

Abstract
Many human diseases are associated with mutations causing protein misfolding and aggregation in the endoplasmic reticulum (ER). ER-associated degradation (ERAD) is a principal quality-control mechanism responsible for targeting misfolded ER proteins for cytosolic degradation. However, despite years of effort, the physiological role of ERAD in vivo remains largely unknown. Several recent studies have reported intriguing phenotypes of mice deficient for ERAD function in specific cell types. These studies highlight that mammalian ERAD has been designed to perform a wide-range of cell-type-specific functions in vivo in a substrate-dependent manner.
Keywords: endoplasmic reticulum, quality control, protein degradation, Sel1L-Hrd1, mouse models, substrates, physiology
The ER is the major site of protein synthesis and folding in the cell. In mammalian cells, an estimated one third of total proteome enter the ER through a channel known as the translocon (1). These newly synthesized proteins enter the ER in an unfolded state and become modified and folded with the help of a myriad of chaperones and enzymes. Upon reaching a native conformation, they are either transported to various organelles or released from cells via the secretory pathway (1). However, protein folding is inherently error prone, with a subpopulation of newly synthesized polypeptides failing to reach a native conformation. Moreover, many proteins containing disease-associated mutations are structurally incapable of reaching a native conformation, thereby becoming ‘permanently” entrapped in the ER (2). Indeed, accumulation of misfolded proteins disrupts ER homeostasis, which has been linked to altered immune response, metabolic regulation, insulin resistance, tumor growth and some degenerative diseases (3, 4).
Three pathways of quality control are integrated to maintain ER homeostasis: unfolded protein response (UPR), ER-associated degradation (ERAD) and autophagy. The latter two degrade misfolded proteins and protein aggregates. Failure to degrade misfolded proteins in the ER triggers UPR (Figure 1), a coordinated response including cytoplasmic-to-nuclear signaling designed to increase ER folding capacity, reduce protein input into the ER, activate ERAD and possibly autophagy. ERAD is responsible for the recognition and clearance of terminally misfolded proteins in the ER (Figure 1), thereby maintaining ER homeostasis. Pointing to its physiological significance, genetic deletion of several components of ERAD including Hrd1, Sel1L and p97 (VCP/CDC48) are embryonic lethal in mice (5-7). Moreover, nearly 70 human diseases have been linked to ERAD (2). While the biochemical processes of ERAD have been well characterized in the last two decades (8-10), the physiological significance of mammalian ERAD at the organismal level remains vague. This is an especially challenging problem given a vast number of different cell types and their crosstalk in mammals. Recent studies using cell type-specific ERAD-deficient animal models have begun to highlight the (patho)physiological significance of ERAD in vivo and to unveil the nature of endogenous substrates in the context of health and disease (11-17).
Figure 1. Two key ER quality control systems in mammals.
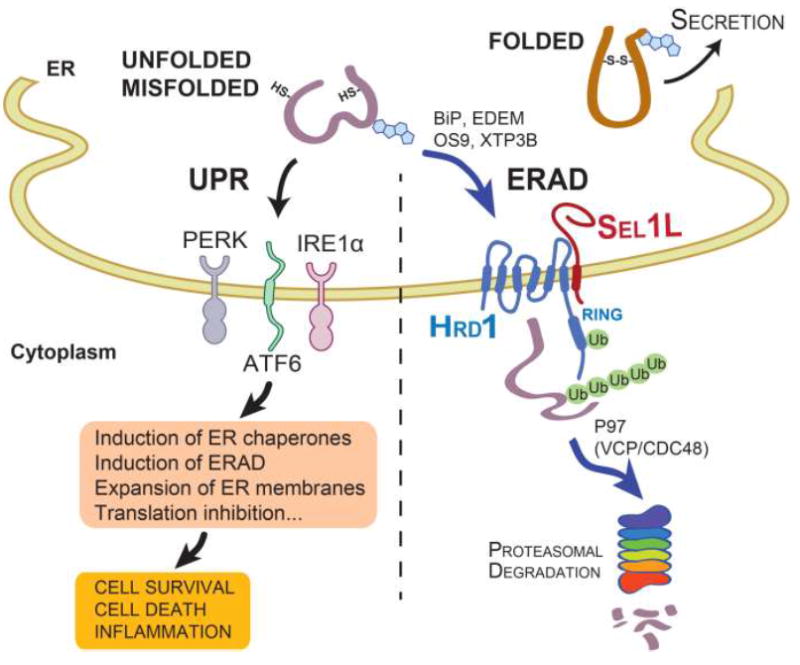
While folded proteins exit the ER, terminally unfolded or misfolded proteins in the ER activate UPR via three sensors IRE1α, PERK and ATF6, which initiate multiple signaling pathways leading to the induction of ER chaperones and ERAD, expansion of ER membranes and translation inhibition. In addition, misfolded proteins can be recruited to the ERAD complex via the activity of various ER chaperones such as BiP, EDEM, OS9 and XTP3B for cytosolic degradation. The Sel1L-Hrd1 protein complex represents the most conserved ERAD complex in mammals. Following retrotranslocation into the cytosol, substrates are ubiquitinated and, with the help of p97 (VCP/CDC48), degraded by the proteasome in the cytosol.
Several recent excellent reviews have focused on the biochemical processes of ERAD in yeast and mammalian cells (8-10). In this review, we focus on recent advances in understanding the nature of specific endogenous ERAD substrates and the physiological role of ERAD in mouse models. These studies underscore that knowledge of ERAD lags behind that of other branches of ER quality control, and that an improved understanding of how ERAD functions in a cell type-specific manner is critically important for understanding the pathogenesis of multiple diseases.
Cell Biology of ERAD
Misfolded proteins are retained in the ER and are in most cases recognized and translocated to the cytosol for proteasomal degradation by the ERAD machinery. One example is the cystic fibrosis transmembrane conductance regulator (CFTR), which is highly misfolding prone (18). Indeed, a majority of newly synthesized wildtype CFTR protein is quickly degraded (19, 20). In addition, in some cases ERAD can recognize and degrade certain proteins that are considered to be properly folded in order to regulate protein concentration. The best-characterized substrate for this is the 3-hydroxy-3-methylglutaryl acetyl-coenzyme-A reductase (HMGCR), a rate-limiting enzyme in sterol biogenesis (21). Indeed, remarkable insights into the molecular mechanisms underlying ERAD have been garnered from investigation of the turnover of resident ER membrane proteins, in yeast and human cells (8, 21-26).
The mechanisms underlying substrate recognition by ERAD remain poorly defined in mammalian cells (1, 27). In general, ERAD substrates can be recognized by maturation tags due to post-translational modifications and/or chaperone associations. Some nascent proteins are glycosylated cotranslationally in the ER, which can be used as a maturation tag to help direct intracellular processing, trafficking and degradation by ERAD (27). Trimming of several terminal mannose residues by ER mannosidases may direct terminally misfolded cargo to the ERAD pathway (28). Misfolded glycoproteins also can be targeted to the ERAD pathway independent of glycosylation signals (29, 30). In both instances, many ER chaperones are likely to participate in the recognition and recruitment of misfolded proteins to the ERAD complex, including BiP/GRP78, ER degradation enhancing α-mannosidase like protein 1 (EDEM1), Osteosarcoma amplified 9 (OS9), and XTP3B (29, 31-36). These chaperones, which can bind to unfolded proteins in a glycan-(in)dependent manner, deliver them to the Sel1L-Hrd1 ERAD complex in preparation for transport to the cytosol.
Among over a dozen E3 ligases in mammalian ERAD, Hrd1 is a principal ER-resident E3 ligase that forms a complex with the ER-resident Sel1L (also known as mammalian Hrd3) – together this complex is responsible for the degradation of a subset of misfolded proteins in the ER (21, 22, 29, 37-40) (Figure 1). Hrd1 protein consists of six transmembrane domains, and its cytoplasmic tail has a catalytic RING finger needed for E3 ligase activity, plus a long proline-rich C terminus. The transmembrane regions of Hrd1 may form a retrotranslocation channel to export ER proteins (41-43). The RING finger domain is strategically positioned in the cytosol to serve at least two distinct purposes. First, Hrd1-dependent autoubiquitination of the RING finger domain gates its own channel function (42). This finding raises the possibility that deubiquitinases might counter the ubiquitination reaction and control the retrotranslocation event as well. Whether autoubiquitination is a general feature that regulates the channel activity of other E3 ubiquitin ligases dedicated to ERAD is unclear at this point. Second, Hrd1 catalyzes ubiquitination of the misfolded substrates once exposed to the cytosol, which in turn are tagged for proteasomal degradation. A recent study showed that another E3 ligase gp78 may act downstream (or in parallel) with the Sel1L-Hrd1 complex to increase the solubility of retrotranslocated protein substrates for proteasomal degradation (44).
The activity of Hrd1 is tightly regulated by many different mechanisms. In addition to transcriptional induction by extracellular signals that are detailed below, two most prominent regulatory mechanisms for Hrd1 ERAD activity are Hrd1 self-ubiquitination (see above) and its interaction with the cofactor Sel1L (38, 39). Sel1L, a type I transmembrane protein with a large luminal domain (45), may form a dimer or oligomer and interact with the N-terminal domain of Hrd1 (46). Sel1L is necessary for the ERAD process for a subset of model substrates (29, 37-40) as well as endogenous substrates including luminal hedgehog (47), transmembrane CD147 (48) and ATF6 (49). However, until recently, the role of Sel1L in the mammalian Hrd1 ERAD complex has been considered dispensable (37, 50, 51). Indeed, like its yeast counterpart Hrd3p (22), the mammalian Sel1L is absolutely required for the stability of Hrd1 (12) and may directly interact with and recruit substrates to Hrd1 (13). Acute loss of Sel1L leads to premature death in adult mice within 3 weeks, with profound pancreatic atrophy; and the loss of Sel1L renders Hrd1 unstable and increases ER stress, promoting pancreatic cell death. Interestingly, loss of Sel1L causes the aggregation of both small and large ribosomal subunits, thereby attenuating protein translation (13). Acute loss of Hrd1 in adult mice also exhibited a similar premature lethality around 2 weeks post-Hrd1 deletion (17), suggesting that Sel1L and Hrd1 act largely in the same pathway and that they are indispensable postnatally in mammals. Of note, a recent study in yeast showed a more direct role of Hrd3p in the stimulation of Hrd1p ubiquitination activity, in addition to Hrd1p stabilization (52). Whether this finding may be applicable in mammals remains to be tested.
Physiological role of ERAD in mouse models
The physiological role of Sel1L-Hrd1 ERAD has been difficult to assess due to the embryonic lethality of Sel1L- or Hrd1-deficient mice (5, 6). The recent generation of cell type-specific ERAD-deficient mouse (and cellular) models has offered a unique opportunity to delineate the significance of ERAD in physiology and disease pathogenesis. These studies have pointed to a cell type-specific requirement of Sel1L-Hrd1 ERAD. Moreover, these studies have identified a number of endogenous substrates for the Sel1L-Hrd1 complex, including not only soluble ER proteins such as OS9, and membrane-bound ER proteins or protein complexes such as EDEM1, IRE1α and pre-B cell receptor (pre-BCR), but even nuclear proteins such as peroxisome proliferator-activated receptor coactivator β (PGC1β), NF-E2-related factor 2 (NRF2) and B lymphocyte-induced maturation protein 1 (BLIMP1). [How Hrd1 recognizes nuclear substrates for proteasome degradation remains unclear, but recent studies indicate that the degradation of inner nuclear membrane proteins requires a distinct protein complex called Asi localized to the inner nuclear membrane in yeast (53, 54)].
Below we discuss the phenotypes of mice with cell type-specific Sel1L or Hrd1 deficiency, leading to the identification of new ERAD substrates. For simplicity, we break our discussion in two major categories, metabolism and immunity. Key findings are summarized in Table 1 and schematically shown in Figure 2.
Table 1.
Cell-type-specific ERAD mouse models and endogenous substrates
| Cell type specificity | Cre strain (source) | Phenotypes | Substrates | References |
|---|---|---|---|---|
|
| ||||
| Global inducible deletion in adult mice | CAGGCre-ER (B6.Cg-Tg(CAG-cre/Esr1*)5A mc/J, JAX 004682) | Progressive loss of body weight and premature death within 2-3 weeks following the deletion of Sel1L or Hrd1 (12, 17); Exocrine pancreatic insufficiency with ER dilation and defects in secretory granules (12). | OS9 and EDEM1: Dramatic accumulation of ER chaperones OS9 and EDEM1 (but not XTP3B) in pancreas (12). | (12, 17) |
|
| ||||
| Adipocytes | Adipoq-Cre (B6;FVB-Tg(Adipoq-cre)1Evdr/J, JAX 010803) | Normal growth rate on low-fat diet up to 16 weeks, no increase of adipocyte apoptosis, and resistant to diet-induced obesity (11, 17), postprandial hypertriglyceridemia associated with intracellular retention of lipoprotein lipase (LPL) (11). | OS9 and LPL (11): Accumulation and stabilization of OS9; Intracellular retention and aggregation of LPL in the ER. PGC1β (17): PGC1β accumulation, leading to increased mitochondrial number and basal energy expenditure. | (11, 17) |
|
| ||||
| Enterocytes | Villin-Cre (B6.SJL-Tg(Vil-cre)997Gum/J, JAX 004586) | Normal growth rate on LFD in both males and females; normal appearance of small and large intestines; apoptosis only increased in DSS-induced colitis (13). Paneth cells lack secretory granules and exhibit ER dilation with elevated cell death under basal conditions (14). | IRE1α (13): Degraded under basal conditions, and degradation is attenuated under UPR; accumulates by over 20-fold in intestinal epithelium in the absence of Sel1L-Hrd1; heterozygosity of IRE1α reduces colitis severity in the DSS model in Sel1L-deficient mice. | (13, 14) |
|
| ||||
| Hepatocytes | Hrd1f/f mice injected i.v. with Cre-encoding adenoviruses | No characterization of liver-specific Hrd1 knockout mice; pharmacological inhibition of Hrd1 (LS-102) reduces the development of liver cirrhosis and CCl4-indcued liver damage in an NRF2-dependent manner. | NRF2: elevated NRF2 protein level in the absence of Hrd1 in vivo and in vitro. | (15) |
| Overexpression of Hrd1 reduces NRF2 protein level. | ||||
|
| ||||
| Dendritic cells | CD11c-Cre (B6.Cg-Tg(Itgax-cre)1-1Reiz/J, JAX 008068) | TLR4 signaling by LPS stimulates the expression of Hrd1. Hrd1 deficiency reduces transcription of MHC class II gene via BLIMP1 and protects the mice against experimental autoimmune encephalomyelitis (EAE). | BLIMP1: in the absence of Hrd1, BLIMP1 accumulates which in turn represses MHC class II expression. | (16) |
|
| ||||
| B cells | CD19-Cre (B6.129P2(C)-Cd19tm1(cre) Cgn/J, JAX 006785) | B-cell-specific Sel1L deficiency blocks the large pre-B cell transition to small pre-B cells due to the persistent pre-BCR signaling, hence dramatically reducing peripheral mature B cells in spleen and lymph nodes. | Pre-BCR: In the absence of ERAD, pre-BCR accumulates and is increased at the cell surface, thereby increasing proliferation and attenuating differentiation. Note that the BCR complex in mature B cells is not degraded by Sel1L-Hrd1 ERAD. | (71) |
Figure 2. Recently identified endogenous substrates by Sel1L-Hrd1 ERAD.
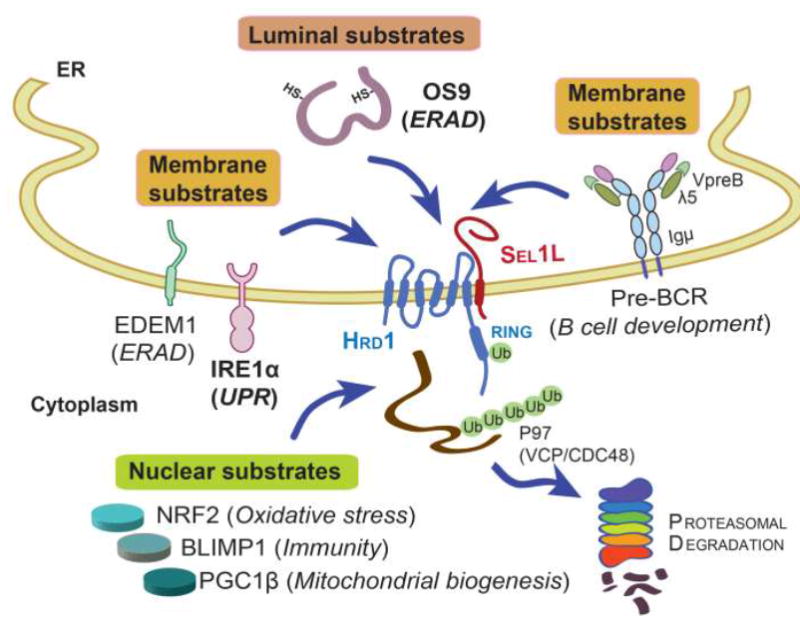
Three types of misfolded proteins, ER luminal (OS9), transmembrane (EDEM1, IRE1α and pre-BCR), and nuclear proteins (NRF2, PGC1β and BLIMP1), have been identified as Sel1L-Hrd1 ERAD substrates using ERAD-deficient mouse models. Substrates in bold (OS9 and IRE1α) have been tested in several different cell types while those in plain fonts have been tested in only one cell type. Substrate-related function is indicated in parentheses. Among all the substrates, the pre-BCR protein complex is unique in that it is consisted of heavy chain Igµ and light chains λ5 and VpreB.
ERAD in metabolism
Adipocytes
Adipocytes store energy in the form of lipid droplets and regulate food intake, systemic energy homeostasis and lipid metabolism via the secretion of many bioactive molecules collectively known as adipokines. Both Sel1L and Hrd1 are induced during cellular differentiation to mature adipocytes (11). To understand the role of Sel1L in mature adipocytes, adipocyte-specific Sel1L-deficient (Sel1LΔadipo) mice were generated by crossing Sel1L floxed mice with mice expressing adiponectin-promoter-driven Cre (11). These mice have comparable body and adipose tissue weights and are largely normal before 12 weeks of age on regular low-energy chow diet. When fed an energy-dense Western diet, they are resistant to diet-induced obesity. Sel1LΔadipo mice also develop postprandial hypertriglyceridemia – a condition with elevated circulating triglyceride levels after a meal – which is caused at least in part by the ER retention and accumulation of lipoprotein lipase (LPL). In the absence of Sel1L, LPL accumulates and aggregates in the ER. A similar observation was made in other LPL-secreting cells such as myocytes and macrophages. Biochemical analysis demonstrated that Sel1L is a cofactor required for the ER exit of LPL to the Golgi complex. When LPL cannot be delivered to the endothelium of adipose tissue, triglycerides in circulating lipoproteins are not efficiently hydrolyzed after feeding, leading to postprandial hypertriglyceridemia. Interestingly, knockdown of Hrd1 in 3T3-L1 adipocytes has no effect on the ER exit of LPL, suggesting a possible ERAD-independent function of Sel1L in LPL maturation in the ER. Whether LPL maturation is controlled by Hrd1 remains to be established in vivo; nevertheless, it is tempting to speculate that a pool of Sel1L might not be physically complexed with Hrd1, enabling it to operate in an Hrd1-independent manner.
Using the same Cre mice, adipocyte-specific Hrd1 knockout mice were generated independently and were found to exhibit a normal growth rate for the first 8 weeks of life, before becoming leaner than their wildtype littermates - similar to the phenotypes of adipocyte-specific Sel1L knockout mice (17). In this instance, the transcriptional coactivator PGC1β directly interacts with Hrd1 and is degraded by the proteasome. Hrd1 deficiency stabilizes PGC1β, thereby enhancing PPARα-mediated transcriptional activation of genes involved in mitochondrial biogenesis and fatty acid oxidation. Indeed, in the absence of Hrd1, mitochondrial number, size and respiratory activity are significantly elevated in adipocytes (17). Injection of LS-102, an Hrd1-specific small molecule inhibitor (55), into wildtype mice activates PGC1β and attenuates body weight gain due to elevated mitochondrial activity and basal energy expenditure (17). Although further studies are required to delineate the role of Hrd1 in lipid and glucose metabolism as well as in other metabolic tissues, Hrd1 in adipose tissue may represent a viable option in the treatment of obesity.
Exocrine pancreas
Global inducible Sel1L knockout mice were recently generated by crossing Sel1L floxed mice with transgenic mice expressing estrogen receptor-Cre recombinase fusion protein driven by the actin promoter (12, 17). The knockout mice develop exocrine pancreatic insufficiency (12) with a dramatic reduction of eosinophilic zymogen granules and increased basophilia-marked anisokaryosis in exocrine pancreas. Protein levels of two major components of acinar cell zymogen granules, pancreatic α-amylase and pancreatic lipase are significantly reduced in the Sel1L-deficient pancreas. Moreover, both OS9 and EDEM1, ER-resident chaperones involved in substrate recruitment to the ERAD machinery (26, 31, 32), are highly elevated in the Sel1L deficient pancreas, without changes in their respective mRNA levels (12). These data suggest that both OS9 and EDEM1 are possible ERAD substrates in vivo. The stabilization of OS9 was later confirmed in Sel1L-deficient adipocytes (11). Degradation of these chaperones most likely represents a self-regulatory mechanism to prevent excessive ERAD activity in the cell.
Enterocytes
Enterocytes form the lining of the intestinal epithelium responsible for nutrient absorption, as well as serving as the first-line of defense (by forming a physical barrier) between the host and gut microbiota. There are several specialized cell subtypes for enterocytes including absorptive enterocytes, highly secretory Paneth and Goblet cells. Unexpectedly, these mice grow normally with largely normal morphologies of epithelium, enterocytes and Goblet cells, with the exception of Paneth cells in which the ER is dilated and secretory granules are much smaller (13, 14). Cell death is elevated only in the crypts of the small intestine but not in the colon under basal conditions (13, 14). When challenged orally with 3% dextran sulfate sodium (DSS) or with the protozoan parasite Toxoplasma Gondii, Sel1L deficient mice exhibit increased susceptibility with massive colitis and edema of the colon (13) and ileitis (14), respectively. These data suggest that Sel1L-Hrd1 ERAD function may be dispensable for normal absorptive function of intestinal epithelium, but is required for the defense against external challenges.
IRE1α is the most highly conserved UPR sensor from yeast to humans, with bifunctional kinase and endoribonuclease activities (56-60) (Figure 1). IRE1α regulates ER folding capacity in response to the accumulation of misfolded proteins in the ER (3, 4). In addition, IRE1α also regulates the expression of Sel1L-Hrd1 ERAD both in yeast (61) and mammalian cells (13, 37, 61-63). In enterocytes, IRE1α accumulates over 20-fold in the absence of Sel1L-Hrd1 ERAD (13). Sel1L-Hrd1 ERAD degrades IRE1α under basal conditions both in vivo and in vitro in a BiP-dependent manner (13). Upon ER stress, IRE1α dissociates from BiP and the ERAD complex, leading to its stabilization and accumulation. Hydrophilic residues in the transmembrane domain of IRE1α are required for IRE1α degradation by Sel1L-Hrd1 ERAD (13). In vivo, IRE1α heterozygosity in intestinal epithelium partially rescues the colitis phenotype of DSS-treated epithelial-specific Sel1L-deficient mice. Further analysis revealed that IRE1α degradation by ERAD occurs in many different cell types, including adipocytes, enterocytes, pancreas, and mouse embryonic fibroblasts (13). This regulatory feedback mechanism of IRE1α by ERAD represents a novel aspect of quality-control that adjusts the capacity of protein folding and disposal in the ER to environmental and (patho-)physiological changes. [Of note, an earlier study showed that, although IRE1α could be ubiquitinated by Hrd1 in vitro, knockdown of Hrd1 by siRNA fails to alter IRE1α protein level in WT fibroblasts (64). The disparity may stem from insufficient knockdown of Hrd1 in vitro].
Hepatocytes
The liver is a key processor and distribution center for energy homeostasis. In cirrhotic livers induced by multiple injections of CCl4, the IRE1α-XBP1 pathway of the UPR is activated, which in turn induces the expression of Hrd1 (15). In liver cirrhosis, NRF2 is a key nuclear transcription factor regulating expression of genes involved in oxidative responses (65). NRF2 is known to be degraded through cytosolic E3 ligases such as Keap1/Cul3 (66) and Skp1-Cul1-F-bpx (SCF)-β-TrCP (SCFβ-TrCP) complexes (67) in a redox-dependent fashion. Using mice in which floxed Hrd1 was deleted in liver after i.v. injection of adenoviruses carrying Cre recombinase, it was shown recently that Hrd1 targets NRF2 for proteasomal degradation (15). Hrd1 deficiency in the cirrhotic liver results in NRF2 accumulation and elevated NRF2 signaling, thereby attenuating oxidative stress and liver cirrhosis. Moreover, inhibition of Hrd1 activity using the small molecule inhibitor LS-102 also protects the mice against liver cirrhosis. This interesting study shows the implications for tissue-specific targeting of Hrd1 as a potential therapeutic approach to hepatotoxcity.
Moreover, a recent genome-wide association study identified Sel1L as a candidate gene for variance in serum total bile acid concentrations (68). In this study, authors compared the sensitivity to atherosclerosis in two different mouse strains A/J and C57BL/6J fed a high-fat/cholesterol/cholate atherogenic Paigen diet [a diet commonly used to examine the sensitivity of different strains to atherosclerosis by inducing sustained hypercholesterolemia (69)]. The effect of Sel1L on serum bile acids is mediated through the suppression of hepatocyte nuclear factor 1A/4A (HNF1A/4A) gene expression, two known modifiers of bile acid transporters, independently of UPR. While the overall finding from the genome-wide association analysis is very interesting, the underlying molecular mechanisms by which Sel1L regulates bile acids and the expression of HFN1A/4A remains unclear. Future studies using liver-specific Sel1L or Hrd1 deficient mice will be required to establish a causal relationship between Sel1L, Hrd1 and serum bile acid levels, and to address whether this is related to ERAD.
ERAD in Immunity
B cells
B lymphocytes secrete antibodies that function in the humoral component of adaptive immunity. To reach the antibody-producing mature stage, B cell precursors undergo a stepwise differentiation process in the bone marrow (BM) from pro-B cells to cycling large pre-B cells, resting small pre-B cells, and then to immature B cells (70). Indeed, B lymphocyte development requires the complicated yet well-defined and developmentally-restricted expression of many growth factors and cell surface-associated proteins. Using B cell specific Sel1L knockout mice driven by CD19-Cre, a recent study showed that Sel1L-Hrd1 ERAD deficiency in B cells regulates the transition from the large pre-B cell to small pre-B cell stage (71). The effect of ERAD on B cell development is independent of ER stress or CHOP expression. Rather, ERAD regulates the turnover and signaling of a key developmental marker: pre-BCR, consisted of an IgL-chain-like structure known as the surrogate light chain paired with Igμ heavy chain. In the absence of Sel1L-Hrd1, the pre-BCR protein complex is stabilized and accumulates in large pre-B cells, leading to enhanced pre-B cell proliferation and a block of further B cell differentiation. Sel1L-Hrd1 ERAD does not seem to affect either mature B cell function or plasma cell differentiation (71). How ERAD distinguishes the pre-BCR complex in developing B cells vs. the BCR complex in mature B cells (which share the common heavy chain Igμ) remains an open question.
Dendritic cells
Dendritic cells are key antigen presenting cells derived from bone marrow, or differentiated from monocytes under inflammatory conditions. Dendritic cells are important for the induction of antigen-specific immunity against pathogens, as well as the maintenance of immune tolerance to self antigens (70). Hrd1 is transcriptionally induced in response to LPS activation of the TLR4 signaling pathway, which in turn regulates the gene transcription of MHC class II via a key transcription factor BLIMP1 (16). Hrd1 targets BLIMP1 for proteasomal degradation. BLIMP1 transcriptionally represses the expression of MHC class II (72). In the absence of Hrd1, BLIMP accumulates, which reduces surface MHC class II levels and the priming of CD4+ T cells. By contrast, Hrd1 has no effect on the expression nor stability of MHC class I in dendritic cells, and hence no effect on MHC class I-mediated activation of CD8+ T cells. Pointing to a critical role of dendritic cell Hrd1 in disease pathogenesis, loss of Hrd1 in dendritic cells protects mice from an autoimmune condition known as experimental autoimmune encephalomyelitis (72). Importantly, Hrd1 deficiency has no effect on cell survival and death in dendritic cells, indicating that its activity is substrate-specific rather than a global effect of ER stress on cell death.
Concluding Remarks and Future Perspectives
Although ERAD is the only known mechanism in the cell dedicated exclusively to the disposal of misfolded proteins in the ER, its function controls (and is controlled by) the other quality control systems such as UPR and autophagy. Moreover, ERAD function may directly or indirectly influence gene transcription and protein translation as discussed above. Hence, the physiological function of cell type-specific ERAD components in vivo yields phenotypes that might not be predicted simply from studies of those same components in simpler cell culture models. This physiological understanding has been made possible using cell type-specific ERAD-deficient mice. These studies demonstrated that ERAD regulates many fundamental cellular processes in various cell types via mediating the turnover of specific substrates (Figure 2), rather than simply via the activation of ER stress responses. Certainly, in some cell types with a diverse secretome and large secretory demand such as pancreatic exocrine cells (12) and Paneth cells (14), ERAD-deficient cells undergo cell death when the burden of misfolded substrates becomes insurmountable. However, it should be emphasized that other cell types examined to date including adipocytes, enterocytes, developing B cells and dendritic cells seem to survive well under basal conditions with chronic ER stress. Therefore, chronic ER stress often associated with (patho-)physiological conditions may not necessarily trigger a cascade of deleterious changes, such as inflammation and/or cell death in vivo as proposed (73) – as cells have an adaptive capacity to reset ER homeostasis (more below). Therefore, we believe, one should not automatically adopt the commonly reported assumption that (patho)physiological perturbation of ER homeostasis will definitely lead to insurmountable ER stress followed by inflammation and/or cell death. Before making such conclusions, one should carefully quantitate the stress level in the ER by analyzing the phosphorylation state of UPR sensors including IRE1α and PERK and their immediate downstream signaling events (74, 75), rather than relying exclusively on the level of a subset of ER chaperones (such as BiP) as an ER stress indicator. A deeper understanding of cell type-specific ERAD in pathophysiological settings may reveal key insights into many human diseases where protein misfolding and incomplete turnover lies at the root cause. Three major disease areas that need further exploration are cancer (76), neurodegeneration and aging. Genetic studies in lower organisms indicate that the fidelity of protein quality control breaks down as the organism ages (77), but it is unclear if this is also true for ERAD in aging mammals and other disease settings.
Secretory capacity of the cell varies from organ to organ and may fluctuate according to metabolic needs and nutrient status. Unlike pancreatic exocrine cells (12) and Paneth cells (14) where ERAD deficiency causes cell death, other cells such as adipocytes (11, 17), enterocytes (13), dendritic cells (16), and developing B cells (71) seem able to tolerate ERAD deficiency and chronic ER stress under basal conditions. This is likely a result of cellular adaptation due to other compensatory mechanisms including, but not limited to, UPR-mediated regulation of overall ER capacity, alternative ERAD pathways, autophagy, and/or a combination of all. How cells handle the accumulation of certain substrates and survive without functional Sel1L-Hrd1 ERAD should be considered on a case-by-case basis for different substrates and cell types. In the next several years, more cell- and tissue- type specific knockout mice will certainly emerge, which will undoubtedly identify more specific processes and signaling pathways that are regulated by ERAD. We believe that ERAD deficient models represent an excellent window into the study of human diseases associated with defects in protein folding and ER protein turnover.
There is also a need for further investigation into the role of Sel1L-Hrd1 in human diseases caused by both loss- and gain-of-function mutations in proteins that encounter the classical ERAD machinery described in this review. Many disease-associated mutant proteins identified to date arise from point mutations, some of which cause protein misfolding, ER retention, protein aggregation and/or degradation. A few well-known examples include proinsulin in the syndrome of Mutant INS gene-induced Diabetes of Youth (MIDY) (78, 79), pro-opiomelanocortin (POMC) in early-onset obesity (80), pro-vasopressin (pro-AVP) in autosomal dominant form of diabetes insipidus (81, 82), and thyroglobulin in congenital hypothyroidism (83). Most of the mutations affect the folding and maturation of nascent proteins that transverse the secretory pathway. In the gain-of-toxic-function mutations, a dominant negative behavior in the ER is commonly seen as a result of mispairing of disulfide bonds that may engage both mutant and wildtype oligomerization partners (84). The Sel1L-Hrd1 ERAD complex participate in the turnover of mutant proinsulin in the ER (85), but the extent to which it is involved in the clearance of other disease-associated mutants requires further studies. We predict that many fundamental questions about ERAD under pathophysiological conditions will be addressed in the next few years (see Outstanding Questions), including the specific engagement of ERAD machinery in the disposal of disease-associated mutant proteins. The extent to which activating selective components of the ERAD machinery could be a therapeutic target, such as to partially block the dominant-negative effect of misfolded proteins and thereby inhibit disease progression, offers an exciting new challenge at the interface of cell biology and medicine.
OUTSTANDING QUESTIONS.
What are the physiological function and significance of Sel1L-Hrd1 ERAD in cell type-specific manner at the whole-organism level? Does ERAD function regulate other importance cellular processes such as lipid droplet biogenesis, mitochondrial function and etc? What kind of proteins are ERAD substrates?
What is the pathological significance of Sel1L-Hrd1 ERAD in human diseases? Human diseases are often associated with protein misfolding and aggregation. Are mutant proteins simply resistant to ERAD quality control or does the fidelity of ERAD break down in the face of protein aggregation?
How does the crosstalk between ERAD and other quality-control processes such as UPR and autophagy affect ER homeostasis, and health and disease? What is the mechanism underlying cellular adaption to ERAD deficiency? Do UPR and autophagy play complementary roles in vivo in ERAD-deficient cells?
How many ERAD machineries are there in mammalian cells and what are their functions? More than a dozen ERAD complexes composed of different E3 ubiquitin ligases have been identified, and their importance and relevance in vivo remains largely unclear.
TRENDS.
ERAD is the principal stress management mechanism used to clear both misfolded and normal proteins from the ER for cytosolic proteasomal degradation.
The protein complex consisting of the E3 ligase Hrd1 and its cofactor Sel1L is the most conserved branch of ERAD.
Sel1L-Hrd1 ERAD plays important roles in regulating ER homeostasis, metabolism and immunity in a cell type-specific manner.
Several proteins including the UPR sensor IRE1α and pre-B cell receptor (pre-BCR) have recently been identified as endogenous ERAD substrates.
Acknowledgments
The work in authors’ laboratory is supported by NIH R01GM113188, R01DK105393, Juvenile Diabetes Research Foundation 1-SRA-2014-251-Q-R, and American Diabetes Association (ADA) 1-12-CD-04 (L.Q.), 1R01AI083252 and 1R01AI064296 (B.T.), R01DK48280 and R01DK40344 (P.A.), R01 DK111174 and University of Michigan Protein Folding Diseases Initiative (L.Q., P.A. and B.T). L.Q. is the recipient of the Junior Faculty and Career Development Awards from ADA.
GLOSSARY
- Endoplasmic reticulum (ER)
the cellular organelle where synthesis and folding of secretory and membrane proteins take place.
- ER-associated degradation (ERAD)
a highly conserved protein quality-control mechanism in the cell that retrotranslocate misfolded ER proteins for cytosolic proteasomal degradation. The Sel1L-Hrd1 protein complex, consisting of the ER-resident E3 ubiquitin ligase Hrd1 and its cofactor Sel1L protein (Hrd1p and Hrd3p in yeast, respectively), is the most conserved ERAD machinery.
- E3 ubiquitin ligase
a protein that catalyzes the transfer of ubiquitin from E2 ubiquitin-conjugating enzyme to its protein substrate.
- Proteasome
a multi-protein complex responsible for protein degradation in the cell, in a process also known as proteolysis.
- Quality control
a system that ensures the quality of a process or product.
- ERAD substrates
Proteins that are recognized and targeted for proteasomal degradation by the ERAD machinery.
- Unfolded protein response (UPR)
A key cellular quality control mechanism identified at the late 1980s and 1990s. It involves the sensing of misfolded proteins in the ER by distinct ER-resident protein sensors including IRE1α, PERK and ATF6, and the subsequent initiation of downstream signaling events leading to the adaptation of ER homeostasis and/or cell death.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Braakman I, Hebert DN. Protein folding in the endoplasmic reticulum. Cold Spring Harbor perspectives in biology. 2013;5:a013201. doi: 10.1101/cshperspect.a013201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guerriero CJ, Brodsky JL. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol Rev. 2012;92:537–576. doi: 10.1152/physrev.00027.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 4.Sha H, et al. Stressed out about obesity: IRE1alpha-XBP1 in metabolic disorders. Trends Endocrinol Metab. 2011;22:374–381. doi: 10.1016/j.tem.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yagishita N, et al. Essential role of synoviolin in embryogenesis. J Biol Chem. 2005;280:7909–7916. doi: 10.1074/jbc.M410863200. [DOI] [PubMed] [Google Scholar]
- 6.Francisco AB, et al. Deficiency of suppressor enhancer lin12 1 like (SEL1L) in mice leads to systemic endoplasmic reticulum stress and embryonic lethality. J Biol Chem. 2010;285:13694–13703. doi: 10.1074/jbc.M109.085340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muller JM, et al. Targeted deletion of p97 (VCP/CDC48) in mouse results in early embryonic lethality. Biochem Biophys Res Commun. 2007;354:459–465. doi: 10.1016/j.bbrc.2006.12.206. [DOI] [PubMed] [Google Scholar]
- 8.Ruggiano A, et al. Quality control: ER-associated degradation: protein quality control and beyond. J Cell Biol. 2014;204:869–879. doi: 10.1083/jcb.201312042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christianson JC, Ye Y. Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat Struct Mol Biol. 2014;21:325–335. doi: 10.1038/nsmb.2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stevenson J, et al. Endoplasmic Reticulum-Associated Degradation and Lipid Homeostasis. Annu Rev Nutr. 2016 doi: 10.1146/annurev-nutr-071715-051030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sha H, et al. The ER-associated degradation adaptor protein Sel1L regulates LPL secretion and lipid metabolism. Cell Metab. 2014;20:458–470. doi: 10.1016/j.cmet.2014.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun S, et al. Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc Natl Acad Sci U S A. 2014;111:E582–591. doi: 10.1073/pnas.1318114111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun S, et al. IRE1a is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nat Cell Biol. 2015;17:1546–1555. doi: 10.1038/ncb3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun S, et al. Epithelial Sel1L is required for the maintenance of intestinal homeostasis. Mol Biol Cell. 2016;27:483–490. doi: 10.1091/mbc.E15-10-0724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu T, et al. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014;28:708–722. doi: 10.1101/gad.238246.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang H, et al. Hrd1-mediated BLIMP-1 ubiquitination promotes dendritic cell MHCII expression for CD4 T cell priming during inflammation. J Exp Med. 2014;211:2467–2479. doi: 10.1084/jem.20140283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fujita H, et al. The E3 ligase synoviolin controls body weight and mitochondrial biogenesis through negative regulation of PGC-1beta. EMBO J. 2015;34:1042–1055. doi: 10.15252/embj.201489897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ward CL, et al. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995;83:121–127. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- 19.Ward CL, Kopito RR. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J Biol Chem. 1994;269:25710–25718. [PubMed] [Google Scholar]
- 20.Lukacs GL, et al. Conformational maturation of CFTR but not its mutant counterpart (delta F508) occurs in the endoplasmic reticulum and requires ATP. EMBO J. 1994;13:6076–6086. doi: 10.1002/j.1460-2075.1994.tb06954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hampton RY, et al. Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol Biol Cell. 1996;7:2029–2044. doi: 10.1091/mbc.7.12.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gardner RG, et al. Endoplasmic reticulum degradation requires lumen to cytosol signaling. Transmembrane control of Hrd1p by Hrd3p. J Cell Biol. 2000;151:69–82. doi: 10.1083/jcb.151.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bays NW, et al. Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat Cell Biol. 2001;3:24–29. doi: 10.1038/35050524. [DOI] [PubMed] [Google Scholar]
- 24.Gardner RG, et al. In vivo action of the HRD ubiquitin ligase complex: mechanisms of endoplasmic reticulum quality control and sterol regulation. Mol Cell Biol. 2001;21:4276–4291. doi: 10.1128/MCB.21.13.4276-4291.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kopito RR. Biosynthesis and degradation of CFTR. Physiol Rev. 1999;79:S167–173. doi: 10.1152/physrev.1999.79.1.S167. [DOI] [PubMed] [Google Scholar]
- 26.Olzmann JA, et al. The Mammalian Endoplasmic Reticulum-Associated Degradation System. Cold Spring Harbor perspectives in biology. 2013;5 doi: 10.1101/cshperspect.a013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tannous A, et al. N-linked sugar-regulated protein folding and quality control in the ER. Semin Cell Dev Biol. 2015;41:79–89. doi: 10.1016/j.semcdb.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thibault G, Ng DT. The endoplasmic reticulum-associated degradation pathways of budding yeast. Cold Spring Harbor perspectives in biology. 2012;4 doi: 10.1101/cshperspect.a013193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Christianson JC, et al. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat Cell Biol. 2008;10:272–282. doi: 10.1038/ncb1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ninagawa S, et al. Forcible destruction of severely misfolded mammalian glycoproteins by the non-glycoprotein ERAD pathway. J Cell Biol. 2015;211:775–784. doi: 10.1083/jcb.201504109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhamidipati A, et al. Exploration of the topological requirements of ERAD identifies Yos9p as a lectin sensor of misfolded glycoproteins in the ER lumen. Mol Cell. 2005;19:741–751. doi: 10.1016/j.molcel.2005.07.027. [DOI] [PubMed] [Google Scholar]
- 32.Kim W, et al. Yos9p detects and targets misfolded glycoproteins for ER-associated degradation. Mol Cell. 2005;19:753–764. doi: 10.1016/j.molcel.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 33.Molinari M, et al. Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science. 2003;299:1397–1400. doi: 10.1126/science.1079474. [DOI] [PubMed] [Google Scholar]
- 34.Oda Y, et al. EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science. 2003;299:1394–1397. doi: 10.1126/science.1079181. [DOI] [PubMed] [Google Scholar]
- 35.Plemper RK, et al. Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature. 1997;388:891–895. doi: 10.1038/42276. [DOI] [PubMed] [Google Scholar]
- 36.Cormier JH, et al. EDEM1 recognition and delivery of misfolded proteins to the SEL1L-containing ERAD complex. Mol Cell. 2009;34:627–633. doi: 10.1016/j.molcel.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Christianson JC, et al. Defining human ERAD networks through an integrative mapping strategy. Nat Cell Biol. 2012;14:93–105. doi: 10.1038/ncb2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mueller B, et al. SEL1L, the homologue of yeast Hrd3p, is involved in protein dislocation from the mammalian ER. J Cell Biol. 2006;175:261–270. doi: 10.1083/jcb.200605196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lilley BN, Ploegh HL. Multiprotein complexes that link dislocation, ubiquitination, and extraction of misfolded proteins from the endoplasmic reticulum membrane. Proc Natl Acad Sci U S A. 2005;102:14296–14301. doi: 10.1073/pnas.0505014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mueller B, et al. SEL1L nucleates a protein complex required for dislocation of misfolded glycoproteins. Proc Natl Acad Sci USA. 2008;105:12325–12330. doi: 10.1073/pnas.0805371105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carvalho P, et al. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell. 2010;143:579–591. doi: 10.1016/j.cell.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baldridge RD, Rapoport TA. Autoubiquitination of the Hrd1 Ligase Triggers Protein Retrotranslocation in ERAD. Cell. 2016;166:394–407. doi: 10.1016/j.cell.2016.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stein A, et al. Key steps in ERAD of luminal ER proteins reconstituted with purified components. Cell. 2014;158:1375–1388. doi: 10.1016/j.cell.2014.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang T, et al. gp78 functions downstream of Hrd1 to promote degradation of misfolded proteins of the endoplasmic reticulum. Mol Biol Cell. 2015;26:4438–4450. doi: 10.1091/mbc.E15-06-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sundaram M, Greenwald I. Suppressors of a lin-12 hypomorph define genes that interact with both lin-12 and glp-1 in Caenorhabditis elegans. Genetics. 1993;135:765–783. doi: 10.1093/genetics/135.3.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jeong H, et al. Crystal structure of SEL1L: Insight into the roles of SLR motifs in ERAD pathway. Sci Rep. 2016;6:20261. doi: 10.1038/srep20261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen X, et al. Processing and turnover of the Hedgehog protein in the endoplasmic reticulum. J Cell Biol. 2011;192:825–838. doi: 10.1083/jcb.201008090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tyler RE, et al. Unassembled CD147 is an endogenous endoplasmic reticulum-associated degradation substrate. Mol Biol Cell. 2012;23:4668–4678. doi: 10.1091/mbc.E12-06-0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Horimoto S, et al. The Unfolded Protein Response Transducer ATF6 Represents a Novel Transmembrane-type Endoplasmic Reticulum-associated Degradation Substrate Requiring Both Mannose Trimming and SEL1L Protein. J Biol Chem. 2013;288:31517–31527. doi: 10.1074/jbc.M113.476010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iida Y, et al. SEL1L protein critically determines the stability of the HRD1-SEL1L endoplasmic reticulum-associated degradation (ERAD) complex to optimize the degradation kinetics of ERAD substrates. J Biol Chem. 2011;286:16929–16939. doi: 10.1074/jbc.M110.215871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Williams JM, et al. The ERdj5-Sel1L complex facilitates cholera toxin retrotranslocation. Mol Biol Cell. 2013;24:785–795. doi: 10.1091/mbc.E12-07-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vashistha N, et al. Direct and essential function for Hrd3 in ER-associated degradation. Proc Natl Acad Sci U S A. 2016;113:5934–5939. doi: 10.1073/pnas.1603079113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Foresti O, et al. Quality control of inner nuclear membrane proteins by the Asi complex. Science. 2014;346:751–755. doi: 10.1126/science.1255638. [DOI] [PubMed] [Google Scholar]
- 54.Khmelinskii A, et al. Protein quality control at the inner nuclear membrane. Nature. 2014;516:410–413. doi: 10.1038/nature14096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yagishita N, et al. RING-finger type E3 ubiquitin ligase inhibitors as novel candidates for the treatment of rheumatoid arthritis. Int J Mol Med. 2012;30:1281–1286. doi: 10.3892/ijmm.2012.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mori K, et al. A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell. 1993;74:743–756. doi: 10.1016/0092-8674(93)90521-q. [DOI] [PubMed] [Google Scholar]
- 57.Nikawa J, Yamashita S. IRE1 encodes a putative protein kinase containing a membrane-spanning domain and is required for inositol phototrophy in Saccharomyces cerevisiae. Mol Microbiol. 1992;6:1441–1446. doi: 10.1111/j.1365-2958.1992.tb00864.x. [DOI] [PubMed] [Google Scholar]
- 58.Cox JS, et al. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell. 1993;73:1197–1206. doi: 10.1016/0092-8674(93)90648-a. [DOI] [PubMed] [Google Scholar]
- 59.Tirasophon W, et al. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes & Development. 1998;12:1812–1824. doi: 10.1101/gad.12.12.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang XZ, et al. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 1998;17:5708–5717. doi: 10.1093/emboj/17.19.5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Travers KJ, et al. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- 62.Yoshida H, et al. A time-dependent phase shift in the mammalian unfolded protein response. Dev Cell. 2003;4:265–271. doi: 10.1016/s1534-5807(03)00022-4. [DOI] [PubMed] [Google Scholar]
- 63.Kaneko M, et al. A different pathway in the endoplasmic reticulum stress-induced expression of human HRD1 and SEL1 genes. FEBS Lett. 2007;581:5355–5360. doi: 10.1016/j.febslet.2007.10.033. [DOI] [PubMed] [Google Scholar]
- 64.Gao B, et al. Synoviolin promotes IRE1 ubiquitination and degradation in synovial fibroblasts from mice with collagen-induced arthritis. EMBO Rep. 2008;9:480–485. doi: 10.1038/embor.2008.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009;34:176–188. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 66.Kobayashi A, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rada P, et al. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol. 2011;31:1121–1133. doi: 10.1128/MCB.01204-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu W, et al. Genome-wide association study in mice identifies loci affecting liver-related phenotypes including Sel1l influencing serum bile acids. Hepatology. 2016;63:1943–1956. doi: 10.1002/hep.28495. [DOI] [PubMed] [Google Scholar]
- 69.Paigen B. Genetics of responsiveness to high-fat and high-cholesterol diets in the mouse. Am J Clin Nutr. 1995;62:458S–462S. doi: 10.1093/ajcn/62.2.458S. [DOI] [PubMed] [Google Scholar]
- 70.Janeway CA, Jr, et al. Immunobiology: The Immune System in Health & Disease. 5. Vol. 7. Garland Publishing Inc.; 2001. pp. 1–35. [Google Scholar]
- 71.Ji Y, et al. The Sel1L-Hrd1 Endoplasmic Reticulum-Associated Degradation Complex Manages a Key Checkpoint in B Cell Development. Cell reports. 2016;16:2630–2640. doi: 10.1016/j.celrep.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim SJ, et al. Regulation of dendritic cell activation by microRNA let-7c and BLIMP1. J Clin Invest. 2013;123:823–833. doi: 10.1172/JCI64712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hotamisligil G. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang L, et al. A Phos-tag-based method reveals the extent of physiological endoplasmic reticulum stress. PLos ONE. 2010;5:e11621. doi: 10.1371/journal.pone.0011621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Qi L, et al. Detecting and quantitating physiological endoplasmic reticulum stress. Meth Enzymol. 2011;490:137–146. doi: 10.1016/B978-0-12-385114-7.00008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim H, et al. Endoplasmic reticulum quality control in cancer: Friend or foe. Semin Cancer Biol. 2015;33:25–33. doi: 10.1016/j.semcancer.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.David DC, et al. Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol. 2010;8:e1000450. doi: 10.1371/journal.pbio.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu M, et al. Proinsulin disulfide maturation and misfolding in the endoplasmic reticulum. J Biol Chem. 2005;280:13209–13212. doi: 10.1074/jbc.C400475200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Weiss MA. Proinsulin and the genetics of diabetes mellitus. J Biol Chem. 2009;284:19159–19163. doi: 10.1074/jbc.R109.009936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Creemers JW, et al. Mutations in the amino-terminal region of proopiomelanocortin (POMC) in patients with early-onset obesity impair POMC sorting to the regulated secretory pathway. J Clin Endocrinol Metab. 2008;93:4494–4499. doi: 10.1210/jc.2008-0954. [DOI] [PubMed] [Google Scholar]
- 81.Birk J, et al. Dominant pro-vasopressin mutants that cause diabetes insipidus form disulfide-linked fibrillar aggregates in the endoplasmic reticulum. J Cell Sci. 2009;122:3994–4002. doi: 10.1242/jcs.051136. [DOI] [PubMed] [Google Scholar]
- 82.Friberg MA, et al. Degradation of wild-type vasopressin precursor and pathogenic mutants by the proteasome. J Biol Chem. 2004;279:19441–19447. doi: 10.1074/jbc.M310249200. [DOI] [PubMed] [Google Scholar]
- 83.Di Jeso B, Arvan P. Thyroglobulin From Molecular and Cellular Biology to Clinical Endocrinology. Endocr Rev. 2016;37:2–36. doi: 10.1210/er.2015-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Haataja L, et al. Disulfide Mispairing During Proinsulin Folding in the Endoplasmic Reticulum. Diabetes. 2016 doi: 10.2337/db15-1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.He K, et al. PDI reductase acts on Akita mutant proinsulin to initiate retrotranslocation along the Hrd1/Sel1L-p97 axis. Mol Biol Cell. 2015;26:3413–3423. doi: 10.1091/mbc.E15-01-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]