

Abstract
Accumulating epidemiological studies have implicated a strong link between age associated metabolic diseases and cancer, though direct and irrefutable evidence is missing. In this review, we discuss the connection between Warburg effects and tumorigenesis, as well as adaptive responses to environment such as circadian rhythms on molecular pathways involved in metabolism. We also review the central role of the sirtuin family of proteins in physiological modulation of cellular processes and age-associated metabolic diseases. We also provide a macroscopic view of how the circadian rhythm affects metabolism and may be involved in cell metabolism reprogramming and cancer pathogenesis. The aberrations in metabolism and the circadian system may lead to age-associated diseases directly or through intermediates. These intermediates may be either mutated or reprogrammed, thus becoming responsible for chromatin modification and oncogene transcription. Integration of circadian rhythm and metabolic reprogramming in the holistic understanding of metabolic diseases and cancer may provide additional insights into human diseases.
Keywords: Age-associated diseases, circadian rhythm, metabolic reprogramming, sirtuin, tumorigenesis, Warburg effect
1. Introduction
Modern society faces age associated health care problems globally. Age-associated abnormality in metabolic processes and functions lead to propensity to cancer and an array of metabolic diseases posing a huge challenge to human health. Metabolic diseases include obesity, diabetes and cardiovascular disease. In recent years, epidemiological studies have suggested a surprising association between metabolic diseases and an increased risk of multiple types of cancer, and have recommended precautionary measures [1-3] (Table 1). For example, a molecular mechanism of tumorigenesis in obesity has been proposed, involving secretion of adipokines (leptin, adiponectin and increased inflammatory cytokines) and secondary effects of obesity that increase anabolic pathways, such as insulin-insulin growth factor (IGF) signaling [4]. However, the biological mechanisms remain poorly understood, which hinders the development of therapeutic strategies for cancer prevention and treatment in patients with metabolic diseases.
Table 1.
The recent epidemiological studies relating metabolic disease and cancer risk.
| Metabolic disease | Cancer risk | Country/Population | Reference |
|---|---|---|---|
| Hyperglycemia | Renal cell and liver cancer in men; Endometrial and pancreatic cancers in women are increased | Europe; Taiwan | [161, 162] |
| Diabetes | Colon, liver, pancreatic, endometrial and kidney, esophagus, rectum (F), stomach (F), thyroid (F), brain (F), lung (F), bladder, biliary tract and ovary cancer risks are increased | Sweden; Netherlands; Australia; African Americans; Native Hawaiians, Japanese Americans; Taiwan, Italy; USA; Japan; Hong Kong; Netherlands; Canada | [161-175] |
| Metabolic syndrome | Colorectal neoplasm; Prostate cancer; liver cancer risks are increased | Taiwan; Canada; England | [177-179] |
| Obesity | Esophageal, thyroid, liver, biliary tract, colorectal, ovary, gastric, breast, prostate, lung cancer risks are increased | USA; Europe; Japan; African American; Australia; Italy | [180-185] |
| Metformin use | Reduced breast, prostate, colorectal cancer risk and mortality, increased survival with gastric cancer | Canada; USA; Denmark; Taiwan; Korea | [186-191] |
At the cellular level, metabolism provides essential energy and substrates for maintenance of cellular function, support of cell growth and stimulation of cell proliferation. Reprogramming of glucose metabolism, represented by enhanced glycolysis and glucose uptake, has been considered as a hallmark of cancer in recent years [5]. Further, the abnormal metabolism in cancer cell may have a crucial role in the discovery of new therapeutic targets. In addition to the tendency of cancer cells to accumulate glutamine, acetate has also been highlighted as a new carbon source for consumption during cancer cell metabolism [6]. The disproportionate usage of these fuel sources illustrates a metabolic adaptation to allow tumor proliferation and is a consequence of metabolic reprogramming. The hypotheses proposed to decipher this reprogramming include oncogenes driving the aberration of specialized metabolic pathways, metabolic enzyme mutations and epigenetic modifications.
In biological systems, in addition to metabolism, circadian rhythms can also adapt to environmental change, and display conservative modulation in response to day/night, feed/starvation, warm/cold, and so on [7]. The latest report indicates that longer exposure to light fluctuates circadian rhythms and induces detrimental effects on biological processes[8]. And even use of electronic devices that emit light, could be enough to affect the circadian clock [9]. An intertwinement between circadian rhythms and metabolic diseases has also been proposed [10]. The emerging evidence indicates that the intrinsic clock is tightly coupled with different aspects of metabolic processes [11]. Endocrine factors that mediate metabolism are also orchestrated by the circadian clock, disorders of which influence physiological parameters such as blood pressure, as well as endocrine secretion, cell cycle, DNA repair etc. [12]. Based on this knowledge, chronotherapy has been applied for treatment of various diseases, for example in anticancer treatment, leading to optimized therapy plans for cancer patients [13]. The complex interactions between circadian rhythms, metabolism and human diseases are expected to shed light on the study of tumorigenesis and aging, which may have association with behavior, nutrient supplementation and energy consumption.
2. Initialization of Warburg effect
Almost a hundred years ago, Otto Warburg demonstrated how cancer cells predominately consume glucose through aerobic glycolysis, generating lactic acid even in the presence of oxygen [14]. The process observed in cancer cells result in glycolytic rates up to two hundred-fold higher than that in normal cells. Warburg postulated that dysfunctional mitochondria were the main cause of this change, and has been later called Warburg effect [15]. This interpretation was widely accepted by Warburg’s contemporaries and evolved in this century into a theory of adaptation to hypoxic conditions in pre-malignant lesions [16]. However, the root of this typical cancer metabolism has been examined further by scientists recently and the complexity underlying the mechanism of the Warburg effect is being unraveled [17, 18].
It appears that cell reprogramming may take place in cancer cells, and is responsible for metabolic change. Mounting evidence shows that oncogene/tumor suppressors are involved in this process [19, 20]. For example, the oncogene Myc and hypoxia-inducible factor 1 (HIF-1) have been reported to regulate most of the glycolytic enzymes that lead to enhanced glycolysis. In addition to the secondary effects on cancer cells, driven by metabolic reprogramming, enhanced anabolic metabolism would also appear in environments rich in nutrients and growth factors [21, 22]. More promisingly, in recent years, metabolic enzymes and metabolites have been reported to be involved in regulating gene transcription in addition to their roles in metabolism, for example oncoprotein (PKM2) and oncometabolite (2HG). This control of oncogene and tumor suppressor transcription by metabolic enzymes will be discussed in following sections.
3. Cell proliferation and metabolism are tightly constrained by each other
Using unbiased screening for cDNA that can immortalize mouse embryonic fibroblasts (MEFs), scientists have identified two kinds of glycolytic enzymes, phosphoglycerate mutase (PGM) and glucosephosphate isomerase (GPI), which can enhance glycolysis, allowing uncontrolled proliferation and escaping Ras-induced arrest [23]. This observation led to a hypothesis that glycolytic activation contributes to immortalization of cells. Following this, another group of scientists published a study in Science that demonstrated a 4- to 5-hour metabolic cycle (alternating between glycolysis and respiration) in yeast cultured in a nutrient-limited medium, which mimics growth conditions in the wild. DNA replication occurred only in the glycolytic phase [24]. Moreover, disruption of DNA checkpoint kinase, which links the cell cycle to the circadian rhythm, will lead to synchrony between the metabolic cycle and the cell division cycle. This also shed light on the associations among metabolism, the circadian rhythm and cell proliferation. The circadian clock has been proposed as a bridge between metabolism and cancer [25].
That cell proliferation and metabolism are tightly constrained with each other give us a clue about the mediators involved in this situation. This point can be illustrated with the following examples: when a cell is proliferating, a mediator is needed to “tell” the metabolism that glycolysis is an appropriate method to choose for now. In contrast, when the cell begins glycolysis to prepare the substrates for macromolecular synthesis, a signal can be transferred to the cell cycle with “ready” messages. Therefore, it is not difficult to understand that oncogenes and tumor suppressors can regulate metabolic pathways. For instance, a key function of Myc in tumor cells is to promote utilization of glutamine in order to provide an extra nitrogen and carbon source to fulfill rapid proliferation of the cells, and the function of tumor suppressor p53 is mediated by nutrient deprivation that increases the expression of p53 isoform and relocalization of region-binding protein 1 (SMAR) [26]. In contrast, the metabolic enzymes Akt and AMP-activated protein kinase (AMPK) are now considered as oncogene and tumor suppressor, respectively. Furthermore, reports have revealed common regulatory pathways that are shared between metabolism and proliferation in cancer cells [20]. Some groups targeting glutamine metabolism have demonstrated positive effects against tumor proliferation [27]. Therefore, question can be raised: can metabolic status gate cell cycle entry (Fig. 1)?
Figure 1. The metabolic cycle gates cell cycle entry.
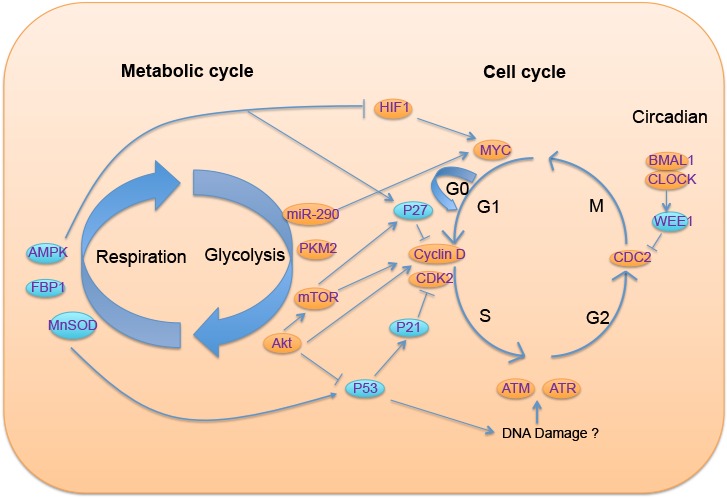
On the left is the metabolic cycle with two phases, glycolysis and respiration; different colors of circular icon represent either suppressors (blue) or oncogenes in tumorigenesis (orange); on the right is the cell cycle, in which the regulatory relationship of mediators between these two cycles have been illustrated.
4. New advances in metabolism and cancer---glycolytic factors tuning tumorigenesis
The explanation of initialization of the Warburg effect can no longer be satisfied with the demands of rapid anabolic biosynthesis [18]. Evidence is mounting that metabolic pathways and signal transduction are reciprocally regulated, rather than as separate entities. This means not only that oncogenic signaling is regulated by increased nutrient uptake and disorders of cell metabolism, but also that the signal transduction pathway can be modified by metabolic status and has effects on cell physiology. This pathway of “metabolism gates cell cycle entry” involves: (a). the nutrient-sensor, AMP-activated protein kinase, AMPK [28] and anabolic pathways PI3K/Akt [29]; (b). Availability of “oncometabolites”, the metabolites involved in protein modification, such as acetyl-CoA, 2-hydroxyglutarate (2-HG) [30]; and (c). Glycolytic factors (enzymes, miRNA) that serve as mediators with multiple functions in both cell metabolism and tumorigenesis.
4.1 Anabolic pathways driving metabolic disorders in tumorigenesis
In a normal cell, growth, proliferation and differentiation are precisely controlled by a series of extra- and intracellular factors. Among them, growth factors directly signal cells to increase nutrient uptake and enhance anabolic metabolism [31]. Under the condition that growth factor signaling has been constitutively activated through an appropriate receptor, increased anabolic metabolism is observed in transformed cancer cells. Influenced by this, deregulation of several metabolic pathways has been proposed to be involved in tumorigenesis.
Many experiments have directly explored the effects of energy expenditure on tumor progression. Feeding Drosophila a high sugar diet led to enhanced tumor growth; a similar result was not found when the flies were fed a high-fat diet [32]. The tumorigenesis associated with a high-carbohydrate diet requires the insulin/PI3K pathway, which has been evaluated by both PI3K gene modification and pharmacologic approaches. Moreover, it has been found, via Wingless-mediated insulin receptor upregulation in a Ras/Src tumor model, that tissues are in fact insulin sensitive rather than resistant. This provides evidence that, although sugar might promote insulin resistance, it will also enhance tumor formation in an insulin/PI3K-dependent manner [32]. The activation of the PI3K/Akt/mTOR pathway has already been discussed as an oncogenic factor that has multiple effects, including provoking the Warburg effect (Fig. 2). Akt can phosphorylate and activate ATP-citrate lyase (ACLY), which is responsible for converting citrate back to acetyl-CoA in the cytoplasm. Owing to this process, it can prevent a cytosolic accumulation of citrate that negatively regulates glycolysis and maintains the intracellular level of acetyl-CoA. By inducing Gcn5p/SAGA-catalyzed histone acylation, which eventually influences transcription of growth factor, acetyl-CoA represents a critical metabolic intermediate and carbon source in biological processes that may also be involved in tumorigenesis [33]. Thus, cancer cells harbor an abnormal tendency to metabolize glutamine and acetate as carbon sources, which has been highlighted in recent years [6].
Fig 2. Enhanced anabolic PI3K/Akt pathway in a cancer cell.
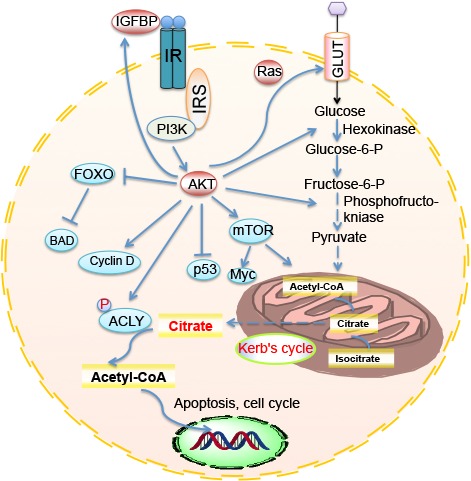
PI3K/Akt/mTOR pathways are involved in 1. Glycolysis; 2. Cell growth; 3. Cell cycle; 4. Cell survival.
With regard to metabolic enzymes, studies have demonstrated the pivotal role of ACLY in cell proliferation and tumorigenesis in vitro and in vivo [34, 35]; it is also required for histone acylation in response to growth factor stimulation [36]. Higher expression of ACLY was indeed found in human lung adenocarcinoma [37]. Inhibition of ACLY resulted in cell growth arrest both in vitro and in vivo, showing its possible role in cancer therapy. Moreover, it has been found that, in addition to PI3K, another ligase (Skp2-SCF) can also regulate glycolysis and tumorigenesis through ubiquitination of Akt [38]. This testifies the core position of the Akt node in controlling the metabolic balance and normal physiology of cells by controlling a series of downstream signals, from cell growth (mTOR) and the cell cycle (cyclin D) to cell apoptosis (Bad) [3, 39]. In addition to ACLY, another Akt-activated glycolytic enzyme, hexokinase, responsible for phosphorylating glucose, has been demonstrated to be essential for nuclear repression of HXK1 (hexokinase) and GLK1 (glucokinase) in yeast and to modulate apoptosis through interaction with Bad [40, 41]. These are not the only examples of the multifaceted functions of a glycolytic enzyme [42], and more new evidence to testify that these glycolytic factors are involved in tumorigenesis remains to be explored.
Furthermore, it has been reported that Akt regulates the expression of insulin-like growth factor binding protein-3 (IGFBP-3) in human non-small cell lung cancer (NSCLC) cells [43]. IGFBP-3 induces G1 cell cycle arrest and apoptosis in several cancer cell lines, including human NSCLC [44]. The alteration of this protein by oncogenic Akt can be considered as a new cancer therapeutic target. Lastly, it has been reported that Akt activation is a key determinant of histone acetylation in tumor cells [45], which provides direct evidence of epigenetic alternation for a particular metabolic enzyme. This observation was also confirmed in human glioma and prostate cancer, in which AcH4 is associated with pAKT levels. In summary, enhanced anabolic effects centered on Akt appear to be a reliable pathway of abnormal metabolism caused by cancer.
AMPK, the nutrient/energy-sensor, plays a crucial role in various biological processes, and has been reviewed as a “metabolic checkpoint” [46]. It controls the ATP/AMP ratio and anabolic metabolism. Lack of AMPK promotes the Warburg effect and increases HIF-1a expression, which eventually causes lymphoma [47]. Because HIF-1 is downstream factor of mTORC1, it emphasizes the control of AMPK in oncogenic mTORC1 [48]. The most recent studies provide new insight that an alternative nitrogen source may regulate AMPK in controlling mTORC1 signaling, which might constitute a complement of AMPK/mTOR signaling in the metabolic response [49]. The yeast ortholog of AMPK has been shown to play a role in the regulation of global histone acylation and the amount of acetyl-CoA [50]. In mammals, AMPK activation is directly associated with H2B phosphorylation at serine 36 [51]. Recent findings suggest that AMPK directly phosphorylates O-linked β-N-acetylglucosamine (O-GlcNAc) transferase and suppresses histone H2B O-GlcNAcylation [52]. Importantly, AMPK can also directly phosphorylate p53 on serine and regulate the cyclin D inhibitor p27 by phosphorylation of Thr198, leading to indirect control of the cell cycle [53]. In addition, AMPK is also connected with cell autophagy and polarity, as reviewed recently [54]. Collectively, the role of AMPK as a tumor suppressor is well reported, and it cooperates with Akt, which can in turn phosphorylates and activates mTORC1, provides a representative anabolic pathway that is involved in cancer pathogenesis. Abnormal metabolic status may activate or inactivate one of these mediators linked with cellular metabolism and eventually lead to cancer.
4.2 Tumorigenesis through metabolic enzyme mutation
Through whole genome sequencing, scientists have identified mutations of isocitrate dehydrogenase 1 (IDH1, cytoplasm) and IDH2 (mitochondria) in gliomas and acute myeloid leukemia [55, 56]; the mutants have an enzymatic property of producing 2HG from alpha-ketoglutarate. The mutations are correlated with global histone methylation [57]; unexpectedly, 2HG metabolites inhibited histone demethylases, thus are considered to be oncometabolites [58]. Strikingly, pyruvate kinase (PK), the glycolytic enzyme that catalyzes transformation of phosphoenolpyruvate (PEP) to pyruvate, has been reported to be involved in tumorigenesis in its M2 isoform [59] (Fig. 3). In mammals, there are four isoforms of PK (M1, M2, L, and R) expressed in a tissue-specific manner; PKM2 is mainly expressed in fetal and cancer cells [60]. The PKM1 and PKM2 isoforms are produced by alternative splicing of the pre-messenger RNA on the PKM2 gene [61]. When PKM2 is replaced with PKM1 in lung cancer cell lines by shRNA, the tumor formation in nude xenograft mice was reduced following decreased glycolytic metabolism, which confirmed the indispensable role of PKM2 in the Warburg effect and tumor growth [62]. This regulation could be a consequence of the function of PKM2 as a co-activator of HIF-1 [63]. As noted by Lu’s group, activation of epidermal growth factor receptor (EGFR) can induce the translocation of PKM2, wherein K433 of PKM2 binds to β-catenin at c-Src-phosphorylated Y333 and then assists recruitment at the CCND1 promoter and histone 3 acetylation, leading to cyclin D1 transcription. These researchers next revealed that this acetylation is required for PKM2 phosphorylation on histone H3 at threonine and subsequent expression of cyclin D1 and c-Myc, both of which are involved in cell proliferation and tumorigenesis as protein kinases [64-66]. This mechanism of EGFR induction of PKM2 may act through PKCε- and NF-κB-dependent activation [67]. Other evidence showed that PKM2 could phosphorylate mitosis checkpoint protein Bub3 at Y207, which is responsible for the spindle-assembly checkpoint and accurate chromosome segregation involved in EGF receptor activation during the induction of brain tumors [68].
Figure 3.
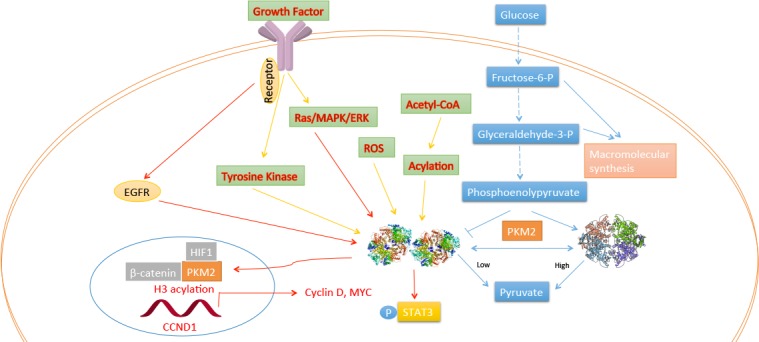
Regulation of PKM2 and its function in the nucleus.
Intriguingly, inactive PKM2 dimer can be regulated by transduction signaling, leading to Warburg effect and tumorigenesis, as demonstrated in recent years (Fig. 3). PKM2 has two forms, an active tetramer form (with high ligand affinity) and inactive dimer form which is prevented from binding to the cofactor fructose-1,6-bisphosphate (FBP) by tyrosine kinase signaling [59]. Hence, high glucose stimulates the acetylation of PKM2 on lysine 305, leading to a decrease in its active form. Meanwhile, its acetylation has been shown to be responsible for nuclear localization and tumorigenesis [69, 70]. Modification of PKM2 by phosphorylation and sumoylation can also lead to this translocation into the nucleus [66, 71]. Moreover, intracellular reactive oxygen species (ROS) can inhibit PKM2 by Cys358 oxidation which impairs the ontogenetic function of PKM2 [72]. The regulation by PKM2 enables the cell to adopt environmental changes, and its activation leads to nuclear protein kinase function.
There is evidence suggesting that fructose-1, 6-bisphosphatase 1 (FBP1) enzyme antagonizes glycolytic flux and is usually decreased in cancer cells. Pan-metabolomic analysis has been used to show that the FBP1 protein was inhibited in both clear cell renal cell carcinoma (ccRCC) tumors and hepatocellular carcinomas; moreover, this disappearance was not regulated by HIF activation and exhibited the tumor-suppressive functions of FBP1. Finally, these researchers showed that HIF enzymatic activity could be directly inhibited by FBP1 through interaction with the HIF-α inhibitory domain [73]. These non-enzymatic functions of FBP1 implicate the metabolic regulation of hypoxic responses in tumor formation. In addition, a type I transmembrane protein, MUC1, was also reported to enhance glucose uptake and glycolysis by occupying a multiple gene promoter. It can also facilitate recruitment of HIF-1α on gene promoters which control glycolytic metabolism, thus being responsible for tumor metabolic changes [74]. Other glycolytic factors, summarized in Table 2, include the glycolytic enzyme ENO1 which has a similar gene sequence to Myc promoter-binding protein-1 (MBP-1) in chromosome 1 and eventually leads to transcriptional repression of the oncogene c-myc [75]. 6-Phosphofructo-2-kinase (PFKFB3), expression of which can be stimulated by insulin, is localized in the nucleus and controls expression of cell cycle proteins such as cyclin-dependent kinase (Cdk)-1, cyclin D3 and p27 [76, 77].
Table 2.
Non-metabolic functions of glycolytic factors
| Name | Cancer | Metabolism | Modification | Reference |
|---|---|---|---|---|
| Lysine demethylase (LSD1) | Overexpressed in hepatocellular carcinoma | Glycolytic activity; Decreases mitochondrial metabolism genes | Methylate histone H3 at Lysine 4 in the promoter region | [192] |
| miR-122 microRNA | Breast cancer-secreted | Regulating the glycolytic enzyme PKM; Glucose uptake | Regulates glucose consumption in distant organs, including brain and lungs, and increases the incidence of metastasis | [92] |
| miR-290 miRNAs | Promotes pluripotency in PSCs | Up-regulates glycolytic enzymes Pkm2 and Ldha, stimulates glycolysis | miR-290 targets Mbd2, a reader for methylated CpGs, unregulated Myc | [193] |
| MnSOD-deficient mice | Skin carcinogenesis | Increased aerobic glycolysis | Increased uncoupling proteins (UCPs); p53 | [194] |
| Fructose-1,6-bisphosphatase (FBP1) | FBP1 was suppressed in kidney tumours | FBP1 controls cell proliferation, glycolysis and the pentose phosphate pathway | Inhibits nuclear HIF function via direct interaction with the HIF inhibitory domain | [73] |
| 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (pFKFB3) | Colon carcinoma | Glycolytic enzyme Induced by insulin |
Regulates autophagy; increases cyclin-dependent kinase (Cdk)-1, Cdc25C, and cyclin D3; decreased the expression of the cell cycle inhibitor p27 | [142] [195] [76] [77] |
| Type I transmembrane protein (MUC) | Pancreatic adenocarcinoma | Enhances glycolytic activity; enhances in vivo glucose uptake | MUC1 facilitates and stabilizes recruitment of HIF-1α and p300 on glycolytic gene promoters in a hypoxia-dependent manner | [74] |
| ENO1 (alpha-enolase) | Pancreatic cancer | Glycolytic enzyme | Alternative splicing form of ENO1, transcriptionally represses MYC | [75] |
These discoveries provide crucial elements that show how the downstream cascade can interfere with upstream signaling homeostasis and provide a good example of how glycolytic factors are involved in gene transcription and tumor progression.
4.3 MicroRNAs serve as glycolytic regulators in tumorigenesis
MicroRNAs (miRNAs) are small non-coding RNA molecules, which are 17-24 nucleotides in length and which target sites on the 3′-untranslated regions of messenger RNA (mRNA) to provide post-transcriptional modification [77]. In recent years, they have been widely used as diagnostic markers for various cancers, including lung, ovarian and pancreatic cancers [78-81]. Their oncogenic and tumor suppressive roles have been described as intertwined with typical molecular pathways that are regulated by Myc, Ras and p53 [82]. Overexpression of miR-21 in cancer cells will up regulate HIF-1α and activation of the AKT and ERK pathways through targeting of PTEN [83]. In addition to miR-21, other microRNAs have roles in regulating HIF-1 and other oncogenes and tumor suppressors [84-87]. More interestingly, miRNAs that are reduced in tumors, when delivered by viral vectors, can serve as a therapeutic agent that suppresses tumorigenesis [88]. Among them, the microRNA let-7 tumor suppressor has surprisingly showed regulation of glucose metabolism through the Lin28/let-7 axis, targeting multiple components of the PI3K-mTOR pathway involving impaired glucose tolerance and insulin resistance [89]. Moreover, let-7a was found recently to be able to reprogram cancer metabolism by decreasing anabolic enzymes and upregulation oxidative phosphorylation genes [90]. In addition, miR-378 inhibited the expression of two PGC-1β partners, ERRγ and GABPA, leading to metabolic shift of cancer cells [91]. Most recently, miR-122 secreted by breast cancer cells was reported to be responsible for reprogramming glucose metabolism in remote organs in order to increase metastasis [92]. As circulating microRNAs are cosidered to be biomarkers for cancer prognosis and to modulate cell function, these characteristics identify them as emerging intercellular messengers and they may have a crucial role in tumorigenesis.
5. New advances in the circadian clock and metabolism
In addition to metabolism, another system can also sense the “outliers” of the environment: the circadian rhythm, which orchestrates the 24-hour day and night light cycle based on Earth’s gravitational field, has been proposed as a connection between metabolism and cancer [25]. However, its role in cancer cell transformation in response to metabolic cues has been underestimated for a long time. The circadian clock comprises a central clock, located in the hypothalamic suprachiasmatic nucleus (SCN), and peripheral clocks, both of which regulate various important biological processes. Importantly, circadian clock genes are expressed in almost all central nerve system (CNS) and peripheral tissues and may be responsible for autonomous cell alteration in different external environments. Because the circadian rhythm is intertwined with the metabolic rhythm, we hypothesize that disturbance of circadian rhythm will in turn activate cell-autonomous transformation to generate the Warburg effect. This could be significant because metabolic disease and cancer occur in multiple organs in the human body. More evidence about metabolism and the circadian rhythm in that has emerged in recent years is reviewed below.
5.1 Sirtuins at the crossroad that connects cancer, metabolism and circadian clock
Sirtuins are a NAD+-dependent protein family composed of seven protein members that occur in different compartments of living cells (SIRT3, SIRT4, SIRT5, located in mitochondria; SIRT1, SIRT6, SIRT7, in the nucleus; and SIRT2, mainly in the cytoplasm). Similar to their divergent locations, various enzymatic activities have also been demonstrated, ranging from lysine deacetylase and ADP-ribosyltransferase to deacylase[93]. SIRT1, the best understood sirtuin protein, requires NAD+ as a cofactor for its activity and has been noted in tumorigenesis, owing to its biological role in maintaining genome integrity and DNA repair. In murine models involving knockout or overexpression of different sirtuin genes, direct evidence has been found that defective sirtuin function may either promote or suppress cancer formation [94]. Although the precise role of sirtuin in tumorigenesis is not fully understood, it can be inferred that: a. as an advanced regulatory node in metabolism, cancer and aging, the maintenance of SIRT1 is fundamental in normal cell function; b. the deacetylation of histone and non-histone proteins by SIRT1 may also be an approach for cell reprogramming. Here, we demonstrate that the circadian clock can either work together with sirtuin to control chromatin remodeling, or affect their activity to contribute to disease pathogenesis.
The circadian clock is controlled by a set of core clock genes. There are two heterodimeric transcription factors, CLOCK and BMAL1, that control rhythmic expression of many genes including its negative regulators, period (per1 and per2) and cryptochrome (cry1 and cry2). As shown by previous studies, the CLOCK itself has histone and BMAL1 acetyl transferase activity that is implicated in its regulatory functions in chromatin remodeling [95, 96]. Given that SIRT1 deacetylase activity is oscillating according to circadian rhythm, detailed research has demonstrated that SIRT1 directly interacts with CLOCK to control histone acetylation and regulate BMAL1 acetylation at Lys537 [97]. In addition, SIRT1 is also known to deacetylate PER2 and determine its stability. Supplementation with a series of SIRT1 activators, including SRT1720, has been found to repress clock genes both in vitro and in vivo and to have a chromatin modification effect which was SIRT1-dependent [98]. However, the activity of SIRT1 is dependent on its cosubstrate NAD+, which senses intracellular energy metabolism. NAMPT (nicotinamide phosphoribosyl-transferase), which catalyzes a rate-limiting step in the NAD+ salvage pathway, is regulated by CLOCK/BMAL1 transcription [99]. This pivotal finding has built a bridge between the classical transcription circadian loop and an enzymatic feedback loop, and has further demonstrated that metabolic disorder will interface with chromatin modification by the SIRT1-circadian system through the key metabolic regulator NAD+. Knockdown of CLOCK or BMAL1 induces hepatic insulin resistance, as demonstrated in a previous report. The authors found that CLOCK/BMAL1 are required for circadian expression of hepatic SIRT1, via binding to the SIRT1 promoter and controlling its expression [100]. This is how circadian oscillation influences metabolism through SIRT1 (Fig. 4). In contrast, environmental changes, including in metabolism, can also disrupt clock function through SIRT1. A report gives direct evidence that tobacco consumption leads to lung inflammation via SIRT1-dependent BMAL1 mediation, and this mutation was observed following chronic and acute exposure to tobacco in mice [101].
Figure 4.
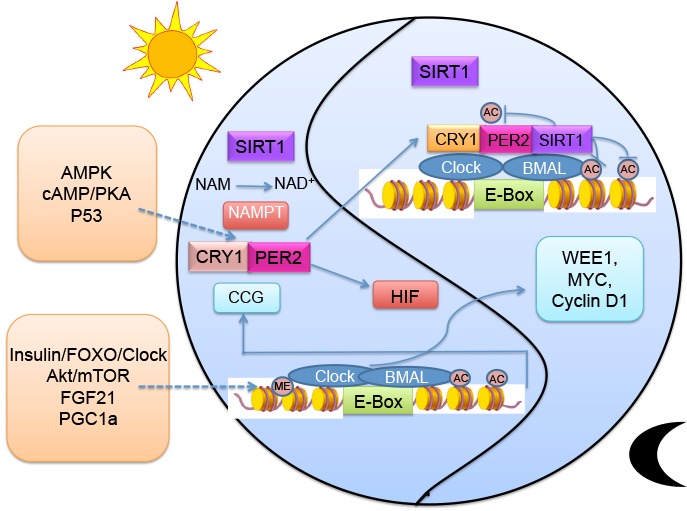
The transcriptional and post-translational loop of circadian systems
SIRT6 and SIRT3 have also been implicated in involvement with the circadian clock. The activity of SIRT3 in mitochondria is also mediated by the concentration of NAD+. As the circadian system involves rhythmic control of its rate-limiting enzyme NAMPT, as described above, it has also been found that the clock may regulate SIRT3 activity through the concentration of NAD+ and drive mitochondrial oxidative metabolism [102]. Once the circadian clock has been disrupted, the capacity for mitochondrial oxidative ability seems to be impaired, as detected by measurement of the oxygen consumption rate (OCR). The data indicate that Bmal1 mutants have defects in β-oxidation in addition to problems with pyruvate entry into the TCA cycle. This is a critical clue from which we can deduce the involvement of glycolysis and mitochondrial defects in cancer [103]. However, this is an indirect implication, and direct evidence needs to be obtained in future studies. Recent data also show that SIRT6 has different functions in regulating the CLOCK/BMAL1 network and also controls distinct hepatic circadian genes [104]. SIRT6 governs CLOCK/BMAL1 and recruitment of SREBP1 to circadian gene promoters and specifically regulates hepatic circadian transcription, which is correlated with downstream modulators of fatty acid metabolism [105].
Accumulating evidence has demonstrated that sirtuin may function either as oncogenes or tumor suppressors, influencing cell fate and metabolism during their modulation in cancer [106]. In this case, we provide details of interactions between sirtuin and the circadian clock via NAD+ concentration, demonstrating a more complicated situation in which sirtuin are involved in metabolic disorders and cancer. In addition, recent work has also found other rhythmic epigenetic modifications of the circadian clock, such as feedback actions of the PER complex on H3K9 di- and trimethylation [107]. In addition, histone methyltransferase mixed lineage leukemia 3 is controlled by the clock, which modulates over 100 epigenetic circadian outputs [108], demonstrating that the circadian clock alone is also important for sustaining temporal tissue physiology.
5.2 The circadian rhythm regulates metabolism and integrates nutrient signaling
Time-restricted feeding in mice will improve metabolic disease, even when they are fed a high-fat diet [109]. This provides a macroscopic view of how the circadian rhythm affects metabolism and may be involved in cell metabolism reprogramming and cancer pathogenesis. More evidence of how controlled feeding times influence the metabolism has been revealed [110]. Liver-specific deletion of Bmal1 in mice will lead to disrupted hepatic glucose regulatory genes and hypoglycemia [111]. Mice deficient in RORγ, another circadian gene, exhibit improvement of insulin sensitivity and glucose tolerance because of reduced hepatic gluconeogenesis [112]. This molecule can also occupy the citrate synthase promoter and regulate the expression of this key enzyme providing citrate derived acetyl-CoA [113]. This influence of lipogenesis is also indicated by another finding that binding of RORγ to ROREs controlled the transcription of lipid metabolic genes, such as insulin-induced gene 2a, Elovl3, Insig2a and sterol 12α-hydroxylase [114]. In clock mutant mice, the phenotype of impaired cholesterol metabolism is also observed and is involved in promotion of atherosclerosis [115]. Reviews have summarized the influence of the circadian clock on bile acid synthesis, lipogenesis, cardiovascular function, inflammation and, most importantly, glucose homeostasis [116]. Bioinformatics analysis has shown that most key rate-limiting enzymes involved in metabolism exhibit circadian rhythmicity, including those acting in glycolysis, gluconeogenesis, glycogenolysis, fatty acid synthesis, triglyceride storage, and cholesterol biosynthesis [117]. However, direct evidence of how the circadian systems regulate initialization of the Warburg effect is still lacking, although the clock indeed influences several curial metabolic enzymes and is an important factor in metabolic functions.
In turn, metabolic cues can also re-tune the performance of circadian oscillation. First, the nutrient sensor AMPK can phosphorylate the clock component cryptochrome 1 (CRY1), leading to its destabilization and rhythmic alteration [118]. In addition, casein kinases, also important modulators of circadian rhythms, can be phosphorylated by AMPK at Ser389 and thereby degrade PER2 [119]. This effect could also be observed after injection with metformin, but it could not be replicated in AMPK alpha2 knock-out mice. This regulatory function of AMPK in the circadian rhythm provides a striking example of nutrient signaling coupled with the human clock. Akt signaling was also found to alter the circadian rhythm in the SCN to lengthen rest-activity behavior [120]. Moreover, p53 binds directly to a response element in the PER2 promoter and hinders its expression. Mice deficient in p53 have a shorter, unstable period length under a light pulse [121]. The rhythmically expressed energy regulator PGC-1α stimulates the expression of Bmal1 and Rev-erbα through activation of orphan nuclear receptors and leads to a requirement for cell-autonomous clock reprogramming [122]. The direct connection between energy expenditure and the circadian cycle was demonstrated in 2013: feeding of a high-calorie diet influenced the circadian transcriptome and metabolome via impaired BMAL1 recruitment to its targeting chromatin sites and rhythmic recruitment of transcriptional factor PPARγ [123]. Through transcriptional remodeling, nutrients are directly linked to the circadian metabolome.
Post-transcriptional modification is also very important in circadian systems. Deacetylation-acetylations, described previously, are one possible functional biological modification that has the potential to influence the regulation of the circadian clock and is involved in metabolism. In one report, UBE3A disrupted oscillations by binding to BMAL1 and degrading it in a ubiquitin ligase-dependent manner [124]. In addition, the participation of histone methylation in the circadian clock has been investigated recently [108]. The occurrence of non-histone RNA methylation dependent on circadian control testified that m6A-RNA methylation sites are sufficient to generate a circadian rhythm that is sensitive to the amount of SAM (S-adenosyl-L-methionine) and SAH (S-adenosyl-L-homocysteine) [125]. This research was highlighted by several recent reviews [126, 127]. In an analogy with metabolic regulation, microRNA can also mediate circadian systems in response to environment alteration; for example, miR-219 was found to interact directly with the CLOCK-BMAL1 dimer, affecting the circadian pacemaker [128]. By facilitating phosphorylation in a GSK3β-dependent manner, the glucose sensor O-GlcNAcylation was shown to regulate clock and periodic protein transcriptional activities [129]. This promoter, being a nutrient sensor, shows how nutrients, especially glucose, fine-tune the circadian system and this provides strong evidence that transduction signaling is integrated with higher systems. Interestingly, fibroblast growth factor 21 (FGF21) and insulin also show the potential to the influence circadian rhythm [130-132]. The insulin-FOXO3-Clock signaling cascade has been described in recent years; it shows a mediating function in hepatic metabolism and oxidative sensitivity. This overlap of transduction signaling with metabolism provides a link by which outliers are integrated in both systems and play very important roles in the associations between metabolism, circadian rhythms and cancer.
5.3 Tissue-specific action of the circadian clock and its relation to cancer
Only 5%-10% of oscillatory transcripts are common between two given tissues (SCN, liver and muscle are involved), despite the fact that they share a highly conserved control system [133]. Using genetic techniques, scientists have observed cell type-specific clock gene knockdown phenotypes in 3T3-L1 adipocytes, MMH-D3 hepatocytes and other cell lines [134]. This involvement of CLOCK/BMAL1 may activate different sets of target genes in various tissues, as demonstrated in Drosophila [135]. Recent work done by Alexander’s group identified sequence motifs associated with CLOCK/BMAL1 binding sites unique to different parts of the body. One motif, the GATA factor binding site, has been revealed to be responsible for appropriate tissue-specific gene expression, thus providing a comprehensive picture of a clock gene selectively activating target gene transcription [136, 137]. More studies are necessary to identify binding sites of clocks in specific tissues, in order to analyze their effect on tissue-specific gene expression. Furthermore, tissue-specific actions have been observed in cell cycle control through cyclin-dependent kinase network, providing new insight into tumorigenesis in different organs [138]. The circadian influence on local physiology involves different types of epigenetic modification and may eventually cause different diseases, for example metabolic disease involving metabolic organs and cancers involving other organs. Like the example of clock protein DEC1 and DEC2, which is induced by CLOCK/BMAL1, control of metabolism and behavior by clock output is involved with hypoxia responses and carcinogenesis in a tissue-specific manner [139].
In the human population, CLOCK variants have been found to be associated with breast cancer risk, and CLOCK promoter hyper methylation will reduce breast cancer risk [140]. Clock is an independent risk factor for cancer; it is involved in control of cell proliferation, the DNA damage response, cellular senescence and the inflammatory response, in addition to metabolic homeostasis [141]. Overexpression of Bmal1 can increase the anticancer drug sensitivity of colorectal cancer [142]. Per2 mutation increased RAS-mediated oncogenic transformation and significantly changed the expression of the cell cycle genes p21 and cyclin D [66]. Per1 is being investigated in association with unfolded protein response (UPR), which has a role in tumorigenesis [143]. Interestingly, a recent report described that HCD feeding before tumor induction in mice was defected with ER-induced UPR [144], again showing the possible regulatory link between metabolism and the circadian clock in cancer onset. More specific molecular mechanisms for involvement of the clock in cancer require exploration, especially those acting with metabolism and the Warburg effect.
6. “Outliers” are also taken a role in aging process
Metabolic diseases and cancer are all age-associated diseases, with metabolism and circadian clock directly connecting with aging process. Decades ago, it was suggested that life-span was dependent on the rate of metabolic expenditure and oxidative damage [145]. Animals with calories restriction exhibit extension of average life span and lower oxidative damage rate. The balance between mitochondrial oxidant production and antioxidant can reflect mitochondrial respiration rate. Mitochondrial redox generation has long been implicated in the aging process [146]. Interestingly, treatment with metformin, a biguanide drug to treat type 2 diabetes, increases C. elegans life span by altering folate and methionine metabolism. Nevertheless, metformin had no effect on extending the life span of worms with 1% glucose supplement. The main target of metformin, AMPK, can activate SKN-1 that is essential in longevity [147]. As a regulator of metabolic homeostasis, sestrins are also described to have anti-aging function through AMPK-TORC1 axis [148]. These evidences imply that altered longevity are related to metabolic perturbations. Indeed, there is increased glycolysis in senescent fibroblasts, suggesting metabolic shift is also attributed to senescent cells and AMPK, which act as a sensor. AMPK can phosphorylate p53 or inhibits p21 or p16 transcription [149]. On the other hand, NAD+ and Sirt1 function together to regulate metabolism and circadian clock. Meanwhile, NAD+ level declined upon aging and restoring NAD+ could ameliorate many age-associated diseases[150]. Most recently, it has been found that NADase (CD38) expression was increased in multiple organs. CD38 deficient mice have higher level of NAD+ and are protected from aging related diseases. However, knockout of both CD38 and SIRT3 abolished the protective effects[151]. As NAD+ dependent deacetylases, increased sir2 is linked with extension of life span in worms; in mammals, deficiency of SIRT1 in MEF cells are resistant to senescence. Besides, Sirt6 deficiency will lead to age-related degeneration [152]. Accumulating evidence emphasized the central role of sirtuin between aging and metabolism.
Circadian clock has also been associated with control of aging. Disruption of circadian rhythm can accelerate aging. Resetting the clock accompanied with healthy diet increases longevity. Experimentally, deficient of BMAL1 will lead to premature aging and decreased life span because BMAL1 negatively control of mTORC1 signaling [153]. In the SCN, Sirt1 positively regulates genes controlling circadian clock and is reported to decline with aging [154]. Circadian gene per2 and sirt1 display a negative reciprocal relationship to regulate aging [155]. In sum, the interconnected clock and metabolism that incorporate transduction signaling, can in turn affect human aging process.
7. Prospective view
Accumulated evidence testifies that there are tight correlations between aging, cancer and metabolism [46, 156]. In this review, we have interpreted the association of these elements and included the concept of the circadian clock, stating that metabolism and the circadian rhythm may act together to contribute to tumorigenesis (Fig. 5). This review also provides some evidence that non-metabolic functions of glycolytic enzymes may be involved in tumorigenesis, and further research should be conducted in this area owing to its important roles in linking metabolism and cancer. Every metabolite is rhythmically circulated in human systems and their oscillation in both space and time may eventually destroys systemic homeostasis and causes physiological disorders. This effect occurs through multiple facets of interaction between the circadian clock and metabolism. Moreover, according to our findings in liver-specific SIRT1 knockout (Sirt1-LKO) mice, hepatic sirt1 deficiency will lead to hepatic glucose over-production, increasing circulatory glucose (hyperglycemia) and insulin resistance. Further understanding of the function of sirt1 in glucose metabolism has delineated a pathway in which sirt1 positively regulates transcription of Rictor and triggers phosphorylation of Akt at S473 and Foxo1 at S253. This is followed by negative regulation of the transcription of G6pase (glucose-6-phosphatase) and Pepck (phosphoenolpyruvate carboxykinase), which are responsible for gluconeogenesis [157]. Surprisingly, at a late stage in the study of sirt1 LKO mice aged over one year, lung tumors were observed by both gross observation and histological examination (unpublished data). It gives direct evidence between these two diseases. The circadian clock has a multilayer link with lung disease. It can regulate E-box mediated circadian activation of Nrf2 which uses antioxidant effects to modulate pulmonary fibrosis [158]. It regulates the inflammatory response in lung injury and also regulates glucocorticoid hormones involved in pulmonary inflammation [159, 160]. More evidence that metabolic disease leads to lung cancer is required; it may act through both metabolic reprogramming and disorder of the circadian system. From the review, we point out that all living organisms are immersed in more than one dimension in the universe. Consideration of both the quantity and the temporal challenge to the system might allow more precise deconstruction of a particular problem and leads to the truth, avoid practical mistakes. The integration of the circadian rhythm and metabolism with diseases is an example.
Figure 5.
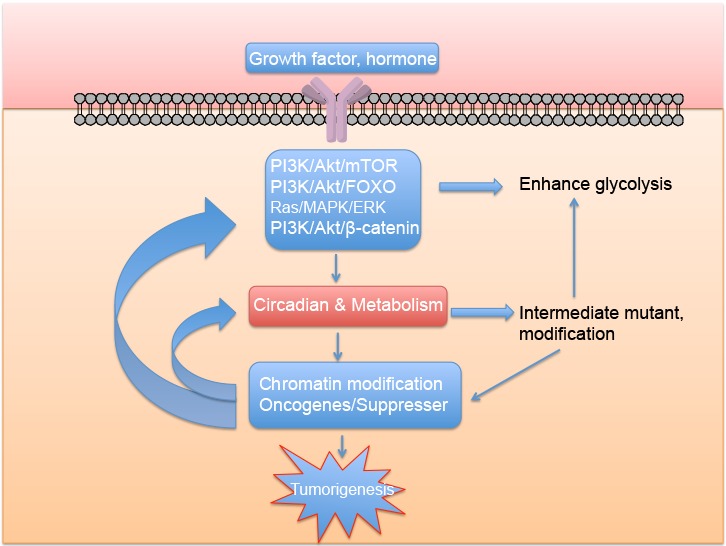
Proposed mechanism relating metabolic disease and cancer
Contributor Information
Yiwei Cao, Faculty of Health Science, University of Macau, Macau, China.
Rui-Hong Wang, Faculty of Health Science, University of Macau, Macau, China.
References
- [1].Calle EE, Kaaks R (2004). Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer, 4: 579-591 [DOI] [PubMed] [Google Scholar]
- [2].Calle EE, Thun MJ (2004). Obesity and cancer. Oncogene, 23: 6365-6378 [DOI] [PubMed] [Google Scholar]
- [3].Gallagher EJ, LeRoith D (2011). Diabetes, cancer, and metformin: connections of metabolism and cell proliferation. Ann N Y Acad Sci, 1243: 54-68 [DOI] [PubMed] [Google Scholar]
- [4].Khandekar MJ, Cohen P, Spiegelman BM (2011). Molecular mechanisms of cancer development in obesity. Nat Rev Cancer, 11: 886-895 [DOI] [PubMed] [Google Scholar]
- [5].Yeung S, Pan J, Lee M-H (2008). Roles of p53, MYC and HIF-1 in regulating glycolysis—the seventh hallmark of cancer. Cell Mol Life Sci, 65: 3981-3999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lyssiotis CA, Cantley LC (2014). Acetate Fuels the Cancer Engine. Cell, 159: 1492-1494 [DOI] [PubMed] [Google Scholar]
- [7].Reppert SM, Weaver DR (2002). Coordination of circadian timing in mammals. Nature, 418: 935-941 [DOI] [PubMed] [Google Scholar]
- [8].Lucassen EA, Coomans CP, van Putten M, de Kreij SR, van Genugten JH, Sutorius RP, et al. (2016). Environmental 24-hr Cycles Are Essential for Health. Curr Biol, 26(14):1843-53 [DOI] [PubMed] [Google Scholar]
- [9].Chang A-M, Aeschbach D, Duffy JF, Czeisler CA (2015). Evening use of light-emitting eReaders negatively affects sleep, circadian timing, and next-morning alertness. Proc Natl Acad Sci U S A, 112: 1232-1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Turek FW, Joshu C, Kohsaka A, Lin E, Ivanova G, McDearmon E, et al. (2005). Obesity and metabolic syndrome in circadian Clock mutant mice. Science, 308: 1043-1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Huang W, Ramsey KM, Marcheva B, Bass J (2011). Circadian rhythms, sleep, and metabolism. J Clin Invest, 121: 2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gamble KL, Berry R, Frank SJ, Young ME (2014). Circadian clock control of endocrine factors. Nat Rev Endocrinol, 10: 466-475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Innominato PF, Lévi FA, Bjarnason GA (2010). Chronotherapy and the molecular clock: Clinical implications in oncology. Adv Drug Deliv Rev, 62: 979-1001 [DOI] [PubMed] [Google Scholar]
- [14].Warburg O (1925). Iron, the oxygen-carrier of respiration-ferment. Science, 61: 575-582 [DOI] [PubMed] [Google Scholar]
- [15].Warburg O (1956). On respiratory impairment in cancer cells. Science, 124: 269-270 [PubMed] [Google Scholar]
- [16].Gatenby RA, Gillies RJ (2004). Why do cancers have high aerobic glycolysis? Nat Rev Cancer, 4: 891-899 [DOI] [PubMed] [Google Scholar]
- [17].Gao P, Sun L, He X, Cao Y, Zhang H (2012). MicroRNAs and the Warburg effect: new players in an old arena. Curr Gene Ther, 12: 285-291 [DOI] [PubMed] [Google Scholar]
- [18].Ward PS, Thompson CB (2012). Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer cell, 21: 297-308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Vander Heiden MG, Cantley LC, Thompson CB (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science, 324: 1029-1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fritz V, Fajas L (2010). Metabolism and proliferation share common regulatory pathways in cancer cells. Oncogene, 29: 4369-4377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kupfer SR, Underwood LE, Baxter RC, Clemmons DR (1993). Enhancement of the anabolic effects of growth hormone and insulin-like growth factor I by use of both agents simultaneously. J Clin Invest, 91: 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Fryburg DA, Jahn LA, Hill SA, Oliveras DM, Barrett EJ (1995). Insulin and insulin-like growth factor-I enhance human skeletal muscle protein anabolism during hyperaminoacidemia by different mechanisms. J Clin Invest., 96: 1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G, et al. (2005). Glycolytic enzymes can modulate cellular life span. Cancer Res, 65: 177-185 [PubMed] [Google Scholar]
- [24].Chen Z, Odstrcil EA, Tu BP, McKnight SL (2007). Restriction of DNA replication to the reductive phase of the metabolic cycle protects genome integrity. Science, 316: 1916-1919 [DOI] [PubMed] [Google Scholar]
- [25].Sahar S, Sassone-Corsi P (2009). Metabolism and cancer: the circadian clock connection. Nat Rev Cancer, 9: 886-896 [DOI] [PubMed] [Google Scholar]
- [26].Khan D, Katoch A, Das A, Sharathchandra A, Lal R, Roy P, et al. (2015). Reversible induction of translational isoforms of p53 in glucose deprivation. Cell Death Differ, 22(7):1203-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Korangath P, Teo WW, Sadik H, Han L, Mori N, Huijts CM, et al. (2015). Targeting Glutamine Metabolism in Breast Cancer with Aminooxyacetate. Clin Cancer Res, 21(14):3263-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hardie DG (2011). AMPK and autophagy get connected. EMBO J, 30: 634-635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zoncu R, Efeyan A, Sabatini DM (2011). mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol, 12: 21-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wellen KE, Thompson CB (2012). A two-way street: reciprocal regulation of metabolism and signalling. Nat Rev Mol Cell Biol, 13: 270-276 [DOI] [PubMed] [Google Scholar]
- [31].Cross M, Dexter TM (1991). Growth factors in development, transformation, and tumorigenesis. Cell, 64: 271-280 [DOI] [PubMed] [Google Scholar]
- [32].Hirabayashi S, Baranski TJ, Cagan RL (2013). Transformed Drosophila cells evade diet-mediated insulin resistance through wingless signaling. Cell, 154: 664-675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cai L, Sutter BM, Li B, Tu BP (2011). Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol Cell, 42: 426-437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bauer DE, Hatzivassiliou G, Zhao F, Andreadis C, Thompson CB (2005). ATP citrate lyase is an important component of cell growth and transformation. Oncogene, 24: 6314-6322 [DOI] [PubMed] [Google Scholar]
- [35].Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, et al. (2005). ATP citrate lyase inhibition can suppress tumor cell growth. Cancer cell, 8: 311-321 [DOI] [PubMed] [Google Scholar]
- [36].Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB (2009). ATP-citrate lyase links cellular metabolism to histone acetylation. Science, 324: 1076-1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Migita T, Narita T, Nomura K, Miyagi E, Inazuka F, Matsuura M, et al. (2008). ATP Citrate Lyase: Activation and Therapeutic Implications in Non-Small Cell Lung Cancer. Cancer Res, 68: 8547-8554 [DOI] [PubMed] [Google Scholar]
- [38].Chan C-H, Li C-F, Yang W-L, Gao Y, Lee S-W, Feng Z, et al. (2012). The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell, 149: 1098-1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Luo J, Manning BD, Cantley LC (2003). Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell, 4: 257-262 [DOI] [PubMed] [Google Scholar]
- [40].Rodriguez A, De La Cera T, Herrero P, Moreno F (2001). The hexokinase 2 protein regulates the expression of the GLK1, HXK1 and HXK2 genes of Saccharomyces cerevisiae. Biochem J, 355: 625-631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Pastorino JG, Shulga N, Hoek JB (2002). Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem, 277: 7610-7618 [DOI] [PubMed] [Google Scholar]
- [42].Kim J-w, Dang CV (2005). Multifaceted roles of glycolytic enzymes. Trends Biochem Sci, 30: 142-150 [DOI] [PubMed] [Google Scholar]
- [43].Jin Q, Lee H-J, Min H-Y, Smith JK, Hwang SJ, Whang YM, et al. (2014). Transcriptional and posttranslational regulation of insulin-like growth factor binding protein-3 by Akt3. Carcinogenesis, 35: 2232-2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wu C, Liu X, Wang Y, Tian H, Xie Y, Li Q, et al. (2013). Insulin-like factor binding protein-3 promotes the G1 cell cycle arrest in several cancer cell lines. Gene, 512: 127-133 [DOI] [PubMed] [Google Scholar]
- [45].Lee JV, Carrer A, Shah S, Snyder NW, Wei S, Venneti S, et al. (2014). Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab, 20: 306-319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Green DR, Galluzzi L, Kroemer G (2014). Metabolic control of cell death. Science, 345: 1250256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, et al. (2013). AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab, 17: 113-124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Green AS, Chapuis N, Lacombe C, Mayeux P, Bouscary D, Tamburini J (2011). LKB1/AMPK/mTOR signaling pathway in hematological malignancies: from metabolism to cancer cell biology. Cell Cycle, 10: 2115-2120 [DOI] [PubMed] [Google Scholar]
- [49].Davie E, Forte GM, Petersen J (2015). Nitrogen Regulates AMPK to Control TORC1 Signaling. Curr Biol, 25(4):445-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zhang M, Galdieri L, Vancura A (2013). The yeast AMPK homolog SNF1 regulates acetyl coenzyme A homeostasis and histone acetylation. Mol Cell Biol, 33: 4701-4717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Bungard D, Fuerth BJ, Zeng P-Y, Faubert B, Maas NL, Viollet B, et al. (2010). Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation. Science, 329: 1201-1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Xu Q, Yang C, Du Y, Chen Y, Liu H, Deng M, et al. (2014). AMPK regulates histone H2B O-GlcNAcylation. Nucleic Acids Res, 42(9):5594-604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Shackelford DB, Shaw RJ (2009). The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer, 9: 563-575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mihaylova MM, Shaw RJ (2011). The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol, 13: 1016-1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. (2009). Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med, 361: 1058-1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Parsons DW, Jones S, Zhang X, Lin JC-H, Leary RJ, Angenendt P, et al. (2008). An integrated genomic analysis of human glioblastoma multiforme. Science, 321: 1807-1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. (2012). IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature, 483: 474-478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, et al. (2011). The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep, 12: 463-469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Hitosugi T, Kang S, Vander Heiden MG, Chung T-W, Elf S, Lythgoe K, et al. (2009). Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal, 2: ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Mazurek S (2007) Pyruvate kinase type M2: a key regulator within the tumour metabolome and a tool for metabolic profiling of tumours. Ernst Schering Found Symp Proc, (4):99-124 [DOI] [PubMed] [Google Scholar]
- [61].Noguchi T, Inoue H, Tanaka T (1986). The M1-and M2-type isozymes of rat pyruvate kinase are produced from the same gene by alternative RNA splicing. J Biol Chem, 261: 13807-13812 [PubMed] [Google Scholar]
- [62].Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, et al. (2008). The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature, 452: 230-233 [DOI] [PubMed] [Google Scholar]
- [63].Luo W, Hu H, Chang R, Zhong J, Knabel M, O'Meally R, et al. (2011). Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell, 145: 732-744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, et al. (2011). Nuclear PKM2 regulates [bgr]-catenin transactivation upon EGFR activation. Nature, 480: 118-122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, et al. (2012). PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell, 150: 685-696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F, et al. (2012). ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat Cell Biol, 14: 1295-1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Yang W, Xia Y, Cao Y, Zheng Y, Bu W, Zhang L, et al. (2012). EGFR-induced and PKCε monoubiquitylation-dependent NF-κB activation upregulates PKM2 expression and promotes tumorigenesis. Mol Cell, 48: 771-784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Jiang Y, Li X, Yang W, Hawke DH, Zheng Y, Xia Y, et al. (2014). PKM2 regulates chromosome segregation and mitosis progression of tumor cells. Mol Cell. 53: 75-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, et al. (2011). Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell, 42: 719-730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Lv L, Xu Y-P, Zhao D, Li F-L, Wang W, Sasaki N, et al. (2013). Mitogenic and oncogenic stimulation of K433 acetylation promotes PKM2 protein kinase activity and nuclear localization. Mol Cell, 52: 340-352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Spoden GA, Morandell D, Ehehalt D, Fiedler M, Jansen-Dürr P, Hermann M, et al. (2009). The SUMO-E3 ligase PIAS3 targets pyruvate kinase M2. J Cell Biochem, 107: 293-302 [DOI] [PubMed] [Google Scholar]
- [72].Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang J-k, Shen M, et al. (2011). Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science, 334: 1278-1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Li B, Qiu B, Lee DS, Walton ZE, Ochocki JD, Mathew LK, et al. (2014). Fructose-1, 6-bisphosphatase opposes renal carcinoma progression. Nature, 513: 251-255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Chaika NV, Gebregiworgis T, Lewallen ME, Purohit V, Radhakrishnan P, Liu X, et al. (2012). MUC1 mucin stabilizes and activates hypoxia-inducible factor 1 alpha to regulate metabolism in pancreatic cancer. Proc Natl Acad Sci U S A, 109: 13787-13792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Feo S, Arcuri D, Piddini E, Passantino R, Giallongo A (2000). ENO1 gene product binds to the c-myc promoter and acts as a transcriptional repressor: relationship with Myc promoter-binding protein 1 (MBP-1). FEBS letters, 473: 47-52 [DOI] [PubMed] [Google Scholar]
- [76].Yalcin A, Clem BF, Simmons A, Lane A, Nelson K, Clem AL, et al. (2009). Nuclear targeting of 6-phosphofructo-2-kinase (PFKFB3) increases proliferation via cyclin-dependent kinases. J Biol Chem, 284: 24223-24232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Riera Ls, Manzano A, Navarro-Sabaté A, Perales JC, Bartrons R (2002). Insulin induces PFKFB3 gene expression in HT29 human colon adenocarcinoma cells. BBA-MolCell Res, 1589: 89-92 [DOI] [PubMed] [Google Scholar]
- [78].Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, Yi M, et al. (2006). Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell, 9: 189-198 [DOI] [PubMed] [Google Scholar]
- [79].Taylor DD, Gercel-Taylor C (2008). MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol Oncol, 110: 13-21 [DOI] [PubMed] [Google Scholar]
- [80].Dillhoff M, Liu J, Frankel W, Croce C, Bloomston M (2008). MicroRNA-21 is overexpressed in pancreatic cancer and a potential predictor of survival. J Gastrointest Surg, 12: 2171-2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Calin GA, Croce CM (2006). MicroRNA-cancer connection: the beginning of a new tale. Cancer Res, 66: 7390-7394 [DOI] [PubMed] [Google Scholar]
- [82].Kent O, Mendell J (2006). A small piece in the cancer puzzle: microRNAs as tumor suppressors and oncogenes. Oncogene, 25: 6188-6196 [DOI] [PubMed] [Google Scholar]
- [83].Liu L-Z, Li C, Chen Q, Jing Y, Carpenter R, Jiang Y, et al. (2011). MiR-21 induced angiogenesis through AKT and ERK activation and HIF-1α expression. PLoS One, 6: e19139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Cascio S, D'Andrea A, Ferla R, Surmacz E, Gulotta E, Amodeo V, et al. (2010). miR-20b modulates VEGF expression by targeting HIF-1α and STAT3 in MCF-7 breast cancer cells. J Cell Physiol, 224: 242-249 [DOI] [PubMed] [Google Scholar]
- [85].Lei Z, Li B, Yang Z, Fang H, Zhang G-M, Feng Z-H, et al. (2009). Regulation of HIF-1α and VEGF by miR-20b tunes tumor cells to adapt to the alteration of oxygen concentration. PLoS One, 4: e7629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Ghosh G, Subramanian IV, Adhikari N, Zhang X, Joshi HP, Basi D, et al. (2010). Hypoxia-induced microRNA-424 expression in human endothelial cells regulates HIF-α isoforms and promotes angiogenesis. J Clin Invest, 120: 4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].He L, He X, Lim LP, De Stanchina E, Xuan Z, Liang Y, et al. (2007). A microRNA component of the p53 tumour suppressor network. Nature, 447: 1130-1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Kota J, Chivukula RR, O'Donnell KA, Wentzel EA, Montgomery CL, Hwang H-W, et al. (2009). Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell, 137: 1005-1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Zhu H, Shyh-Chang N, Segrè AV, Shinoda G, Shah SP, Einhorn WS, et al. (2011). The Lin28/let-7 axis regulates glucose metabolism. Cell, 147: 81-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Serguienko A, Grad I, Wennerstrøm AB, Meza-Zepeda LA, Thiede B, Stratford EW, et al. (2015). Metabolic reprogramming of metastatic breast cancer and melanoma by let-7a microRNA. Oncotarget, 6: 2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Eichner LJ, Perry M-C, Dufour CR, Bertos N, Park M, St-Pierre J, et al. (2010). miR-378* mediates metabolic shift in breast cancer cells via the PGC-1β/ERRγ transcriptional pathway. Cell Metab, 12: 352-361 [DOI] [PubMed] [Google Scholar]
- [92].Fong MY, Zhou W, Liu L, Alontaga AY, Chandra M, Ashby J, et al. (2015). Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat Cell Biol, 17: 183-194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Poulose N, Raju R (2015). Sirtuin regulation in aging and injury. BBA Mol Basis Dis, 1852: 2442-2455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Roth M, Chen W (2014). Sorting out functions of sirtuins in cancer. Oncogene, 33: 1609-1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Doi M, Hirayama J, Sassone-Corsi P (2006). Circadian regulator CLOCK is a histone acetyltransferase. Cell, 125: 497-508 [DOI] [PubMed] [Google Scholar]
- [96].Hirayama J, Sahar S, Grimaldi B, Tamaru T, Takamatsu K, Nakahata Y, et al. (2007). CLOCK-mediated acetylation of BMAL1 controls circadian function. Nature, 450: 1086-1090 [DOI] [PubMed] [Google Scholar]
- [97].Nakahata Y, Kaluzova M, Grimaldi B, Sahar S, Hirayama J, Chen D, et al. (2008). The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell, 134: 329-340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Bellet MM, Nakahata Y, Boudjelal M, Watts E, Mossakowska DE, Edwards KA, et al. (2013). Pharmacological modulation of circadian rhythms by synthetic activators of the deacetylase SIRT1. Proc Natl Acad Sci U S A, 110: 3333-3338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone-Corsi P (2009). Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science, 324: 654-657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Zhou B, Zhang Y, Zhang F, Xia Y, Liu J, Huang R, et al. (2014). CLOCK/BMAL1 regulates circadian change of mouse hepatic insulin sensitivity by SIRT1. Hepatology, 59: 2196-2206 [DOI] [PubMed] [Google Scholar]
- [101].Hwang J-W, Sundar IK, Yao H, Sellix MT, Rahman I (2014). Circadian clock function is disrupted by environmental tobacco/cigarette smoke, leading to lung inflammation and injury via a SIRT1-BMAL1 pathway. FASEB J, 28: 176-194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Peek CB, Affinati AH, Ramsey KM, Kuo H-Y, Yu W, Sena LA, et al. (2013). Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science, 342: 1243417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Wang X, Peralta S, Moraes CT (2013). Mitochondrial alterations during carcinogenesis: a review of metabolic transformation and targets for anticancer treatments. Adv Cancer Res, 119: 127-160 [DOI] [PubMed] [Google Scholar]
- [104].Xi Y, Chen D (2014). Partitioning the circadian clock. Science, 345: 1122-1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Masri S, Rigor P, Cervantes M, Ceglia N, Sebastian C, Xiao C, et al. (2014). Partitioning circadian transcription by SIRT6 leads to segregated control of cellular metabolism. Cell, 158: 659-672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Chen Y, Fu L, Wen X, Wang X, Liu J, Cheng Y, et al. (2014). Sirtuin-3 (SIRT3), a therapeutic target with oncogenic and tumor-suppressive function in cancer. Cell Death Dis, 5: e1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Duong HA, Weitz CJ (2014). Temporal orchestration of repressive chromatin modifiers by circadian clock Period complexes. Nat. Struct. Mol Biol, 21: 126-132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Valekunja UK, Edgar RS, Oklejewicz M, van der Horst GT, O’Neill JS, Tamanini F, et al. (2013). Histone methyltransferase MLL3 contributes to genome-scale circadian transcription. Proc Natl Acad Sci U S A, 110: 1554-1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Hatori M, Vollmers C, Zarrinpar A, DiTacchio L, Bushong EA, Gill S, et al. (2012). Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab, 15: 848-860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Adamovich Y, Rousso-Noori L, Zwighaft Z, Neufeld-Cohen A, Golik M, Kraut-Cohen J, et al. (2014). Circadian clocks and feeding time regulate the oscillations and levels of hepatic triglycerides. Cell Metab, 19: 319-330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Lamia KA, Storch K-F, Weitz CJ (2008). Physiological significance of a peripheral tissue circadian clock. Proc Natl Acad Sci U S A, 105: 15172-15177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Takeda Y, Kang HS, Freudenberg J, DeGraff LM, Jothi R, Jetten AM (2014). Retinoic acid-related orphan receptor γ (RORγ): a novel participant in the diurnal regulation of hepatic gluconeogenesis and insulin sensitivity. PLoS genetics, 10: e1004331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Crumbley C, Wang Y, Banerjee S, Burris TP (2012). Regulation of expression of citrate synthase by the retinoic acid receptor-related orphan receptor α (RORα). PLoS One, 7: e33804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Takeda Y, Kang HS, Lih FB, Jiang H, Blaner WS, Jetten AM (2014). Retinoid acid-related orphan receptor γ, RORγ, participates in diurnal transcriptional regulation of lipid metabolic genes. Nucleic Acids Res, 42(16):10448-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Pan X, Jiang X-C, Hussain MM (2013). Impaired cholesterol metabolism and enhanced atherosclerosis in clock mutant mice. Circulation, 113002885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Bass J, Takahashi JS (2010). Circadian integration of metabolism and energetics. Science, 330: 1349-1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Rudic RD, McNamara P, Reilly D, Grosser T, Curtis A-M, Price TS, et al. (2005). Bioinformatic analysis of circadian gene oscillation in mouse aorta. Circulation, 112: 2716-2724 [DOI] [PubMed] [Google Scholar]
- [118].Lamia KA, Sachdeva UM, DiTacchio L, Williams EC, Alvarez JG, Egan DF, et al. (2009). AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science, 326: 437-440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Um JH, Yang S, Yamazaki S, Kang H, Viollet B, Foretz M, et al. (2007). Activation of 5′-AMP-activated kinase with diabetes drug metformin induces casein kinase Iϵ (CKIϵ)-dependent degradation of clock protein mPer2. J Biol Chem, 282: 20794-20798 [DOI] [PubMed] [Google Scholar]
- [120].Zheng X, Sehgal A (2010). AKT and TOR signaling set the pace of the circadian pacemaker. Curr Biol, 20: 1203-1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Miki T, Matsumoto T, Zhao Z, Lee CC (2013). p53 regulates Period2 expression and the circadian clock. Nat Commun, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Liu C, Li S, Liu T, Borjigin J, Lin JD (2007). Transcriptional coactivator PGC-1&agr; integrates the mammalian clock and energy metabolism. Nature, 447: 477-481 [DOI] [PubMed] [Google Scholar]
- [123].Eckel-Mahan KL, Patel VR, de Mateo S, Orozco-Solis R, Ceglia NJ, Sahar S, et al. (2013). Reprogramming of the circadian clock by nutritional challenge. Cell, 155: 1464-1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Gossan NC, Zhang F, Guo B, Jin D, Yoshitane H, Yao A, et al. (2014). The E3 ubiquitin ligase UBE3A is an integral component of the molecular circadian clock through regulating the BMAL1 transcription factor. Nucleic Acids Res, 42: 5765-5775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Fustin J-M, Doi M, Yamaguchi Y, Hida H, Nishimura S, Yoshida M, et al. (2013). RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell, 155: 793-806 [DOI] [PubMed] [Google Scholar]
- [126].Lokody I (2014). Gene regulation: RNA methylation regulates the circadian clock. Nat Rev Genet, 15: 3-3 [DOI] [PubMed] [Google Scholar]
- [127].Hastings MH (2013). m(6)A mRNA Methylation: A New Circadian Pacesetter. Cell, 155: 740-741 [DOI] [PubMed] [Google Scholar]
- [128].Bartok O, Kyriacou CP, Levine J, Sehgal A, Kadener S (2013). Adaptation of molecular circadian clockwork to environmental changes: a role for alternative splicing and miRNAs. Pro Bio Sci, 280: 20130011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Kaasik K, Kivimäe S, Allen JJ, Chalkley RJ, Huang Y, Baer K, et al. (2013). Glucose sensor O-GlcNAcylation coordinates with phosphorylation to regulate circadian clock. Cell Metab, 17: 291-302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Bookout AL, de Groot MH, Owen BM, Lee S, Gautron L, Lawrence HL, et al. (2013). FGF21 regulates metabolism and circadian behavior by acting on the nervous system. Nat Med, 19: 1147-1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Chaves I, van der Horst GT, Schellevis R, Nijman RM, Koerkamp MG, Holstege FC, et al. (2014). Insulin-FOXO3 signaling modulates circadian rhythms via regulation of clock transcription. Curr Biol, 24: 1248-1255 [DOI] [PubMed] [Google Scholar]
- [132].Zheng X, Yang Z, Yue Z, Alvarez JD, Sehgal A (2007). FOXO and insulin signaling regulate sensitivity of the circadian clock to oxidative stress. Proc Natl Acad Sci U S A, 104: 15899-15904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Masri S, Sassone-Corsi P (2010). Plasticity and specificity of the circadian epigenome. Nat Neurosci, 13: 1324-1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Ramanathan C, Xu H, Khan SK, Shen Y, Gitis PJ, Welsh DK, et al. (2014). Cell type-specific functions of period genes revealed by novel adipocyte and hepatocyte circadian clock models. PLoS genetics, 10: e1004244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Abruzzi KC, Rodriguez J, Menet JS, Desrochers J, Zadina A, Luo W, et al. (2011). Drosophila CLOCK target gene characterization: implications for circadian tissue-specific gene expression. Genes Dev, 25: 2374-2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Meireles-Filho AC, Bardet AF, Yáñez-Cuna JO, Stampfel G, Stark A (2014). Cis-regulatory requirements for tissue-specific programs of the circadian clock. Curr Biol, 24: 1-10 [DOI] [PubMed] [Google Scholar]
- [137].Menet JS, Hardin PE (2014). Circadian clocks: the tissue is the issue. Curr Biol, 24: R25-R27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Pagano M, Jackson PK (2004). Wagging the dogma: tissue-specific cell cycle control in the mouse embryo. Cell, 118: 535-538 [DOI] [PubMed] [Google Scholar]
- [139].Kato Y, Kawamoto T, Fujimoto K, Noshiro M (2013). DEC1/STRA13/SHARP2 and DEC2/SHARP1 Coordinate Physiological Processes, Including Circadian Rhythms in Response to Environmental Stimuli. Curr Top Dev Biol, 110: 339-372 [DOI] [PubMed] [Google Scholar]
- [140].Hoffman AE, Yi C-H, Zheng T, Stevens RG, Leaderer D, Zhang Y, et al. (2010). CLOCK in breast tumorigenesis: genetic, epigenetic, and transcriptional profiling analyses. Cancer Res, 70: 1459-1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Kettner NM, Katchy CA, Fu L (2014). Circadian gene variants in cancer. Ann Intern Med, 46: 208-220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Zeng Z-l, Luo H-y, Yang J, Wu W-j, Chen D-l, Huang P, et al. (2014). Overexpression of the circadian clock gene Bmal1 increases sensitivity to oxaliplatin in colorectal cancer. Clin. Cancer Res, 20: 1042-1052 [DOI] [PubMed] [Google Scholar]
- [143].Pluquet O, Dejeans N, Bouchecareilh M, Lhomond S, Pineau R, Higa A, et al. (2013). Posttranscriptional Regulation of PER1 Underlies the Oncogenic Function of IREα. Cancer Res, 73: 4732-4743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Ramadori G, Konstantinidou G, Venkateswaran N, Biscotti T, Morlock L, Galié M, et al. (2015). Diet-Induced Unresolved ER Stress Hinders KRAS-Driven Lung Tumorigenesis. Cell Metab, 21: 117-125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Sohal R, Allen R (1986). Relationship between oxygen metabolism, aging and development. Free Radic Biol Med, 2: 117-160 [Google Scholar]
- [146].Sohal RS, Ku H-H, Agarwal S, Forster MJ, Lal H (1994). Oxidative damage, mitochondrial oxidant generation and antioxidant defenses during aging and in response to food restriction in the mouse. Mech Ageing Dev, 74: 121-133 [DOI] [PubMed] [Google Scholar]
- [147].Cabreiro F, Au C, Leung K-Y, Vergara-Irigaray N, Cochemé HM, Noori T, et al. (2013). Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell, 153: 228-239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Lee JH, Budanov AV, Karin M (2013). Sestrins orchestrate cellular metabolism to attenuate aging. Cell Metab, 18: 792-801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Wiley CD, Campisi J (2016). From Ancient Pathways to Aging Cells—Connecting Metabolism and Cellular Senescence. Cell Metab, 23: 1013-1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Imai S-i, Guarente L (2014). NAD+ and sirtuins in aging and disease. Trends Cell Biol, 24: 464-471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Camacho-Pereira J, Tarragó MG, Chini CC, Nin V, Escande C, Warner GM, et al. (2016). CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab, 23: 1127-1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, et al. (2006). Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell, 124: 315-329 [DOI] [PubMed] [Google Scholar]
- [153].Khapre RV, Kondratova AA, Patel S, Dubrovsky Y, Wrobel M, Antoch MP, et al. (2014). BMAL1-dependent regulation of the mTOR signaling pathway delays aging. Aging, 6: 48-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Chang H-C, Guarente L (2013). SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell, 153: 1448-1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Wang R-H, Zhao T, Cui K, Hu G, Chen Q, Chen W, et al. (2016). Negative reciprocal regulation between Sirt1 and Per2 modulates the circadian clock and aging. Sci Rep, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Carrer A, Wellen KE (2015). Metabolism and epigenetics: a link cancer cells exploit. Curr Opin Biotechnol 34: 23-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Wang R-H, Kim H-S, Xiao C, Xu X, Gavrilova O, Deng C-X (2011). Hepatic Sirt1 deficiency in mice impairs mTorc2/Akt signaling and results in hyperglycemia, oxidative damage, and insulin resistance. J Clin Invest, 121: 4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Pekovic-Vaughan V, Gibbs J, Yoshitane H, Yang N, Pathiranage D, Guo B, et al. (2014). The circadian clock regulates rhythmic activation of the NRF2/glutathione-mediated antioxidant defense pathway to modulate pulmonary fibrosis. Genes Dev, 28: 548-560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Gibbs J, Ince L, Matthews L, Mei J, Bell T, Yang N, et al. (2014). An epithelial circadian clock controls pulmonary inflammation and glucocorticoid action. Nat Med, 20: 919-926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Haspel JA, Chettimada S, Shaik RS, Chu J-H, Raby BA, Cernadas M, et al. (2014). Circadian rhythm reprogramming during lung inflammation. Nat Commun, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Stocks T, Bjørge T, Ulmer H, Manjer J, Häggström C, Nagel G, et al. (2015). Metabolic risk score and cancer risk: pooled analysis of seven cohorts. Int J Epidemiol: 44: 1353-1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Bosetti C, Rosato V, Li D, Silverman D, Petersen G, Bracci P, et al. (2014). Diabetes, antidiabetic medications, and pancreatic cancer risk: an analysis from the International Pancreatic Cancer Case-Control Consortium. Ann Oncol, 25: 2065-2072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Liu X, Hemminki K, Försti A, Sundquist K, Sundquist J, Ji J (2015). Cancer risk in patients with type 2 diabetes mellitus and their relatives. Int J Cancer, 137: 903-910 [DOI] [PubMed] [Google Scholar]
- [164].Peeters PJ, Bazelier MT, Leufkens HG, de Vries F, De Bruin ML (2014). The Risk of Colorectal Cancer in Patients With Type 2 Diabetes: Associations With Treatment Stage and Obesity. Diabetes Care, 38: 495-502 [DOI] [PubMed] [Google Scholar]
- [165].Harding JL, Shaw JE, Peeters A, Cartensen B, Magliano DJ (2015). Cancer Risk Among People With Type 1 and Type 2 Diabetes: Disentangling True Associations, Detection Bias, and Reverse Causation. Diabetes Care, 38: 264-270 [DOI] [PubMed] [Google Scholar]
- [166].Setiawan VW, Hernandez BY, Lu SC, Stram DO, Wilkens LR, Le Marchand L, et al. (2014). Diabetes and Racial/Ethnic Differences in Hepatocellular Carcinoma Risk: The Multiethnic Cohort. J Natl Cancer Inst, 106: dju326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Lin C-C, Chiang J-H, Li C-I, Hsieh T-F, Liu C-S, Lin W-Y, et al. (2014). Independent and joint effect of type 2 diabetes and gastric and hepatobiliary diseases on risk of pancreatic cancer risk: 10-year follow-up of population-based cohort. Br J Cancer, 111: 2180-2186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Luo J, Beresford S, Chen C, Chlebowski R, Garcia L, Kuller L, et al. (2014). Association between diabetes, diabetes treatment and risk of developing endometrial cancer. Br J Cancer, 111: 1432-1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Sekikawa A, Fukui H, Maruo T, Tsumura T, Okabe Y, Osaki Y (2014). Diabetes mellitus increases the risk of early gastric cancer development. Eur J Cancer, 50: 2065-2071 [DOI] [PubMed] [Google Scholar]
- [170].Tsilidis KK, Allen NE, Appleby PN, Rohrmann S, Nöthlings U, Arriola L, et al. (2015). Diabetes mellitus and risk of prostate cancer in the European Prospective Investigation into Cancer and Nutrition. Int J Cancer, 136: 372-381 [DOI] [PubMed] [Google Scholar]
- [171].Kong AP, Yang X, Luk A, Ma RC, So WY, Ozaki R, et al. (2014). Severe hypoglycemia identifies vulnerable patients with type 2 diabetes at risk for premature death and all-site cancer: the Hong Kong diabetes registry. Diabetes Care, 37: 1024-1031 [DOI] [PubMed] [Google Scholar]
- [172].Eijgenraam P, Heinen M, Verhage B, Keulemans Y, Schouten L, van den Brandt P (2013). Diabetes type II, other medical conditions and pancreatic cancer risk: a prospective study in The Netherlands. Br J Cancer, 109: 2924-2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Colmers IN, Majumdar SR, Yasui Y, Bowker SL, Marra CA, Johnson JA (2013). Detection Bias and Overestimation of Bladder Cancer Risk in Type 2 Diabetes A matched cohort study. Diabetes Care, 36: 3070-3075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Schlesinger S, Aleksandrova K, Pischon T, Jenab M, Fedirko V, Trepo E, et al. (2013). Diabetes mellitus, insulin treatment, diabetes duration, and risk of biliary tract cancer and hepatocellular carcinoma in a European cohort. Ann Oncol, 24: 2449-2455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Yang W, Shu X, Gao J, Li H, Cai H, Yang G, et al. (2013). Prospective evaluation of type 2 diabetes mellitus on the risk of primary liver cancer in Chinese men and women. Ann. Oncol, 4(6):1679-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Koh W, Wang R, Jin A, Yu M, Yuan J (2013). Diabetes mellitus and risk of hepatocellular carcinoma: findings from the Singapore Chinese Health Study. Br J Cancer, 108: 1182-1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Chiu H-M, Lee Y-C, Tu C-H, Chang L-C, Hsu W-F, Chou C-K, et al. (2014). Effects of Metabolic Syndrome and Findings from Baseline Colonoscopies on Occurrence of Colorectal Neoplasms. Clin Gastroenterol Hepatol, 13: 1134-1142 [DOI] [PubMed] [Google Scholar]
- [178].Bhindi B, Locke J, Alibhai SM, Kulkarni GS, Margel DS, Hamilton RJ, et al. (2015). Dissecting the association between metabolic syndrome and prostate cancer risk: analysis of a large clinical cohort. Eur Urol, 67: 64-70 [DOI] [PubMed] [Google Scholar]
- [179].Dyson J, Jaques B, Chattopadyhay D, Lochan R, Graham J, Das D, et al. (2014). Hepatocellular cancer: the impact of obesity, type 2 diabetes and a multidisciplinary team. J Hepatol, 60: 110-117 [DOI] [PubMed] [Google Scholar]
- [180].Duggan C, Onstad L, Hardikar S, Blount PL, Reid BJ, Vaughan TL (2013). Association between markers of obesity and progression from Barrett's esophagus to esophageal adenocarcinoma. Clin Gastroenterol Hepatol, 11: 934-943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Kitahara CM, Platz EA, Freeman LEB, Hsing AW, Linet MS, Park Y, et al. (2011). Obesity and thyroid cancer risk among US men and women: a pooled analysis of five prospective studies. Cancer Epidemiol Biomarkers Prev, 20: 464-472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Schlesinger S, Aleksandrova K, Pischon T, Fedirko V, Jenab M, Trepo E, et al. (2013). Abdominal obesity, weight gain during adulthood and risk of liver and biliary tract cancer in a European cohort. Int J Cancer, 132: 645-657 [DOI] [PubMed] [Google Scholar]
- [183].Anderson GL, Neuhouser ML (2012). Obesity and the risk for premenopausal and postmenopausal breast cancer. Cancer Prev Res, 5: 515-521 [DOI] [PubMed] [Google Scholar]
- [184].De Nunzio C, Albisinni S, Freedland SJ, Miano L, Cindolo L, Agrò EF, et al. (2013) Abdominal obesity as risk factor for prostate cancer diagnosis and high grade disease: a prospective multicenter Italian cohort study. Urol Oncol, 31: 997-1002 [DOI] [PubMed] [Google Scholar]
- [185].Mungo B, Zogg CK, Hooker CM, Yang SC, Battafarano RJ, Brock MV, et al. (2015). Does obesity affect the outcomes of pulmonary resections for lung cancer? A National Surgical Quality Improvement Program analysis. Surgery,157(4): 792-800 [DOI] [PubMed] [Google Scholar]
- [186].Chlebowski RT, McTiernan A, Wactawski-Wende J, Manson JE, Aragaki AK, Rohan T, et al. (2012). Diabetes, metformin, and breast cancer in postmenopausal women. J Clin Oncol, 30: 2844-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Margel D, Urbach DR, Lipscombe LL, Bell CM, Kulkarni G, Austin PC, et al. (2013). Metformin use and all-cause and prostate cancer-specific mortality among men with diabetes. J Clin Oncol, 31: 3069-3075 [DOI] [PubMed] [Google Scholar]
- [188].Preston MA, Riis AH, Ehrenstein V, Breau RH, Batista JL, Olumi AF, et al. (2014). Metformin use and prostate cancer risk. Eur Urol, 66: 1012-1020 [DOI] [PubMed] [Google Scholar]
- [189].Tseng C-H (2014). Metformin significantly reduces incident prostate cancer risk in Taiwanese men with type 2 diabetes mellitus. Eur J Cancer, 50: 2831-2837 [DOI] [PubMed] [Google Scholar]
- [190].Sehdev A, Shih YCT, Vekhter B, Bissonnette MB, Olopade OI, Polite BN (2014). Metformin for primary colorectal cancer prevention in patients with diabetes: A case-control study in a US population. Cancer, 121: 1071-1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Lee C-k, Jung M, Jung I, Heo SJ, Jeong YH, An JY, et al. (2015). Cumulative Metformin Use and Its Impact on Survival in Gastric Cancer Patients After Gastrectomy. Ann Surg, 263: 96-102 [DOI] [PubMed] [Google Scholar]
- [192].Sakamoto A, Hino S, Nagaoka K, Anan K, Takase R, Matsumori H, et al. (2015). Lysine demethylase LSD1 coordinates glycolytic and mitochondrial metabolism in hepatocellular carcinoma cells. Cancer Res, 75: 1445-1456 [DOI] [PubMed] [Google Scholar]
- [193].Cao Y, Guo WT, Tian S, He X, Wang XW, Liu X, et al. (2015). miR-290/371-Mbd2-Myc circuit regulates glycolytic metabolism to promote pluripotency. EMBO J, e201490441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Xu Y, Miriyala S, Fang F, Bakthavatchalu V, Noel T, Schell D, et al. (2014). Manganese superoxide dismutase deficiency triggers mitochondrial uncoupling and the Warburg effect. Oncogene, 34: 4229-4237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Bando H, Atsumi T, Nishio T, Niwa H, Mishima S, Shimizu C, et al. (2005). Phosphorylation of the 6-phosphofructo-2-kinase/fructose 2, 6-bisphosphatase/PFKFB3 family of glycolytic regulators in human cancer. Clin Cancer Res, 11: 5784-5792 [DOI] [PubMed] [Google Scholar]