

Abstract
Shiga toxins belong to a family of structurally and functionally related toxins serving as the main virulence factors for pathogenicity of the Shiga toxin-producing Escherichia coli (STEC) associating with Hemolytic uremic syndrome (HUS). At present, there is no effective treatment or prevention for HUS. The aim of the present study was to find conserved regions within the amino acid sequences of Stx1, Stx2 (Shiga toxin) and their variants. In this regard, In-silico identification of conformational continuous B cell and T-cell epitopes was performed in order to introduce new potential vaccine candidates. 93–100% Homology was observed in Stx1 and its variants. In Stx2 and its variants, 69–100% homology was shown. By sequence alignment with Stx1 and Stx2, 54% homology was detected. T-cell epitope identification in Stx1A and Stx2A epitopes with highest binding affinity for each HLA (human leukocyte antigen) was demonstrated with 100% identity among all Stxs. B-cell epitope prediction was resulted in finding of four common epitopes between Stxs. In silico analysis of Stxs was resulted to identification of new peptide targets that could be used in development of new epitope vaccine candidates or in immunodiagnostic tests.
Keywords: EHEC, HUS, Vaccine, Protein analysis, T-cell epitope, B-cell epitope
Background
Enterohaemorrhagic Escherichia coli (EHEC) strains are food-borne zoonotic pathogens that can cause of sporadic outbreaks, globally (Garcia-Angulo et al. 2013). EHEC can cause severe gastroenteritis, hemorrhagic colitis and in some cases, Hemolytic uremic syndrome (HUS) due to the production of Shiga toxins (Stxs) (Basu and Tumer 2015). HUS is a life-threatening infection, with a high mortality rate among children and old people. O157:H7 EHEC is the most common serotype associated with outbreaks, worldwide. Ruminants, especially cattle are considered as a reservoir for EHEC O157:H7 and fecal shedding leads to food contamination. Studies in cattle from high prevalence countries demonstrated carriage ranging from <1% to more than 30% (Garcia-Angulo et al. 2013).
Stxs are a group of genetically and structurally related exotoxins expressed by Shigella dysenteriae serotype 1 and some of the Shiga toxin-producing Escherichia coli (STEC) serotypes. Stxs are also known as VT (Verotoxin) due to the sensitivity of Vero cells to them (Pal 2015). STEC strains produce Stxs categorized as Stx1, Stx2 and also variants of either toxin. Stx1 variants are Stx1, Stx1c, Stx1d and Stx2 variants are Stx2, Stx2c, Stx2d, Stx2e, Stx2f and Stx2g. E. coli strains expressing Stx2 are potentially more virulent and they are more frequently associated with HUS (Pal 2015). The Stx genes in E. coli strains are coded with active or cryptic lambdoid bacteriophages and they are highly expressed upon activation of the lytic cycle of the phage (Mauro and Koudelka 2011; Johannes and Römer 2010). Stx family has an AB5 molecular structure, composed of five identical B subunits (7.7 kDa each) or binding domains and one enzymatically active A subunit (32 kDa). The B subunits are responsible for the attachment of holotoxin (AB5) to glycolipid receptor globotriaosylceramide (Gb3, or CD77) on the surface of host cells (Pal 2015; Johannes and Römer 2010). The A subunits are ribosome inactivating proteins (RIPs) that catalyze the irreversible damage in ribosome by modifying the large rRNA and finally inhibiting protein synthesis of host cells (Basu and Tumer 2015).
At present, there is no effective treatment or prevention for HUS. Administration of certain antibiotics has been shown to enhance the risk of HUS development. On the other hand, passively administered toxin-specific antibodies could effectively prevent toxin-mediated diseases. Therefore, several Stx2-specific monoclonal antibodies have been developed and many of them neutralized the activity of Stx2 in vitro or in vivo. In fact, Stx2 is much stronger than Stx1 in HUS infection. In addition, the essential virulence factor of EHEC, Stxs has been used for vaccination by induction of immunity responses to prevent intoxication. Primary studies demonstrated that vaccination with inactive Stx-derivatives could effectively induce the production of neutralizing antibodies and in some cases, induce protection against toxemia in animal models (Garcia-Angulo et al. 2013). Moreover, a number of studies aimed to evaluate the immunogenicity of the purified B subunit or its derivative, 2S protein. The results of the studies have been showed that the vaccines induced antibodies are able to neutralize the cytotoxic activity of both Stxs (Garcia-Angulo et al. 2013).
Therefore, finding protective B and T cell epitopes can be an effective approach for the development of a new antimicrobial vaccine by using bioinformatics tools. Additionally, identification of B-cell epitopes can lead to the development of therapeutics neutralizing antibody against microbes or their toxins. In fact, prediction of Stxs B-cell and T-cell epitopes with high affinity for antibodies MHC-II (Major histocompatibility complex) may lead to the introduction of a new vaccine candidate against STEC infections.
The purpose of the current study was to find conserved epitope regions within the Stx1, and Stx2 amino acid sequences in A and B subunits of their variants. Moreover, in silico identification of conformational continuous B-cell and T-cell epitopes have been performed in order to introduce a new potential vaccine candidate.
Methods
Stx1 and stx2 sequence analysis
The E. coli and Shigella full-length sequences of Stx1 and Stx2 subunits were obtained from GenBank and aligned using multiple sequence alignment software (http://workbench.sdsc.edu/) (Higgins et al. 1992). The identical fragments within protein sequences of A and B subunits were considered as the conserved areas.
Stx1 and stx2 structures analysis
The location of signal peptides within the stx1 and stx2 subunits was determined using SignalP server (Petersen et al. 2011). Surface accessibility, hydrophilicity and antigenicity of the Stx1 and Stx2 subunits were also determined using Immune Epitope Data Base (IEDB) analysis resource (http://www.iedb.org).
Secondary structure prediction
The secondary structure of the Stx1 and Stx2 subunits was predicted using GOR4 server (https://npsa-prabi.ibcp.fr/cgi-bin/npsa) (Sen et al. 2005).
Tertiary structure prediction
In fact, the 3D structure of the Stx1 and Stx2f are not available from protein data bank, Stx1 and Stx2f subunits protein modeling was performed using I-TASSER server (http://zhanglab.ccmb.med.umich.edu/I-TASSER) (Zhang 2008).
Validation and analysis of the 3D models
Energy minimization for the 3D models was performed using Swiss-PDB Viewer 4.1 software. Analysis of the 3D models was made using protein structure analysis (ProSa) server (https://prosa.services.came.sbg.ac.at/prosa.php) (Wiederstein and Sippl 2007) and Ramachandran Plot Analysis resource (RAMPAGE) (Lovell et al. 2002). The Z-score (overall model quality) and energy plots were created by ProSa server.
Alignment of the 3D structures was performed using Swiss-PDB Viewer and TM-align server (Zhang and Skolnick 2005) to calculate RMS (Root-mean-square deviation) and TM (Transactional memory) scores between models, respectively. These scores were represented the conformational differences between two proteins of interest. TM-score of Stx2 and Stx2f structures was calculated using TM-align server.
Prediction of antigenic B-cell epitopes
Bcepred server (http://www.imtech.res.in/-raghava/bcepred) was used for prediction of continuous B-cell epitopes (20mers) of the Stx1 and Stx2 subunits sequences (EL-Manzalawy et al. 2008a). Prediction of conformational B-cell epitope from protein 3D structures was performed using Elipro server (Ponomarenko et al. 2008).
Prediction of antigenic T-cell epitope
The location of human MHC II epitopes within the Stx1 and Stx2 subunits sequences were predicted by IEDB analysis resource (Wang et al. 2010).
Prediction of allergenicity of the proteins
Prediction of allergenicity of the Stx1 and Stx2 protein sequences has been done using AlgPred tool (Saha and Raghava 2006).
Results
Stxs protein sequences analysis
The sequences of Stx1, Stx2 and their variants were aligned using clustal W tool. The results have been summarized in the (Table 1). In A and B subunits of Stx1, 100% homology was shown between E. coli and Shigella. However, in A and B subunits of Stx2 between E. coli and Shigella 56% and 51% homology was detected, respectively. Additionally, the lowest percentage of sequence similarity with Stx2 was demonstrated in the Stx2f variant. The conformational homology with TM-score between Stx2 and Stx2f was found to predict the tertiary structure of this variant.
Table 1.
Alignment of the Stx1, Stx2 and their variants sequences using clustal W tool
| Stx1 | Stx2 | Stx1d | Stx2c | Stx2d | Stx2e | Stx2f | Stx2g | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | B | A | B | A | B | A | B | A | B | A | B | A | B | A | B | |
| Stx1 | 100 | 94–97 | 54 | 51 | – | – | – | – | – | – | – | – | – | – | ||
| Stx1c | 97 | 94 | – | – | 93 | 94 | – | – | – | – | – | – | – | – | – | – |
| Stx1d | 93 | 96 | – | – | – | – | – | – | – | – | – | – | – | – | – | |
| Shigella | 100 | 100 | 56 | 51 | – | – | – | – | – | – | – | – | – | – | – | – |
| Stx2 | 54 | 51 | 98–100 | 98–100 | – | – | – | – | – | – | – | – | – | – | – | – |
| Stx2c | – | – | 99 | 96 | – | – | – | – | 98 | 97 | 93 | 87 | 69 | 81 | 94 | 93 |
| Stx2d | – | – | 99 | 96 | – | – | 98 | 97 | – | – | 94 | 87 | 70 | 81 | 95 | 93 |
| Stx2e | – | – | 93 | 87 | – | – | 93 | 87 | 94 | 87 | – | – | 70 | 91 | 93 | 90 |
| Stx2f | – | – | 69 | 82 | – | – | 69 | 81 | 70 | 81 | 70 | 91 | – | – | 69 | 82 |
| Stx2g | – | – | 95 | 94 | – | – | 94 | 93 | 95 | 93 | 93 | 90 | 69 | 82 | – | – |
Analysis of Stxs protein structures
In order to predict the 3D model of the toxins, the location of the signal peptides were determined using SignalP server. The cleavage sites of A and B subunits were between (amino acids) aa22–aa23 and aa20–aa21, respectively. The results predicting the surface accessibility, hydrophilicity and antigenicity of the stx1 and stx2 subunits were also summarized in the (Table 2).
Table 2.
The surface accessibility, hydrophilicity and antigenicity of the Stx1 and Stx2 subunits epitopes prediction
| Subunit | Prediction parameter | Start | End | Epitope | Maximum score |
|---|---|---|---|---|---|
| Stx1B subunit | Surface accessibility | 28 | 38 | KVEYTKYNDDD | 5.916 |
| Hydrophilicity | 32 | 39 | TKYNDDDTFT | 6.571 | |
| Antigenicity | 56 | 64 | LQSLLLSAQ | 1.149 | |
| Stx1A subunit | Surface accessibility | 79 | 87 | IDPEEGRFN | 4.677 |
| Hydrophilicity | 64 | 70 | DSGTGDN | 7.157 | |
| Antigenicity | 251 | 274 | INAILGSVALILNCHHHASRVARM | 1.184 | |
| Stx2B subunit | Surface accessibility | 24 | 34 | DGKEYWTSRWN | 4.863 |
| hydrophilicity | 52 | 60 | KSSTCESGS | 5.675 | |
| Antigenicity | 35 | 43 | LQPLLQSAQ | 1.122 | |
| Stx2A subunit | Surface accessibility | 274 | 280 | VNEESQP | 4.876 |
| Hydrophilicity | 275 | 281 | NEESQPE | 6.429 | |
| Antigenicity | 250 | 274 | ISAILGTVAVILNCHHQGARSVRAV | 1.241 |
Secondary structure prediction
Prediction of Stxs secondary structures has performed using GOR4 server (Fig. 1). The results are summarized in (Table 3).
Fig. 1.
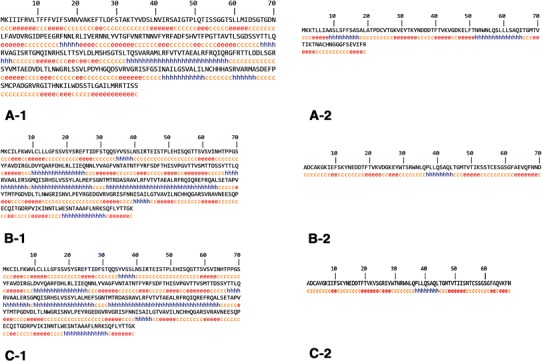
Secondary structure prediction of Stxs using GOR4 server. A-1, B-1 and C-1, are Stx1 A, Stx2 and Stx2f (a) subunits structure and A-2, B-2 and C-2, are Stx1, Stx2, and Stx2f (b) subunits respectively
Table 3.
Secondary structure analysis of Stxs
| Subunit | Alpha helix (%) | Extended strand (%) | Random coil (%) |
|---|---|---|---|
| Stx1A subunit | 17.46 | 30.79 | 51.75 |
| Stx1B subunit | 25.84 | 35.96 | 38.2 |
| Stx2A subunit | 31.97 | 24.76 | 43.26 |
| Stx2B subunit | 11.43 | 31.43 | 57.14 |
| Stx2f A subunit | 32.6 | 22.88 | 44.51 |
| Stx2f B subunit | 11.76 | 32.35 | 55.88 |
Tertiary structures prediction and analysis
Tertiary structure of Stx1 and Stx2f were predicted using I-TASSER server. Analysis of the models has been done using ProSa server after energy minimization of the 3D models by SPDBV software (Figs. 2, 3, 4, 5). Evaluation of residues in Ramachandran plot was presented in Table 4. The Z-score of the tertiary models were within the range of scores typically found for native proteins of the similar size. Moreover, the C-score of the models demonstrated that the models are predicted with a high confidence (Table 4). Calculating the TM-score of Stx2 and Stx2f structures using TM-align server was indicated that they are in the same folding (TM-score 0.99) because TM scores between 0.5 and 1 is representative of same protein fold.
Fig. 2.
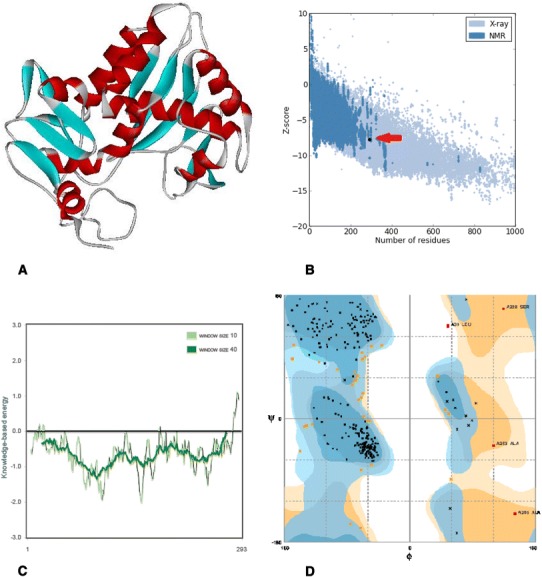
In silico prediction and analysis of the tertiary structure of Stx1 subunit A. a The best model predicted by I-TASSER tool. b Z-plot of the best model (the arrow shows the position of the model). c Knowledge-based energy plot of the best model (low energy = stability). d Ramachandran plot of the best model
Fig. 3.
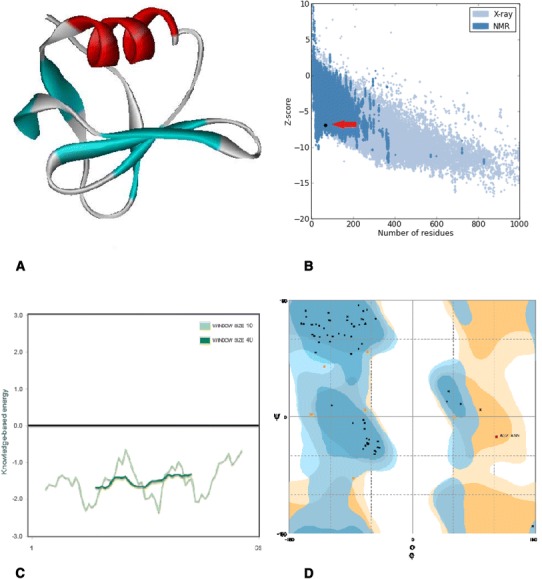
In silico prediction and analysis of the tertiary structure of Stx1 subunit B. a The best model predicted by I-TASSER tool. b Z-plot of the best model (the arrow shows the position of the model). c Knowledge-based energy plot of the best model (low energy = stability). d Ramachandran plot of the best model
Fig. 4.
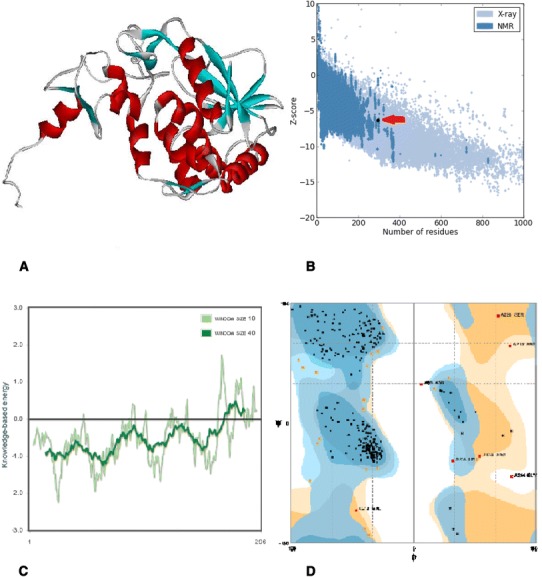
In silico prediction and analysis of the tertiary structure of Stx2f subunit A. a The best model predicted by I-TASSER tool. b Z-plot of the best model (the arrow shows the position of the model). c Knowledge-based energy plot of the best model (low energy = stability). d Ramachandran plot of the best model
Fig. 5.
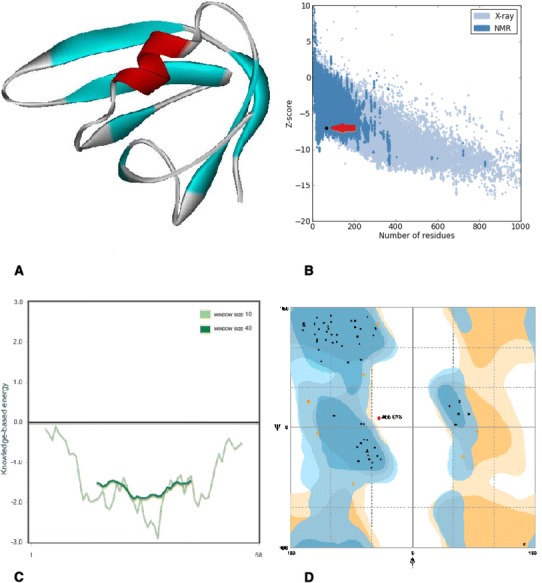
In silico prediction and analysis of the tertiary structure of Stx2f subunit B. a The best model predicted by I-TASSER tool. b Z-plot of the best model (the arrow shows the position of the model). c Knowledge-based energy plot of the best model (low energy = stability). d Ramachandran plot of the best model
Table 4.
Tertiary structure analysis of Stx1 and Stx2f
| Model | C-score | TM-score | RMSD | Z-score | Evaluation of residues n Ramachandran plot (%) | ||
|---|---|---|---|---|---|---|---|
| Favored | Allowed | Outlier | |||||
| Stx1 A subunit | 1.15 | 0.87 | 3.8 | −7.79 | 87.6 | 11 | 1.4 |
| Stx1 B subunit | 1.57 | 0.93 | 0.5 | −6.92 | 89.6 | 9 | 1.5 |
| Stx2f A subunit | 0.96 | 0.84 | 4.2 | −6.32 | 91.5 | 6.1 | 2.4 |
| Stx2f B subunit | 1.56 | 0.93 | 0.5 | −7.06 | 87.9 | 10.6 | 1.5 |
Prediction of B-cell epitopes
Prediction of continuous epitopes was performed using BCpred tool (Table 5). Conformational B-cell epitopes were predicted using Elipro tool (Table 6). The strongest B- cell epitopes were selected according to the criteria based on cutoff values for BCpred and Elipro which were >0.8 and >0.5, respectively.
Table 5.
- Prediction of linear B-cell epitopes
| Toxin | Subunit | Epitope | Start–end | Score | Toxin | Subunit | Epitope | Start–end | Score |
|---|---|---|---|---|---|---|---|---|---|
| Stx1 | A | GTGDNLFAVDVRGIDPEEGR | 66–86 | 0.987 | Stx2 | A | TGDRPVIKINNTLWESNTAA | 285–305 | 0.999 |
| RFRQIQRGFRTTLDDLSGRS | 192–210 | 0.939 | ETAPVYTMTPGDVDLTLNWG | 206–226 | 0.986 | ||||
| GISRTGMQINRHSLTTSYLD | 144–164 | 0.895 | SQGTTSVSVINHTPPGSYFA | 54–74 | 0.951 | ||||
| RMASDEFPSMCPADGRVRGI | 273–293 | 0.876 | HHQGARSVRAVNEESQPECQ | 264–284 | 0.936 | ||||
| DFSHVTFPGTTAVTLSGDSS | 116–136 | 0.736 | VPGVTTVSMTTDSSYTTLQR | 122–142 | 0.868 | ||||
| VLPEYRGEDGVRVGRISFNN | 230–250 | 0.796 | |||||||
| LDVYQARFDHLRLIIEQNNL | 79–99 | 0.744 | |||||||
| B | ITGMTVTIKTNACHNGGGFS | 65–85 | 0.950 | B | KVDGKEYWTSRWNLQPLLQS | 22–42 | 0.926 |
Table 6.
Prediction of conformational B-cell epitopes
| Toxin | Subunit | Residues | Score |
|---|---|---|---|
| Stx1 | A | T290, I291, S292, S293 | 0.976 |
| G25, T26, P27, L28, Q29, T30, I31, S32, S33, G34, G35, T36, S37, L39, M40, P211, D212, Y213, H214, G215, Q216, D217, S218, R220, G222, G227, S228, I229, N230, H245, A246, S247, R248, V249, A250, R251, M252, A253, S254, D255, E256, S259, P262, A263, D264, G265, R266, V267, R268, G269, I270, T271, H272, N273, K274, I275, L276, W277, S279, S280, T281, L282, G283, A284, I285, L286, M287, R288, R289 | 0.704 | ||
| F7, S8, T9, A10, K11, T12, D15, G56, I57, D58, P59, E60, E61, G62, R63, F64, N65, N66, N83, R84, T85, N86, N87, V88, Y90, R91, F92, A93, S96, H97, V98, F100, P101, G102, T103, T104, A105, V106, T107, L108, S109, G110, V120, A121, G122, I123, S124, T126, G127, M128, Q129, I130, N131, H133, T136, T137, L140, D141, M143, S144, H145, S146, G147, T148, S149, L150, T151, Q152, S153, R179, T180, T181, D183, D184, L185, S186, G187, R188, S189, Y190, V191, M192 | 0.665 | ||
| D42, S43, G44, T45, G46, D47, N48, N241, C242, H243, H244 | 0.533 | ||
| B | K33, Y34, N35, D36, D37, D38, T39 | 0.798 | |
| K3, T4, L5, I7, A8, A9, S10, L11, S12, F13, F14, S15, A16, S17, A18, L19, A20, T21, P22, T74, N75, A76, C77, H78, N79, G80, G81, G82, E85 | 0.606 | ||
| K28, V29, I65, T66, G67, M68 | 0.596 | ||
| Stx2 | A | K288, S289, Q290, F291, L292, Y293, T294, T295, G296, K297 | 0.977 |
| E29, H30, Y212, R213, G214, E215, D216, G217, R266, P267, V268, I269, K270, I271, N272, N273, T274, L275, W276, S278, N279, A281, A282, A283, F284, L285, N286, R287 | 0.771 | ||
| S8, AT9, Q10, Q11, H43, T44, P45, P46, G47, S48, G56, L57, D58, V59, Y60, Q61, A62, R63, F64, D65, H66, Q73, N74, N83, T84, A85, T86, N87, T88, Y90, R91, F92, S93, T96, H97, I98, S99, V100, P101, G102, V103, T104, T105, V106, S107, M108, T109, T110, D111, R119, V120, A121, A122, L123, E124, S126, G127, M128, Q129, I130, S131, R132, H133, V136, S137, L140, A141, M143, E144, F145, S146, G147, N148, T149, M150, T151, R152, D153, R156, Q180, A181, L182, S183, E184, T185, A186, P187, V188, T190 | 0.666 | ||
| T26, P27, L28, I31, S32, Q33, G34, T35, T36, S37, V38 | 0.610 | ||
| C260, Q261, I262, T263, G264, D265 | 0.601 | ||
| B | K12, Y13, N14, E15, D16, D17, T18, S31, R32, W33, A63 | 0.710 | |
| K7, N34, P37, Q40, S41, L44, T45, G46, M47, Q66, N68, N69, D70 | 0.700 | ||
| A1, D2, S53, S54, T55, C56, E57, S58, G59, S60 | 0.687 |
Prediction of T-cell epitopes
Prediction of T-cell epitopes was done for identification of epitopes having strong affinity for human MHC-II alleles. The list of epitopes with highest binding affinity for MHC molecules is summarized in Table 7. The strongest predicted Stx1 epitope is 100% conserved in Stx1 and Stx2 sequences. Analysis of physico-chemical properties of this epitope using Protparam tool revealed that the epitope has 1849.1 Da molecular weight and the estimated half-life of the epitope in mammalian cells is 7.2 h and the epitope is considered as stable peptide (II < 40) (Table 8).
Table 7.
Identification of HLA class II epitopes and HLA-epitope docking results
| Toxin | HLA type | Start–end | Epitope* | Binding score | Docking binding energy (kcal/mol) |
|---|---|---|---|---|---|
| Stx1 | HLADRB5*01:01 1 | 187–201 | TAEALRFRQIQRGFR | 0.11 | −873.6 |
| HLADPA1*01:03/DPB1*02:01 | 151–165 | QINRHSLTTSYLDLM | 0.28 | −528.9 | |
| HLADQA1*05:01/DQB1*02:01 | 179–193 | AMLRFVTVTAEALRF | 0.76 | −542.6 | |
| Stx2 | HLADRB3*01:01 | 296–310 | TLWESNTAAAFLNRK | 0.01 | −807.6 |
| HLADPA1*01/DPB1*04:01 | 109–123 | NTFYRFSDFTHISVP | 0.15 | −463.4 | |
| HLADQA1*05:01/DQB1*02:01 | 179–193 | AVLRFVTVTAEALRF | 0.74 | −548.1 |
Table 8.
Analysis of physico-chemical properties of predicted T-cell epitopes
| Epitope* | Molecular weight | Theoretical pI | Formula | Total number of atoms | Half-life (h) | Instability index* |
|---|---|---|---|---|---|---|
| TAEALRFRQIQRGFR | 1849.1 | 12 | C81H133N29O21 | 264 | 7.2 | 29.35 |
| QINRHSLTTSYLDLM | 1792.0 | 6.74 | C77H126N22O25S1 | 251 | 0.8 | 22.17 |
| AMLRFVTVTAEALRF | 1725.0 | 9.64 | C79H129N21O20S1 | 250 | 4.4 | 23.69 |
| EFSGNTMTRDASRAV | 1641.7 | 6.17 | C66H108N22O25S1 | 222 | 1 | 8.93 |
| TLWESNTAAAFLNRK | 1721.9 | 8.41 | C77H120N22O23 | 242 | 7.2 | 23.30 |
| NTFYRFSDFTHISVP | 1831.0 | 6.74 | C86H119N21O24 | 250 | 1.4 | 59.47 (US) |
| AVLRFVTVTAEALRF | 1693.0 | 9.64 | C79H129N21O20 | 249 | 4.4 | 23.69 |
* Instability index less than 40 is considered as stable
The best predicted Stx2 epitope was 100% conserved in Stx2 sequences but not in stx1 sequences. However, the HLADQA1* 05:01/DQB1*02:01 epitope was 100% identical in Stx1 and stx2 sequences. Analysis of its physico-chemical properties showed that this 1693 Da peptide has 4.4 h half life in mammalian cells and it is also stable (Table 8).
Docking of predicted T-cell epitopes to MHC molecules
Molecular docking was performed using Hex program to analyze the interaction of the best T-cell epitopes with MHC II molecules. Best binding epitopes were docked with HLADRB3*01:01, HLADRB5*01:01, HLADPA1*01:03/DPB1*02:01, HLADPA1*01/DPB1*04:01 and HLADQA1* 05:01/DQB1*02:01.
Binding energy resulting from docking simulation has summarized in Table 7.
Prediction of allergenicity of Stxs
AlgPred tool was used to predict the allergenicity of the Stxs sequence (Threshold = 0.4). The results demonstrated that A subunit of Stx1 and Stx2 (with scores of −1.6 and −1.2, respectively) were not allergens. However, B subunit of Stx1 and Stx2 were predicted to have allergens potential with scores of 0.5 and 0.8, respectively.
Discussion
Even though many strategies have been introduced to prevent HUS, effective therapeutics against Stxs infections are still in great demand. Antibiotics therapy is relatively dangerous since it can trigger subsequent bacterial increase and release more stxs that eventually raise the risk of severe clinical complications. Many efforts performed to find protective antigens and develop new vaccine candidates against HUS (Mukherjee et al. 2002; Cheng et al. 2009). Stxs can directly contribute to the EHEC pathogenesis because they have capability for eliciting protection against EHEC infections by inducing neutralizing antibodies. Previous study by Marcato et al. revealed that recombinant Stx2B subunit conjugated to keyhole limpet hemocyanin was able to elicit a protective immunity against Stx2 holotoxin in mice. The results of the study carried out by Ran et al. (2008) indicated that fusion protein consist of Stx2B subunit linked to the B subunit of E. coli heat-labile enterotoxin (LT-B) could elicit strong immunogenicity (Cheng et al. 2009).
In the present study, we aimed in silico analysis of the Stxs protein sequences and structures to find new potential vaccine candidates. With regard to the fact that conserved regions of the special antigen are good targets for developing vaccines, Firstly, amino acid sequences of Stx1 and Stx2 variants were analyzed by using alignment tools to find conserved areas among each Stx variants and also between Stx1 and Stx2. Our results demonstrated that Stx1, Stx1c and Stx1d have 93–100% homology. Except for Stx2 and Stx2f (showing 69% identity), Stx2 and other variants have 93–100% homology. Sequence alignment of Stx1 and Stx2 was shown that they have 54% homology. Since, the tertiary structure of Stx1 and Stx2f is not available, the 3D structures of these toxins were predicted using I-TASSER server. Models analysis was done using Prosa and SPDBV to show that both models are in the range of natural proteins of the same size. Calculating the TM-score of Stx2 and Stx2f 3D structures and also analysis of the secondary structure of both proteins demonstrated that both proteins are in almost the same fold and they are structurally identical.
Prediction of potential antigen allergenicity in the therapeutic proteins is realized as an important factor in vaccine therapy and a significant parameter by WHO (World health organization) (Saha and Raghava 2006). Prediction of allergenicity of Stxs by AlgPred was indicated that Stxs B subunits are potentially allergenic. Therefore, their application as vaccine candidate could be limited to epitope vaccines. On the other hand, the improved knowledge of antigen recognition at molecular level has contributed to the development of designed epitope vaccines. The major purpose of epitope vaccine design is to synthesize the identified B-cell and T-cell epitopes that are immunodominant and can elicit specific immune responses. Therefore, B-cell epitopes in conjugation with T-cell epitopes can increase the immunogenicity of the target molecule. Peptide vaccines have advantages for their chemical stability, absence of infectious potential, and ease of construction and production (Sette and Fikes 2003; Correia et al. 2014).
In order to find potential vaccine candidates, prediction of B-cell and T-cell epitopes was performed using bioinformatics tools. Presently, there are many techniques available for experimental identification of B-cell epitopes. Computational techniques offer a fast scalable cost-effective approach for: (1) predicting B-cell epitopes, (2) focusing experimental investigations, and (3) improving our knowledge of antigen–antibody interactions (EL-Manzalawy and Honavar 2010). Although a large majority of B-cell epitopes are discontinuous, epitope prediction methods have primarily focused on identifying linear B-cell epitopes that have crucial role in protection (EL-Manzalawy et al. 2008b). Linear B-cell epitope has vast application in the area of antibody production, immunodiagnostics, epitope-based vaccine design and selective immunization of therapeutic proteins (Singh et al. 2013).
In the present study, the results of linear Stx1A, Stx1B, stx2A and Stx2B B-cell epitopes prediction was indicated that 66-GTGDNLFAVDVRGIDPEEGR-86, 65-ITGMTVTIKTNACH NGGGFS-85, 285-TGDRPVIKINNTLWESNTAA-305, and 22-KVDGKEYWTSR WNLQPLLQS-42 are the strongest epitopes, respectively. Moreover, a computational investigation was shown that epitopes 144-GISRTGMQINRHSLTTSYLD-164, 116-DFSHVTF PGTTAVTLSGDSS-136, 230-VLPEYRGEDGVRVGRISFNN-250, and 79-LDVYQARFD HLRLIIEQNNL-99 are within common regions between Stx1A and Stx2A. Therefore, they have potential of being used in epitope vaccines targeting both Stxs. Prediction of conformational B-cell epitopes using Elipro was resulted to identify strong epitopes that can be used in immunodiagnostic tests and antibody production. The most probable Stx1A and Stx2A conformational epitopes are amino acids from 312 to 317 and amino acids from 288 to 297, respectively. These epitopes could be used in epitope vaccine design or immunodiagnostic tests. The strongest Stx1B and Stx2B epitopes are amino acids from 33 to 39 and amino acids from 14 to 19, respectively that could be used in antibody production against Stx receptor attachment. Previously, Smith et al. reported three regions within the Stx1B sequence, and via them 13C4 monoclonal antibody binds to the B subunit and neutralizes the cytotoxic and lethal activities of Stx1 (Nakao et al. 1999). Their findings indicated that three segments of 13C4 monoclonal antibody epitope are amino acids from 21–26, 45–52 and 74–81 with critical asparagine residue at position 55 that are necessary for binding of the 13C4 monoclonal antibody. Another critical epitope segment has been predicted by our study as a strong conformational B-cell epitope. Consequently, this epitope could serve as a candidate for antibody production against Stx1 B subunit.
On the other hand, EHEC immunity requires humoral and T helper cell responses. Generally, analysis of the binding affinity of antigenic peptides to MHC molecules is the essential object when predicting immunogenic epitopes (Patronov and Doytchinova 2010; Betts and Russell 2003). Identification of epitopes with high binding affinity for MHC class II was performed using IEDB server. Our results show that Stx1A and Stx2A proposed epitopes with highest binding affinity for each HLA super types bear 100% identity among all Stx1A and Stx2A sequences. According to the total free energy reported in our docking outputs the strongest Stx1A and Stx2A T-cell epitopes were shown the best interaction affinity to the related HLA super types. Additionally, these epitopes have the longest half-life in the mammalian cells. On the other hand, although the best predicted Stx1A T-cell epitope 187-TAEALRFRQIQRGFR-201 has 93% identity with the Stx2A sequence, it has not been identified as T-cell epitope in Stx2A. The amino acid substitution of glutamic acid by glycine in Stx2A may explain the difference (Betts and Russell 2003; Aftabuddin and Kundu 2007). Glutamic acid is a polar negatively hydrophilic charged amino acid while glycine is an aliphatic neutral amino acid. Moreover, they have different sizes and hydropathy scores. Glycine is a unique amino acid containing hydrogen as its side chain. Glycine also affects the protein structure since it has destabilized α-helices (Krause et al. 2000). Regarding secondary structure prediction of Stxs (Fig. 1), presence of glycine in position 199 of Stx1A has changed the type of the structure from helix to random coil. Furthermore, Stx1A epitope 179-AMLRFVTVTAEALRF-193 and Stx2A epitope 179-AVLRFVTVTAEALRF-193 was shown 93% identity indicating that it is a universal epitope. Substitution of methionine, 180 in Stx1A by valine 180 in Stx2A is the only amino acid residue substitution in the predicted universal epitope. Although, methionine and valine have different sizes and hydropathy scores, they are both aliphatic hydrophobic amino acids. Therefore, the amino acid substitution in the predicted epitope may not remarkably modify the properties of the epitope to alter its universality (Betts and Russell 2003; Aftabuddin and Kundu 2007).
In conclusion, a new peptide targets was identified by in silico analysis that could be used in development of new epitope vaccine candidates or in immunodiagnostic tests.
Compliance with ethical standards
Conflict of interest
The authors do not have any conflict of interest.
References
- Aftabuddin MD, Kundu S. Hydrophobic, hydrophilic and charged amino acid networks within protein. Biophys J. 2007;93(1):225–231. doi: 10.1529/biophysj.106.098004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu D, Tumer NE. Do the A subunits contribute to the differences in the toxicity of Shiga toxin 1 and Shiga toxin 2? Toxins. 2015;7(5):1467–1485. doi: 10.3390/toxins7051467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betts MJ, Russell RB. Amino acid properties and consequences of substitutions. In: Barnes MR, Gray IC, editors. Bioinformatics for geneticists. Amsterdam: Wiley; 2003. [Google Scholar]
- Cheng Y, Feng Y, Luo P, Gu J, Yu S, Zhang WJ, Liu YQ, Wang QX, Zou QM, Mao XH. Fusion expression and immunogenicity of EHEC EspA-Stx2Al protein: implications for the vaccine development. J Microbiol. 2009;47(4):498–505. doi: 10.1007/s12275-009-0116-8. [DOI] [PubMed] [Google Scholar]
- Correia BE, Bates JT, Loomis RJ, Baneyx G, Carrico C, Jardine JG, Rupert P, Correnti C, Kalyuzhniy O, Vittal V, Connell MJ, Stevens E, Schroeter A, Chen M, Macpherson S, Serra AM, Adachi Y, Holmes MA, Li Y, Klevit RE, Graham BS, Wyatt RT, Baker D, Strong RK, Crowe JE, Jr, Johnson PR, Schief WR. Proof of principle for epitope-focused vaccine design. Nature. 2014;507(7491):201–206. doi: 10.1038/nature12966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EL-Manzalawy Y, Honavar V. Recent advances in B-cell epitope prediction methods. Immunome Res 3. 2010;6(Suppl 2):S2. doi: 10.1186/1745-7580-6-S2-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EL-Manzalawy Y, Dobbs D, Honavar V. Predicting linear B-cell epitopes using string kernels. J Mol Recognit. 2008;21(4):243–255. doi: 10.1002/jmr.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EL-Manzalawy Y, Dobbs D, Honavar V. Predicting flexible length linear B-cell epitopes. Comput Syst Bioinf Conf. 2008;7:121–132. [PMC free article] [PubMed] [Google Scholar]
- Garcia-Angulo VA, Kalita A, Torres AG. Advances in the development of enterohemorrhagic Escherichia coli vaccines using murine models of infection. Vaccine. 2013;31(32):3229–3235. doi: 10.1016/j.vaccine.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins DG, Bleasby AJ, Fuchs R. CLUSTAL V: improved software for multiple sequence alignment. Comput Appl Biosci. 1992;8(2):189–191. doi: 10.1093/bioinformatics/8.2.189. [DOI] [PubMed] [Google Scholar]
- Johannes L, Römer W. Shiga toxins—from cell biology to biomedical applications. Nat Rev Microbiol. 2010;8(2):105–116. doi: 10.1038/nrmicro2279. [DOI] [PubMed] [Google Scholar]
- Krause E, Bienert M, Schmieder P, Wenschuh H. The Helix-destabilizing propensity Scale of D-amino acids: the influence of side chain steric effects. J Am Chem Soc. 2000;122(20):4865–4870. doi: 10.1021/ja9940524. [DOI] [Google Scholar]
- Lovell SC, Davis IW, Arendall WB, III, de Bakker PIW, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins. 2002;50(3):437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- Mauro SA, Koudelka GB. Shiga toxin: expression, distribution, and its role in the environment. Toxins (Basel) 2011;3(6):608–625. doi: 10.3390/toxins3060608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee J, Chios K, Fishwild D, Hudson D, O’Donnell S, Rich SM, Donohue-Rolfe A, Tzipori A. Human Stx2- specific monoclonal antibodies prevent systemic complications of Escherichia coli O157:H7 infection. Infect Immun. 2002;70(2):612–619. doi: 10.1128/IAI.70.2.612-619.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao H, Kiyokawa N, Fujimoto J, Yamasaki S, Takeda T. Monoclonal antibody to Shiga toxin 2 which blocks receptor binding and neutralizes cytotoxicity. Infect Immun. 1999;67(11):5717–5722. doi: 10.1128/iai.67.11.5717-5722.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal P. Molecular aspects of effect of Shiga toxin in humans—a review. Int Lett Nat Sci. 2015;7:78–89. doi: 10.18052/www.scipress.com/ILNS.34.78. [DOI] [Google Scholar]
- Patronov A, Doytchinova I. T-cell epitope vaccine design by immunoinformatics. Open Biol. 2010;3(1):120139. doi: 10.1098/rsob.120139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8(10):785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- Ponomarenko JV, Bui H, Li W, Fusseder N, Bourne PE, Sette A, Peters B. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinf. 2008;9:514. doi: 10.1186/1471-2105-9-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran XQ, Wang HZ, Liu JJ, Li S, Wang JF. The immunogenicity of fusion protein linking the carboxyl terminus of the B subunit of Shiga toxin 2 to the B subunit of E. coli heat-labile enterotoxin. Vet Microbiol. 2008;127(1–2):209–215. doi: 10.1016/j.vetmic.2007.08.021. [DOI] [PubMed] [Google Scholar]
- Saha S, Raghava GPS. AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucl Acid Res. 2006;34(Web Server issue):W202–W209. doi: 10.1093/nar/gkl343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen TZ, Jernigan RL, Garnier J, Kloczkowski A. GOR V server for protein secondary structure prediction. Bioinformatics. 2005;21(11):2787–2788. doi: 10.1093/bioinformatics/bti408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sette A, Fikes J. Epitope-based vaccines: an update on epitope identification, vaccine design and delivery. Curr Opin Immunol. 2003;15(4):461–470. doi: 10.1016/S0952-7915(03)00083-9. [DOI] [PubMed] [Google Scholar]
- Singh H, Ansari HR, Raghava GPS. Improved method for linear B-cell epitope prediction using antigen’s primary sequence. PLoS One. 2013;8(5):e62216. doi: 10.1371/journal.pone.0062216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Sidney J, Kim Y, Sette A, Lund O, Nielsen M, Peters B. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinf. 2010;11:568. doi: 10.1186/1471-2105-11-568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucl Acid Res. 2007;35(Web Server issue):W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinf. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on TM-score. Nucl Acid Res. 2005;33(7):2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]