

Abstract
Colo-Rectal Cancer is a common cancer worldwide with 5–10% cases being hereditary. Familial Adenomatous Polyposis (FAP) syndrome is due to germline mutations in the APC or rarely MUTYH gene. NTHL1, POLD1, POLE have been recently reported in previously unexplained FAP cases. Unlike the Caucasian population, FAP phenotype and its genotypic associations have not been widely studied in several geoethnic groups. We report the first FAP cohort from South Asia and the only non-Caucasian cohort with comprehensive analysis of APC, MUTYH, NTHL1, POLD1, POLE genes. In this cohort of 112 individuals from 53 FAP families, we detected germline APC mutations in 60 individuals (45 families) and biallelic MUTYH mutations in 4 individuals (2 families). No NTHL1, POLD1, POLE mutations were identified. Fifteen novel APC mutations and a new Indian APC mutational hotspot at codon 935 were identified. Eight very rare FAP phenotype or phenotypes rarely associated with mutations outside specific APC regions were observed. APC genotype-phenotype association studies in different geo-ethnic groups can enrich the existing knowledge about phenotypic consequences of distinct APC mutations and guide counseling and risk management in different populations. A stepwise cost-effective mutation screening approach is proposed for genetic testing of south Asian FAP patients.
Introduction
Inherited predisposition is seen in 5–10% of all colorectal cancers (CRC). Major forms of hereditary colorectal cancer include the non-polyposis Lynch syndrome and the Familial Adenomatous Polyposis (FAP) syndrome1. Colorectal polyposis syndromes are characterized by multiple adenomatous or hamartomatous polyps and account for about 1% of all CRC cases. The adenomatous polyposis syndromes with high risk of colorectal cancer include the autosomal dominant Familial Adenomatous Polyposis (FAP [MIM: 175100]); and the autosomal recessive MUTYH associated polyposis (MAP [MIM: 608456]) syndrome. Recently two new entities have been described – the autosomal recessive NTHL1 associated polyposis (NAP [MIM: 616415])2 and the autosomal dominant polymerase proofreading-associated polyposis (PPAP) syndrome due to mutations in POLD1 [MIM: 174761] and POLE [MIM: 174762] genes3, 4.
FAP is characterized by the early onset of hundreds to thousands of adenomatous polyps throughout the colon and rectum with over 90% risk of development of carcinoma in one or more of the polyps1. FAP is caused by germline mutation in the APC gene5. APC is an integral part of the wnt-signalling mechanism and regulates the proliferation of colonic epithelial cells6. APC mutation carriers also have an increased risk of developing small bowel, upper gastrointestinal and papillary thyroid carcinoma as well as childhood medulloblastoma and hepatoblastoma7. Benign manifestations like congenital hypertrophy of the retinal pigment epithelium (CHRPE), desmoid tumors, osteomas and dental anomalies are also common7. Correlation between the location of mutations in APC gene (genotype) and the clinical phenotype in terms of the number of polyps, age of onset of polyps and CRC and distinct extracolonic manifestations is well described8. An attenuated variant of FAP (AFAP) due to mutations in 5′ or 3′ end of the APC gene, is characterized by polyps not exceeding 100 and late age of onset1, 5. Up to 10% of FAP cases in whom APC mutation is not identified, there is bi-allelic germline mutation in the MUTYH gene5. Unlike FAP, the MUTYH associated polyposis has a lower polyp burden which rarely exceeds 1005, 9. Comprehensive genetic analysis of APC and MUTYH fails to identify underlying gene mutation in 10–20% of FAP cases2, 4, 10, 11 and only a small proportion of these are explained by the recently described NAP and PPAP syndromes2, 4.
Current knowledge regarding the spectrum of APC gene mutation, mutational hotspots and the genotype phenotype correlations is derived mainly from studies in Caucasian cohorts5, 8, 12. In recent years, studies from other geo-ethnic groups have identified several novel APC genotypes, phenotypes and genotype-phenotype associations10, 13–16. The underlying reason for differences in phenotypic associations has not been investigated but may be due to difference in the underlying genetic background or dietary habits17, 18. APC genotype-phenotype association studies in different geo-ethnic groups can enrich the existing knowledge about phenotypic consequences of distinct APC mutations and guide counseling and risk management in different populations. This is the first FAP cohort being reported from South Asia and the only non-Caucasian cohort with comprehensive molecular genetic analysis of all the five adenomatous polyposis associated genes (APC, MUTYH, NTHL1, POLD1 and POLE).
Results
The 53 unrelated Indian FAP families reported here represent the diverse regions and religions of the Indian subcontinent with 15 hailing from northern, 15 from eastern, 14 from western and 9 from southern states of India and belonging to Hindu (46), Muslim (2), Christian (3) and Jain (2) religions. Of the 53 probands, 25 had no family history of polyposis or cancer suggesting a de novo mutation. The remaining 28 probands reported a family history of polyposis with or without CRC or other extracolonic manifestations. All the probands had classical polyposis except three AFAP cases with <100 adenomatous polyps. Through Sanger sequencing and MLPA of APC and MUTYH genes, 45 families were found to harbor deleterious germline mutation in the APC gene (35 distinct mutations) and 2 families with bi-allelic MUTYH gene mutation. With extended testing of family members, a total of 60 carriers of APC mutation and 4 carriers of bi-allelic MUTYH mutations were identified. In a combined analysis in 60 APC mutation carriers and their 58 untested relatives with FAP associated cancer or benign manifestation, the phenotypic features observed were 79 CRC, 5 upper GI cancers, 3 thyroid cancer, 2 brain tumors, 13 desmoid tumors/fibromatosis. CHRPE was noted in 14/34 APC mutation carriers for whom fundus examination details were available.
Mutation spectrum
Of the 35 distinct APC mutations described in Table 1 and Fig. 1, 15 (43%) were novel mutations not previously described in the literature or the InSiGHT database. Vast majority of the mutations were truncating (17 frameshift & 14 nonsense), 2 splice site and 2 large genomic rearrangements (LGR). All the mutations were between codons 197 to 1538. The proximal exon 15 harbored 24 (69%) of all the mutations. A 5 base pair deletion at codon 1309 (c.3927_3931delAAAGA) was the most frequent mutation, identified in 7 unrelated families. A 2 base pair deletion affecting the same codon 1309 (c.3925_3927delGA) was identified in an additional family. Codon 1061 mutation (c.3183_3187delACAAA) was identified in 4 families. Interestingly, 4 distinct truncating mutations at codon 935 occurred due to 4 different nucleotide alterations (c.2804dupA, c.2805_2815del11, c.2805 C > A and c.2802_2805delTTAC) in 4 families. The remaining 28 mutations were rare and identified in one family each. The APC LGRs identified were a duplication of the Promoter1B identified in two families and deletion of exons 9–13 in one family. In 2 of the 3 AFAP cases, biallelic MUTYH mutations were identified. A homozygous MUTYH mutation E466X (now E480X) was identified in a South Indian Tamil AFAP patient with 40 polyps and CRC. Compound heterozygous MUTYH mutations R241W and G286E were identified in a case with less than 100 polyps. In the 6 APC and MUTYH mutation negative cases with classical FAP phenotype, sequencing of the entire coding region of NTHL1 gene and the exonuclease domain of POLD1 gene (exons 6–13) and POLE gene (exons 9–14) did not identify any mutation.
Table 1.
Spectrum of APC mutations in Indian FAP cohort.
| Sr. No | Nucleotide change | Exon | Consequence | Type of mutation | No. of families with this mutation | Reported in InSiGHT* database or novel |
|---|---|---|---|---|---|---|
| 1 | c.589delA | 5 | p.R197Efs*8 | Frameshift | 1 | Novel |
| 2 | c.706 C > T | 6 | p.Q236* | Nonsense | 1 | Reported |
| 3 | c.694 C > T | 6 | p.R232* | Nonsense | 1 | Reported |
| 4 | c.1620dupA | 12 | p.Q541Tfs*19 | Frameshift | 1 | Reported |
| 5 | c.1690C > T | 13 | p.R564* | Nonsense | 1 | Reported |
| 6 | c.1779G > A | 14 | p.W593* | Nonsense | 1 | Reported |
| 7 | c.1861dupA | 14 | p.T621Nfs*13 | Frameshift | 1 | Reported |
| 8 | c.2274delA | 15 | p.A759Pfs*2 | Frameshift | 1 | Novel |
| 9 | c.2802_2805delTTAC | 15 | p.Y935Ifs*19 | Frameshift | 1 | Reported |
| 10 | c.2804dupA | 15 | p.Y935* | Frameshift | 1 | Reported |
| 11 | c.2805_2815del11 | 15 | p.Y935* | Frameshift | 1 | Novel |
| 12 | c.2805 C > A | 15 | p.Y935* | Nonsense | 1 | Reported |
| 13 | c.2828 C > G | 15 | p.S943* | Nonsense | 1 | Reported |
| 14 | c.3183_3187del5 | 15 | p.Q1062* | Frameshift | 4 | Reported |
| 15 | 3259_3260delCT | 15 | p.L1087Qfs*31 | Frameshift | 1 | Novel |
| 16 | c.3298dupT | 15 | p.S1100Ffs*19 | Frameshift | 1 | Novel |
| 17 | c.3358 G > T | 15 | p.G1120* | Nonsense | 1 | Reported |
| 18 | c.3682 C > T | 15 | p.Q1228* | Nonsense | 1 | Reported |
| 19 | c.3815 C > A | 15 | p.S1272* | Nonsense | 1 | Novel |
| 20 | c.3925-3926delGA | 15 | p.E1309Lfs*5 | Frameshift | 1 | Reported |
| 21 | c.3927_3931del5 | 15 | p.E1309Dfs*4 | Frameshift | 7 | Reported |
| 22 | c.4012 C > T | 15 | p.Q1338* | Nonsense | 1 | Reported |
| 23 | c.4037 C > G | 15 | p.S1346* | Nonsense | 1 | Novel |
| 24 | c.4202_4203delTT | 15 | p.I1401Sfs*7 | Frameshift | 1 | Novel |
| 25 | c.4216 C > T | 15 | p.Q1406* | Nonsense | 1 | Reported |
| 26 | c.4285 C > T | 15 | p.Q1429* | Nonsense | 1 | Novel |
| 27 | c.4387_4394dup8 | 15 | p. S1465Rfs*11 | Frameshift | 1 | Novel |
| 28 | 4446delT | 15 | p.P1483Qfs*24 | Frameshift | 1 | Novel |
| 29 | c.4463 T > G | 15 | p.L1488* | Nonsense | 1 | Reported |
| 30 | c.4529delG | 15 | p.S1510Tfs*13 | Frameshift | 1 | Novel |
| 31 | c.4612_4613delGA | 15 | p.E1538Ifs*5 | Frameshift | 1 | Reported |
| 32 | IVS14 + 1 G > A | — | — | Splice site | 1 | Reported |
| 33 | IVS14 + 2 T > C | — | — | Splice site | 1 | Novel |
| 34 | Deletion of Exons 9-13 | — | — | LGR | 1 | Novel |
| 35 | Duplication of promoter 1B | — | — | LGR | 2 | Novel |
Figure 1.
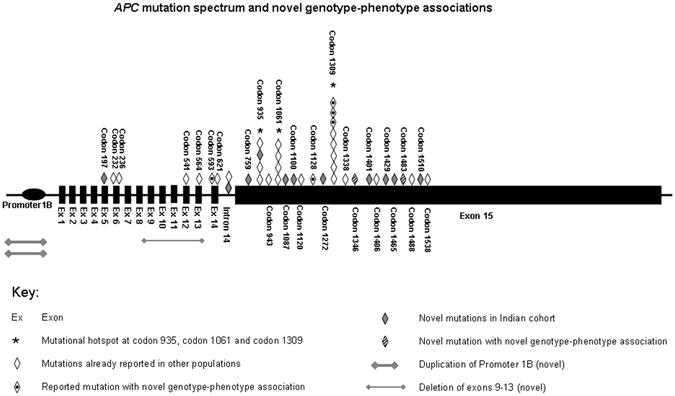
APC mutation spectrum and novel genotype-phenotype associations. The mutation distribution shows clustering of two thirds of all APC mutations in proximal Exon 15, with three Indian mutational hotspots (codon 935, 1061 and 1309) contributing to one third of all APC mutations. Large number of novel APC mutations (n = 15) and few novel genotype phenotype associations for codon 1228, 1346 and 1483 mutations.
Phenotypic features and rare genotype-phenotype associations
Of the 60 APC mutation carriers, 31 had developed CRC at a mean age of 38.3 years (range18–53 years) in a background of classical polyposis with hundreds to thousands of polyps in all but one case of AFAP with only 50 polyps. In 23 APC carriers, polyposis was diagnosed at a mean age of 32 years (range: 9–60 years) without CRC on endoscopic evaluation or histopathological examination of prophylactic procto-colectomy. In the remaining 6 carriers, colonoscopy was yet to be performed or its details were not available. Six APC carriers developed extracolonic cancers with or without CRC. These included 2 cases with papillary thyroid cancer, 1 case with duodenal cancer, 1 case with intracranial germinoma, 1 case with papillary thyroid carcinoma and duodenal cancer, and 1 case with duodenal cancer and small intestine cancer. One or more benign extracolonic manifestations were identified in 27/60 APC mutation carriers. These included CHRPE (n = 14), desmoid tumor or fibromatosis (n = 13), upper GI polyps (n = 8) and osteomas (n = 3). Eight very rare FAP phenotypes or phenotypes rarely associated with mutations outside specific regions of the APC gene were observed. These include the second reported case of intracranial germ cell tumor in an APC carrier19, absence of profuse polyposis and early onset CRC in 3 of the 7 codon 1309 mutation carriers as is classically described20, attenuated phenotype with only 50 polyps at age 33 years in a codon 593 mutation carrier, desmoid tumor with codon 1228 mutation, papillary thyroid cancer with codon 1346 mutation and most interestingly CHRPE with codon 1483 mutation7, 8.
Discussion
In FAP, the mutation spectrum of APC gene and genotype-phenotype correlations is well characterized for the Caucasian population12, 21–25, and to some extent for the East Asian population15, 26–30. Also, comprehensive molecular characterization of all the 5 known genes has been performed in very limited number of cases, that too only in the Caucasian population. Our study is the first report of a South Asian cohort of 53 FAP families and the only non-Caucasian FAP cohort analysed for all the 5 adenomatous polyposis associated genes.
The wide variation in the reported frequency of germline APC or MUTYH mutations in FAP cohorts from as low as 40–60%23, 31, 32 to as high as 75–94%10, 24, 33 is due to the stringency in making a syndromic diagnosis or lack of comprehensive genetic analysis. The high mutation detection rate of 89% in our cohort reflects the appropriateness of our clinical characterization for making the syndromic diagnosis and the comprehensive genetic analysis for APC and MUTYH including MLPA.
This study has identified a new Indian mutational hotspot at codon 935 seen in 4 (9%) FAP families. In addition, the two other known hotspot mutations at codons 1309 and 1061 were seen in 18% and 9% families respectively. High frequency of codon 1309 and 1061 mutations worldwide32 is a result of repetitive nucleotides in DNA sequence making it a mutational hotspot. Identification of APC LGR in 3 of the 11 families negative for APC point mutation or small indels and biallelic MUTYH mutation in 2 of the 8 families without APC mutation or LGR mandates its inclusion in comprehensive genetic analysis for south Asian FAP/AFAP cases. The MUTYH mutation E466X (now E480X), previously described in 3 unrelated Indian families living in the UK34 was identified as a homozygous mutation in one of our AFAP case from Tamil Nadu in south India. E466X may thus be a founder MUTYH mutation in Indians, possibly of Tamil ancestry. The founder effect of E466X needs to be confirmed with haplotyping studies and its population frequency can be established in a larger cohort. NTHL1, POLD1 or POLE mutations were not identified in any of the 6 FAP probands negative for APC or MUTYH mutations. This is not surprising as none of these families fulfilled the salient features of PPAP or NAP as described in the literature4, 35.
Of the 35 distinct mutation identified in our cohort, 15 (43%) are novel and not previously reported in Caucasian or other geo-ethnic groups. Moreover eight very rare FAP phenotype or phenotypes rarely associated with mutations outside specific regions of the APC gene were identified. APC genotypes and genotype-phenotype associations rarely or never observed in Caucasian cohorts are being increasingly reported from other geo-ethnic groups10, 14–16, 29. This highlights the need to study different geo-ethnic groups to enrich the global APC mutational spectrum and expand our knowledge of phenotypic associations of distinct APC mutations.
Based on the mutational spectrum and hotspots identified, a pragmatic stepwise genetic testing algorithm is proposed for FAP cases in south Asian countries where genetic testing is not routinely performed due to resource constraints (Fig. 2). Initial screening of three amplicons (15D–15F) harboring the mutational hotspot codons 1309, 1061 and 935 could identify 40% of all APC mutations and sequencing of additional 3 amplicons of exon 15 (15 C, 15 G, 15 H) could identify two thirds of all APC mutations. If no mutation is identified rest of the APC should be screened followed by LGR analysis and MUTYH gene sequencing. Extended testing of other adenomatous polyposis associated genes (NTHL1, POLD1 and POLE) may be considered but the yield is likely to be very low. The present study and few recent reports36 highlight that a significant proportion of FAP cases do not harbor pathogenic mutations in the genes known to be associated with FAP, MAP, NAP, PPAP syndrome. Germline exome sequencing in an adenomatous polyposis cohort has recently reported loss-of-function germline mutations in a few promising candidate genes (DSC2, PIEZO1, ZSWIM7)36 and biallelic mutations in MSH3 gene37. However these recently identified adenomatous polyposis genes are likely to remain under-reported, unless they are tested as single genes or included in multi-gene next generation sequencing (NGS) panels. The currently used multi-gene panels may not be informative as they do not include NTHL1, POLD1 and POLE genes. Therefore there is a need to conduct comprehensive genetic analysis of all the known adenomatous polyposis genes or exome sequencing studies in large pooled cohorts of APC and MUTYH negative adenomatous polyposis cases with detailed phenotypic and geo-ethnicity correlation.
Figure 2.
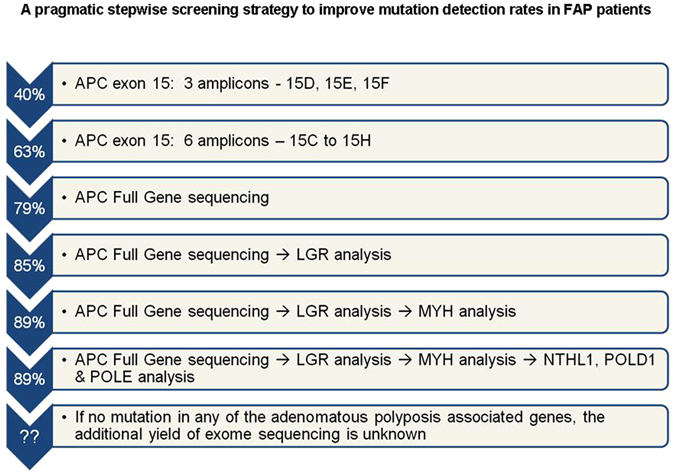
A pragmatic stepwise screening strategy to improve mutation detection rates in FAP patients. Cumulative mutation detection rates with step wise screening of exons/genes most likely to be mutated in south Asian FAP cases. Arrows on left side shows the cumulative mutation detection rates in our cohort achieved after each step. In our cohort, the cumulative mutation detection rate did not change with NTHL1, POLD1 and POLE gene analysis it may increase the detection rate slightly in larger cohorts of APC and MUTYH negative adenomatous polyposis cases from different geo-ethnic background.
In conclusion, the comprehensive investigation of all the five adenomatous polyposis genes in a well characterized Indian FAP cohort confirms the high frequency of APC mutations in classical FAP, MUTYH in AFAP cases and absence of NTHL1, POLD1 and POLE mutations in cases not showing syndromic features of PPAP or NAP. The pragmatic stepwise approach proposed can improve uptake of genetic testing for FAP in south Asian countries. Identification of a large number of novel APC mutations and genotype phenotype associations that are rare in the Caucasian population highlights the need for comprehensive phenotypic characterization and genetic analysis in large FAP cohorts from diverse geo-ethnic backgrounds.
Methods
Patients and Phenotype characterization
The study was conducted on 53 FAP families recruited through Cancer Genetics Clinic at Tata Memorial Centre, Mumbai and Christian Medical College, Vellore; India. The study was approved by the Hospital Ethics Committee of the Tata Memorial Hospital and all participating subjects provided written informed consent. All experiments were carried out in accordance with the approved guidelines and regulations. Syndromic diagnosis of FAP or AFAP was based on the number of adenomatous polyps in the colorectum with or without colorectal cancer. Further phenotypic characterization was done based on colonoscopy, esophago-gastro-duodenoscopy (EGD), computerized tomography of abdomen, thyroid ultrasound and ophthalmic examination. Detailed family history and medical records were taken from all the families reported in this study. Genetic testing was extended on first and second degree relatives if a deleterious germline mutation was identified in the proband. Blood sample was collected from 112 members from these 53 families.
PCR and Sequencing
For germline mutation analysis the complete coding sequence of the APC, MUTYH and NTHL1 genes and the exonuclease domain of POLD1 gene (exons 6–13) and POLE gene (exons 9–14) were amplified by Polymerase Chain Reaction (PCR). Primer sequences and annealing temperatures for PCR used are given in the supplementary Tables S1–S4. PCR products were purified with ExoSAP-IT [USB products, Affymetrix] and sequenced using an ABI 310 Avant, 3500 and 3730 DNA sequencer (Applied Biosystems). All mutations were confirmed by bidirectional sequencing. For most of the cases, the mutations were further reconfirmed on a second independent sample collected after the identification of mutation. InSiGHT database (LOVD) and available literature was used to check if the mutations identified was reported or novel. The mutations identified in our cohort are submitted in the InSiGHT database (www.insight-group.org).
MLPA analysis
If no APC mutation was identified on sequencing, large genomic rearrangement (LGR) in APC and MUTYH gene were evaluated with Multiplex ligation-dependent probe amplification (MLPA) using the SALSA MLPA APC P043 kit [MRC-Holland] as per the instructions provided by the company. The data was analyzed with Coffalyser software. All deletions or duplications identified and all uncertain results were confirmed in at least two independent MLPA reactions.
Electronic supplementary material
Acknowledgements
We thank the Indian Council of Medical Research for funding the project and ACTREC for providing fellowship to Nikhat Khan. We thank the genetic counselors, Nina Bhatnagar, Payal Manek, Gouri Pandit, Ravindra Reddy and Vandana Kembhavi at Cancer Genetics Clinic, TMC. We also appreciate the help extended from Dr. Pradnya Kowtal, Jyoti Patel and Moquitul Haque from Sarin Lab, ACTREC; Sharda Haralkar and Naresh Mahida from Genomics Facility, ACTREC. We acknowledge the cooperation of members of the Gastro-Intestinal Disease Management Group of TMH for referring the patients. We are thankful to all the patients and their families for participating in the study.
Author Contributions
R.S. and N.K. designed the project. N.K. and A.L. performed experiments. R.S., G.A. and M.R. assisted with sample collection and clinical evaluation. N.K. and R.S. wrote the manuscript. All authors approved the final manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-02319-6
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Stoffel EM, et al. Hereditary Colorectal Cancer Syndromes: American Society of Clinical Oncology Clinical Practice Guideline Endorsement of the Familial Risk–Colorectal Cancer: European Society for Medical Oncology Clinical Practice Guideline. Journal of Clinical Oncology. 2015;33:209–217. doi: 10.1200/JCO.2014.58.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weren RD, et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nature Genetics. 2015;47:668–671. doi: 10.1038/ng.3287. [DOI] [PubMed] [Google Scholar]
- 3.Palles C, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nature Genetics. 2013;45(2):136–144. doi: 10.1038/ng.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bellido F, et al. POLE and POLD1 mutations in529 kindred with familial colorectal cancer and/or polyposis: review of reported cases and recommendations for genetic testing and surveillance. Genetics in Medicine. 2016;18(4):325–332. doi: 10.1038/gim.2015.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grover S, et al. Prevalence and phenotypes of APC and MUTYH mutations in patients with multiple colorectal adenomas. JAMA. 2012;308:485–92. doi: 10.1001/jama.2012.8780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Narayan S, Roy D. Role of APC and DNA mismatch repair genes in the development of colorectal cancers. Molecular Cancer. 2003;2:41–55. doi: 10.1186/1476-4598-2-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Groen EJ, et al. Extra-Intestinal Manifestations of Familial Adenomatous Polyposis. Annals of Surgical Oncology. 2008;15:2439–2450. doi: 10.1245/s10434-008-9981-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nieuwenhuis MH, Vasen HF. Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): A review of the literature. Critical Reviews in Oncology/Hematology. 2007;61:153–161. doi: 10.1016/j.critrevonc.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 9.Vogt S, et al. Expanded extracolonic tumor spectrum in MUTYH-associated polyposis. Gastroenterology. 2009;137:1976–1985. doi: 10.1053/j.gastro.2009.08.052. [DOI] [PubMed] [Google Scholar]
- 10.Cao X, Hong Y, Eu KW, Loi C, Cheah PY. Singapore Familial Adenomatous Polyposis (FAP) Patients with Classical Adenomatous Polyposis but Undetectable APC Mutations Have Accelerated Cancer Progression. American Journal of Gastroenterology. 2006;101:1–8. doi: 10.1111/j.1572-0241.2006.00842.x. [DOI] [PubMed] [Google Scholar]
- 11.Mongin C, et al. Unexplained polyposis: a challenge for geneticists, pathologists and gastroenterologists. Clinical Genetics. 2012;81:38–46. doi: 10.1111/j.1399-0004.2011.01676.x. [DOI] [PubMed] [Google Scholar]
- 12.Friedl W, Aretz S. Familial Adenomatous Polyposis: Experience from a Study of 1164 Unrelated German Polyposis Patients. Hereditary Cancer in Clinical Practice. 2005;3:95–114. doi: 10.1186/1897-4287-3-3-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patel N, et al. A novel APC mutation defines a second locus for Cenani-Lenz syndrome. Journal of Medical Genetics. 2015;52(5):317–321. doi: 10.1136/jmedgenet-2014-102850. [DOI] [PubMed] [Google Scholar]
- 14.Tao H, et al. Identification of 5 novel germline APC mutations and characterization of clinical phenotypes in Japanese patients with classical and attenuated familial adenomatous polyposis. BMC Research Notes. 2010;3:305–313. doi: 10.1186/1756-0500-3-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim DW, et al. Mutation Spectrum of the APC Gene in 83 Korean FAP Families. Human Mutation. 2005;26:281–292. doi: 10.1002/humu.9360. [DOI] [PubMed] [Google Scholar]
- 16.Torrezan GT, et al. A novel SYBR-based duplex qPCR for the detection of gene dosage: detection of an APC large deletion in a familial adenomatous polyposis patient with an unusual phenotype. BMC Medical Genetics. 2012;13:55–61. doi: 10.1186/1471-2350-13-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crabtree MD, et al. Analysis of candidate modifier loci for the severity of colonic familial adenomatous polyposis, with evidence for the importance of the N-acetyl transferases. Gut. 2004;53(2):271–276. doi: 10.1136/gut.2003.015586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cruz-Correa M, et al. Combination treatment with curcumin and quercetin of adenomas in familial adenomatous polyposis. Clinical Gastroenterology and Hepatology. 2006;4(8):1035–1038. doi: 10.1016/j.cgh.2006.03.020. [DOI] [PubMed] [Google Scholar]
- 19.Lima BR, Schoenfield L, Rychwalski PJ. Germinoma presenting as a fourth cranial nerve palsy in a patient with adenomatous polyposis coli (APC) gene mutation. Journal of American Association for Pediatric Ophthalmology. 2011;15(1):71–73. doi: 10.1016/j.jaapos.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 20.Caspari R, et al. Familial adenomatous polyposis: mutation at codon 1309 and early onset of colon cancer. Lancet. 1994;343(8898):629–32. doi: 10.1016/S0140-6736(94)92634-4. [DOI] [PubMed] [Google Scholar]
- 21.Bisgaard ML, Ripa RS, Bulow S. Mutation Analysis of the Adenomatous Polyposis Coli (APC) Gene in Danish Patients with Familial Adenomatous Polyposis (FAP) Human Mutation. 2004;23:522–526. doi: 10.1002/humu.9234. [DOI] [PubMed] [Google Scholar]
- 22.Torrezan GT, et al. Mutational spectrum of the APC and MUTYH genes and genotype–phenotype correlations in Brazilian FAP, AFAP, and MAP patients. Orphanet Journal of Rare Diseases. 2013;8:54–65. doi: 10.1186/1750-1172-8-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Plawski A, Slomski R. APC gene mutations causing familial adenomatous polyposis in Polish patients. Journal of Applied Genetics. 2008;49:407–414. doi: 10.1007/BF03195640. [DOI] [PubMed] [Google Scholar]
- 24.De Rosa M, et al. The Mutation Spectrum of the APC Gene in FAP Patients from Southern Italy: Detection of Known and Four Novel Mutations. Human Mutation. 2003;21:655–656. doi: 10.1002/humu.9151. [DOI] [PubMed] [Google Scholar]
- 25.Vandrovcova J, Stekrova J, Kebrdlova V, Kohoutova M. Molecular Analysis of the APC and MUTYH Genes in Czech Families Affected by FAP or Multiple Adenomas: 13 Novel Mutations. Human Mutation. 2004;23:397–404. doi: 10.1002/humu.9224. [DOI] [PubMed] [Google Scholar]
- 26.Song G, Yuan Y, Zheng F, Yang N. Novel insertion mutation p.Asp610GlyfsX23 in APC gene causes familial adenomatous polyposis in Chinese families. Gene. 2013;516:204–208. doi: 10.1016/j.gene.2012.12.077. [DOI] [PubMed] [Google Scholar]
- 27.Chiang JM, et al. Mutation analysis of the APC gene in Taiwanese FAP families: low incidence of APC germline mutation in a distinct subgroup of FAP families. Familial Cancer. 2010;9:117–124. doi: 10.1007/s10689-009-9292-2. [DOI] [PubMed] [Google Scholar]
- 28.Sheng JQ, et al. APC gene mutations in Chinese familial adenomatous polyposis patients. World Journal of Gastroenterology. 2010;16:1522–1526. doi: 10.3748/wjg.v16.i12.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liao DX, et al. Two Chinese pedigrees for adenomatous polyposis coli: new mutations at codon 1309 and predisposition to phenotypic variations. Familial Cancer. 2014;13(3):361–368. doi: 10.1007/s10689-014-9713-8. [DOI] [PubMed] [Google Scholar]
- 30.Wei SC, et al. Genetic Analysis of the APC Gene in Taiwanese Familial Adenomatous Polyposis. Journal of Biomedical Sciences. 2004;11:260–265. doi: 10.1007/BF02256569. [DOI] [PubMed] [Google Scholar]
- 31.Gavert N, et al. Molecular Analysis of the APC Gene in 71 Israeli Families: 17 Novel Mutations. Human Mutation. 2002;19:664–670. doi: 10.1002/humu.9037. [DOI] [PubMed] [Google Scholar]
- 32.Gomez-Fernandez N, et al. Molecular analysis of the APC and MUTYH genes in Galician and Catalonian FAP families: a different spectrum of mutations? BMC Medical Genetics. 2009;10:57–68. doi: 10.1186/1471-2350-10-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rivera B, et al. Clinical and genetic characterization of classical forms of familial adenomatous polyposis: a Spanish population study. Annals of Oncology. 2011;22:903–909. doi: 10.1093/annonc/mdq465. [DOI] [PubMed] [Google Scholar]
- 34.Jones S, et al. Biallelic germline mutations in MUTYH predispose to multiple colorectal adenoma and somatic G:C–>T:A mutations. Human Molecular Genetics. 2002;11:2961–2967. doi: 10.1093/hmg/11.23.2961. [DOI] [PubMed] [Google Scholar]
- 35.Rivera B, Castellsague E, Bah I, van Kempen LC, Foulkes WD. Biallelic NTHL1 Mutations in a Woman with Multiple Primary Tumors. New England Journal of Medicine. 2015;37:1985–1986. doi: 10.1056/NEJMc1506878. [DOI] [PubMed] [Google Scholar]
- 36.Spier I, et al. Exome sequencing identifies potential novel candidate genes in patients with unexplained colorectal adenomatous polyposis. Familial Cancer. 2016;15:281–288. doi: 10.1007/s10689-016-9870-z. [DOI] [PubMed] [Google Scholar]
- 37.Adam R, et al. Exome Sequencing Identifies Biallelic MSH3 Germline Mutations as a Recessive Subtype of Colorectal Adenomatous Polyposis. American Journal of Human Genetics. 2016;99(2):337–351. doi: 10.1016/j.ajhg.2016.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.