before vs. after treatment by paired t‐test.
Figure 2.
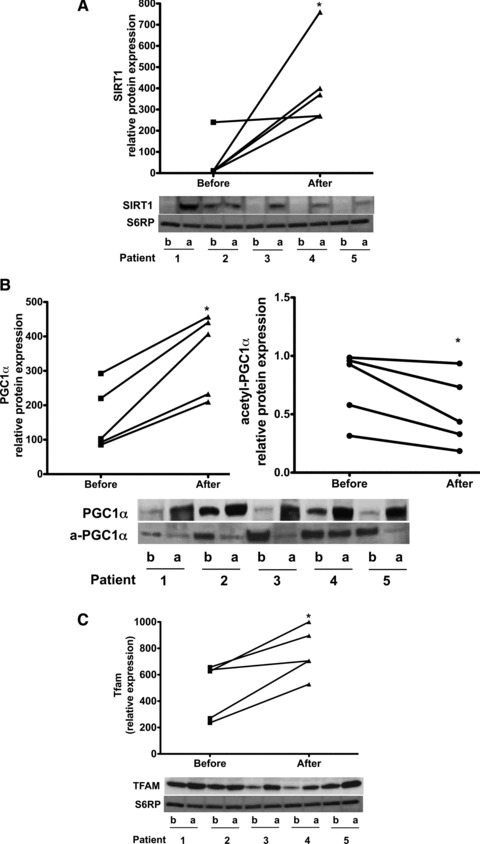
Comparison of changes observed in protein levels before (b) and after (a) treatment with Epi‐rich cocoa in: (A) SIRT1, (B) PGC1α (including its acetylated form) and (C); Tfam. PGC1α was immunoprecipitated with an antibody against total PGC1α and the blot probed with an anti‐acetyl‐lysine antibody to detect protein acetylation (a‐). Western blots were normalized against S6RP. Significant changes (*p < 0.05) were observed in all endpoints by paired t‐test.
Figure 3.
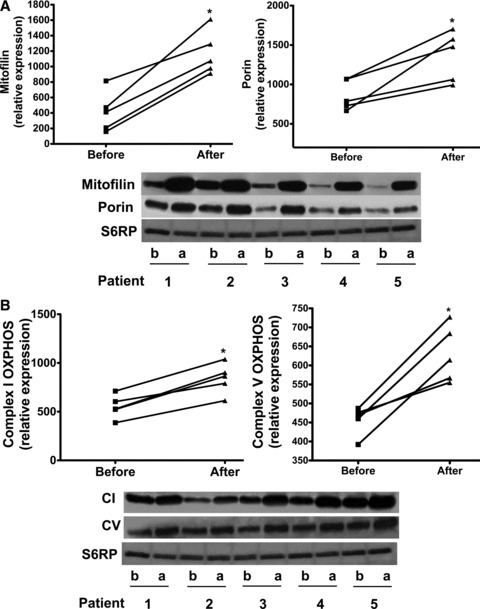
Comparison of changes observed in protein levels before (b) and after (a) treatment with Epi‐rich cocoa in: (A) Mitofilin and porin and, (B) complex I and complex V. Western blots were normalized against S6RP. Significant changes (*p < 0.05) were observed in all endpoints by paired t‐test.
Figure 4.
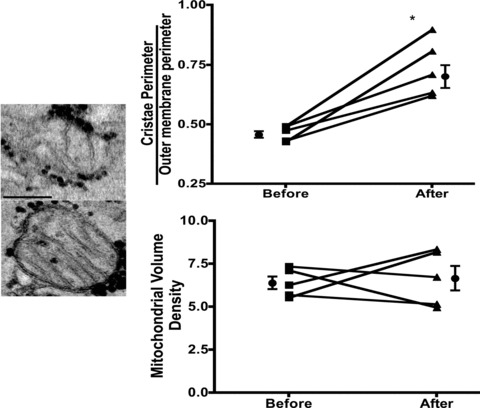
Microphotographs illustrating representative changes observed in mitochondrial structure of one patient before and after treatment with Epi‐rich cocoa. Plots summarize changes observed in mitochondrial cristae abundance and volume density in all patients before and after Epi‐rich cocoa treatment. *p < 0.05 by paired t‐test.
Discussion
Novel observations derived from this study indicate that in HF and DM2 patients, there is a remarkable deficiency in cristae abundance and overall disarray in mitochondrial structure that is accompanied by apparent prominent reductions in SkM protein levels of several molecular markers of mitochondrial biogenesis. The levels of molecules suspected to play important roles in the upstream stimulation of biogenesis including those of nNOS, p‐nNOS (i.e., phosphorylation status), and cGMP were low compared to the quantities after cocoa treatment. Similarly, more downstream markers in some cases were either not detectable as in SIRT1, barely detectable as in PGC1α or decreased as in Tfam. Mitochondrial components of cristae and outer membrane structure (mitofilin, porin) and function (complex I and V) also were low compared to the quantities after cocoa treatment. These results correlate with severe reductions in mitochondria cristae and because cristae are where oxidative phosphorylation occurs, it is reasonable to assume that the capacity to produce ATP is constrained.
Several results derived from this study are also first in kind, including the fact that treatment of patients with Epi‐rich cocoa for 3 months increased markers of SkM mitochondrial biogenesis. The acetylation of PGC1α was decreased with treatment suggesting a functionally enhanced biogenesis stimulus. Mitofilin and porin levels increased. These effects correlated with a striking 59% increase in mitochondrial cristae abundance and higher levels of complex I and V proteins.
The impetus driving this preliminary study was our recent results using mice gavaged with 1 mg/kg of Epi BID for 15 days. 4 Results indicated that components of oxidative phosphorylation complexes, nNOS, p‐nNOS, mitofilin, porin, and Tfam were significantly higher in hindlimb and cardiac muscles with Epi treatment. SkM mitochondrial volume and cristae abundance also increased. In this proof of concept trial, we aimed to provide a dose of Epi, via cocoa consumption, which was in the range for that used in our animal studies. The duration of treatment took into account the severe nature of the patient's pathology where a longer stimulus may be required to achieve the desired effect.
Drexler et al. (1992) reported that patients with severe HF develop ultrastructural abnormalities in SkM including a loss of approximately 20% in mitochondrial volume and cristae surface area. 1 These changes correlated with depressed oxidative capacity of muscle and thus, may relate to the impaired exercise capacity of HF patients. The loss of cristae surface area in our patients before treatment was apparently more severe, as values (∼0.5) are notably lower versus those reported in normal subjects. 1 Therapies aimed at ameliorating or even reversing these structural alterations would be desired as they may improve the quality of life of patients and potentially limit some of the deleterious effects of HF. One tested strategy is exercise/endurance training. In HF patients, exercise training seemed safe, increased mitochondrial inner surface membrane, improved exercise capacity, helped to attenuate abnormal HF pathophysiology and yielded a modest reduction in clinical events. 7 However, the use of exercise as a means to improve mitochondria structure can be problematic in patients with physical limitations thus, the need to identify alternate and safe strategies that trigger such effects.
Similar to HF, disruption of mitochondria in SkM is evident in insulin‐resistant subjects years before they develop DM2. 2 Exercise capacity is known to be reduced in DM2 patients. 8 There are controversial reports indicating possible decreases in the number of SkM intermyofibrillar mitochondria in DM2 patients. 9 , 10 What seems to be clear, are the positive effects that exercise has on mitochondrial density and enzyme activity in DM2 patients 11 whereas those of thiazolidinediones are at best, inconclusive. 12 , 13
There is a dearth of published science pertaining to the nature of changes observed in markers of SkM mitochondria biogenesis as it relates to either changes in protein levels and/or activity in either HF or DM2 patients. Essentially, reports have been limited to the evaluation of changes in SkM gene expression of DM2 patients, where reductions are observed in PGC1α/β and nuclear respiratory factor 1 (NRF‐1) levels. 2 Interestingly, whereas treatment of DM2 patients with rosiglitazone increased PGC1α SkM mRNA, no changes were observed in protein levels. 13
The results provided in this study of baseline and posttreatment changes in mediators of biogenesis in patients with HF and DM2 are without precedent. The stimulatory effects of Epirich cocoa on cristae density are consistent with increased markers of mitochondrial biogenesis. The absence of an effect on total mitochondrial volume density may suggest that the biogenesis response was incomplete. Whether this relates to the Epi dose, the duration of treatment, or a more complex issue in signaling of biogenesis in this cohort of patients with profound illness is unclear. Future studies would need to explore these possibilities.
Limitations
Although we hypothesize that Epi is primarily responsible for the changes observed, this study cannot rule out the possibility that other compounds in cocoa contribute to mitochondrial effects. Also, this study was not designed and does not provide evidence linking changes in mitochondrial structure with muscle function. Furthermore, this proof‐of‐concept study did not include the use of a placebo treatment or control subjects (healthy).
Conclusions
In this proof of concept study, we demonstrate major derangements in mitochondrial structure in a profoundly ill cohort of patients with HF and DM2. Promising restorative effects on these mitochondrial parameters are observed using inexpensive and safe dietary Epi‐rich cocoa products. Results warrant the implementation of controlled studies using Epi‐rich cocoa (or pure Epi) that aim to translate improved mitochondrial structure into enhanced cardiac and/or SkM muscle function.
Funding Sources
This study was supported by NIH AT4277, HL43617, P60‐MD000220 grants to Dr. Villarreal. Dr. Taub is supported by an American College of Cardiology/Merck Fellowship and a research grant from The Hershey Company. Part of the work was performed at the National Center for Microscopy and Imaging Research supported by NIH RR004050.
Disclosures
None.
Acknowledgments
We would like to thank Dr. Arrash Fard, Dr. Navaid Iqbal, and Paulette Gabbai‐Saldate for their assistance with patient recruitment and follow‐up.
References
- 1. Drexler H, Riede U, Munzel T, Konig H, Funke E, Just H. Alterations of skeletal muscle in chronic heart failure. Circulation. 1992; 85(5): 1751–1759. [DOI] [PubMed] [Google Scholar]
- 2. Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Konahe I, Costello M, R Saccone, al et. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci USA. 2003; 100(14): 8466–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nisoli E, Clementi E, Carruba MO, Moncada S. Defective mitochondrial biogenesis: a hallmark of the high cardiovascular risk in the metabolic syndrome? Circ Res. 2007; 100(6): 795–806. [DOI] [PubMed] [Google Scholar]
- 4. Nogueira L, Ramirez‐Sanchez I, Perkins G, Murphy A, Taub P, Ceballos G, Villarreal F, Hogan M, Malek M. (‐)‐Epicatechin enhances fatigues resistance and oxidative capacity in mouse muscle. Journal of Physiology. 2011; 589(18): 4615–4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Corti R, Flammer AJ, Hollenberg NK, Luscher TF. Cocoa and cardiovascular health. Circulation. 2009; 119(10): 1433–1441. [DOI] [PubMed] [Google Scholar]
- 6. Mostofsky E, Levitan EB, Wolk A, Mittleman MA. Chocolate intake and incidence of heart failure: a population‐based prospective study of middle‐aged and elderly women. Circ Heart Fail 2010; 3(5): 612–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Keteyian SJ, Fleg JL, Brawner CA, Pina IL. Role and benefits of exercise in the management of patients with heart failure. Heart Fail Rev. 2010; 15(6): 523–530. [DOI] [PubMed] [Google Scholar]
- 8. Regensteiner JG, Sippel J, McFarling ET, Wolfel EE, Hiatt WR. Effects of non‐insulin‐dependent diabetes on oxygen consumption during treadmill exercise. Med Sci Sports Exerc. 1995; 27(5): 661–667. [PubMed] [Google Scholar]
- 9. Nielsen J, Mogensen M, Vind BF, Sahlin K, Hojlund K, Schroder HD, Ortenblad N. Increased subsarcolemmal lipids in type 2 diabetes: effect of training on localization of lipids, mitochondria, and glycogen in sedentary human skeletal muscle. Am J Physiol Endocrinol Metab. 2010; 298(3): E706–E713. [DOI] [PubMed] [Google Scholar]
- 10. Chomentowski P, Coen PM, Radikova Z, Goodpaster BH, Toledo FG. Skeletal muscle mitochondria in insulin resistance: differences in intermyofibrillar versus subsarcolemmal sub‐populations and relationship to metabolic flexibility. J Clin Endocrinol Metab. 2011; 96(2): 494–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Toledo FG, Menshikova EV, Ritov VB, Azuma K, Radikova Z, DeLany J, Kelley DE. Effects of physical activity and weight loss on skeletal muscle mitochondria and relationship with glucose control in type 2 diabetes. Diabetes. 2007; 56(8): 2142–2147. [DOI] [PubMed] [Google Scholar]
- 12. Pagel‐Langenickel I, Schwartz DR, Arena RA, Minerbi DC, Johnson DT, Waclawiw MA, Cannon RO 3rd, RS Balaban, Tripodi DJ, Sack MN. A discordance in rosiglitazone mediated insulin sensitization and skeletal muscle mitochondrial content/activity in Type 2 diabetes mellitus. Am J Physiol Heart Circ Physiol. 2007; 293(5): H2659–H2666. [DOI] [PubMed] [Google Scholar]
- 13. Mensink M, Hesselink MK, Russell AP, Schaart G, Sels JP, Schrauwen P. Improved skeletal muscle oxidative enzyme activity and restoration of PGC‐1 alpha and PPAR beta/delta gene expression upon rosiglitazone treatment in obese patients with type 2 diabetes mellitus. Int J Obes (Lond). 2007; 31 (8): 1302–1310. [DOI] [PubMed] [Google Scholar]