

SUMMARY
We describe a novel Solid-phase Reversible Sample-Prep (SRS) platform, which enables rapid sample preparation for concurrent proteome and N-glycome characterization by mass spectrometry. SRS utilizes a uniquely functionalized, silica-based bead that has strong affinity toward proteins with minimal-to-no affinity for peptides and other small molecules. By leveraging the inherent size difference between, SRS permits high-capacity binding of proteins, rapid removal of small molecules (detergents, metabolites, salts, etc.), extensive manipulation including enzymatic and chemical treatments on beads-bound proteins, and easy recovery of N-glycans and peptides. The efficacy of SRS was evaluated in a wide range of biological samples including single glycoprotein, whole cell lysate, murine tissues, and human urine. To further demonstrate the SRS platform, we coupled a quantitative strategy to SRS to investigate the differences between DU145 prostate cancer cells and its DIAPH3-silenced counterpart. Our previous studies suggested that DIAPH3 silencing in DU145 prostate cancer cells induced transition to an amoeboid phenotype that correlated with tumor progression and metastasis. In this analysis we identified distinct proteomic and N-glycomic alterations between the two cells. Intriguingly, a metastasis-associated tyrosine kinase receptor ephrin-type-A receptor (EPHA2) was highly upregulated in DIAPH3-silenced cells, indicating underling connection between EPHA2 and DIAPH3. Moreover, distinct alterations in the N-glycome were identified, suggesting a cross-link between DIAPH3 and glycosyltransferase networks. Overall, SRS is an enabling universal sample preparation strategy that is not size limited and has the capability to efficiently prepare and clean peptides and N-glycans concurrently from nearly all sample types. Conceptually, SRS can be utilized for the analysis of other posttranslational modifications, and the unique surface chemistry can be further transformed for high-throughput automation. The technical simplicity, robustness, and modularity of SRS make it a highly promising technology with great potential in proteomic-based research.
Keywords: Reversible Solid-phase, Sample-prep, glycoprotein, membrane protein, Urine, Plasma, proteome, N-glycome
Introduction
Sample preparation is critical for shotgun-based proteomics studies. Current strategies can be classified into either solid- or solution-phase approaches, based on whether proteins or proteases are immobilized. Classic “in-gel” digestion is an early version of solid-phase strategies, with the advantage of being amenable to all protein types, but the disadvantage of unpredictable loss of “in-gel trapped peptides” (1). Alternatively, the solution-phase strategy FASP (Filter-Aided Sample Preparation) has become particularly prevalent because of its broad capabilities and ease of use (1). When combined with lectins, FASP has the ability to isolate lectin-specific glycopeptides (2). Other spin-filter workflows can separate released N-glycans from deglycosylated proteins without the use of lectins by acidification (3). Even with these advances, the throughput of a spin filter workflow is limited by the size of the filter, and the speed and capacity of the centrifuge. Furthermore, automated high-throughput workflows based on spin-filters are technically challenging (3).
Beyond the proteome, interrogating the most abundant post-translational modification (PTM), N-glycosylation, may be critical in deciphering numerous physiological and pathological processes (4). Releasing heterogeneous and hydrophilic glycan-moieties from proteins enables the identification of formerly occupied sites and the potential quantification of the degree of glycosite occupation (3, 5). An ideal sample preparation strategy would: 1) be simple and non-tedious; 2) be amenable to all biological samples, including membrane proteins; 3) minimize sample transfers to avoid sample loss; 4) yield peptide and PTM products, such as N-glycans ready for subsequent analysis; and 5) be adaptable to automated high-throughput. To meet these challenges, we developed a novel Solid-phase Reversible Sample-prep (SRS) strategy based on uniquely functionalized silica-based beads that have strong non-specific affinity towards proteins (or large polypeptides) with minimal-to-no affinity for peptides and other small molecules. Utilizing this novel, size-based chemical affinity approach, proteins are immobilized onto beads and then fully recovered upon proteolytic digestion.
EXPERIMENTAL PROCEDURES
Materials and Reagents
Human serum IgG, Bovine fetuin, dithiothreitol (DTT), iodoacetamide (IAA), dimethyl sulfoxide, methylamine hydrochloride, 4-methylmorpholine, sodium hydroxide, cellulose (medium fibrous), iodomethane, 2,5-dihydroxybenzoic acid (DHB), 1-butanol, HPLC grade of ethanol, 2-amniobenzoic acid, and 2-[13C6]-aminobenzoic acid were purchased from Sigma-Aldrich (St. Louis, MO). PNGase F (glycerol-free) was purchased from New England Biolabs (Ipswich, MA). Sequencing grade trypsin was obtained from Promega (Madison, WI). H218O (98%) was obtained from Rotem (Arava, Israel). The uniquely-functionalized silica beads (10 μm) were synthesized in-house (EMD Millipore, Billerica, MA). Urine was obtained from one healthy donor under an institutional review board-approved protocol. Kidney, bladder, and colon were isolated from C57BL/6 laboratory mice.
DU145 Cell culture and Immofluorescence of EPHA2
The generation of DU145 prostate cancer cells stably expressing control (DU145Ctrl) shRNA or shRNAs against DIAPH3 (knock-down, DU145KD) was described previously (6). Both DU145Ctrl and DU145KD cells were maintained in DMEM high glucose (Gibco) supplemented with 10% fetal bovine serum (Valley Biomedical) and with 0.2<Image> g/mL puromycin (Invitrogen). Cells were seeded into 24-well plates at 50,000 cells per well. After overnight attachment, cells were fixed with 4% paraformaldehyde (Electron Microscopy Sciences), permeabilized in 0.1% Triton X-100, and blocked in 1% goat serum in 0.2% BSA/PBS. After overnight incubation at 4°C with EphA2 primary antibody (Cell Signaling, D4A2 XP Rabbit mAb, 1:100 dilution), cells were washed, incubated with Cy3 anti-rabbit secondary antibody (1:1000 dilution), and mounted in Vectashield containing DAPI. Samples were imaged with identical exposure times on a Zeiss Axioplan 2 microscope using a 63× PlanApo 1.4 objective, Axiocam camera, and Axiovision software.
Cytosolic (CP), membrane-enriched (MP), and whole lysate (WCL) preparation
Both DU145Ctrl and DU145KD cells were seeded on to 100 mm tissue culture plates (Corning), at a density of 4 × 106 cells per plate, to achieve a total of 12 × 106 cells per plate. After 24h, plates were washed twice with ice cold PBS, and then gently scraped into 2 mL Eppendorf vial using ice cold PBS (1×) supplemented with 2 mM EDTA (1 mL/plate). Cells were pelleted by centrifugation at 14,000 rpm, at 4°C for 15 min, and the supernatant were discarded. The pellets were re-suspended in 1 mL of PBS (1×) (containing of 2% SDS and 1 mM CaCl2) and transferred to a 5 mL glass homogenizer tube. The cells were homogenized by 12 reverse strokes at 1800 rpm on a Glass-Col Homogenizer Motor Drives (Terre Haute, IN). After homogenization, the lysates were centrifuged for 20 min at 14000 rpm and the insoluble pellet was discarded. The supernatants (Whole Cell Lysate, WCL) were stored at −80 °C for further use.
Membrane-enriched proteins (MP) were prepared using a modified procedure. Alternatively, the cell pellets were homogenized in 1 mL of buffer M (50 mM HEPES pH 7.4, 10 mM NaCl, 5 mM MgCl2, and 0.1 mM EDTA) as opposed to 1 mL of PBS containing of 2% SDS and 1 mM CaCl2. Cells were centrifuged for 20 min at 14000 rpm, and the supernatant consisted primarily cytosolic and soluble proteins (CP). The pellet was re-suspended in 1 mL of PBS with 2% SDS and 1 mM CaCl2. The sample was centrifuged at 14000 rpm for 15 min at 10 °C. The insoluble was discarded, and the supernatant containing the solubilized membrane-enriched proteins was kept.
Tissue samples were frozen using liquid nitrogen. Once frozen, larger murine tissues were cut into small pieces and manually ground into powder. Buffer M (50 mM HEPES pH 7.4, 10 mM NaCl, 5 mM MgCl2, and 0.1 mM EDTA) was used to suspend and transfer the powder into a 5 mL glass homogenizer tube. After homogenization, the membrane-enriched protein fractions from tissues were prepared similarly to those from cells. Protein concentrations were measured by the BCA assay in triplicate on a Nanodrop 2000c spectrometer (Thermo Scientific, Wilmington, DE).
Western blotting of DU145Ctrl and DU145KD Cells
10 μg of each cell fraction (WCL, CP, and MP), for EPHA2 and β-tubulin visualization, were aliquoted and suspended in 1× lamelli buffer. Proteins were then resolved on 10% SDS-PAGE gels and subsequently transferred to nitrocellulose for 1.5h at 4°C. After a 1 -hr block in 5% BSA in 1×TBS-Tween and three 15 min washes in 1×TBS-Tween, membranes were incubated with the appropriate primary antibodies (1:1000 dilutions in 1×TBS-Tween + 2% BSA) overnight at 4°C with constant rocking. Membranes were then washed three times, 15 min each, with 1×TBS-Tween, incubated at room temperature with the appropriate HRP-conjugated antibody, washed again three times at 15 min per wash, and visualized with West Pico ECL reagent. All lanes within a given antibody condition were imaged simultaneously.
SRS Workflow
Protein Immobilization and Pre-treatment
Aliquots of 10 mg of dry SRS beads (~25 μL of physical volume) were set as the default amount of starting material for each experiment except of other notifications. All SRS experiments were carried out in a benchtop thermomixer (Eppendorf ThermoMixer C) with constant shaking (1400 rpm). The SRS beads were rinsed twice with 150 μL of PBS (1X) prior to use. All protein samples were dissolved in PBS (1X), or in 2% SDS in PBS if SDS was required for solubility. Samples were then added to the SRS beads and kept at constant shaking for a period of 20 min at room temperature. 100% acetonitrile was immediately added to samples dissolved with 2% SDS in PBS to reach a final concentration of 70% acetonitrile. The addition of organic acetonitrile was necessary to offset the effect of SDS and to enable effective binding of proteins onto the surface of the SRS beads. Unbound proteins were collected from the supernatant after centrifugation and measured by BCA assay. Next, 150 μL of 8M urea in PBS was added to the sample vial. Reduction and alkylation were carried out with 10 μL of 0.5M DTT at room temperature (45 min), then followed by 15 μL of 0.5M IAA in a dark environment (45 min). Excess reagents and other small molecules were removed by three washes with 150 μL of 8M urea. In select cases, an additional wash with 80% acetonitrile was performed to ensure the complete removal of SDS prior to PNGase F exposure. For highly complex background samples (e.g. urine) additional washes (3–4 times) with 100 mM ammonium bicarbonate buffer (ABC) were conducted.
SRS De-N-glycosylation and the collection of N-glycans
After reduction and alkylation on beads, the buffer was changed to 50 mM ABC. If N-glycosites were part of the study, H218O was used to prepare the ABC buffer. PNGase F (~1 μL of enzyme per 100 μg of protein) was introduced, and the vials were incubated for a period of 12 to 20 hrs at 37 °C. After de-N-glycosylation, released N-glycans in the supernatant were readily separated from the beads by centrifugation. Additional rinsing with 150 μL of 0.1% formic acid (twice) was performed to fully recover any remaining free N-glycans. The glycan fractions were combined and completely dried in a speed-vacuum.
SRS Proteolytic Digestion
After deglycosylation, the SRS beads were suspended in 150 μL of 50 mM ammonium bicarbonate buffer (prepared in normal H2O). Trypsin (trypsin: protein = 1: 50 by weight) was added and samples were incubated at 37 °C for 6 hours. Additional fresh trypsin was added after 6 hours. After digestion, tryptic peptides in the supernatant were collected after centrifugation. Furthermore, 150 μL of 20% and 70% acetonitrile were sequentially applied to recover any remaining peptides. All peptide fractions were combined and stored at −20 °C for future use. Under selected cases, intact proteins can be stripped off the beads by incubating the proteins-bound SRS beads into 100 mM Tris HCl (pH 7.5) with 2% SDS at 70 °C for 10 min.
Mass spectrometric analysis of permethylated N-glycans by MALDI-MS
Without further purification, the collected N-glycans were directly permethylated according to the solid-phase permethylation procedure(7). The matrix solution was prepared by dissolving 10 mg of 2,5-dihydroxybenzoic acid in a volume of 1 mL of 50% methanol containing 1 mM sodium acetate. Glycans were spotted directly onto a stainless steel MALDI plate and mixed with an equal volume of matrix solution (0.5 – 1 μL). MALDI-MS was carried out on an MDS SCIEX 4800 (Applied Biosystems, Carlsbad, CA) using the interactive mode. The external calibration was performed using the ProteoMass Peptide MALDI-MS calibration kit (Sigma-Aldrich, St. Louis, MO). MS data were processed using Data Explorer 4.9 (Applied Biosystems, Carlsbad, CA).
Quantitative comparison of DU145KD vs. DU145Ctrl N-Glycome by LC-MS
Quantitative comparison of the N-Glycomes of DU145Ctrl and DU145KD was performed according to our published DRAG protocol (8). In short, without any additional purification, free N-glycans from the same amount of MP of both cell types were respectively derivatized with 2-AA and 2-13[C6]-AA, using a methanol-based condition (9). After derivatization, the respective 2-AA and 2-13[C6]-AA modified samples were pooled and purified by a hand-packed cellulose protocol (8). The 2-AA modified N-glycans were further subjected to methylamidation modification. The final products were purified by second cellulose SPE. The derivatized N-glycans were further reconstituted in 500 μL of water, and passed through a 0.2 μM syringe filter to remove particles (Pall Life Science, Port Washington, NY). The derivatized N-glycans were then analyzed on a Q-Exactive mass spectrometer (Thermo Scientific, Waltham, MA) connected to an autosampler and nanoflow HPLC pump (Eksigent, Dublin, CA). The reverse-phase columns were packed in-house by using Magic C18 particles (3 μm, 200 Å; Michrom Bioresource), and PicoTip Emitters (New Objective). Buffer A was 0.2% formic acid, buffer B was acetonitrile and 0.2% formic acid, and loading buffer was 5% formic acid with 5% acetonitrile. The modified glycans were eluted from 10% to 50% of buffer B in a 10 min linear gradient. The mass spectrometer was operated in a full MS mode (350–1800 m/z) with automatic gain control (AGC) target of 1 × 106 ions and a maximum ion transfer (IT) of 100 ms. Source ionization parameters were as follows: spray voltage, 2.2 KV; capillary temperature, 200 °C; and s-lens level, 50.0.
Mass spectrometric analysis of peptides
All tryptic peptides were further desalted by C18 tips prior to LC-MS/MS analyses. The peptides were also analyzed by Q-Extractive instrument and similar LC-MS/MS setting as described above for LC-MS N-glycans analysis. The peptides were eluted with a 120 min linear gradient. The mass spectrometer was operated in a Top 10 data dependent mode with automatic switching between MS and MS/MS. Full scan MS mode (350–1800 m/z) was operated at a resolution of 70 000 with automatic gain control (AGC) target of 1 × 106 ions and a maximum ion transfer (IT) of 20 ms. Ions selected for MS/MS were subjected to the following parameters: resolution 17 500; AGC 1 × 105 ions; maximum IT 80 ms; 2.0 m/z isolation window; normalized collision energy 28.0; underfill ratio 1.0%, and dynamic exclusion of 30.0 s. All samples were run with duplicate injections.
Database searching and validation
All peptide LC-MS/MS data were analyzed using the Thermo Proteome Discoverer (1.3.0.339) software platform, searched against the UniProtKB/Swiss-Prot database (Homo sapiens, released in 2012_07) containing both forward and reverse protein sequences using the Mascot search engine (v 2.3) (10). One miscleavage per peptide was allowed and mass tolerances were set at 10 ppm for precursor and 30 mmu for fragment ions. The search included fixed modification of carbamidomethylation on cysteine, and variable modifications: oxidation on methionine, deamidation (O16) on both asparagine and glutamine, and O18-incorporated deamidation on asparagine. The false discovery rate (FDR) was set at 1% at the peptide level by searching the same data set against the decoy database. A minimum of two peptides per protein were required for the identification of a protein. A protein group was treated as a single entity with one representative protein (accession number).
N-glycosite determination
A deglycopeptide was assigned by meeting two criteria simultaneously (3): 1) the peptide sequence possessed the N-glycosylation consensus motif (Asn-XXX-Ser/Thr, in which XXX is any amino acid except proline); and 2) the specific asparagine residue within that motif was identified as the 18O-incorporated deamidation derivative (N, +2.9982 Da) (3). The N-glycosites were determined by those asparagine residues characterized as 18O-incorporated deamidated aspartic acid within deglycopeptides. Glycoproteins in this study were defined as those proteins identified by at least one unique deglycopeptide.
Quantitative Proteomic Comparison of DU145Ctrl vs. DU145KD
LC-MS/MS settings for the label-free strategy was nearly identical to the qualitative runs as described above, except that no dynamic exclusion was employed during the course of data-dependent acquisition. Approximately 1300 unique proteins were characterized per cell line with a combination of two parallel MS replicates (data not shown). The spectral counts analysis was performed using Scaffold (v 4.3.2, Proteome Software Inc. Portland, OR). For improved quantification, only those proteins with more than 10 PSM (peptide spectral match) in all four dataset (two MS replicates for each cell lines) were chosen and only those PSMs without any modification were considered. The protein tubulin β-5 chain (TBB5) was used for normalization. Thus, a total of 888 proteins were quantified by this methodology (Supplementary materials).
RESULTS
The SRS platform
In the SRS platform, proteins are initially bound to the beads (20 min), then washed (15–20 min), and then fully recovered by proteolytic digestion for LC-MS/MS analysis (5 min) (Figure 1). Upon immobilization, protein samples can be easily processed (e.g. impurity removal, buffer exchanges, PTM removal and capture) with minimal sample loss. Optional enzymatic or chemical treatments can be integrated to capture a specific PTM of interest or to modify the SRS-bound proteins or glycan moieties prior to proteolytic digestion(11). veness is directly dependent on the accessibility of the innermost core GlcNAc within a N-glycan chain. SRS significantly decreased the associated time for processing and handling, and for glycan or peptide recovery. Although the associated enzyme reactions are governed by their underlying biochemical principles, both PNGase F and trypsin digestions could be alternatively accelerated using microwave irradiation(12, 13). As shown in Figure 1, H2O18-based buffer can be used with PNGase F to identify the N-glycosite by incorporating one O18-atom onto the former N-glycosylated asparagine (Asn to O18-Asp) (14).
FIG. 1.
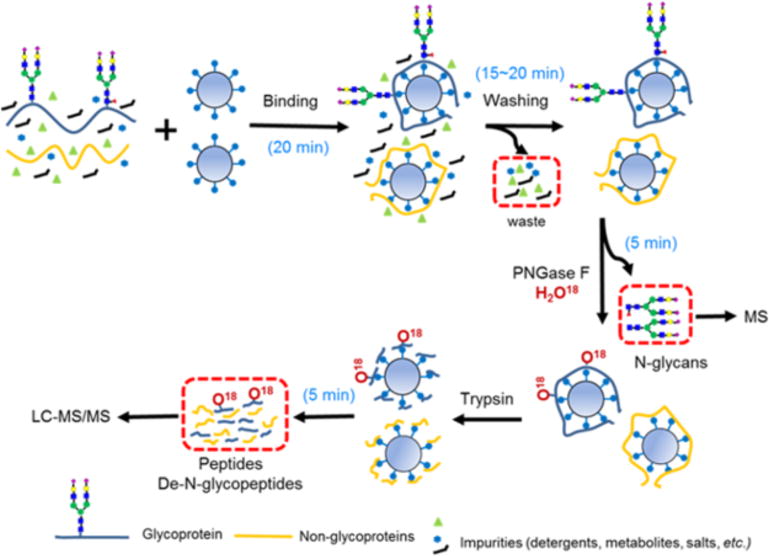
The SRS workflow separates and captures purified free N-glycans and tryptic peptides for downstream mass spectrometry (MS) analyses. SRS has high binding capacity with all biological samples, including membrane-enriched proteins solubilized in 2% SDS. Non-protein compounds such as metabolites, reagents, salts, and detergents are rapidly removed in minutes. In this workflow, samples undergo sequential enzymatic de-N-glycosylation to release and capture N-glycans, followed by proteolytic digestion to recover peptides. H2O18 is used to label the original sites of N-glycosylation. The handling times are reported, excluding the time for enzymatic reactions.
Evaluation of SRS efficacy
The SRS beads were found to have high binding capacity toward most biological samples, even polypeptides. For example, 1 mg of horse heart cytochrome C, a small and hydrophilic polypeptide, bound to 10 mg of beads (dry weight) (Fig. 2a). High binding capacities were observed for a broad range of samples irrespective of their complexity and nature, including single glycoproteins (bovine fetuin, serum IgG), human urine, whole cell lysates, tissue, and enriched membrane proteins solubilized in 2% SDS (Table 1). Urinary proteins were effectively purified in minutes, even though urine is a notoriously challenging analyte because of its inherent metabolites, endogenous peptides, oligosaccharides, and other non-protein contaminants (Fig. 2b). This timescale provides a marked advantage over current solution and filter-based protocols (3, 15).
FIG. 2.
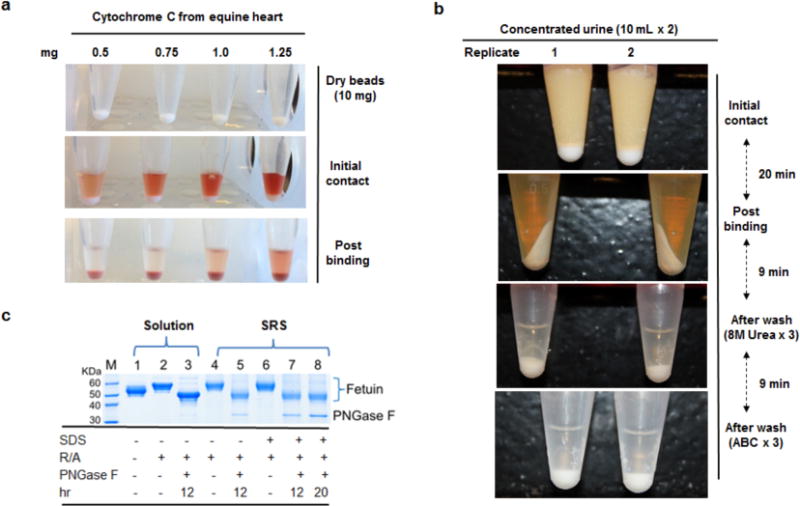
The evaluation of SRS beads on protein binding, buffer washing and enzyme accessibility. (a) Nearly 1 mg of horse heart cytochrome C was bound to 10 mg of dry beads, demonstrating excellent binding of a small and hydrophilic polypeptide. (b) After protein immobilization, efficient and rapid removal of urinary non-protein components was achieved with a rapid two-stage wash. (c) SRS bound bovine fetuin was completely reduced and alkylated, and de-N-glycosylated with PNGase F, achieving results comparable to the classic solutionphase procedure (R/A: reduction and alkylation of disulfide bonds). The intact bovine fetuin was recovered by a mild stripping protocol (Experimental section).
Table 1.
The binding capacity of SRS beads for different biological samples including: single protein, whole cell lysate, membrane-enriched fractions from cells, and murine tissues.
| Sample Types | Starting Amount (μg) | Unbound Amount (μg) | Bound proteins ratio (%) |
|---|---|---|---|
| Bovine fetuin (A) | 300 | 26.9 | 91% |
| Bovine fetuin (B) | 300 | 22.2 | 93% |
| Bovine fetuin (C)* | 300 | 27 | 91% |
| Bovine fetuin (D)* | 300 | 25 | 92% |
| Human serum IgG (A) | 300 | 18 | 94% |
| Human serum IgG (B) | 300 | 13 | 96% |
| Whole Lysate, DU145Ctrl (A)* | 500 | 28.7 | 94% |
| Whole Lysate, DU145Ctrl (B)* | 500 | 25.6 | 95% |
| Membrane-enriched DU145Ctrl (A)* | 300 | 29 | 90% |
| Membrane-enriched DU145Ctrl (B)* | 300 | 35 | 88% |
| Membrane-enriched DU145KD (A)* | 300 | 31 | 90% |
| Membrane-enriched DU145KD (B)* | 300 | 28 | 91% |
| Membrane-enriched colon (A)* | 400 | 39 | 90% |
| Membrane-enriched colon (B)* | 400 | 34 | 92% |
| Membrane-enriched colon (C)* | 400 | 48 | 88% |
| Membrane-enriched colon (D)* | 400 | 40 | 90% |
| Membrane-enriched bladder (A)* | 300 | 28 | 91% |
| Membrane-enriched bladder (B)* | 300 | 28 | 91% |
| Membrane-enriched bladder (C)* | 300 | 31 | 90% |
| Membrane-enriched kidney (A)* | 300 | 22 | 93% |
| Membrane-enriched kidney (B)* | 300 | 26 | 91% |
| Membrane-enriched kidney (C)* | 300 | 33 | 89% |
Murine kidney, bladder, and colon were isolated from C57BL/6 laboratory mice. The default solvent to dissolve protein samples was PBS buffer (1X), unless otherwise noted. An aliquot of 10 mg of dry beads was used for each experiment. (* Indicates samples dissolved in PBS (1X) containing 2% SDS). The protein concentrations were measured by BCA assay.
To further evaluate the compliant nature of SRS protein immobilization, the efficiency of enzymatic deglycosylation was tested on bovine fetuin in the presence or absence of 2% SDS as a surrogate measure of immobilized protein accessibility (Fig. 2c). Fetuin +/− 2% SDS was SRS-immobilized, and then underwent reduction and alkylation of disulfide residues followed by deglycosylation with PNGase F. Intact fetuin was fully recovered utilizing a mild intact protein release protocol (Experimental procedures). All disulfide bonds within fetuin were completely reduced and alkylated on the beads (Lanes 4 and 6). Irrespective of SDS, complete deglycosylation of SRS fetuin (Lanes 5, 7, and 8) was achieved, and digestion time was comparable to classic solution-phase deglycosylation (Lane 3). Both SRS and solution phase N-glycans were completely extracted, and demonstrated near-identical peak patterns by MALDI-MS (Supplementary Fig. 1). Diverse biological samples were further tested. These included human serum IgG, human urine, whole cell lysates (DU145), and murine tissue lysates (Supplementary Fig. 2–5, and Supplementary Tables 1–2). For all samples, highly comparable N-glycan spectra were observed among their respective biologic and technical replicates, demonstrating the pronounced reproducibility of SRS platform for N-glycome analysis.
After extraction of N-glycans, SRS-immobilized proteins underwent tryptic digestion to recover peptides (Fig. 1). Employing a simple three-stage extraction procedure, peptides were efficiently recovered for all samples (86 – 94%, Supplementary Table 3). Utilizing bovine fetuin as a reference protein, repeated ion chromatography detected nearly all expected peptides except those that were either too large or too small for MS analysis (Supplementary Fig. 6, and Supplementary Table 4). Proteomic and glycoproteomic analyses of complex samples such as cell lysates, murine tissue, and human urine specimens demonstrated appropriate proteome characterization using a standard LC-MS/MS pipeline (Supplementary Fig. 7 and Supplementary Table 5). Multiple technical and biological replicates demonstrated a high degree of reproducibility, further highlighting the potential of SRS for complex proteomic studies. Overall, the above experiments demonstrated numerous advantages of the SRS platform including: 1) high binding capacity toward most protein samples; 2) efficacy of chemical reduction and alkylation on beads-bound proteins; 3) efficiency of enzymatic de-N-glycosylation and proteolytic digestion; and 4) easy recover of released N-glycans and digested peptides.
Proteomic and Glycomic Comparison of DU145 prostate cancer cells
Having confirmed the utility of the SRS methodology, we next employed SRS to perform an exploratory hypothesis-generating study. We previously reported that in DU145 prostate cancer cells, DIAPH3 silencing induced transition to an amoeboid phenotype, prompting migration, invasion, and pulmonary colonization; in prostate cancer patients, genomic loss of DIAPH3 correlated with tumor progression and metastasis (6). We utilized the SRS technology to investigate the proteomic and N-glycomic alterations of the highly aggressive amoeboid tumor cells arising from DIAPH3 silencing in DU145 (DU145KD) as compared to its un-modified counterpart (DU145Ctrl).
N-glycans and tryptic peptides from the membrane-enriched fractions of both cell types were collected and characterized (Fig. 3a). N-glycomes were initially compared on MALDI-MS, revealing that DU145KD had relatively higher levels of large carbohydrate species than DU145Ctrl (Supplementary Fig. 8 and Supplementary Table 6). To accurately measure N-glycome differences, we next incorporated an isotope labeling strategy with LC-MS profiling: DRAG (Fig. 3b)(8). Dramatic quantitative differences were observed among five specific subgroups of N-glycans, which were grouped by their specific stages within the N-glycosylation biosynthetic pathway (16) (Fig. 3c). DIAPH3 knockdown resulted in decreased expression of high-mannose (green), complex-type biantennary (light green), and complex-type triantennary (purple) N-glycans. In contrast, expression of large and complex-type tetraantennary N-glycans (pink) was increased, in agreement with results from MALDI-MS. Intriguingly; a subgroup of complex-type N-glycans with several uncapped HexNAc residues (blue) was specifically enriched in DU145KD cells. The results of these N-glycome analyses uncovered a previously unappreciated link between DIAPH3 and glycosyltransferase networks, further confirming that alterations in N-glycans may be highly relevant to cancer metastasis (4).
FIG. 3.
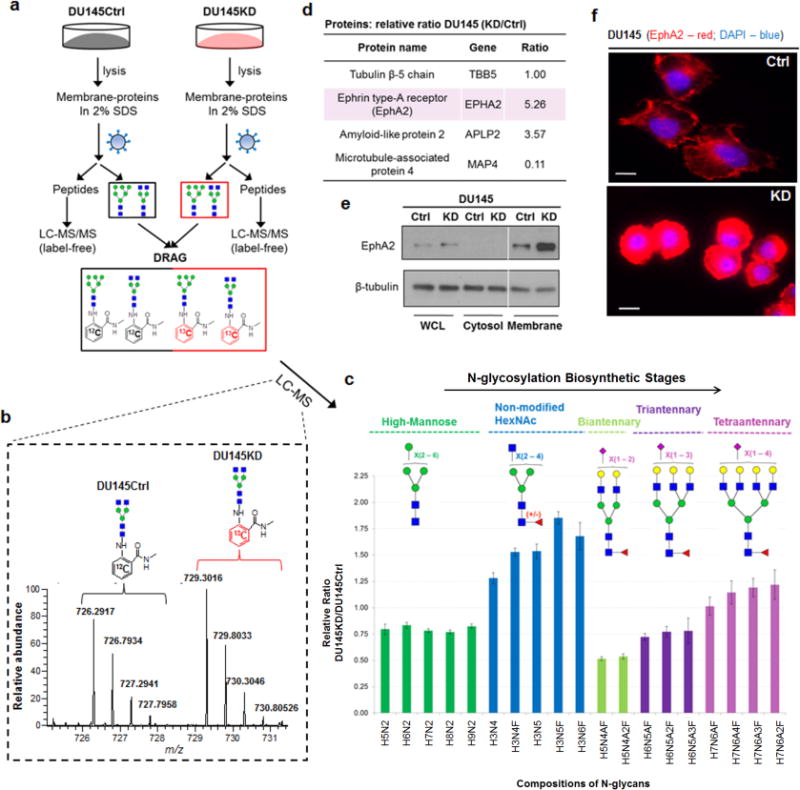
(a) SRS was employed to quantitatively compare the proteome and N-glycome of DIAPH3-silenced (amoeboid, DU145KD) and non-silenced (control, DU145Ctrl) human prostate cancer cell lines. Released N-glycans were compared by LC-MS utilizing a pair of light and heavy isotopes incorporated tags with a 6 Da mass difference (DRAG)(8). Proteins were quantitatively compared by spectral counting. (b) A representative LC-MS spectrum used to calculate the relative ratio of the same N-glycan species (Hex3HexNAc4) identified in both cell lines. (c) Distinct quantitative ratios were determined among five specific N-glycan subgroups: high-mannose (green), complex-type with unmodified non-reducing end HexNAc (blue), biantennary with terminal sialic acid (light green), triantennary with terminal sialic acid (purple), and tetraantennary with terminal sialic acid (pink). All putative topologies were proposed based on the mammalian N-glycosylation biosynthetic pathway and their m/z values (<10 ppm). Selected compositions were further manually validated by MS/MS fragmentations. H: hexose; N: N-acetylhexosamine; A: N-acetylneuraminic acid (Neu5Ac); F: fucose. (d) Differential expression of selected proteins identified by SRS from DU145Ctrl and DU145KD cell lines. Additional validation of EphA2 expression differences by immunoblotting (e), and immunofluorescence (f). Scale bar, 10μm.
Approximately 900 proteins were quantitatively compared between DU145Ctrl and DU145KD (Supplemental materials). Several intriguing candidates were identified (Fig. 3d). For example, Ephrin-type-A receptor (EPHA2) is a tyrosine kinase receptor up-regulated in multiple cancers, including prostate, breast, liver, colon and melanoma (17); EphA2 up-regulation has been correlated with metastasis and tumor progression (18). Furthermore, EPHA2 up-regulation evokes an amoeboid transition (18, 19). EPHA2 was highly upregulated in DU145KD cells, as confirmed by both immunoblotting and immuno-fluorescence imaging (Fig. 3e and 3f). Use of SRS resulted in the identification of a previously unappreciated connection between EPHA2, DIAPH3, and amoeboid-mediated invasion (6).
Discussion
Solid phase strategies are attractive because of their technical ease. Various solid-phase materials have been used in current glycoproteomics research, such as hydrazide beads (5) and lectin columns(14). These approaches are typically used to enrich a subset of the glycoproteome to facilitate the identification of low-abundant glycopeptides and N-glycosites. Unlike the SRS protocol, hydrazide based protocols will not capture the non-glycosylated version of the glycopeptide which can result in a quantitation bias of the glycoprotein. Although lectin protocols can be very effective, they are inherently selective. Recently, the GIG method (Glycoprotein immobilization for glycan extraction) was introduced (11). GIG utilizes Aminolink beads to covalently bind glycoproteins by reductive amination, thereby allowing for removal of N-glycans. We performed a similar protocol by covalently binding glycoproteins to NHS-activated magnetic beads (EMD Millipore) (data not shown). Although the NHS and GIG protocols are both simple and effective strategies to capture N-glycans, their covalent nature leads to incomplete recovery of bead bound peptides, and typically require harsh decoupling conditions that can be detrimental to protein characterization or quantitation. Conversely, the SRS protocol manipulates the size difference between proteins and peptides to bind proteins in a non-covalent manner, but yet allowed for extensive protein manipulation, and comprehensive sequential N-glycan and peptide recovery without selective enrichment.
Overall, SRS is an enabling universal sample preparation strategy with the capability to efficiently prepare and clean peptides and N-glycans concurrently from almost all sample types, including single glycoproteins, body fluids, tissue, cell lysates, and membrane-enriched proteins. Unlike other methods, SRS is not sample size limited. Conceptually, a variety of enzymatic and chemical treatments could also be employed into the SRS strategy: such as on-bead exoglycosidase digestion and sialic acid derivatization (11, 20). Importantly, since peptides are fully recoverable, the SRS strategy may be feasible for many other PTM analyses such as ADP-ribosylation on asparagine and glutamate residues (21), or palmitoylation on cysteine residues (22) as long as the integrity of the protein backbone are not affected by these treatments. As a chemically modified surface, SRS is not limited by sample size, can be magnetized, and is easily integrated into a high-throughput automated workflow (e.g. column or plate platform). The technical simplicity and robustness of SRS make it a highly promising technology with great potential in proteomic-based research.
Supplementary Material
Acknowledgments
We thank Dr. Rosalyn Adam for providing murine tissues, Dr. Hanno Steen and Dr. Judith Steen for mass spectrometry assistance, and Jiyoung Choi for technical assistance. This work was supported by an internal grant from the Technology and Innovation Development Office (Boston Children’s Hospital).
Abbreviations List
- SRS
Solid-phase Reversible Sample-Prep
- N-Glycosite
N-glycosylation site
- Deglycopeptide
de-N-glycosylated peptide
- WCL
Whole cell lysate
- CP
Cytosolic proteins
- MP
Membrane-enriched proteins
- DU145Ctrl
Non-modified DU145 cell
- DU145KD
DIAPH3 silenced DU145 cell
Footnotes
AUTHOR CONTRIBUTIONS
H.Z. and R.S.L. designed the chemistry for the SRS beads and developed the experimental protocol. V.J and D.B. synthesized the beads. S.K. and H.Z. carried out all experiments. S.M. and M.R.F. provided reagents and biological analyses of DU145 cells. H.Z. and R.S.L. analyzed data and interpreted the results. H.Z. and R.S.L. wrote the manuscript, and all authors contributed to the final version.
COMPETING FINANCIAL INTERESTS
Boston Children’s Hospital has filed a patent application covering the invention described in this report.
References
- 1.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 2.Zielinska DF, Gnad F, Wisniewski JR, Mann M. Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell. 2010;141:897–907. doi: 10.1016/j.cell.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 3.Zhou H, Froehlich JW, Briscoe AC, Lee RS. The GlycoFilter: a simple and comprehensive sample preparation platform for proteomics, N-glycomics and glycosylation site assignment. Mol Cell Proteomics. 2013;12:2981–2991. doi: 10.1074/mcp.M113.027953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hart GW, Copeland RJ. Glycomics hits the big time. Cell. 2010;143:672–676. doi: 10.1016/j.cell.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang H, Li XJ, Martin DB, Aebersold R. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat Biotechnol. 2003;21:660–666. doi: 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
- 6.Zhang HR, Xu JJ, Chen HY. Electrochemiluminescence ratiometry: a new approach to DNA biosensing. Anal Chem. 2013;85:5321–5325. doi: 10.1021/ac400992u. [DOI] [PubMed] [Google Scholar]
- 7.Kang P, Mechref Y, Klouckova I, Novotny MV. Solid-phase permethylation of glycans for mass spectrometric analysis. Rapid Commun Mass Spectrom. 2005;19:3421–3428. doi: 10.1002/rcm.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou H, Warren PG, Froehlich JW, Lee RS. Dual Modifications Strategy to Quantify Neutral and Sialylated N-Glycans Simultaneously by MALDI-MS. Analytical chemistry. 2014;86:6277–6284. doi: 10.1021/ac500298a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anumula KR, Dhume ST. High resolution and high sensitivity methods for oligosaccharide mapping and characterization by normal phase high performance liquid chromatography following derivatization with highly fluorescent anthranilic acid. Glycobiology. 1998;8:685–694. doi: 10.1093/glycob/8.7.685. [DOI] [PubMed] [Google Scholar]
- 10.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 11.Yang S, Li Y, Shah P, Zhang H. Glycomic analysis using glycoprotein immobilization for glycan extraction. Anal Chem. 2013;85:5555–5561. doi: 10.1021/ac400761e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou H, Briscoe AC, Froehlich JW, Lee RS. PNGase F catalyzes de-N-glycosylation in a domestic microwave. Anal Biochem. 2012;427:33–35. doi: 10.1016/j.ab.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vaezzadeh AR, Deshusses JM, Waridel P, Francois P, Zimmermann-Ivol CG, Lescuyer P, Schrenzel J, Hochstrasser DF. Accelerated digestion for high-throughput proteomics analysis of whole bacterial proteomes. J Microbiol Methods. 2010;80:56–62. doi: 10.1016/j.mimet.2009.10.019. [DOI] [PubMed] [Google Scholar]
- 14.Kaji H, Saito H, Yamauchi Y, Shinkawa T, Taoka M, Hirabayashi J, Kasai K, Takahashi N, Isobe T. Lectin affinity capture, isotope-coded tagging and mass spectrometry to identify N-linked glycoproteins. Nat Biotechnol. 2003;21:667–672. doi: 10.1038/nbt829. [DOI] [PubMed] [Google Scholar]
- 15.Vaezzadeh AR, Briscoe AC, Steen H, Lee RS. One-step sample concentration, purification, and albumin depletion method for urinary proteomics. J Proteome Res. 2010;9:6082–6089. doi: 10.1021/pr100924s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stanley P, Schachter H, Taniguchi N. N-Glycans. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Essentials of Glycobiology. 2nd. Cold Spring Harbor; NY: 2009. [PubMed] [Google Scholar]
- 17.Taddei ML, Parri M, Angelucci A, Onnis B, Bianchini F, Giannoni E, Raugei G, Calorini L, Rucci N, Teti A, Bologna M, Chiarugi P. Kinase-dependent and -independent roles of EphA2 in the regulation of prostate cancer invasion and metastasis. The American journal of pathology. 2009;174:1492–1503. doi: 10.2353/ajpath.2009.080473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taddei ML, Parri M, Angelucci A, Bianchini F, Marconi C, Giannoni E, Raugei G, Bologna M, Calorini L, Chiarugi P. EphA2 induces metastatic growth regulating amoeboid motility and clonogenic potential in prostate carcinoma cells. Molecular cancer research : MCR. 2011;9:149–160. doi: 10.1158/1541-7786.MCR-10-0298. [DOI] [PubMed] [Google Scholar]
- 19.Giannoni E, Taddei ML, Parri M, Bianchini F, Santosuosso M, Grifantini R, Fibbi G, Mazzanti B, Calorini L, Chiarugi P. EphA2-mediated mesenchymal-amoeboid transition induced by endothelial progenitor cells enhances metastatic spread due to cancer-associated fibroblasts. J Mol Med (Berl) 2013;91:103–115. doi: 10.1007/s00109-012-0941-9. [DOI] [PubMed] [Google Scholar]
- 20.Shah P, Yang S, Sun S, Aiyetan P, Yarema KJ, Zhang H. Mass spectrometric analysis of sialylated glycans with use of solid-phase labeling of sialic acids. Anal Chem. 2013;85:3606–3613. doi: 10.1021/ac3033867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, Wang J, Ding M, Yu Y. Site-specific characterization of the Asp- and Glu-ADP-ribosylated proteome. Nat Methods. 2013;10:981–984. doi: 10.1038/nmeth.2603. [DOI] [PubMed] [Google Scholar]
- 22.Yang W, Di Vizio D, Kirchner M, Steen H, Freeman MR. Proteome scale characterization of human S-acylated proteins in lipid raft-enriched and non-raft membranes. Mol Cell Proteomics. 2010;9:54–70. doi: 10.1074/mcp.M800448-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.