

Abstract
Precuneus (PreC) cortex is affected with amyloid plaques early in Alzheimer’s disease (AD), and this pathology may be associated with alterations in PreC synapses and cognitive impairment. We quantified the spinophilin-immunoreactive (ir) dendritic spine density and the intensity of spinophilin immunofluorescence, the latter as a measure of relative protein levels of spinophilin, in PreC lamina III from 33 subjects with clinical diagnoses of no cognitive impairment (NCI), mild cognitive impairment (MCI), mild-moderate AD (mAD), or severe AD (sAD). Both measures of spinophilin were lower in mAD and sAD compared with NCI. The MCI group had higher protein levels of spinophilin compared with mAD and sAD, and higher spinophilin-ir dendritic spine density compared with sAD. Lower spinophilin-ir dendritic spine density and relative protein levels of spinophilin were associated with greater amyloid beta (Aβ) plaque burden, detected with a derivative of Pittsburgh compound-B (6-CN-PiB), and worse cognitive performance. Clinical onset of AD is marked by the loss of PreC spinophilin-ir dendritic spines that is related to Aβ pathology and may contribute to cognitive symptoms early in the disease.
Keywords: Amyloid, Default mode network, Episodic memory, Mild cognitive impairment, Synapse
1. Introduction
Alzheimer’s disease (AD) is a chronic neurodegenerative disorder with a clinical presentation of dementia and distinct neuropathology hallmarks including amyloid-beta (Aβ) plaques and neurofibrillary tangles (NFTs) (Montine et al., 2012). While the pathologic significance of plaques and tangles is an ongoing debate, loss of neocortical synapses has been widely considered the best structural and neurochemical correlate of cognitive decline in AD (DeKosky and Scheff, 1990; Terry et al., 1991). However, the involvement of specific synaptic compartments (e.g., presynaptic or postsynaptic) and the time of onset of such changes in relation to clinical disease progression are not well defined. Electron microscopy (EM) and biochemical studies demonstrated that the hippocampus undergoes synaptic loss in clinically mild AD (mAD) and even in some people with mild cognitive impairment (MCI, prodromal AD) (Scheff et al., 2006) as well as cognitively normal people with AD pathology (preclinical AD) (Scheff et al., 2016). Whether a similar vulnerability characterizes select neocortical regions remains unclear.
Positron emission tomography imaging studies identified precuneus (PreC) and posterior cingulate cortex as neocortical areas exhibiting functional impairment and amyloid accumulation early in AD, even before clinical disease onset (Buckner et al., 2005; Mintun et al., 2006; Sperling et al., 2009, 2010). These regions are components of the default mode network (DMN) which exhibits task-independent decreases in metabolic activity during attention-demanding, goal-directed behavior, but returns to a default state of activity during rest (Raichle et al., 2001). The pathologic involvement of DMN regions in preclinical and symptomatic AD (Becker et al., 2011; Buckner et al., 2005; Koch et al., 2015) is associated with disruption of connectivity within this network as well as with regions involved in sensorimotor processing, and, relevant to the current study, impairment of memory retrieval (Elman et al., 2016; Myers et al., 2014; Sheline et al., 2010). Disrupted episodic memory, one of the earliest symptoms of AD (Hodges, 2000), likely results from impaired connectivity between the PreC and the hippocampus (Sheline et al., 2010) or within the DMN itself (Buckner et al., 2005, 2008). At the ultrastructural level, the PreC and posterior cingulate display significant synaptic loss in mAD but not in MCI (Scheff et al., 2013, 2015). Interestingly, complementary biochemical studies of the posterior cingulate cortex in the same cohort revealed that MCI subjects had significantly reduced levels of presynaptic (synaptophysin, synapsin-1) and postsynaptic (PSD-95, SAP-97) markers, and that these measures correlated with the levels of [H-3]PiB binding to fibrillar Aβ (Scheff et al., 2015). Whether changes in select synaptic compartments precede the loss of synapses in PreC, similar to what was observed in the posterior cingulate cortex, is unknown.
In the present study, we utilized unbiased stereological principles and quantitative fluorescence confocal microscopy to evaluate the postsynaptic structures immunoreactive (ir) to spinophilin, a protein highly enriched in dendritic spines that regulates their formation and function, and a marker of dendritic spine alterations in human cortex in health and disease (Sweet et al., 2009), in PreC lamina III from 33 elderly individuals who died with a clinical diagnosis of no cognitive impairment (NCI), MCI, mAD, or severe AD (sAD). Lamina III was chosen as it gives rise to corticocortical neuronal projections which are considered selectively vulnerable in AD (reviewed in: Hof, 2001). The second goal of the study was to test the hypothesis that along the clinical stages of AD, alterations of spinophilin-ir dendritic spines in the PreC are associated with amyloid pathology and impaired episodic memory function and global cognition.
2. Materials and methods
2.1. Standard protocol approvals, registrations, and patient consents
The study was approved by Rush University Medical Center and the University of Pittsburgh’s Committee for Oversight of Research and Clinical Training Involving the Dead. Written informed consent for research and autopsy was obtained for all subjects in the study.
2.2. Subjects
The 33 cases (age range 79–94; 15 males and 18 females) examined in the study included 25 participants in the Rush Religious Orders Study (RROS), a longitudinal clinicopathologic study of aging and AD in retired Catholic clergy (Bennett et al., 2002), and 8 cases from the University of Pittsburgh Alzheimer’s Disease Research Center (ADRC). Diagnosis of AD was made using standard diagnostic criteria (McKhann et al., 1984). MCI was defined as impairments on neuropsychological testing, without a diagnosis of dementia by the examining neurologist (Bennett et al., 2002), criteria similar to those used at other centers (Morris and Price, 2001; Petersen et al., 1999). Using these criteria, and the last clinical evaluation within 12 months before death, the RROS cases were assigned to 3 clinical groups: NCI (n = 8, Mini-Mental State Examination [MMSE] = 28.1 ± 1.4), MCI (n = 9, MMSE = 26.8 ± 2.6), and mAD (n = 8, MMSE = 22.7 ± 2.9, Table 1). The 8 Pittsburgh Alzheimer’s Disease Research Center cases were clinically classified as severe AD (MMSE = 6 ± 3.9, Table 1). Neuropathology workup included immunohistochemical analyses of Aβ, p-tau, α-synuclein, and TDP-43, as well as H&E and Bielschowsky silver staining. Cases with non-AD pathology (e.g., stroke, Parkinson’s disease and hippocampal sclerosis) were excluded from the study. One sAD case had scarce alpha synuclein pathology in parietal cortex. Neuropathologic diagnosis was based on the National Institute on Aging (NIA)–Reagan Institute (RI) criteria (NIA-RI) (Consensus, 1997), recommendations of the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD (Mirra et al., 1991)), and Braak staging of NFTs (Braak and Braak, 1991). Application of the new NIA-Alzheimer’s Association guidelines (Hyman et al., 2012; Montine et al., 2012) to the examined cohorts is ongoing at the time of publication. All cases were deidentified and randomly assigned a unique identifier that was used throughout the study. Investigators were blinded to case demographics and diagnosis.
Table 1.
Demographic, clinical, and neuropathological characteristics by clinical diagnosis category
| Demographic,
clinicaland neuropathological characteristics |
Clinical diagnosis |
p value | Pairwise comparison | ||||
|---|---|---|---|---|---|---|---|
| NCI (N=8) | MCI (N=9) | mAD (N=8) | sAD (N=8) | Total (N=33) | |||
| Age (y) at death | |||||||
| Mean ± SD | 86.2 ± 4.6 | 86.1 ± 5.1 | 89.1 ± 3.9 | 83.3 ± 2.7 | 86.1 ± 4.5 | – | |
| (Range) | (81.3–92.8) | (79.4–94) | (83.1–94.5) | (79.0–88.0) | (79.0–94.5) | 0.086a | |
| Number (%) of males | 4 (50.0%) | 2 (22.2%) | 3 (37.5%) | 6 (75.0%) | 15 (45.5%) | 0.212b | – |
| MMSE | |||||||
| Mean ± SD | 28.1 ± 1.4 | 26.8 ± 2.6 | 22.7 ± 2.9 | 6.0 ± 3.9 | 21.1 ± 9.3 | 0.0001c | (NCI, MCI) > mAD > sAD |
| (Range) | (26–30) | (22–29) | (17–28) | (1–12) | (1–30) | ||
| PMI, h | |||||||
| Mean ± SD | 5.5 ± 2.1 | 6.2 ± 3.1 | 5.1 ± 2.5 | 8.2 ± 4.0 | 6.3 ± 3.1 | 0.19a | – |
| (Range) | (3.2–8.6) | (2.8–11.5) | (1.5–8.2) | (3.0–14.0) | (1.5–14.0) | ||
| Braak scores | |||||||
| 0 | 0 | 0 | 0 | 0 | 0 | ||
| I/II | 1 | 2 | 0 | 0 | 3 | 0.001c | (NCI, MCI, mAD) < sAD |
| III/IV | 7 | 4 | 6 | 1 | 18 | ||
| V/VI | 0 | 3 | 2 | 7 | 12 | ||
| CERAD diagnosis | |||||||
| No AD | 3 | 1 | 0 | 0 | 4 | ||
| Possible | 0 | 1 | 0 | 0 | 1 | 0.002c | NCI < (mAD, sAD) |
| Probable | 4 | 5 | 4 | 0 | 13 | MCI < sAD | |
| Definite | 1 | 2 | 4 | 8 | 15 | ||
| NIA-RI diagnosis | |||||||
| No AD | 0 | 0 | 0 | 0 | 0 | ||
| Low | 4 | 3 | 0 | 0 | 7 | 0.0001c | (NCI, MCI, mAD) < sAD |
| Intermediate | 4 | 5 | 6 | 0 | 15 | ||
| High | 0 | 1 | 2 | 8 | 11 | ||
Key: CERAD, Consortium to Establish a Registry for Alzheimer’s Disease; mAD, mild Alzheimer’s disease; MCI, mild cognitive impairment; MMSE, Mini-Mental State Examination; NCI, no cognitive impairment; NIA-RI, National Institute on Aging-Reagan Institute; PMI, post-mortem interval; sAD, severe Alzheimer’s disease.
Anova test, with Bonferroni correction for multiple comparisons.
Fisher’s exact test.
Kruskal-Wallis test, with Bonferroni correction for multiple comparisons.
2.3. Tissue preparation, immunohistochemistry, and histofluorescence
At autopsy, PreC was dissected and immersion fixed in 4% paraformaldehyde made in 0.1-M phosphate buffer (pH 7.2) for 48–72 hours at 4°C. Following fixation, samples were cryoprotected in 10% glycerol with 2% dimethyl sulfoxide in 0.1-M phosphate buffer at 4 °C for 2 days, followed by a solution of 20% glycerol and 2% dimethyl sulfoxide. PreC sections were cut on a freezing sliding microtome at 40-μm thickness and stored in cryoprotectant at −20 °C until processed.
2.3.1. Spinophilin immunofluorescence
To minimize potential variations across multiple sets of tissue sections processed using the immunofluorescence procedure, each batch of processed tissue sections included an equal number of cases from each clinical diagnostic group. Batch variation was also monitored by repeating sections from select cases in all batches. Spinophilin immunofluorescence was performed on 3 sections per case, as previously described (Sweet et al., 2009). Briefly, the free-floating tissue sections of PreC were incubated in a polyclonal rabbit anti-spinophilin antibody (Millipore, Temecula, CA, #AB5669 (Sweet et al., 2009)) for 96 hours at 4 °C. Specificity and selectivity of the spinophilin antibody was described in our previous work (Sweet et al., 2009). Following primary antibody incubation, sections were incubated for 24 hours at 4 °C in biotinylated donkey anti-rabbit secondary antibody (1:200; Jackson, West Grove, PA, #711-066-152), followed by a 24-hours incubation at 4 °C with a Cy5-streptavidin conjugate (1:500; Jackson, #016-170-084). A fluorescent Nissl substance counterstain (Neurotrace, ThermoFisher, Waltham, MA, # N21480) was used to determine the cortical lamina borders (Fig. 1A). Sections were mounted onto charged slides (Superfrost Plus, ThermoFisher) and air dried for 1 hour. After rehydrating the sections for 10 minutes in distilled water (to reduce Z-axis tissue shrinkage and therefore increase counting accuracy by enhancing the ability to resolve objects in the Z-axis) (Konopaske et al., 2007), the slides were coverslipped with Vectashield (Vector, Burlingame, CA, #H-1400).
Fig. 1.
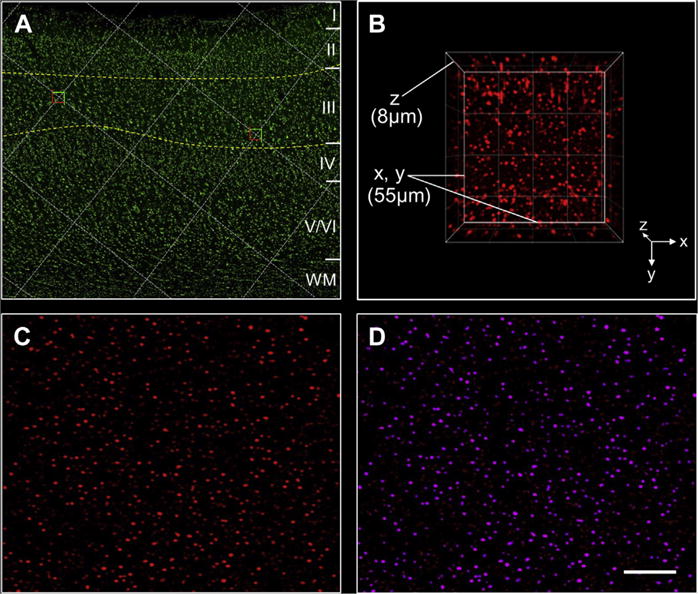
Image collection and processing. (A) A section of PreC processed using fluorescent Nissl histology illustrates cortical laminae I to VI, delineation of lamina III, and selection of sampling sites at grid intersections within the outlined area; (B) a 3-dimensional view of an image stack of spinophilin immunofluorescence in PreC lamina III with X, Y, and Z axes indicated; (C) a single-plane image showing spinophilin-ir puncta after camera background subtraction, deconvolution, and Gaussian filter transformation; (D) examples of masked (selected) spinophilin-ir puncta (purple). Each selected puncta is an “object” with measured attributes such as intensity obtained using Slidebook software. Scale bar = 400 μm for A and 20 μm for C and D. Abbreviation: PreC, precuneus.
2.3.2. 6-CN-PiB histofluorescence
Spinophilin immunostained sections were overstained with 6-CN-PiB (a highly fluorescent derivative of PiB) to reveal amyloid plaque pathology as described previously (Ikonomovic et al., 2008). Briefly, sections were incubated in 10-mM 6-CN-PiB for 45 minutes, dipped 3 times in potassium phosphate buffer (0.1-M, pH 7.4), followed by a 1-minute differentiation in potassium phosphate buffer, then coverslipped with Fluoromount (Electron Microscopy Services, Hatfield, PA).
2.4. Confocal microscopy and sampling guided by stereological principles
2.4.1. Image collection
Image collection was carried out using an Olympus BX51WI upright microscope (Olympus, Center Valley, PA) equipped with an Olympus DSU spinning disk confocal, a super-corrected Olympus Plan Apo N 1.42 N.A. oil immersion objective, an ORCA-R2 CCD camera (Hamamatsu, Bridgewater, NJ), MBF CX9000 front-mounted digital camera (MicroBrightField, Inc, Williston, VT), a BioPrecision2 XYZ motorized stage with linear XYZ encoders (Ludl Electronic Products, Ltd, Hawthorne, NY), excitation and emission filter wheels (Ludl Electronic Products, Ltd), a Sedat Quad 89,000 filter set (Chroma Technology Corp., Bellows Falls, VT), and a Lumen 220 metal halide lamp (Prior Scientific, Rockland, MA). The microscope was controlled using Stereo Investigator (Micro-BrightField) and SlideBook (Intelligent Imaging Innovations, Denver, CO) software.
Using Stereo Investigator software, a sampling area within lamina III of each PreC section was first outlined at low magnification (1.25X objective) based upon fluorescent Nissl-marked cytoarchitecture (Fig. 1A). A sampling grid was then randomly rotated and applied to the sampling area (Fig. 1A). Five to 8 confocal sampling sites were generated for each section and approximately 20 sites per subject were identified for analysis using systematic uniform random sampling. At each sampling site, tissue thickness was measured. Image stacks were collected with a step size of 0.25 μm between Z-axis planes in the stack, starting from 8 μm below the tissue surface closest to the cover glass and stepping up until the tissue surface was reached, yielding an 8-μm thick (Z-axis depth) stack comprising 32 individual 2-dimensional planes. Image planes were collected using a 512 × 512 pixel (approximately 55 × 55 μm, Fig. 1B) area cutoff. Exposure time during image stack acquisition was held constant across all sections and cases. Guard zones of 10 pixels were applied around all edges in the X and Y dimensions of each stack, and 10 Z-axis planes starting 10 planes below the cover glass were included in analysis, as antibody penetration was uniform (puncta counts and intensities were uniform) across these Z-axis depths (10 planes × 0.25 μm step size = 2.50 μm dissector height).
2.4.2. Image postprocessing
Image stacks were postprocessed using SlideBook and Automation Anywhere software (Automation Anywhere, Inc, San Jose, CA). Intensity levels attributable to electrical fluctuations of the camera sensor (camera background) were subtracted from the 647-nm channel (used to visualize Cy5 fluorescence). Images were then processed using a blind deconvolution algorithm (AutoQuant; Media Cybernetics, Rockville, MD). To improve edge detection during image segmentation, for each image stack 2 new channels were made by convolving the 647 channel with a Gaussian function of standard deviations σ = 2 and σ = 0.7. The channel transformation with the larger standard deviation (σ = 2) was then subtracted from the one with the smaller standard deviation (σ = 0.7) to make a new channel. Fig. 1C shows a camera background subtracted, deconvolved, and Gaussian filter–transformed image.
The deconvolved and edge-enhanced image stack was subject to intensity segmentation coupled with morphologic selection using an iterative masking approach (Fish et al., 2008) in which the initial intensity threshold was determined in advance by the experimenter, and with each subsequent iteration the intensity threshold was increased. After each segmentation step, the mask objects were size-gated (0.03–0.5 μm3) to select spinophilin-ir puncta and merged with the mask generated in the previous segmentation step. Using this approach, individual spinophilin-ir puncta throughout each image stack were represented by a masked object (Fig. 1D). Data on puncta mean intensity were extracted from these image stacks using the generated mask that identified the objects of interest. Density of spinophilin-ir dendritic spines was determined by dividing the total number of mask objects counted per each case (Q) by the total volumes, which is the product of the number of sampling sites per case (CF), the area of the counting frame at each site in μm2 (2823), and the dissector height (2.5 μm) multiplied by the microtome block advance (40 μm) divided by the measured tissue section thickness at each site (tq) to correct for tissue shrinkage (Dorph-Petersen et al., 2001), as follows:
2.5. Estimation of lamina III thickness
PreC lamina III thickness was measured using ImageJ (ImageJ, U. S. National Institutes of Health, Bethesda, MD) and images taken from each Nissl-stained section using a 1.25 × lens. Three sections per case were used for quantification.
2.6. Plaque load analysis
Percent area coverage for 6-CN-PiB–labeled Aβ plaques was determined using 3 randomly spaced 10× microscopic fields in each of the 3 sections from the same area/lamina (PreC lamina III) in the cases used for spinophilin quantification. Percent area values were obtained by dividing 6-CN-PiB–labeled area by the total area sampled.
2.7. Statistical methods
Primary outcome measures were density and intensity of spinophilin-ir dendritic spines and the thickness of PreC lamina III. Predictor variables were comprised of demographic, clinical, and neuropathologic factors. Demographic, clinical, diagnostic neuropathology, and 6-CN-PiB plaque load data were compared among the clinically defined groups using the Kruskal-Wallis test, Mann-Whitney U test, Fisher exact test or ANOVA with Bonferroni correction for multiple comparisons. Associations of dendritic spine measures with demographic and clinical characteristics, neuropathology scores, and 6-CN-PiB plaque load data were assessed by Spearman rank correlation. The level of statistical significance was set at 0.05 (2-sided).
3. Results
3.1. Case demographics and clinical and neuropathologic characteristics
Clinical groups were comparable by age, gender, and postmortem delay (Table 1). The MCI group included amnestic MCI (aMCI, n = 3) and non-amnestic MCI (non-aMCI, n = 6) cases which were combined in the analyses. The 4 clinical groups differed significantly by MMSE (Table 1), with sAD having significantly lower MMSE scores compared with NCI, MCI, and mAD. The mAD cases had significantly lower MMSE scores than NCI and MCI, whereas the latter 2 groups were not statistically different (Table 1). The same group differences were observed when the 4 clinical categories were compared by episodic memory, perceptual speed, and visuospatial ability composite scores (not shown). The clinical groups were also significantly different when compared by neuropathology status, including Braak stage, CERAD scores, and NIA-RI diagnosis (Table 1). The sAD group had more advanced pathology by Braak stage and NIA-Reagan diagnosis than the NCI, MCI, and mAD groups, whereas the latter 3 groups did not differ significantly among themselves. CERAD scores were more severe in mAD and sAD groups compared with the NCI, and in the sAD group compared with the MCI, whereas the MCI group did not differ significantly from the NCI and mAD groups (Table 1). 6-CN-PiB–positive Aβ plaque load (percent area coverage) in the PreC was similar in NCI (median = 0.01) and MCI groups (median = 0.87), whereas in mAD (median = 1.81) and sAD (median = 7.08) groups it was significantly higher than in the NCI group (U = 1.0, p < 0.01, for both comparisons, Mann-Whitney U test). The sAD group, but not mAD group, also had higher plaque load compared with the MCI group (U = 9.0, p < 0.01, Mann-Whitney U Test). Higher 6-CN-PiB plaque load in the PreC correlated with more severe CERAD scores (r = −0.68, p < 0.01, n = 33), Braak stage (r = 0.75, p < 0.01, n = 33), and NIA-RI diagnosis (r = −0.75, p < 0.01, n = 33), and with poorer performance on MMSE (r = −0.69, p < 0.01, n = 33) and episodic memory scores (r = −0.71, p < 0.01, n = 25).
3.2. PreC lamina III spinophilin-ir dendritic spines across clinical groups
Spinophilin-ir dendritic spines appeared as bright puncta (Fig. 2A) with reduced densities immediately surrounding and within amyloid plaques (Fig. 2B and C). Both dendritic spine density and relative protein levels of spinophilin differed across clinical groups (F [3,29] = 13.94, p < 0.001 for density and F [3,29] 13.78, p < 0.001 for relative protein levels, ANOVA). Relative spinophilin protein levels were significantly lower in mAD and sAD compared with NCI and MCI groups (Fig. 3A). Spinophilin-ir dendritic spine density was lower in mAD and sAD groups compared with the NCI group, and in the sAD group compared with the MCI group (Fig. 3B). There were no significant differences in the 2 spinophilin measures between the mAD and sAD or between the NCI and MCI groups. PreC lamina III thickness was not statistically different among the 4 clinical groups (F [3,29] = 0.49, p = 0.699, ANOVA).
Fig. 2.
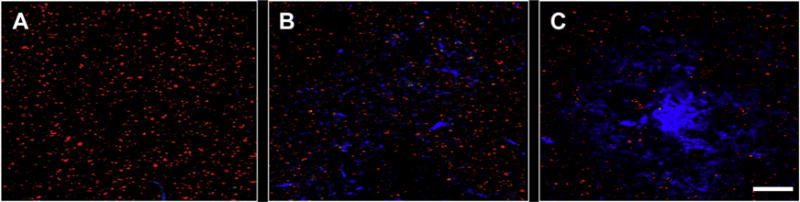
Spinophilin-ir dendritic spines (red fluorescence) in relation to Aβ deposits labeled using 6-CN-PiB (blue fluorescence) in PreC lamina III from a mAD subject. (A) Relatively uniform distribution of dendritic spines in an amyloid-free region; (B) a slight decrease (compared to A) in spinophilin-ir dendritic spine density in an area with diffuse amyloid deposits; (C) markedly decreased spinophilin-ir dendritic spine density within and surrounding a cored amyloid plaque. Scale bar = 20 μm. Abbreviations: Aβ, amyloid beta; mAD, mild Alzheimer disease; PreC, precuneus; spinophilin-ir, spinophilin-immunoreactive.
Fig. 3.
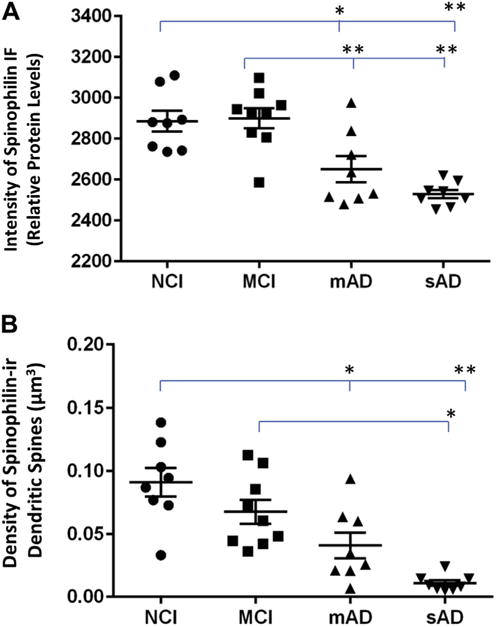
Scatterplots of spinophilin immunofluorescence (IF) intensity as a measure of relative protein levels (A) and density of spinophilin-ir dendritic spines (B) in PreC lamina III from cases in 4 clinical diagnostic groups of NCI, MCI, mAD, and sAD. The means of each group were compared using one-way ANOVA (mean SEM). *p < 0.05; **p < 0.01. Abbreviations: ANOVA, analysis of variance; mAD, mild Alzheimer’s disease; MCI, mild cognitive impairment; NCI, no cognitive impairment; PreC, precuneus; sAD, severe Alzheimer’s disease; spinophilin-ir, spinophilin-immunoreactive.
3.3. Associations of PreC lamina III dendritic spine density and relative protein levels of spinophilin with clinical and neuropathology variables
Across all cases, lower spinophilin relative protein levels were associated with lower MMSE (r = 0.69, p < 0.01, n = 33) (Fig. 4A) and episodic memory (r = 0.54, p < 0.01, n = 25) test scores. A similar relationship was observed between dendritic spine density and MMSE (r = 0.80, p < 0.01, n = 33) (Fig. 4B) as well as episodic memory (r = 0.47, p < 0.05, n = 25) test scores. Lower dendritic spine density and relative protein levels of spinophilin also correlated with more advanced pathology by Braak stage (r = −0.37, p < 0.05 and r = −0.53, p < 0.01, respectively, n = 33), NIA-RI diagnosis (r = −0.47, p < 0.01 and r = −0.59, p < 0.01, respectively, n = 33), and 6-CN-PiB–positive fibrillar Aβ plaque burden in the PreC (r = −0.49, p < 0.01 and r = −0.57, p < 0.01, respectively, n = 33). Lower spinophilin-ir dendritic spine density, but not relative protein levels of spinophilin, correlated with more severe CERAD scores (r = −0.55, p < 0.01, n = 33).
Fig. 4.
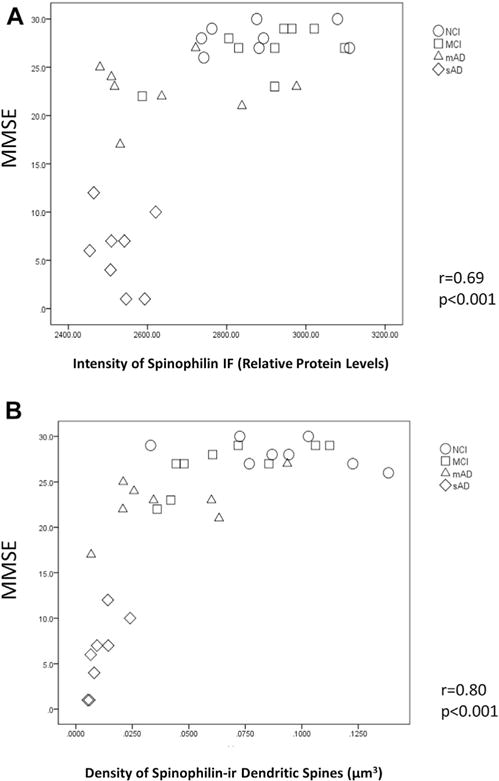
Correlations between Mini-Mental State Examination (MMSE) scores and spinophilin immunofluorescence intensity as an indication of relative protein levels (A) and density of spinophilin-ir dendritic spines (B) in PreC lamina III from clinical diagnostic groups of NCI, MCI, mAD and sAD. Abbreviations: mAD, mild Alzheimer disease; MCI, mild cognitive impairment; NCI, no cognitive impairment; PreC, pre-cuneus; sAD, severe Alzheimer’s disease; spinophilin-ir, spinophilin-immunoreactive.
4. Discussion
Using unbiased stereology principles, we performed a quantitative confocal microscopic analysis of spinophilin-ir dendritic spines in PreC lamina III from cases in clinically defined groups of NCI, MCI, mAD, and sAD. Two major findings emerged. First, PreC spinophilin-ir dendritic spine density and relative protein levels of spinophilin were significantly lower in both mAD and sAD groups compared with the NCI group. In the MCI group, PreC spinophilin measurements were similar to the NCI group, but were higher than the sAD group (both dendritic spine density and relative protein levels of spinophilin) and the mAD group (relative protein levels of spinophilin only). Second, across all cases in the study lower levels of the 2 dendritic spine measures correlated significantly with greater amyloid plaque pathology in the PreC and poorer performance on MMSE and episodic memory tests. Thus, loss of dendritic spines in the PreC occurs at the clinical onset of AD, and may be related to regional amyloid burden.
Our observations contribute to a growing body of literature addressing alterations in cortical synapses at clinically defined stages of AD. Studies of PreC in AD reported loss of total synapse numbers at the ultrastructural level (Scheff et al., 2013) and loss of postsynaptic protein PSD-95 (Gylys et al., 2004). With several notable exceptions (Sweet et al., 2016), loss of postsynaptic proteins was also reported in other cortical areas in AD. Using Western blot analysis, loss of the postsynaptic protein drebrin was reported in the superior frontal, superior temporal, and visual cortex as well as the hippocampus in mAD (Counts et al., 2006, 2012). Interestingly, relative to NCI, MCI cases had lower drebrin protein levels in the superior temporal cortex but higher in the superior frontal cortex (Counts et al., 2006), indicative of region-specific synaptic plasticity changes in MCI (DeKosky et al., 2002). Selective regional plasticity could explain the early impairment of temporal cortical functions subserving memory and language compared with preserved frontal cortical executive function during the initial clinical stages of AD. Based on the current and previous (Scheff et al., 2013) findings, the PreC does not appear to undergo synaptic plasticity changes in MCI.
Interestingly, we found that compared with MCI, mAD cases displayed significantly lower PreC relative protein levels of spinophilin, whereas dendritic spine density was lower but did not reach statistical significance. In contrast, both measures were lower in the sAD group compared with the MCI group. Lower spinophilin-ir dendritic spine density likely reflects reduced numbers of dendritic spines per region, as reported previously (Tang et al., 2004). Lower spinophilin immunofluorescence intensity, on the other hand, may reflect reduced amount of spinophilin protein per dendritic spine, as has been reported in confocal microscopy studies of receptor proteins (Gazzaley et al., 1996), or altered spine morphology (Feng et al., 2000). Given these assumptions, the present findings suggest that during the clinical transition from MCI to mAD, the PreC undergoes significant changes in protein content and/or morphology of dendritic spines, which precedes the loss of dendritic spines in this region. Further deterioration occurs in sAD where spinophilin protein deficit is accompanied by the structural loss of dendritic spines. Thus, dendritic spine loss in the PreC occurs incrementally across clinical diagnostic categories, a conclusion supported by our observation of a significant association between lower PreC dendritic spines and worsened performance on the neuropsychological tests relevant to PreC function (e.g., episodic memory, MMSE). These correlations in the PreC are similar to observations in the prefrontal cortex (PFC), where total numbers of spinophilin-ir dendritic spines correlated significantly with lower MMSE scores (Akram et al., 2008). However, in contrast to our study, Akram et al., reported a correlation of PFC spinophilin-ir spine measures with Braak staging for NFTs, but not with Aβ deposition. This discrepancy may be due to different cortical regions examined (PreC/PFC) or differences in the cortical lamina analyzed (e.g., lamina III in the current study vs. laminae II–VI (Akram et al., 2008)).
The stability of PreC lamina III dendritic spine measures in MCI in the current study is in agreement with an electron microscopy study reporting preserved total synapse counts in the same region/lamina in aMCI cases from the University of Kentucky Alzheimer’s Disease Center and from the RROS population (Scheff et al., 2013). These outcomes could reflect the presence of substantial amyloid pathology in a subset of NCI cases. In the current study, NCI and MCI groups had similar neuropathology status (CERAD, Braak, and NIA-Reagan) and 6-CN-PiB pathology burden. In this regard, the presence of neuritic Aβ plaques in some of our NCI cases (see CERAD scores in Table 1) indicates preclinical AD (Pike et al., 2007; Price et al., 2009) and although still cognitively normal these cases could have some synapse loss, particularly within and at the periphery of Aβ plaques (Fig. 2). In support of this idea, previous studies in transgenic mouse models of AD amyloidosis and in human AD autopsy tissue revealed that dendritic spines are reduced in the vicinity of Aβ plaques (Kirkwood et al., 2013; Koffie et al., 2009; Spires et al., 2005), possibly due to toxic effects of oligomeric Aβ surrounding the plaques (Kirkwood et al., 2013; Koffie et al., 2009, 2012; Lacor et al., 2007; Selkoe, 2002; Spires et al., 2005), or to the combined effects of fibrillar and nonfibrillar Aβ (Kirkwood et al., 2013). Interestingly, loss of synapses is prominent around classic (mature) Aβ plaques but not diffuse Aβ plaques (Masliah et al., 1990). This could partially explain the correlation we observed between lower spinophilin-ir dendritic spines and greater Aβ plaque load visualized by 6-CN-PiB, which labels mature neuritic plaques more prominently than diffuse plaques (Ikonomovic et al., 2008) (see Fig. 2B). Future studies should address the relative contributions of cored/neuritic versus diffuse plaques and oligomeric versus fibrillar Aβ to presynaptic and postsynaptic dysfunctions along the clinical stages of AD.
It could be argued that the reduction of spinophilin-ir dendritic spines observed in the current study is due to a disease-related exacerbation of an aging effect on synapses. Several studies reported altered synaptic density, dendritic arborization, and spine numbers in aged humans, rhesus monkeys, and rats (Allard et al., 2012; Cupp and Uemura, 1980; de Brabander et al., 1998; Duan et al., 2003; Henstridge et al., 2016; Jacobs et al., 1997; Kabaso et al., 2009; Markham and Juraska, 2002; Scheff and Price, 2006; Uemura, 1980); however, these changes appear to be restricted to select regions such as the frontal cortex and the hippocampus (reviewed in Grill and Riddle, 2002; Hof and Morrison, 2004; Uylings and de Brabander, 2002). Studies of spinophilin were conducted in aging rodents in regions other than associational cortex and produced inconsistent results. For example, Calhoun et al. (2008) found no changes in rat hippocampal spinophilin protein with aging, whereas an age-related decrease in spinophilin protein was observed in the striatum (Brown et al., 2005). Our cohort of aged individuals with NCI, MCI, and AD was not optimal for examining an association between age and PreC spinophilin levels, due to a narrow age range (79.0–94.5 years). Thus, studies of cohorts with younger human subjects are required to answer this important question.
There are several technical considerations associated with the current study. Since dendritic spine density was calculated by counting spinophilin-ir puncta, the observed changes in dendritic spine density may partially reflect concurrent changes in spinophilin immunoreactivity. Future studies could address this issue by simultaneously examining 2 dendritic markers in the same samples; for example, by combining spinophilin immunofluorescence with phalloidin, a fluorescent histologic marker of spines. Another consideration is the small number of aMCI cases which precluded separate statistical analyses of aMCI and non-aMCI groups. When the 2 groups were assessed separately, relative protein levels of spinophilin were lower in mAD and sAD compared with non-aMCI, but not aMCI. The sAD group also had lower dendritic spine density compared with the non-aMCI, but not aMCI. These preliminary observations indicate a more advanced process of dendritic spine changes in aMCI, and need to be confirmed in greater numbers of cases.
Taken together, our results indicate that the loss of spinophilin-ir dendritic spines in the PreC marks the onset of clinical AD, and correlates with cognitive status along clinical disease progression. Our results also provide support for a relationship between PreC dendritic spine loss and PiB-positive amyloid burden, which could aid in the interpretation of amyloid positron emission tomography and tractography imaging studies. These findings indicate that in amyloid-burdened cognitively normal people and those with MCI, stimulation of excitatory neurotransmission in PreC circuits, either by pharmacotherapy or cognitive exercise, could potentially delay the onset of clinical AD. The relationship between PreC dendritic spine changes and tau pathology remains to be investigated using more refined immunohistochemical analyses in addition to Braak staging. Further studies examining additional protein components of presynaptic and postsynaptic structure will provide greater insight into the role of PreC pathology in cognitive decline during AD progression.
Acknowledgments
This work was supported by National Institute on Aging grants P01AG014449, P01AG025204, R01AG052528 and R01AG043375, and Veterans Health Administration grant BX000452. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, the Department of Veterans Affairs, or the United States Government. The authors wish to thank Lan Shao and Jason Newman for technical assistance. The authors are indebted to participants in the Rush Religious Orders Study (P30AG010161) and the University of Pittsburgh Alzheimer’s Disease Research Center (AG05133).
Footnotes
Disclosure statement
Z. M., E. E. A., A. Y. R., K. N. F., and R. A. S. declare no conflict of interest. E. J. M. has served as consultant to NeuroPhage Pharmaceuticals, Inc. M. D. I. has served as consultant to and has received research grants from GE Healthcare.
References
- Akram A, Christoffel D, Rocher AB, Bouras C, Kovari E, Perl DP, Morrison JH, Herrmann FR, Haroutunian V, Giannakopoulos P, Hof PR. Stereologic estimates of total spinophilin-immunoreactive spine number in area 9 and the CA1 field: relationship with the progression of Alzheimer’s disease. Neurobiol Aging. 2008;29:1296–1307. doi: 10.1016/j.neurobiolaging.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allard S, Scardochio T, Cuello AC, Ribeiro-da-Silva A. Correlation of cognitive performance and morphological changes in neocortical pyramidal neurons in aging. Neurobiol Aging. 2012;33:1466–1480. doi: 10.1016/j.neurobiolaging.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker JA, Hedden T, Carmasin J, Maye J, Rentz DM, Putcha D, Fischl B, Greve DN, Marshall GA, Salloway S, Marks D, Buckner RL, Sperling RA, Johnson KA. Amyloid-beta associated cortical thinning in clinically normal elderly. Ann Neurol. 2011;69:1032–1042. doi: 10.1002/ana.22333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DA, Wilson RS, Schneider JA, Evans DA, Beckett LA, Aggarwal NT, Barnes LL, Fox JH, Bach J. Natural history of mild cognitive impairment in older persons. Neurology. 2002;59:198–205. doi: 10.1212/wnl.59.2.198. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Brown AM, Deutch AY, Colbran RJ. Dopamine depletion alters phosphorylation of striatal proteins in a model of Parkinsonism. Eur J Neurosci. 2005;22:247–256. doi: 10.1111/j.1460-9568.2005.04190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, Andrews-Hanna JR, Schacter DL. The brain’s default network: anatomy, function, and relevance to disease. Ann N Y Acad Sci. 2008;1124:1–38. doi: 10.1196/annals.1440.011. [DOI] [PubMed] [Google Scholar]
- Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, Sheline YI, Klunk WE, Mathis CA, Morris JC, Mintun MA. Molecular, structural, and functional characterization of Alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005;25:7709–7717. doi: 10.1523/JNEUROSCI.2177-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun ME, Fletcher BR, Yi S, Zentko DC, Gallagher M, Rapp PR. Age-related spatial learning impairment is unrelated to spinophilin immunoreactive spine number and protein levels in rat hippocampus. Neurobiol Aging. 2008;29:1256–1264. doi: 10.1016/j.neurobiolaging.2007.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consensus. Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s disease. Neurobiol Aging. 1997;18(4 Suppl):S1–S2. [PubMed] [Google Scholar]
- Counts SE, He B, Nadeem M, Wuu J, Scheff SW, Mufson EJ. Hippocampal drebrin loss in mild cognitive impairment. Neuro Degener Dis. 2012;10:216–219. doi: 10.1159/000333122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counts SE, Nadeem M, Lad SP, Wuu J, Mufson EJ. Differential expression of synaptic proteins in the frontal and temporal cortex of elderly subjects with mild cognitive impairment. J Neuropathol Exp Neurol. 2006;65:592–601. doi: 10.1097/00005072-200606000-00007. [DOI] [PubMed] [Google Scholar]
- Cupp CJ, Uemura E. Age-related changes in prefrontal cortex of Macaca mulatta: quantitative analysis of dendritic branching patterns. Exp Neurol. 1980;69:143–163. doi: 10.1016/0014-4886(80)90150-8. [DOI] [PubMed] [Google Scholar]
- de Brabander JM, Kramers RJ, Uylings HB. Layer-specific dendritic regression of pyramidal cells with ageing in the human prefrontal cortex. Eur J Neurosci. 1998;10:1261–1269. doi: 10.1046/j.1460-9568.1998.00137.x. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Ikonomovic MD, Styren SD, Beckett L, Wisniewski S, Bennett DA, Cochran EJ, Kordower JH, Mufson EJ. Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann Neurol. 2002;51:145–155. doi: 10.1002/ana.10069. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alz-heimer’s disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- Dorph-Petersen KA, Nyengaard JR, Gundersen HJ. Tissue shrinkage and unbiased stereological estimation of particle number and size. J Microsc. 2001;204:232–246. doi: 10.1046/j.1365-2818.2001.00958.x. [DOI] [PubMed] [Google Scholar]
- Duan H, Wearne SL, Rocher AB, Macedo A, Morrison JH, Hof PR. Age-related dendritic and spine changes in corticocortically projecting neurons in macaque monkeys. Cereb Cortex. 2003;13:950–961. doi: 10.1093/cercor/13.9.950. [DOI] [PubMed] [Google Scholar]
- Elman JA, Madison CM, Baker SL, Vogel JW, Marks SM, Crowley S, O’Neil JP, Jagust WJ. Effects of beta-amyloid on resting state functional connectivity within and between networks reflect known patterns of regional vulnerability. Cereb Cortex. 2016;26:695–707. doi: 10.1093/cercor/bhu259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Yan Z, Ferreira A, Tomizawa K, Liauw JA, Zhuo M, Allen PB, Ouimet CC, Greengard P. Spinophilin regulates the formation and function of dendritic spines. Proc Natl Acad Sci U S A. 2000;97:9287–9292. doi: 10.1073/pnas.97.16.9287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish KN, Sweet RA, Deo AJ, Lewis DA. An automated segmentation methodology for quantifying immunoreactive puncta number and fluorescence intensity in tissue sections. Brain Res. 2008;1240:62–72. doi: 10.1016/j.brainres.2008.08.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazzaley AH, Weiland NG, McEwen BS, Morrison JH. Differential regulation of NMDAR1 mRNA and protein by estradiol in the rat hippocampus. J Neurosci. 1996;16:6830–6838. doi: 10.1523/JNEUROSCI.16-21-06830.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grill JD, Riddle DR. Age-related and laminar-specific dendritic changes in the medial frontal cortex of the rat. Brain Res. 2002;937:8–21. doi: 10.1016/s0006-8993(02)02457-5. [DOI] [PubMed] [Google Scholar]
- Gylys KH, Fein JA, Yang F, Wiley DJ, Miller CA, Cole GM. Synaptic changes in Alzheimer’s disease: increased amyloid-beta and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am J Pathol. 2004;165:1809–1817. doi: 10.1016/s0002-9440(10)63436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henstridge CM, Pickett E, Spires-Jones TL. Synaptic pathology: a shared mechanism in neurological disease. Ageing Res Rev. 2016;28:72–84. doi: 10.1016/j.arr.2016.04.005. [DOI] [PubMed] [Google Scholar]
- Hodges JR. Memory in the dementias. In: Tulving E, Craik FIM, editors. The Oxford Handbook of Memory. Oxford University Press; New York: 2000. pp. 441–459. [Google Scholar]
- Hof P. Regional and laminar patterns of selective neuronal vulnerability in Alzheimer’s disease. In: Hof P, Mobbs CV, editors. Functional Neurobiology of Aging. Academic Press; New York: 2001. [Google Scholar]
- Hof PR, Morrison JH. The aging brain: morphomolecular senescence of cortical circuits. Trends Neurosci. 2004;27:607–613. doi: 10.1016/j.tins.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters HV, Montine TJ. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s Demen. 2012;8:1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomovic MD, Klunk WE, Abrahamson EE, Mathis CA, Price JC, Tsopelas ND, Lopresti BJ, Ziolko S, Bi W, Paljug WR, Debnath ML, Hope CE, Isanski BA, Hamilton RL, DeKosky ST. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain. 2008;131(Pt 6):1630–1645. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs B, Driscoll L, Schall M. Life-span dendritic and spine changes in areas 10 and 18 of human cortex: a quantitative Golgi study. J Comp Neurol. 1997;386:661–680. [PubMed] [Google Scholar]
- Kabaso D, Coskren PJ, Henry BI, Hof PR, Wearne SL. The electrotonic structure of pyramidal neurons contributing to prefrontal cortical circuits in macaque monkeys is significantly altered in aging. Cereb Cortex. 2009;19:2248–2268. doi: 10.1093/cercor/bhn242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood CM, Ciuchta J, Ikonomovic MD, Fish KN, Abrahamson EE, Murray PS, Klunk WE, Sweet RA. Dendritic spine density, morphology, and fibrillar actin content surrounding amyloid-beta plaques in a mouse model of amyloid-beta deposition. J Neuropathol Exp Neurol. 2013;72:791–800. doi: 10.1097/NEN.0b013e31829ecc89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch K, Myers NE, Gottler J, Pasquini L, Grimmer T, Forster S, Manoliu A, Neitzel J, Kurz A, Forstl H, Riedl V, Wohlschlager AM, Drzezga A, Sorg C. Disrupted intrinsic networks link amyloid-beta pathology and impaired cognition in prodromal Alzheimer’s disease. Cereb Cortex. 2015;25:4678–4688. doi: 10.1093/cercor/bhu151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffie RM, Hashimoto T, Tai HC, Kay KR, Serrano-Pozo A, Joyner D, Hou S, Kopeikina KJ, Frosch MP, Lee VM, Holtzman DM, Hyman BT, Spires-Jones TL. Apolipoprotein E4 effects in Alzheimer’s disease are mediated by synaptotoxic oligomeric amyloid-beta. Brain. 2012;135(Pt 7):2155–2168. doi: 10.1093/brain/aws127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, Hyman BT, Spires-Jones TL. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A. 2009;106:4012–4017. doi: 10.1073/pnas.0811698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopaske GT, Dorph-Petersen KA, Pierri JN, Wu Q, Sampson AR, Lewis DA. Effect of chronic exposure to antipsychotic medication on cell numbers in the parietal cortex of macaque monkeys. Neuropsychopharmacology. 2007;32:1216–1223. doi: 10.1038/sj.npp.1301233. [DOI] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markham JA, Juraska JM. Aging and sex influence the anatomy of the rat anterior cingulate cortex. Neurobiol Aging. 2002;23:579–588. doi: 10.1016/s0197-4580(02)00004-0. [DOI] [PubMed] [Google Scholar]
- Masliah E, Terry RD, Mallory M, Alford M, Hansen LA. Diffuse plaques do not accentuate synapse loss in Alzheimer’s disease. Am J Pathol. 1990;137:1293–1297. [PMC free article] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA work group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, Klunk WE, Mathis CA, DeKosky ST, Morris JC. [11C]PIB in a non-demented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67:446–452. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer’s disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Trojanowski JQ, Vinters HV, Hyman BT, National Institute on Aging-Alzheimer’s Association National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 2012;123:1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC, Price JL. Pathologic correlates of nondemented aging, mild cognitive impairment, and early-stage Alzheimer’s disease. J Mol Neurosci. 2001;17:101–118. doi: 10.1385/jmn:17:2:101. [DOI] [PubMed] [Google Scholar]
- Myers N, Pasquini L, Gottler J, Grimmer T, Koch K, Ortner M, Neitzel J, Muhlau M, Forster S, Kurz A, Forstl H, Zimmer C, Wohlschlager AM, Riedl V, Drzezga A, Sorg C. Within-patient correspondence of amyloid-beta and intrinsic network connectivity in Alzheimer’s disease. Brain. 2014;137(Pt 7):2052–2064. doi: 10.1093/brain/awu103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol. 1999;56:303–308. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- Pike KE, Savage G, Villemagne VL, Ng S, Moss SA, Maruff P, Mathis CA, Klunk WE, Masters CL, Rowe CC. Beta-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer’s disease. Brain. 2007;130(Pt 11):2837–2844. doi: 10.1093/brain/awm238. [DOI] [PubMed] [Google Scholar]
- Price JL, McKeel DW, Jr, Buckles VD, Roe CM, Xiong C, Grundman M, Hansen LA, Petersen RC, Parisi JE, Dickson DW, Smith CD, Davis DG, Schmitt FA, Markesbery WR, Kaye J, Kurlan R, Hulette C, Kurland BF, Higdon R, Kukull W, Morris JC. Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol Aging. 2009;30:1026–1036. doi: 10.1016/j.neurobiolaging.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raichle ME, MacLeod AM, Snyder AZ, Powers WJ, Gusnard DA, Shulman GL. A default mode of brain function. Proc Natl Acad Sci U S A. 2001;98:676–682. doi: 10.1073/pnas.98.2.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Ansari MA, Mufson EJ. Oxidative stress and hippocampal synaptic protein levels in elderly cognitively intact individuals with Alzheimer’s disease pathology. Neurobiol Aging. 2016;42:1–12. doi: 10.1016/j.neurobiolaging.2016.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Price DA. Alzheimer’s disease-related alterations in synaptic density: neocortex and hippocampus. J Alzheimer’s Dis. 2006;9(3 Suppl):101–115. doi: 10.3233/jad-2006-9s312. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Ansari MA, Roberts KN, Schmitt FA, Ikonomovic MD, Mufson EJ. Synaptic change in the posterior cingulate gyrus in the progression of Alzheimer’s disease. J Alzheimer’s Dis. 2015;43:1073–1090. doi: 10.3233/JAD-141518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, Mufson EJ. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging. 2006;27:1372–1384. doi: 10.1016/j.neurobiolaging.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, Roberts KN, Ikonomovic MD, Mufson EJ. Synapse stability in the precuneus early in the progression of Alzheimer’s disease. J Alzheimer’s Dis. 2013;35:599–609. doi: 10.3233/JAD-122353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Sheline YI, Raichle ME, Snyder AZ, Morris JC, Head D, Wang S, Mintun MA. Amyloid plaques disrupt resting state default mode network connectivity in cognitively normal elderly. Biol Psychiatry. 2010;67:584–587. doi: 10.1016/j.biopsych.2009.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Dickerson BC, Pihlajamaki M, Vannini P, LaViolette PS, Vitolo OV, Hedden T, Becker JA, Rentz DM, Selkoe DJ, Johnson KA. Functional alterations in memory networks in early Alzheimer’s disease. Neuromolecular Med. 2010;12:27–43. doi: 10.1007/s12017-009-8109-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Laviolette PS, O’Keefe K, O’Brien J, Rentz DM, Pihlajamaki M, Marshall G, Hyman BT, Selkoe DJ, Hedden T, Buckner RL, Becker JA, Johnson KA. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron. 2009;63:178–188. doi: 10.1016/j.neuron.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT, Bacskai BJ, Hyman BT. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005;25:7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet RA, Henteleff RA, Zhang W, Sampson AR, Lewis DA. Reduced dendritic spine density in auditory cortex of subjects with schizophrenia. Neuropsychopharmacology. 2009;34:374–389. doi: 10.1038/npp.2008.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet RA, MacDonald ML, Kirkwood CM, Ding Y, Schempf T, Jones-Laughner J, Kofler J, Ikonomovic MD, Lopez OL, Garver ME, Fitz NF, Koldamova R, Yates NA. Apolipoprotein E*4 (APOE*4) genotype is associated with altered levels of glutamate signaling proteins and synaptic coexpression networks in the prefrontal cortex in mild to moderate Alzheimer disease. Mol Cell Proteomics. 2016;15:2252–2262. doi: 10.1074/mcp.M115.056580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Janssen WG, Hao J, Roberts JA, McKay H, Lasley B, Allen PB, Greengard P, Rapp PR, Kordower JH, Hof PR, Morrison JH. Estrogen replacement increases spinophilin-immunoreactive spine number in the prefrontal cortex of female rhesus monkeys. Cereb Cortex. 2004;14:215–223. doi: 10.1093/cercor/bhg121. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Uemura E. Age-related changes in prefrontal cortex of Macaca mulatta: synaptic density. Exp Neurol. 1980;69:164–172. doi: 10.1016/0014-4886(80)90151-x. [DOI] [PubMed] [Google Scholar]
- Uylings HB, de Brabander JM. Neuronal changes in normal human aging and Alzheimer’s disease. Brain Cogn. 2002;49:268–276. doi: 10.1006/brcg.2001.1500. [DOI] [PubMed] [Google Scholar]