

Abstract
Magnesium is essential to the proper functioning of numerous cellular processes. Magnesium ion (Mg2+) deficits, as reflected in hypomagnesemia, can cause neuromuscular irritability, seizures and cardiac arrhythmias. With normal Mg2+ intake, homeostasis is maintained primarily through the regulated reabsorption of Mg2+ by the thick ascending limb of Henle’s loop and distal convoluted tubule of the kidney. Inadequate reabsorption results in renal Mg2+ wasting, as evidenced by an inappropriately high fractional Mg2+ excretion. Familial renal Mg2+ wasting is suggestive of a genetic cause, and subsequent studies in these hypomagnesemic families have revealed over a dozen genes directly or indirectly involved in Mg2+ transport. Those can be classified into four groups: hypercalciuric hypomagnesemias (encompassing mutations in CLDN16, CLDN19, CASR, CLCNKB), Gitelman-like hypomagnesemias (CLCNKB, SLC12A3, BSND, KCNJ10, FYXD2, HNF1B, PCBD1), mitochondrial hypomagnesemias (SARS2, MT-TI, Kearns–Sayre syndrome) and other hypomagnesemias (TRPM6, CNMM2, EGF, EGFR, KCNA1, FAM111A). Although identification of these genes has not yet changed treatment, which remains Mg2+ supplementation, it has contributed enormously to our understanding of Mg2+ transport and renal function. In this review, we discuss general mechanisms and symptoms of genetic causes of hypomagnesemia as well as the specific molecular mechanisms and clinical phenotypes associated with each syndrome.
Keywords: Magnesium, Homeostasis, Hereditary, Kidney, Distal convoluted tubule, Thick ascending limb of Henle’s loop
Introduction
Magnesium is a vital element for the human body and is involved in numerous biological processes. It is the second-most abundant intracellular cation (Mg2+) in the human body and is crucial for the function of over 600 enzymes and regulation of the activity of several ion channels, as well as for stabilization of negatively charged molecules such ATP, ADP, RNA and DNA (reviewed in [1]). In order to constantly suffice the body’s requirements for this ion, there is a significant storage capacity for Mg2+: an adult human body usually contains about 24 g of Mg2+ at any one time [1]. Blood serum only contains a fraction of this, with normal serum Mg2+ concentrations [Mg2+] ranging from 0.70 to 1.1 mM, which translates to about 60 mg in total. Even though only two-thirds of this is biologically active (the ionized fraction), total serum Mg2+ concentrations are still used in daily practice as a measurement of the total Mg2+ status of a patient. Accordingly, hypomagnesemia is defined as a serum [Mg2+] < 0.70 mM (<1.7 mg/dL) and hypermagnesemia as a serum [Mg2+] > 1.1 mM (>2.5 mg/dL). A shortage of Mg2+ can have direct consequences, some well-established, others less clear, but it is also associated with several other diseases. Direct consequences or symptoms that might arise from hypomagnesemia are variable in severity and may correlate to the extent and duration of the Mg2+ shortage, ranging from leg cramps and tiredness to seizures, coma and eventually death (Table 1). In addition, (severe) hypomagnesemia may have further consequences during pregnancy, as suggested by findings that a Mg2+-deficient diet in pregnant mice was able to induce fetal malformations [2]. Conversely, supplementation with magnesium sulfate (MgSO4) during pregnancy is a treatment for pre-eclampsia [3], suggesting a role for a relative shortage of Mg2+ in this disease too. Lastly, some diseases, such as Parkinson’s disease and diabetes, have merely been associated with low serum Mg2+ concentrations (reviewed in [1]). It is not yet clear, however, whether hypomagnesemia is the cause, a consequence or simply an epiphenomenon in these diseases.
Table 1.
Direct consequences of hypomagnesemia
| Direct consequences of hypomagnesemiaa |
|---|
| Chvostek and Trousseau’s signs |
| Tiredness |
| Generalized weakness |
| Tremor |
| Paresthesias and palpitations |
| Hypokalemia |
| Hypoparathyroidism resulting in hypocalcemia |
| Chondrocalcinosis |
| Failure to thrive (in children) |
| Spasticity and tetany |
| Seizures |
| Electrocardiography changes, including prolonged QT interval (especially with concomitant hypokalemia) |
| Cardiac arrhythmias (especially with concomitant hypokalemia) |
| Basal ganglia calcifications |
| Coma |
| Intellectual disability |
| Death |
aConsequences of hypomagnesemia are presented from top to bottom of table in order of increasing severity
Most of our current knowledge about Mg2+ homeostasis has been obtained by studying the molecular mechanisms through which genetic mutations cause hypomagnesemia. The aim of this review is to provide an update of the currently known genetic defects in Mg2+ homeostasis from a clinical point of view, discussing firmly established knowledge as well as several recently discovered or neglected hereditary hypomagnesemic syndromes.
Maintaining Mg2+ homeostasis
The daily dietary Mg2+ intake recommended by the U.S. Institute of Medicine is dependent on age (Table 2). Approximately 30–50 % of the ingested Mg2+ will be absorbed by the intestine, although this has been reported to increase to 80 % in cases of Mg2+ deficiency. The bulk of the dietary Mg2+ is initially absorbed in the jejunum and ileum via paracellular pathways. The remainder can be absorbed by the colonic epithelium, entering the cells via transient receptor potential melastatin type 6 (TRPM6), an essential ion channel and serine/threonine-protein kinase, and probably exiting the cells at the basolateral side by making use of the sodium (Na+) gradient and the Na+–Mg2+ exchanger cyclinM4 (CNNM4). Mg2+ subsequently enters the bloodstream to be delivered to cells, excreted by the kidneys or stored in bones. The large skeletal stores (50–60 % of total body Mg2+) are in part responsible for keeping the serum Mg2+ concentrations constant (reviewed in [1]).
Table 2.
Recommended dietary allowance of magnesium (Mg2+)
| Age (years) | RDA for males (mg Mg2+/day) | RDA for females (mg Mg2+/day)a |
|---|---|---|
| 0-1 | NA | NA |
| 1–3 | 80 | 80 |
| 4–8 | 130 | 130 |
| 9–13 | 240 | 240 |
| 14–18 | 410 | 360 |
| 19–30 | 400 | 310 |
| >31 | 420 | 320 |
RDA, Recommended dietary allowance; NA, information not available
aFor women during pregnancy the RDA is slightly higher
An even more important role in the regulation of Mg2+ homeostasis has been given to the kidney. After glomerular ultrafiltration, a mere 10–25 % of Mg2+ is reabsorbed by the proximal convoluted tubule (PCT) through paracellular pathways. Next, a further 50–70 % of the filtered Mg2+ is reabsorbed via paracellular pathways in the thick ascending limb of Henle’s loop (TAL), where claudins play a key role in regulating paracellular calcium (Ca2+) and Mg2+ transport [4] (see Fig. 1). Lastly, fine-tuning of the total Mg2+ reabsorption takes place in the distal convoluted tubule (DCT), which absorbs the final 5–10 % via transcellular pathways [5]. Essential for this last step is the apical Mg2+ channel TRPM6, the same channel that is responsible for Mg2+ transport in the large intestine (reviewed in [1]). The activity of this channel and its expression in the membrane are positively regulated through epidermal growth factor (EGF)-activated pathways [6] (see Fig. 1). Finally, also insulin, estrogen, extracellular pH, ATP, oxidative stress and Mg2+ itself are all found to be able to influence the magnitude of Mg2+ transport in the DCT [1]. Towards the end of this last segment, 95–99 % of filtered Mg2+ has been reabsorbed in total, and no further reabsorption takes place beyond this point [7].
Fig. 1.

Reabsorption of the magnesium cation (Mg 2+) in the thick ascending limb of Henle’s loop (TAL) and distal convoluted tubule (DCT). The relevant molecular transport mechanisms of the TAL and DCT are shown. Note that Mg2+ is transported via paracellular pathways into the TAL and via transcellular pathways into the DCT. A more detailed explanation of the molecular transport mechanisms can be found in the text. Black text indicates proteins that are mutated in genetic disorders of Mg2+ homeostasis. Grey text indicates other proteins
Disturbances of Mg2+ homeostasis
Hypomagnesemia is defined as total serum Mg2+ concentrations below 0.70 mM (<1.7 mg/dL), while hypermagnesemia is reserved for concentrations above 1.1 mM (>2.7 mg/dL). Symptomatic hypermagnesemia is rare and mostly induced by excessive use of drugs which contain high amounts of Mg2+, including laxatives and Epsom salts [1]. Hypomagnesemia on the other hand is more frequent [8–10] and can have several distinct causes. First among these is a prolonged general loss of electrolytes, such as during periods of vomiting, diarrhea or malabsorption [11]. Secondly, several genetic disorders are accompanied by hypomagnesemia (see Table 4). Thirdly, albeit rare in the pediatric population, use of alcohol and certain drugs (Table 3; reviewed in [1]) should be considered. Lastly, contemporary food intake is relatively poor in Mg2+ [12] and may therefore contribute to the development of hypomagnesemia.
Table 4.
Genetic causes of hypomagnesemia
| Categories/names of disordersa | Gene | Protein | OMIM catalog number | Inheritance | Renal tubule segment | Plasma Mg2+ concentration (mM)b | Estimated incidence or number of known families/patients | Distinctive findings, other than hypomagnesemiac |
|---|---|---|---|---|---|---|---|---|
| Hypercalciuric hypomagnesemias | Hypercalciuria, nephrocalcinosis | |||||||
| FHHNC type 1 | CLDN16 | Claudin-16 | 248250 | R | TAL | 0.49 | 100s of patients | Polyuria/polydipsia, elevated serum iPTH, renal failure |
| FHHNC type 2 | CLDN19 | Claudin-19 | 248190 | R | TAL | 0.59 | 10s of patients | Same as FHHNC type 1, plus ocular abnormalities |
| ADHH Bartter syndrome type 5 | CASR | CaSR | 601198 | D | TAL | 0.66 | 100s of patients | Hypocalcemia with normal or low PTH |
| Bartter syndrome, type 3 (classical type) | CLCNKB | ClC-Kb | 607634 | R | DCT/TAL | 0.63 | 100s of patients | Gitelman-like phenotype possible, rarely nephrocalcinosis |
| Gitelman-like hypomagnesemias | Hypocalciuria, hypokalemia, metabolic alkalosis | |||||||
| Gitelman syndrome | SLC12A3 | NCC | 263800 | R | DCT | 0.49 | 1:40 000 | Chondrocalcinosis at older age |
| Bartter syndrome, type 4 | BSND | Barttin | 602522 | R | DCT/TAL | 0.60 | 10s of patients | Prenatal complications, renal failure early in life possible |
| EAST syndrome | KCNJ10 | Kir4.1 | 612780 | R | DCT | 0.63 | 26 patients | Sensorineural deafness, seizures, ataxia |
| IDH | FXYD2 | γ-subunit of the Na+-K+-ATPase | 154020 | D | DCT | 0.47 | 3 families (29 patients) | |
| ADTKD/RCAD | HNF1B | HNF1β | 137920 | D | DCT | 0.69 | 1:120 000 | Renal, genital and pancreatic abnormalities and MODY5 in highly variable combination and presentation |
| HPABH4D/RCAD-like | PCBD1 | PCBD1 | 264070 | R | DCT | 0.68 | 23 patients | MODY5-like |
| Mitochondrial hypomagnesemias | Variable | |||||||
| HHH | MT-TI | Mt. tRNAile | 500005 | Mt | DCT? | 0.71 | 1 family (38 patients) | Hypertension and hypercholesterolemia |
| HUPRAS | SARS2 | SARS2 | 613485 | R | TAL? | 0.37 | 2 families (4 patients) | Hyperuricemia, pulmonary hypertension, renal failure and alkalosis |
| KSS | Mitochondrial deletion | – | 530000 | Mt | TAL? | 0.51 | 100s of patients | External ophthalmoplegia, retinopathy and cardiac conduction defects |
| Other hypomagnesemias | Variable | |||||||
| HSH | TRPM6 | TRPM6 | 602014 | R | DCT | 0.20 | 10s of patients | Neonatal presentation with severe hypomagnesemia |
| IRH | EGF | EGF | 611718 | R | DCT | 0.59 | 1 family (2 patients) | Intellectual disability |
| NISBD2 | EGFR | EGFR | 616069 | R | DCT | ? | 1 patient | Severe inflammation of skin and bowel from birth |
| HSMR | CNNM2 | CNNM2 | 613882 | D/R | DCT | 0.50 | 7 families (10 patients) | Intellectual disability, seizures |
| ADH/EA1 | KCNA1 | Kv1.1 | 176260 | D | DCT | 0.37 | 1 family (21 patients) | Episodic myokymia |
| KCS2 | FAM111A | FAM111A | 127000 | D | TAL? | 0.46 | 10s of patients | Impaired skeletal development and hypocalcemic hypoparathyroidism |
aADH, Autosomal dominant hypomagnesemia; ADHH, autosomal dominant hypocalcemia with hypocalciuria; ADTKD, autosomal dominant tubulointerstitial kidney disease; EA1, episodic ataxia type 1; EAST, epilepsy, ataxia, sensorineural deafness and tubulopathy; FHHNC, familial hypomagnesemia with hypocalcemia and nephrocalcinosis; HHH, hypertension, hypercholesterolemia and hypomagnesemia; HPABH4D, hyperphenylalaninemia BH4-deficient; HSH, hypomagnesemia with secondary hypocalcemia; HSMR, hypomagnesemia with seizures and mental retardation; HUPRAS, hyperuricemia, pulmonary hypertension, renal failure and alkalotic syndrome; IDH, isolated dominant hypomagnesemia; IRH, isolated recessive hypomagnesemia; KCS2, Kenny−Chaffey syndrome type 2; KSS, Kearns-Sayre syndrome; NISBD2 neonatal inflammatory skin and bowel disease type 2; RCAD, renal cysts and diabetes; TAL, thick ascending limb of Henle’s loop; DCT distal convoluted tubule
bEstimated average. To convert mM [Mg2+] to mg/dL, multiply by 2.43
ciPTH, Intact parathyroid hormone; MODY5, maturity onset diabetes of the young type 5
Table 3.
Drugs associated with hypomagnesemia
| Drugs associated with hypomagnesemia |
|---|
| Diuretics (furosemide, thiazide) |
| Epidermal growth factor receptor inhibitors (cetuximab) |
| Proton pump inhibitors (all, such as omeprazole) |
| Calcineurin inhibitors (cyclosporin A, tacrolimus) |
| Platinum derivatives (cisplatin, carboplatin) |
| Antimicrobials (aminoglycosides, pentimidine, rapamycin, amphotericin B, foscarnet) |
Diagnosing renal Mg2+ wasting
The most important clinical diagnostic tool for differentiating hypomagnesemia of renal origin from intestinal hypomagnesemia is determination of the fractional excretion of magnesium (FEMg) [13], which can be calculated with the following formula: {([Mg2+]urine × [creatinine]plasma)/ (0.7 [Mg2+]plasma × [creatinine]urine)} × 100 %. The factor of 0.7 is included to adjust the total plasma Mg2+ concentration to the freely filtered fraction. A FEMg of >4 % in a hypomagnesemic patient is consistent with renal Mg2+ wasting, while a patient with a FEMg of <2 % will likely have an extra-renal origin of their hypomagnesemia [13]. However, a FEMg <4 % does not rule out renal Mg2+ wasting. First, a low glomerular filtration rate may result in a lower filtered load of Mg2+. If the absorptive capacity of the kidney for Mg2+ is sufficient to cope with this lower load, the result may be a normal or even low FEMg. By the same mechanism, severe (renal) hypomagnesemia may result in a lower filtered load of Mg2+ and thus a normal or low FEMg. To account for these confounding factors, the serum Mg2+ levels of hypomagnesemic patients should be increased by means of intravenous Mg2+ supplementation before the FEMg is measured [14].
Hereditary hypomagnesemias
The genetic causes of hypomagnesemia are heterogeneous and comprise both recessive and dominant disorders (Table 4). The localization of the responsible genes is firmly established: all known genes encode proteins expressed in the DCT and/or TAL (Fig. 1). Nevertheless, the exact mechanisms at the molecular level remain to be elucidated for many of these diseases. We therefore propose four categories of genetic causes of hypomagnesemia based on similar manifestation, electrolyte abnormalities and localization. The resulting classes presumably also reflect a common pathophysiological mechanism for each group.
Hypercalciuric hypomagnesemias
The hypercalciuric hypomagnesemias are a class of hypomagnesemias in which the ability of the TAL to reabsorb divalent cations is affected. Ca2+ and Mg2+ reabsorption takes place via paracellular pathways in the TAL and is therefore strongly dependent on the lumen positive transepithelial potential difference. Consequently, disruption of this voltage difference or of the integrity of this paracellular pathway will impair both Ca2+ and Mg2+ transport (reviewed in [4]). In two of the four types of Bartter syndrome, the lumen positive transepithelial voltage difference is decreased, while mutations in the CASR, CLDN16 and CLDN19 genes are proposed to interfere with the integrity of the pathway as well as the voltage difference [4]. Compensatory mechanisms in the DCT and other segments, however, may be able to avert hypomagnesemia (in Bartter syndrome type 1 and 2) or hypercalciuria (in Bartter syndrome type 4) [15]. This only leaves Bartter syndrome type 3, which impairs both TAL and DCT function (reviewed in [15]), as meeting the criterion for this group. Clinically, genetic disorders in this group can result in nephrocalcinosis or in chronic kidney disease (CKD), although the incidence and speed of progression differs from one to the other [4, 15].
CLDN16 and CLDN19 (familial hypomagnesemia with hypocalcemia and nephrocalcinosis)
Recessive mutations in CLDN16 (encoding claudin-16) and CLDN19 (encoding claudin-19) are the most frequent cause of hypercalciuric hypomagnesemia [16, 17]. These claudin mutations disrupt the pore selectivity of the tight junction, impairing paracellular Ca2+ and Mg2+ reabsorption in the TAL (reviewed in [18]). Consequently, patients suffer from hypomagnesemia and its associated symptoms, childhood nephrocalcinosis possibly due to the hypercalciuria and polyuria with polydipsia due to additional sodium (Na+) and volume loss [19]. Patients with CLDN19 mutations will also exhibit ocular anomalies [17]. The renal prognosis for both types is poor, with progressive CKD requiring renal replacement therapy typically in the second or third decade of life [20]. The cause of the CKD is unclear, although nephrocalcinosis may be a contributory factor.
CASR gain-of-function (autosomal dominant hypocalcemia with hypercalciuria)
Gain-of-function mutations in the gene encoding the calcium sensing receptor CaSR (CASR) are associated with hypercalciuric hypocalcemia and occasionally with hypomagnesemia [21, 22]. The two large exofacial lobes of the CaSR act as a peritubular Ca2+ and Mg2+ sensor in the TAL and other tissues (reviewed in [23]). Gain-of-function mutations, analogous to higher Ca2+ concentrations, cause the CaSR to suppress salt reabsorption in the TAL and interfere with the claudin-mediated pore selectivity in the tight junctions (reviewed in [23]). Hypercalciuric hypocalcemia with relative hypoparathyroidism is the most important symptom, which, especially when “mistreated” with vitamin D, can lead to nephrocalcinosis in certain cases (reviewed in [24]). Moreover, in patients with more severe gain-of-function of CASR, significant wasting of Mg2+, Na+, potassium (K+) and water can also occur [22]. This has led to the alternative name of Bartter syndrome type V in patients with this severe type of presentation [22].
CLCNKB (Bartter syndrome type III)
Homozygous or compound heterozygous mutations in CLCNKB, which encodes the chloride ion (Cl−) channel ClC-Kb, cause Bartter syndrome type III. ClC-Kb is expressed basolaterally in the TAL and DCT, providing a pathway for (Cl−) to exit the cell. Mutations in the channel therefore interfere with regulation of intracellular chloride levels and the function of NCC (thiazide-sensitive NaCl cotransporter) and NKCC (Na+-K+-Cl− cotransporter) (reviewed in [15, 25] and [26]). Patients with mutations in CLCNKB often present during the first years of life, suffering from a Bartter-like phenotype, including hypercalciuria and loss of Na+, K+ and water. When they grow older, however, a shift to a more Gitelman-like phenotype can be observed, with marked hypocalciuria and hypomagnesemia in addition to the loss of Na+, K+ and water [25, 27]. The CLCNKB gene is thus listed under the hypercalciuric as well as under the Gitelman-like hypomagnesemias.
Gitelman-like hypomagnesemias
The genes from the second group of hypomagnesemias listed in Table 4, the Gitelman-like hypomagnesemias, all encode proteins that are involved in the transport of Na+, K+ and/or Cl− in the DCT. Adequate transcellular Mg2+ reabsorption in the DCT is dependent on the apical membrane potential, which is lumen positive when compared to the cytoplasm (reviewed in [28]). Therefore, Mg2+ reabsorption also depends on the intactness of other ion transport processes in the DCT. Alternatively, it has been proposed that atrophy of the DCT segment is responsible for all symptoms [29], although thiazide diuretics do not cause atrophy of the DCT [30]. Regardless of the mechanism underlying the DCT dysfunction, diseases from this group of hypomagnesemias all lead to increased calcium reabsorption along different nephron segments, proximal as well as distal (reviewed in [29]). This obviously results in hypocalciuria. In addition, the DCT dysfunction leads to fluid loss and a tendency to lower blood pressures despite an activated renin–angiotensin–aldosterone system (due to compensation mechanisms) (reviewed in [31]). Lastly, the relatively increased levels of aldosterone force the collecting duct to secrete potassium in exchange for sodium, leading to hypokalemia, which, in turn, leads to alkalosis. In addition, the combination of hypomagnesemia with hypokalemia observed in this group can give rise to a prolonged QT interval and cardiac arrhythmias [32–34], justifying avoidance of drugs prolonging the QT interval [32].
SLC12A3 (Gitelman syndrome)
With an estimated prevalence of 1:40 000 [31], Gitelman syndrome is the most frequent genetic cause of hypomagnesemia. It is caused by recessive mutations in SLC12A3, the gene encoding the Na+-Cl−-cotransporter (NCC) that is expressed on the apical membrane of the DCT [35] (reviewed in [31]). Symptoms are generally absent in the first years of life and only towards the end of the first decade do patients start to report symptoms [36]. Affected individuals can suffer from a range of hypomagnesemia-related symptoms, such as cramps, paresthesias or even cardiac arrest [32, 37]. In addition, they can suffer from the salt and water wasting that is apparent in most Gitelman-like hypomagnesemias, resulting in polyuria, salt craving and thirst [37]. The mechanism by which Gitelman syndrome causes hypomagnesemia is still not fully understood. One explanation can be found in the atrophy of the DCT that has been observed in a mouse model of Gitelman syndrome [29]. Additionally, a reduced apical membrane potential and a reduction in TRPM6 activation or mobilization could play a role. It may be speculated that the reduced apical membrane potential is caused by increased NHE2 (Na+/H+ exchanger)-mediated Na+ reabsorption in the DCT as a means to compensate for the NCC dysfunction.
BSND (Bartter syndrome type IV)
Barttin, encoded by the BSND gene, is expressed in the ascending thin limb, TAL, DCT and inner ear as a subunit of the ClC-Kb and ClC-Ka Cl− channels (reviewed in [15, 26]). Consequently, patients with recessive mutations in BSND or digenic mutations affecting both ClC-Kb and ClC-Ka will suffer from profound salt wasting in these three tubule segments as well as sensorineural deafness. In addition to complete deafness, Bartter syndrome type IV can be distinguished from the other types of Bartter syndrome by the initial lack of hypercalciuria. In addition, a significant number of patients will develop CKD (reviewed in [15, 26]). Most important for treatment is the adequate supplementation of fluids and sodium directly after birth, much alike all other types of salt-losing tubulopathies with antenatal presentation [38]. Long-lasting treatment with indomethacin can be considered to prevent failure to thrive and decrease renal salt and water wasting, but it should be realized that this treatment will be less effective than in patients with Bartter types I and II [39] and that this drug can cause renal side effects.
KCNJ10 (epilepsy, ataxia, sensorineural deafness and tubulopathy syndrome)
This syndrome, characterized by epilepsy, ataxia, sensorineural deafness and tubulopathy (referred to as EAST syndrome), is a rare recessive genetic disease affecting the K+ channel Kir4.1 encoded by KCNJ10 [40]. This protein is expressed in several tissues, including the central nervous system, inner ear and basolateral side of DCT cells and possibly also TAL cells (reviewed in [41, 42]). In the kidney it forms a basolateral K+ channel that conducts outward K+ currents, thus recycling the K+ imported by the Na+-K+-ATPase. To aid in diagnosis, magnetic resonance imaging of the brain might show subtle changes, especially in the dentate nuclei of the cerebellum [41]. Patients show often pronounced ataxia and obligate sensorineural deafness. Lastly, although intellectual abilities seem to lag behind [43], it is difficult to assess the intelligence of these patients due to deafness and ataxia impairing both verbal and written communication [44].
FXYD2 (isolated dominant hypomagnesemia)
Only three families, all Belgian or Dutch, putatively descendants from a common founder [45], have been reported to carry the hypomagnesemia-causing FXYD2 mutation. The FXYD2 gene encodes the γ-subunit of the Na+-K+-ATPase [46]. The specific dominant mutation causes misrouting of this γ-subunit, thereby preventing the splice variant FXYD2b from assembling with the α- and β-subunit of the Na+-K+-ATPase [46]. Virtually all patients suffer from muscle cramps; additionally, several other hypomagnesemia-related symptoms can occur [45].
HNF1B (autosomal dominant tubulointerstitial kidney disease)
Heterozygous mutations in the HNF1B gene are associated with a multi-system disorder and considered to be the most common genetic cause of congenital anomalies of the kidney and urinary tract (CAKUT) (reviewed in [47, 48]). The HNF1B gene is situated in a region susceptible for genomic rearrangements, resulting in a high frequency of large deletions and de novo gene defects [47]. Mutations in this gene can be detrimental to normal development and function of the kidney, pancreas and genital tract [47], thus giving rise to a highly variable set of symptoms originating from these organs, including CAKUT and maturity onset diabetes in the young (MODY).
Hypomagnesemia is also one of these symptoms, occurring in up to 50 % of affected children [49] and sometimes being the first clinical manifestation [50]. Hypomagnesemia becomes more pronounced with increasing age of the patient and can be missed in affected young children. As a result, HNF1B mutations are the most common cause of genetic hypomagnesemia for pediatric nephrologists. The current view is that HNF1B dysfunction leads to inadequate transcription of the FXYD2 splice-isoform FXYD2a, which encodes the γa-subunit of the Na+-K+-ATPase [51]. The diagnosis is complicated by the large variability in presentation and the lack of a clear genotype–phenotype relationship [47]. If a patient is considered to suffer from HNF1B-ADTKD, the diagnosis should only be rejected if point-mutations as well as deletions and insertions in HNF1B have been properly excluded.
PCBD1 (renal cyst and diabetes-like)
Recessive mutations in PCBD1 had long been identified to be responsible for transient neonatal hyperphenylalaninemia and primapterinuria (HPABH4D) [52], but no other symptoms of this genetic defect had been reported until recently. Re-evaluation of earlier identified patients with this syndrome revealed that hypomagnesemia and MODY5-like diabetes are later manifestations of PCBD1 mutations [53, 54]. This finding also has implications for the importance of looking for alternative genetic diagnoses (including PCBD1 mutations) if screening for mutations of PAH (phenylalanine hydroxylase) is negative after a positive result for the Guthrie test.
The neonatal phenotype can be explained by the failure of PCBD1 to fulfil its role as an enzyme in the metabolism of aromatic acids. The complications appearing later in life are explained by the additional role of PCBD1 as a dimerization factor for HNF1A and HNF1B, regulating their transcriptional activity (reviewed in [53]). This in turn would influence FXYD2 transcription, causing hypomagnesemia. It should be noted, however, that the CAKUT phenotype often observed in HNF1B patients is not seen in PCBD1-disease due to a different expression pattern of these two proteins. Lastly, hypokalemia and hypocalciuria have not been reported in these patients. Still, it is placed here with the Gitelman-like hypomagnesemias based on the pathophysiological mechanism.
Mitochondrial hypomagnesemias
The mitochondrial hypomagnesemias, of which the pathophysiological mechanism is still unexplained, have a highly variable presentation. Their phenotype depends both on the nature of the mutation and the fraction of mitochondria affected in each tissue (reviewed in [55]). Some of the mitochondrial hypomagnesemias are associated with Gitelman-like electrolyte abnormalities [56], while others seem to affect TAL function [57–59]. Physiologically, both have a high energy requirement, although the DCT seems to be a better candidate since it has the most mitochondria [60]. Still needing to be clarified is which function of the mitochondrion is most important in Mg2+ homeostasis: is it the ATP that is necessary to drive the Na+-K+-ATPase and to activate CNNM2 [61]? Or could its role in Ca2+-signaling be key here?
MT-TI, SARS2, POLG1 and mitochondrial deletions/duplications
Impaired mitochondrial function might be associated with hypomagnesemia much more frequently than is currently realized. At least three distinct mitochondrial syndromes are accompanied by hypomagnesemia: (1) deletions in the mitochondrial genome as seen in Kearns–Sayre syndrome [62] (2) recessive mutations in the human gene SARS2 [57] and (3) mutations in the mitochondrial tRNAIle gene MT-TI causing a syndrome with hypertension, hypercholesterolemia and hypomagnesemia [56]. An additional two cases of patients with hypomagnesemia and other mitochondrial diseases have also been identified (mutations in POLG1 and the mitochondrial Pearson’s syndrome [63, 64]). Since checking serum Mg2+ concentrations is not regular clinical practice, it would be interesting to investigate the genuine frequency of hypomagnesemia in patients with mitochondrial disease.
Other hypomagnesemias
The remaining disorders associated with hypomagnesemia are classified in this review as “other hypomagnesemias’” due to their heterogenic nature. One of these is caused by mutations in TRPM6, which encodes the DCT-specific apical Mg2+ transporter TRPM6, resulting in an isolated hypomagnesemia (i.e. no other symptoms except secondary to the hypomagnesemia). Others, such as mutations in EGF and EGFR affect the activity and expression of this channel [6]. FAM111A is the only gene in this group of which the pathophysiological mechanism underlying the hypomagnesemia has received no attention at all.
TRPM6 (hypomagnesemia with secondary hypocalcemia)
Mutations in the gene for the DCT- and colon-specific apical Mg2+ channel, TRPM6, cause the most profound genetic hypomagnesemia [65, 66]. A measured serum Mg2+ concentration as low as 0.2 mM or even lower, as low as immeasurable levels, is not uncommon in these patients [14]. Consequently, patients often present with seizures within the first months of life [14]. A defect in the TRPM6 channel impairs epithelial Mg2+ resorption in the colon and DCT, thereby inhibiting uptake and stimulating wasting of Mg2+, causing significant hypomagnesemia [67]. The secondary hypocalcemia often observed is probably caused by inhibition of the parathyroid gland by the hypomagnesemia, resulting in low levels of parathyroid hormone and eventually leading to hypocalcemia [68].
CNNM2 (hypomagnesemia with seizures and mental retardation)
CNNM2 is most highly expressed in the TAL, DCT and brain [69, 70], which explains the combination of hypomagnesemia with additional neurological symptoms (anatomical abnormalities, seizures and intellectual disability) seen in patients with dominant or sometimes recessive CNNM2 mutations [71]. Although CNNM2 was first thought to be a basolateral Mg2+ transporter itself, it is now thought to fulfill the role of intracellular Mg2+ sensor by undergoing a conformational change upon binding of Mg-ATP [61]. How this conformational change eventually leads to the observed decrease in Mg2+ transport, however, remains unclear, as is the precise basis of the neurological symptoms.
EGF and EGFR (isolated recessive hypomagnesemia)
One mutation in the epidermal growth factor gene (EGF) has been associated with a recessive form of hypomagnesemia with an additional neurological phenotype (including intellectual disability) [72]. The only mutation identified to date interferes with proper trafficking of pro-EGF, which is expressed in TAL and DCT cells, resulting in a decreased peritubular concentration of autocrine EGF [72]. Subsequently, the signaling cascade from the EGF receptor (EGFR) to the Akt-mediated activation of Rac1 is turned off, resulting in a decrease of endomembrane trafficking of TRPM6 to the apical surface and decreased Mg2+ transport [6]. The intellectual disability on the other hand remains unexplained.
Since EGF mutations and cetuximab (an EGFR inhibitor) both cause hypomagnesemia [73], it is not surprising that mutations in EGFR have also been found to cause hypomagnesemia. Only one patient has been diagnosed to date with loss-of-function of the EGFR. This mutation resulted in severe symptoms comparable to those observed with full EGFR blockade, including skin rash, inflammation of the lungs and bowel and hypomagnesemia [74].
KCNA1 (autosomal dominant hypomagnesemia)
Intriguingly, only one mutation (identified with linkage in a large pedigree) in KCNA1 has been associated with hypomagnesemia [75], while all other KCNA1 mutations known to date cause episodic ataxia type 1 without hypomagnesemia (reviewed in [76]). The KCNA1 gene encodes the voltage-gated K+ channel Kv1.1 [75], which is abundantly expressed in certain neurons as well as on the apical membrane of cells in the DCT ([75] and reviewed in [77]). In neurons, the mutations in KCNA1 impair normal repolarization of the membrane potential, resulting in stress-triggered episodes of ataxia and myokymia [77]. Following the same line of reasoning, one might speculate that genetic defects in Kv1.1 might cause hypomagnesemia by depolarizing the apical DCT membrane [78]. However, this theory does not explain why other KCNA1 mutations have not been reported to cause hypomagnesemia. Also, how does a voltage-gated K+-channel like KCNA1, which is closed and thus non-functional at normal apical membrane potential, cause hypomagnesemia in the first place?
FAM111A
Mutations in FAM111A are associated with two distinct dominant diseases: a perinatally lethal disorder called gracile bone dysplasia (GLCEB) and Kenny–Caffey syndrome type 2 (KCS2) [79]. To date, only KCS2 has been associated with hypomagnesemia [80–85]. Other symptoms of KCS2 include severe proportionate short stature, radiological bone anomalies, eye abnormalities and hypocalcemia owing to hypoparathyroidism (reviewed in [82]). FAM111A has been reported to be a host-range restriction factor [86] and shown to be an important component of the cell’s replication machinery near DNA forks, consistent with its proposed role in cancer and cell survival [87–89]. However, it is unclear how this function would link to hypomagnesemia. Alternatively, it might be speculated that its function is related to the function of TBCE [90], the causative gene for the clinically closely related Kenny–Caffey syndrome type 1 [91].
Treatment
Oral or intravenous Mg2+ supplementation is the only treatment available for hypomagnesemia of genetic origin. In the acute situation of a (severely) symptomatically hypomagnesemic patient, intravenous Mg2+ supplementation can be very important. For adults, a dose of 8–12 g of MgSO4 (containing 0.8–1.2 g of Mg2+) in the first 24 h is recommended, followed by 4–6 g/day for 3 or 4 days [92]. For the non-acute setting, oral Mg2+ supplementation of 3 × 120 mg/day is more convenient while being sufficiently effective [93]. Parenteral and oral Mg2+ supplementation is therefore recommended in many of the hypomagnesemias, including Gitelman syndrome, Bartter syndrome, EAST syndrome and those caused by TRPM6 mutations [14, 31, 41]. The resulting rise in serum Mg2+ concentration often alleviates symptoms, such as seizures and secondary hypocalcemia, even though normal Mg2+ values are rarely reached [14, 75]. Further correction of the hypomagnesemia is generally impeded by the gastro-intestinal side effects frequently associated with oral Mg2+ supplementation. Paradoxically, higher doses of oral Mg2+ might even be detrimental because of the resulting diarrhea [31]. Also, attention should be given to the type of oral Mg2+ supplementation given since some preparations have a better bioavailability than others. For this reason, we recommend magnesium chloride or magnesium glycerophospate rather than magnesium oxide or magnesium sulfate for oral Mg2+ supplementation [31].
Since hypokalemia is commonly associated with hypomagnesemia, especially in the Gitelman-like hypomagnesemias, one might consider treatment with the epithelial Na+ channel ENaC or aldosterone blockers. However, care should be taken, especially in the young child, since this treatment will interfere with the distal compensatory salt reabsorption of the kidney [27]. Lastly, seizures are a known result of several genetic hypomagnesemias, as a primary effect of the genetic defect or secondary to the Mg2+ deficiency. In both cases, treatment with anticonvulsants such as valproate or phenobarbital might be beneficial [41].
Summary and future perspectives
In summary, the identification of the different genetic hypomagnesemias has aided our understanding of the important role of the kidney—the TAL and DCT in particular—in maintaining Mg2+ homeostasis. Based on the combination of increased fractional renal Mg2+ excretion with familial occurrence and a variety of additional symptoms, it is possible to recognize cases with a genetic cause and differentiate between them to a certain extent (see Fig. 2). Subsequent characterization of the affected genes and proteins has improved our understanding of which molecular pathways are involved in Mg2+ homeostasis. Hypercalciuric hypomagnesemia is thereby often the presentation of a defect in the TAL, while Gitelman-like and “other” hypomagnesemias are generally localized to the DCT. Treatment still depends on Mg2+ supplementation, but an increased understanding of the pathophysiological mechanisms might make discovery of new treatment opportunities possible in the future.
Fig. 2.
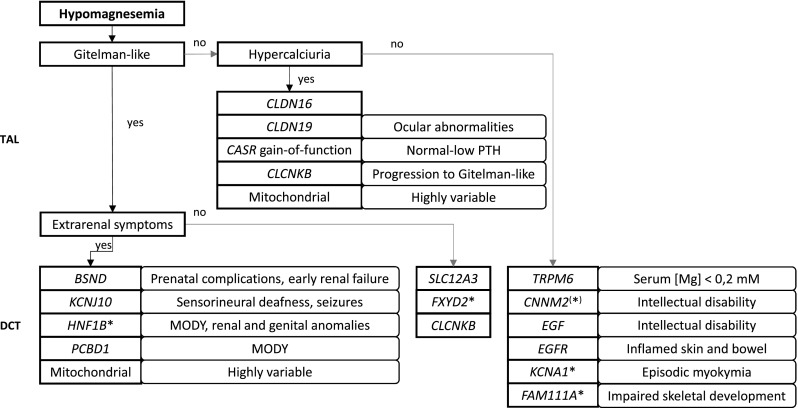
Diagnostic flowchart for a suspected genetic cause of hypomagnesemia. This diagnostic flowchart is primarily provided to give an impression of the clinical characteristics of all known genetic causes of hypomagnesemia. Genetic testing can confirm or reject a diagnosis. Asterisks indicate dominantly inherited disorders
Additional fundamental research might further elucidate the mechanisms of transport and regulation exploited by the DCT and TAL to maintain Mg2+ homeostasis. Not only does the basolateral Mg2+ transporter need to be identified, but the precise pathophysiological mechanisms by which known mutations cause hypomagnesemia also need further clarification. This even holds true for several well-known and thoroughly described hypomagnesemias, such as Gitelman syndrome. After all, some observations cannot be explained by current understanding. The proposed atrophy of the DCT, for example, has been observed in mice with Gitelman syndrome [29], but it is not present in mice on chronic thiazide treatment-induced hypomagnesemia [30]. The proposed decisive role for the membrane potential of the apical membrane is also not completely satisfying, especially since its role is proven for only some of the genetic hypomagnesemias [78].
On the other hand, some pathways linked to hypomagnesemia might be underestimated. The EGF/EGFR pathway and the insulin and estrogen pathways seem to be of significant importance in terms of increasing Mg2+ transport but there is currently little evidence linking them to hypomagnesemic pathology (reviewed in [1]). Additionally, the role of the Na+-K+-ATPase might be more important than currently appreciated. The Na+-K+-ATPase is known to occupy a central position in many of the Gitelman-like hypomagnesemia, while it has the potential of being involved in EGFR activation by means of the Na+-K+-ATPase-Src-kinase complex [94]. The pathophysiology underlying mitochondria-associated diseases might also be of great interest since at least one of the other hypomagnesemias has been reported to be characterized by a vastly diminished size and number of mitochondria in the DCT cells [95]. However, it is not yet clear whether this is a cause, epiphenomenon or result of the hypomagnesemia. Lastly, the recent breakthroughs in identifying new pathways involved in Na+ regulation by the DCT [96] could also shed new light on the transport of other ions in this segment. Identification of these other pathways will hopefully provide decisive evidence on the mechanisms of Mg2+ reabsorption in the kidney and might open new roads to causal treatment of hypomagnesemia.
Acknowledgments
This work was supported by The European Union, FP7 (grant agreement 2012-305608, “European Consortium for High-Throughput Research in Rare Kidney Diseases (EURenOmics)).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflicts of interest.
References
- 1.de Baaij JH, Hoenderop JG, Bindels RJ. Magnesium in man: implications for health and disease. Physiol Rev. 2015;95:1–46. doi: 10.1152/physrev.00012.2014. [DOI] [PubMed] [Google Scholar]
- 2.Schlegel RN, Cuffe JS, Moritz KM, Paravicini TM. Maternal hypomagnesemia causes placental abnormalities and fetal and postnatal mortality. Placenta. 2015;36:750–758. doi: 10.1016/j.placenta.2015.03.011. [DOI] [PubMed] [Google Scholar]
- 3.Hall DG. Serum magnesium in pregnancy. Obstet Gynecol. 1957;9:158–162. doi: 10.1097/00006250-195706000-00019. [DOI] [PubMed] [Google Scholar]
- 4.Hou J, Goodenough DA. Claudin-16 and claudin-19 function in the thick ascending limb. Curr Opin Nephrol Hypertens. 2010;19:483–488. doi: 10.1097/MNH.0b013e32833b7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brunette MG, Vigneault N, Carriere S. Micropuncture study of magnesium transport along the nephron in the young rat. Am J Physiol. 1974;227:891–896. doi: 10.1152/ajplegacy.1974.227.4.891. [DOI] [PubMed] [Google Scholar]
- 6.Thebault S, Alexander RT, Tiel Groenestege WM, Hoenderop JG, Bindels RJ. EGF increases TRPM6 activity and surface expression. J Am Soc Nephrol. 2009;20:78–85. doi: 10.1681/ASN.2008030327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dimke H, Hoenderop JG, Bindels RJ. Hereditary tubular transport disorders: implications for renal handling of Ca2+ and Mg2+ Clin Sci (Lond) 2010;118:1–18. doi: 10.1042/CS20090086. [DOI] [PubMed] [Google Scholar]
- 8.Syedmoradi L, Ghasemi A, Zahediasl S, Azizi F. Prevalence of hypo- and hypermagnesemia in an Iranian urban population. Ann Hum Biol. 2011;38:150–155. doi: 10.3109/03014460.2010.500472. [DOI] [PubMed] [Google Scholar]
- 9.Whang R, Ryder KW. Frequency of hypomagnesemia and hypermagnesemia. Requested vs routine. JAMA. 1990;263:3063–3064. doi: 10.1001/jama.1990.03440220087036. [DOI] [PubMed] [Google Scholar]
- 10.Wong ET, Rude RK, Singer FR, Shaw ST., Jr A high prevalence of hypomagnesemia and hypermagnesemia in hospitalized patients. Am J Clin Pathol. 1983;79:348–352. doi: 10.1093/ajcp/79.3.348. [DOI] [PubMed] [Google Scholar]
- 11.Tong GM, Rude RK. Magnesium deficiency in critical illness. J Intensive Care Med. 2005;20:3–17. doi: 10.1177/0885066604271539. [DOI] [PubMed] [Google Scholar]
- 12.Worthington V. Nutritional quality of organic versus conventional fruits, vegetables, and grains. J Altern Complement Med. 2001;7:161–173. doi: 10.1089/107555301750164244. [DOI] [PubMed] [Google Scholar]
- 13.Elisaf M, Panteli K, Theodorou J, Siamopoulos KC. Fractional excretion of magnesium in normal subjects and in patients with hypomagnesemia. Magnes Res. 1997;10:315–320. [PubMed] [Google Scholar]
- 14.Schlingmann KP, Sassen MC, Weber S, Pechmann U, Kusch K, Pelken L, Lotan D, Syrrou M, Prebble JJ, Cole DE, Metzger DL, Rahman S, Tajima T, Shu SG, Waldegger S, Seyberth HW, Konrad M. Novel TRPM6 mutations in 21 families with primary hypomagnesemia and secondary hypocalcemia. J Am Soc Nephrol. 2005;16:3061–3069. doi: 10.1681/ASN.2004110989. [DOI] [PubMed] [Google Scholar]
- 15.Jeck N, Schlingmann KP, Reinalter SC, Komhoff M, Peters M, Waldegger S, Seyberth HW. Salt handling in the distal nephron: lessons learned from inherited human disorders. Am J Physiol Regul Integr Comp Physiol. 2005;288:R782–795. doi: 10.1152/ajpregu.00600.2004. [DOI] [PubMed] [Google Scholar]
- 16.Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, Casari G, Bettinelli A, Colussi G, Rodriguez-Soriano J, McCredie D, Milford D, Sanjad S, Lifton RP. Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science. 1999;285:103–106. doi: 10.1126/science.285.5424.103. [DOI] [PubMed] [Google Scholar]
- 17.Konrad M, Schaller A, Seelow D, Pandey AV, Waldegger S, Lesslauer A, Vitzthum H, Suzuki Y, Luk JM, Becker C, Schlingmann KP, Schmid M, Rodriguez-Soriano J, Ariceta G, Cano F, Enriquez R, Juppner H, Bakkaloglu SA, Hediger MA, Gallati S, Neuhauss SC, Nurnberg P, Weber S. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet. 2006;79:949–957. doi: 10.1086/508617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu AS. Claudins and the kidney. J Am Soc Nephrol. 2015;26:11–19. doi: 10.1681/ASN.2014030284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weber S, Schneider L, Peters M, Misselwitz J, Ronnefarth G, Boswald M, Bonzel KE, Seeman T, Sulakova T, Kuwertz-Broking E, Gregoric A, Palcoux JB, Tasic V, Manz F, Scharer K, Seyberth HW, Konrad M. Novel paracellin-1 mutations in 25 families with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J Am Soc Nephrol. 2001;12:1872–1881. doi: 10.1681/ASN.V1291872. [DOI] [PubMed] [Google Scholar]
- 20.Konrad M, Hou J, Weber S, Dotsch J, Kari JA, Seeman T, Kuwertz-Broking E, Peco-Antic A, Tasic V, Dittrich K, Alshaya HO, von Vigier RO, Gallati S, Goodenough DA, Schaller A. CLDN16 genotype predicts renal decline in familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J Am Soc Nephrol. 2008;19:171–181. doi: 10.1681/ASN.2007060709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pearce SH, Williamson C, Kifor O, Bai M, Coulthard MG, Davies M, Lewis-Barned N, McCredie D, Powell H, Kendall-Taylor P, Brown EM, Thakker RV. A familial syndrome of hypocalcemia with hypercalciuria due to mutations in the calcium-sensing receptor. N Engl J Med. 1996;335:1115–1122. doi: 10.1056/NEJM199610103351505. [DOI] [PubMed] [Google Scholar]
- 22.Watanabe S, Fukumoto S, Chang H, Takeuchi Y, Hasegawa Y, Okazaki R, Chikatsu N, Fujita T. Association between activating mutations of calcium-sensing receptor and Bartter’s syndrome. Lancet. 2002;360:692–694. doi: 10.1016/S0140-6736(02)09842-2. [DOI] [PubMed] [Google Scholar]
- 23.Alfadda TI, Saleh AM, Houillier P, Geibel JP. Calcium-sensing receptor 20 years later. Am J Physiol Cell Physiol. 2014;307:C221–231. doi: 10.1152/ajpcell.00139.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tyler Miller R. Control of renal calcium, phosphate, electrolyte, and water excretion by the calcium-sensing receptor. Best Pract Res Clin Endocrinol Metab. 2013;27:345–358. doi: 10.1016/j.beem.2013.04.009. [DOI] [PubMed] [Google Scholar]
- 25.Jeck N, Konrad M, Peters M, Weber S, Bonzel KE, Seyberth HW. Mutations in the chloride channel gene, CLCNKB, leading to a mixed Bartter-Gitelman phenotype. Pediatr Res. 2000;48:754–758. doi: 10.1203/00006450-200012000-00009. [DOI] [PubMed] [Google Scholar]
- 26.Hebert SC. Bartter syndrome. Curr Opin Nephrol Hypertens. 2003;12:527–532. doi: 10.1097/00041552-200309000-00008. [DOI] [PubMed] [Google Scholar]
- 27.Kleta R, Bockenhauer D. Bartter syndromes and other salt-losing tubulopathies. Nephron Physiol. 2006;104:p73–80. doi: 10.1159/000094001. [DOI] [PubMed] [Google Scholar]
- 28.McCormick JA, Ellison DH. Distal convoluted tubule. Compr Physiol. 2015;5:45–98. doi: 10.1002/cphy.c140002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loffing J, Vallon V, Loffing-Cueni D, Aregger F, Richter K, Pietri L, Bloch-Faure M, Hoenderop JG, Shull GE, Meneton P, Kaissling B. Altered renal distal tubule structure and renal Na(+) and Ca(2+) handling in a mouse model for Gitelman’s syndrome. J Am Soc Nephrol. 2004;15:2276–2288. doi: 10.1097/01.ASN.0000138234.18569.63. [DOI] [PubMed] [Google Scholar]
- 30.Nijenhuis T, Vallon V, van der Kemp AW, Loffing J, Hoenderop JG, Bindels RJ. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest. 2005;115:1651–1658. doi: 10.1172/JCI24134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knoers NV, Levtchenko EN. Gitelman syndrome. Orphanet J Rare Dis. 2008;3:22. doi: 10.1186/1750-1172-3-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Foglia PE, Bettinelli A, Tosetto C, Cortesi C, Crosazzo L, Edefonti A, Bianchetti MG. Cardiac work up in primary renal hypokalaemia-hypomagnesaemia (Gitelman syndrome) Nephrol Dial Transplant. 2004;19:1398–1402. doi: 10.1093/ndt/gfh204. [DOI] [PubMed] [Google Scholar]
- 33.Malafronte C, Borsa N, Tedeschi S, Syren ML, Stucchi S, Bianchetti MG, Achilli F, Bettinelli A. Cardiac arrhythmias due to severe hypokalemia in a patient with classic Bartter disease. Pediatr Nephrol. 2004;19:1413–1415. doi: 10.1007/s00467-004-1611-0. [DOI] [PubMed] [Google Scholar]
- 34.Topol EJ, Lerman BB. Hypomagnesemic torsades de pointes. Am J Cardiol. 1983;52:1367–1368. doi: 10.1016/0002-9149(83)90611-2. [DOI] [PubMed] [Google Scholar]
- 35.Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, Vaara I, Iwata F, Cushner HM, Koolen M, Gainza FJ, Gitleman HJ, Lifton RP. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet. 1996;12:24–30. doi: 10.1038/ng0196-24. [DOI] [PubMed] [Google Scholar]
- 36.Scholl UI, Dave HB, Lu M, Farhi A, Nelson-Williams C, Listman JA, Lifton RP. SeSAME/EAST syndrome—phenotypic variability and delayed activity of the distal convoluted tubule. Pediatr Nephrol. 2012;27:2081–2090. doi: 10.1007/s00467-012-2219-4. [DOI] [PubMed] [Google Scholar]
- 37.Cruz DN, Shaer AJ, Bia MJ, Lifton RP, Simon DB. Gitelman’s syndrome revisited: an evaluation of symptoms and health-related quality of life. Kidney Int. 2001;59:710–717. doi: 10.1046/j.1523-1755.2001.059002710.x. [DOI] [PubMed] [Google Scholar]
- 38.Azzi A, Chehade H, Deschenes G. Neonates with Bartter syndrome have enormous fluid and sodium requirements. Acta Paediatr. 2015;104:e294–299. doi: 10.1111/apa.12981. [DOI] [PubMed] [Google Scholar]
- 39.Seyberth HW, Schlingmann KP. Bartter- and Gitelman-like syndromes: salt-losing tubulopathies with loop or DCT defects. Pediatr Nephrol. 2011;26:1789–1802. doi: 10.1007/s00467-011-1871-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, Tobin J, Lieberer E, Sterner C, Landoure G, Arora R, Sirimanna T, Thompson D, Cross JH, van’t Hoff W, Al Masri O, Tullus K, Yeung S, Anikster Y, Klootwijk E, Hubank M, Dillon MJ, Heitzmann D, Arcos-Burgos M, Knepper MA, Dobbie A, Gahl WA, Warth R, Sheridan E, Kleta R. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med. 2009;360:1960–1970. doi: 10.1056/NEJMoa0810276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cross JH, Arora R, Heckemann RA, Gunny R, Chong K, Carr L, Baldeweg T, Differ AM, Lench N, Varadkar S, Sirimanna T, Wassmer E, Hulton SA, Ognjanovic M, Ramesh V, Feather S, Kleta R, Hammers A, Bockenhauer D. Neurological features of epilepsy, ataxia, sensorineural deafness, tubulopathy syndrome. Dev Med Child Neurol. 2013;55:846–856. doi: 10.1111/dmcn.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang C, Wang L, Su XT, Lin DH, Wang WH. KCNJ10 (Kir4.1) is expressed in the basolateral membrane of the cortical thick ascending limb. Am J Physiol Renal Physiol. 2015;308(11):F1288–296. doi: 10.1152/ajprenal.00687.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scholl UI, Choi M, Liu T, Ramaekers VT, Hausler MG, Grimmer J, Tobe SW, Farhi A, Nelson-Williams C, Lifton RP. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci USA. 2009;106:5842–5847. doi: 10.1073/pnas.0901749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bandulik S, Schmidt K, Bockenhauer D, Zdebik AA, Humberg E, Kleta R, Warth R, Reichold M. The salt-wasting phenotype of EAST syndrome, a disease with multifaceted symptoms linked to the KCNJ10 K+ channel. Pflugers Arch. 2011;461:423–435. doi: 10.1007/s00424-010-0915-0. [DOI] [PubMed] [Google Scholar]
- 45.de Baaij JH, Dorresteijn EM, Hennekam EA, Kamsteeg EJ, Meijer R, Dahan K, Muller M, van den Dorpel MA, Bindels RJ, Hoenderop JG, Devuyst O, Knoers NV. Recurrent FXYD2 p.Gly41Arg mutation in patients with isolated dominant hypomagnesaemia. Nephrol Dial Transplant. 2015;30:952–957. doi: 10.1093/ndt/gfv014. [DOI] [PubMed] [Google Scholar]
- 46.Meij IC, Koenderink JB, van Bokhoven H, Assink KF, Groenestege WT, de Pont JJ, Bindels RJ, Monnens LA, van den Heuvel LP, Knoers NV. Dominant isolated renal magnesium loss is caused by misrouting of the Na(+), K(+)-ATPase gamma-subunit. Nat Genet. 2000;26:265–266. doi: 10.1038/81543. [DOI] [PubMed] [Google Scholar]
- 47.Clissold RL, Hamilton AJ, Hattersley AT, Ellard S, Bingham C. HNF1B-associated renal and extra-renal disease-an expanding clinical spectrum. Nat Rev Nephrol. 2015;11:102–112. doi: 10.1038/nrneph.2014.232. [DOI] [PubMed] [Google Scholar]
- 48.Bockenhauer D, Jaureguiberry G. HNF1B-associated clinical phenotypes: the kidney and beyond. Pediatr Nephrol. 2015;31:707–714. doi: 10.1007/s00467-015-3142-2. [DOI] [PubMed] [Google Scholar]
- 49.Adalat S, Woolf AS, Johnstone KA, Wirsing A, Harries LW, Long DA, Hennekam RC, Ledermann SE, Rees L, van’t Hoff W, Marks SD, Trompeter RS, Tullus K, Winyard PJ, Cansick J, Mushtaq I, Dhillon HK, Bingham C, Edghill EL, Shroff R, Stanescu H, Ryffel GU, Ellard S, Bockenhauer D. HNF1B mutations associate with hypomagnesemia and renal magnesium wasting. J Am Soc Nephrol. 2009;20:1123–1131. doi: 10.1681/ASN.2008060633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van der Made CI, Hoorn EJ, de la Faille R, Karaaslan H, Knoers NV, Hoenderop JG, Vargas Poussou R, de Baaij JH. Hypomagnesemia as first clinical manifestation of ADTKD-HNF1B: a case series and literature review. Am J Nephrol. 2015;42:85–90. doi: 10.1159/000439286. [DOI] [PubMed] [Google Scholar]
- 51.Ferre S, Veenstra GJ, Bouwmeester R, Hoenderop JG, Bindels RJ. HNF-1B specifically regulates the transcription of the gammaa-subunit of the Na+/K + -ATPase. Biochem Biophys Res Commun. 2011;404:284–290. doi: 10.1016/j.bbrc.2010.11.108. [DOI] [PubMed] [Google Scholar]
- 52.Thony B, Neuheiser F, Kierat L, Blaskovics M, Arn PH, Ferreira P, Rebrin I, Ayling J, Blau N. Hyperphenylalaninemia with high levels of 7-biopterin is associated with mutations in the PCBD gene encoding the bifunctional protein pterin-4a-carbinolamine dehydratase and transcriptional coactivator (DCoH) Am J Hum Genet. 1998;62:1302–1311. doi: 10.1086/301887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ferre S, de Baaij JH, Ferreira P, Germann R, de Klerk JB, Lavrijsen M, van Zeeland F, Venselaar H, Kluijtmans LA, Hoenderop JG, Bindels RJ. Mutations in PCBD1 cause hypomagnesemia and renal magnesium wasting. J Am Soc Nephrol. 2014;25:574–586. doi: 10.1681/ASN.2013040337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Simaite D, Kofent J, Gong M, Ruschendorf F, Jia S, Arn P, Bentler K, Ellaway C, Kuhnen P, Hoffmann GF, Blau N, Spagnoli FM, Hubner N, Raile K. Recessive mutations in PCBD1 cause a new type of early-onset diabetes. Diabetes. 2014;63:3557–3564. doi: 10.2337/db13-1784. [DOI] [PubMed] [Google Scholar]
- 55.Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–1252. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 56.Wilson FH, Hariri A, Farhi A, Zhao H, Petersen KF, Toka HR, Nelson-Williams C, Raja KM, Kashgarian M, Shulman GI, Scheinman SJ, Lifton RP. A cluster of metabolic defects caused by mutation in a mitochondrial tRNA. Science. 2004;306:1190–1194. doi: 10.1126/science.1102521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Belostotsky R, Ben-Shalom E, Rinat C, Becker-Cohen R, Feinstein S, Zeligson S, Segel R, Elpeleg O, Nassar S, Frishberg Y. Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am J Hum Genet. 2011;88:193–200. doi: 10.1016/j.ajhg.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Emma F, Pizzini C, Tessa A, Di Giandomenico S, Onetti-Muda A, Santorelli FM, Bertini E, Rizzoni G. “Bartter-like” phenotype in Kearns-Sayre syndrome. Pediatr Nephrol. 2006;21:355–360. doi: 10.1007/s00467-005-2092-5. [DOI] [PubMed] [Google Scholar]
- 59.Goto Y, Itami N, Kajii N, Tochimaru H, Endo M, Horai S. Renal tubular involvement mimicking Bartter syndrome in a patient with Kearns-Sayre syndrome. J Pediatr. 1990;116:904–910. doi: 10.1016/S0022-3476(05)80648-1. [DOI] [PubMed] [Google Scholar]
- 60.Kriz WK,B. Structural analysis of the rabbit. Kidney. 1979;56(X):126. doi: 10.1007/978-3-642-67147-0. [DOI] [PubMed] [Google Scholar]
- 61.Corral-Rodriguez MA, Stuiver M, Abascal-Palacios G, Diercks T, Oyenarte I, Ereno-Orbea J, de Opakua AI, Blanco FJ, Encinar JA, Spiwok V, Terashima H, Accardi A, Muller D, Martinez-Cruz LA. Nucleotide binding triggers a conformational change of the CBS module of the magnesium transporter CNNM2 from a twisted towards a flat structure. Biochem J. 2014;464:23–34. doi: 10.1042/BJ20140409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harvey JN, Barnett D. Endocrine dysfunction in Kearns-Sayre syndrome. Clin Endocrinol (Oxf) 1992;37:97–103. doi: 10.1111/j.1365-2265.1992.tb02289.x. [DOI] [PubMed] [Google Scholar]
- 63.Giordano C, Powell H, Leopizzi M, De Curtis M, Travaglini C, Sebastiani M, Gallo P, Taylor RW, d’Amati G. Fatal congenital myopathy and gastrointestinal pseudo-obstruction due to POLG1 mutations. Neurology. 2009;72:1103–1105. doi: 10.1212/01.wnl.0000345002.47396.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gilbert RD, Emms M. Pearson’s syndrome presenting with Fanconi syndrome. Ultrastruct Pathol. 1996;20:473–475. doi: 10.3109/01913129609016351. [DOI] [PubMed] [Google Scholar]
- 65.Walder RY, Landau D, Meyer P, Shalev H, Tsolia M, Borochowitz Z, Boettger MB, Beck GE, Englehardt RK, Carmi R, Sheffield VC. Mutation of TRPM6 causes familial hypomagnesemia with secondary hypocalcemia. Nat Genet. 2002;31:171–174. doi: 10.1038/ng901. [DOI] [PubMed] [Google Scholar]
- 66.Schlingmann KP, Weber S, Peters M, Niemann Nejsum L, Vitzthum H, Klingel K, Kratz M, Haddad E, Ristoff E, Dinour D, Syrrou M, Nielsen S, Sassen M, Waldegger S, Seyberth HW, Konrad M. Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family. Nat Genet. 2002;31:166–170. doi: 10.1038/ng889. [DOI] [PubMed] [Google Scholar]
- 67.Voets T, Nilius B, Hoefs S, van der Kemp AW, Droogmans G, Bindels RJ, Hoenderop JG. TRPM6 forms the Mg2+ influx channel involved in intestinal and renal Mg2+ absorption. J Biol Chem. 2004;279:19–25. doi: 10.1074/jbc.M311201200. [DOI] [PubMed] [Google Scholar]
- 68.Anast CS, Mohs JM, Kaplan SL, Burns TW. Evidence for parathyroid failure in magnesium deficiency. Science. 1972;177:606–608. doi: 10.1126/science.177.4049.606. [DOI] [PubMed] [Google Scholar]
- 69.Wang CY, Shi JD, Yang P, Kumar PG, Li QZ, Run QG, Su YC, Scott HS, Kao KJ, She JX. Molecular cloning and characterization of a novel gene family of four ancient conserved domain proteins (ACDP) Gene. 2003;306:37–44. doi: 10.1016/S0378-1119(02)01210-6. [DOI] [PubMed] [Google Scholar]
- 70.Stuiver M, Lainez S, Will C, Terryn S, Gunzel D, Debaix H, Sommer K, Kopplin K, Thumfart J, Kampik NB, Querfeld U, Willnow TE, Nemec V, Wagner CA, Hoenderop JG, Devuyst O, Knoers NV, Bindels RJ, Meij IC, Muller D. CNNM2, encoding a basolateral protein required for renal Mg2+ handling, is mutated in dominant hypomagnesemia. Am J Hum Genet. 2011;88:333–343. doi: 10.1016/j.ajhg.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arjona FJ, de Baaij JH, Schlingmann KP, Lameris AL, van Wijk E, Flik G, Regele S, Korenke GC, Neophytou B, Rust S, Reintjes N, Konrad M, Bindels RJ, Hoenderop JG. CNNM2 mutations cause impaired brain development and seizures in patients with hypomagnesemia. PLoS Genet. 2014;10:e1004267. doi: 10.1371/journal.pgen.1004267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Groenestege WM, Thebault S, van der Wijst J, van den Berg D, Janssen R, Tejpar S, van den Heuvel LP, van Cutsem E, Hoenderop JG, Knoers NV, Bindels RJ. Impaired basolateral sorting of pro-EGF causes isolated recessive renal hypomagnesemia. J Clin Invest. 2007;117:2260–2267. doi: 10.1172/JCI31680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tejpar S, Piessevaux H, Claes K, Piront P, Hoenderop JG, Verslype C, Van Cutsem E. Magnesium wasting associated with epidermal-growth-factor receptor-targeting antibodies in colorectal cancer: a prospective study. Lancet Oncol. 2007;8:387–394. doi: 10.1016/S1470-2045(07)70108-0. [DOI] [PubMed] [Google Scholar]
- 74.Campbell P, Morton PE, Takeichi T, Salam A, Roberts N, Proudfoot LE, Mellerio JE, Aminu K, Wellington C, Patil SN, Akiyama M, Liu L, McMillan JR, Aristodemou S, Ishida-Yamamoto A, Abdul-Wahab A, Petrof G, Fong K, Harnchoowong S, Stone KL, Harper JI, McLean WH, Simpson MA, Parsons M, McGrath JA. Epithelial inflammation resulting from an inherited loss-of-function mutation in EGFR. J Invest Dermatol. 2014;134:2570–2578. doi: 10.1038/jid.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Glaudemans B, van der Wijst J, Scola RH, Lorenzoni PJ, Heister A, van der Kemp AW, Knoers NV, Hoenderop JG, Bindels RJ. A missense mutation in the Kv1.1 voltage-gated potassium channel-encoding gene KCNA1 is linked to human autosomal dominant hypomagnesemia. J Clin Invest. 2009;119:936–942. doi: 10.1172/JCI36948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.D’Adamo MC, Gallenmuller C, Servettini I, Hartl E, Tucker SJ, Arning L, Biskup S, Grottesi A, Guglielmi L, Imbrici P, Bernasconi P, Di Giovanni G, Franciolini F, Catacuzzeno L, Pessia M, Klopstock T. Novel phenotype associated with a mutation in the KCNA1(Kv1.1) gene. Front Physiol. 2014;5:525. doi: 10.3389/fphys.2014.00525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Graves TD, Cha YH, Hahn AF, Barohn R, Salajegheh MK, Griggs RC, Bundy BN, Jen JC, Baloh RW, Hanna MG. Episodic ataxia type 1: clinical characterization, quality of life and genotype-phenotype correlation. Brain. 2014;137:1009–1018. doi: 10.1093/brain/awu012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ellison DH. The voltage-gated K+ channel subunit Kv1.1 links kidney and brain. J Clin Invest. 2009;119:763–766. doi: 10.1172/JCI38835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Unger S, Gorna MW, Le Bechec A, Do Vale-Pereira S, Bedeschi MF, Geiberger S, Grigelioniene G, Horemuzova E, Lalatta F, Lausch E, Magnani C, Nampoothiri S, Nishimura G, Petrella D, Rojas-Ringeling F, Utsunomiya A, Zabel B, Pradervand S, Harshman K, Campos-Xavier B, Bonafe L, Superti-Furga G, Stevenson B, Superti-Furga A. FAM111A mutations result in hypoparathyroidism and impaired skeletal development. Am J Hum Genet. 2013;92:990–995. doi: 10.1016/j.ajhg.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bergada I, Schiffrin A, Abu Srair H, Kaplan P, Dornan J, Goltzman D, Hendy GN. Kenny syndrome: description of additional abnormalities and molecular studies. Hum Genet. 1988;80:39–42. doi: 10.1007/BF00451452. [DOI] [PubMed] [Google Scholar]
- 81.Fanconi S, Fischer JA, Wieland P, Atares M, Fanconi A, Giedion A, Prader A. Kenny syndrome: evidence for idiopathic hypoparathyroidism in two patients and for abnormal parathyroid hormone in one. J Pediatr. 1986;109:469–475. doi: 10.1016/S0022-3476(86)80120-2. [DOI] [PubMed] [Google Scholar]
- 82.Isojima T, Doi K, Mitsui J, Oda Y, Tokuhiro E, Yasoda A, Yorifuji T, Horikawa R, Yoshimura J, Ishiura H, Morishita S, Tsuji S, Kitanaka S. A recurrent de novo FAM111A mutation causes Kenny-Caffey syndrome type 2. J Bone Miner Res. 2014;29:992–998. doi: 10.1002/jbmr.2091. [DOI] [PubMed] [Google Scholar]
- 83.Lee WK, Vargas A, Barnes J, Root AW. The Kenny-Caffey syndrome: growth retardation and hypocalcemia in a young boy. Am J Med Genet. 1983;14:773–782. doi: 10.1002/ajmg.1320140419. [DOI] [PubMed] [Google Scholar]
- 84.Nikkel SM, Ahmed A, Smith A, Marcadier J, Bulman DE, Boycott KM. Mother-to-daughter transmission of Kenny-Caffey syndrome associated with the recurrent, dominant FAM111A mutation p.Arg569His. Clin Genet. 2014;86:394–395. doi: 10.1111/cge.12290. [DOI] [PubMed] [Google Scholar]
- 85.Yorifuji T, Muroi J, Uematsu A. Kenny-Caffey syndrome without the CATCH 22 deletion. J Med Genet. 1998;35:1054. doi: 10.1136/jmg.35.12.1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fine DA, Rozenblatt-Rosen O, Padi M, Korkhin A, James RL, Adelmant G, Yoon R, Guo L, Berrios C, Zhang Y, Calderwood MA, Velmurgan S, Cheng J, Marto JA, Hill DE, Cusick ME, Vidal M, Florens L, Washburn MP, Litovchick L, DeCaprio JA. Identification of FAM111A as an SV40 host range restriction and adenovirus helper factor. PLoS Pathog. 2012;8:e1002949. doi: 10.1371/journal.ppat.1002949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Akamatsu S, Takata R, Haiman CA, Takahashi A, Inoue T, Kubo M, Furihata M, Kamatani N, Inazawa J, Chen GK, Le Marchand L, Kolonel LN, Katoh T, Yamano Y, Yamakado M, Takahashi H, Yamada H, Egawa S, Fujioka T, Henderson BE, Habuchi T, Ogawa O, Nakamura Y, Nakagawa H. Common variants at 11q12, 10q26 and 3p11.2 are associated with prostate cancer susceptibility in Japanese. Nat Genet. 2012;44:426–429. doi: 10.1038/ng.1104. [DOI] [PubMed] [Google Scholar]
- 88.Houston SK, Pina Y, Clarke J, Koru-Sengul T, Scott WK, Nathanson L, Schefler AC, Murray TG. Regional and temporal differences in gene expression of LH(BETA)T(AG) retinoblastoma tumors. Invest Ophthalmol Vis Sci. 2011;52:5359–5368. doi: 10.1167/iovs.10-6321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Singh H, Farouk M, Bose BB, Singh P. Novel genes underlying beta cell survival in metabolic stress. Bioinformation. 2013;9:37–41. doi: 10.6026/97320630009037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Serna M, Carranza G, Martin-Benito J, Janowski R, Canals A, Coll M, Zabala JC, Valpuesta JM. The structure of the complex between alpha-tubulin, TBCE and TBCB reveals a tubulin dimer dissociation mechanism. J Cell Sci. 2015;128:1824–1834. doi: 10.1242/jcs.167387. [DOI] [PubMed] [Google Scholar]
- 91.Parvari R, Hershkovitz E, Grossman N, Gorodischer R, Loeys B, Zecic A, Mortier G, Gregory S, Sharony R, Kambouris M, Sakati N, Meyer BF, Al Aqeel AI, Al Humaidan AK, Al Zanhrani F, Al Swaid A, Al Othman J, Diaz GA, Weiner R, Khan KT, Gordon R, Gelb BD. Mutation of TBCE causes hypoparathyroidism-retardation-dysmorphism and autosomal recessive Kenny-Caffey syndrome. Nat Genet. 2002;32:448–452. doi: 10.1038/ng1012. [DOI] [PubMed] [Google Scholar]
- 92.Topf JM, Murray PT. Hypomagnesemia and hypermagnesemia. Rev Endocr Metab Disord. 2003;4:195–206. doi: 10.1023/A:1022950321817. [DOI] [PubMed] [Google Scholar]
- 93.Gullestad L, Oystein Dolva L, Birkeland K, Falch D, Fagertun H, Kjekshus J. Oral versus intravenous magnesium supplementation in patients with magnesium deficiency. Magnes Trace Elem. 1991;10:11–16. [PubMed] [Google Scholar]
- 94.Haas M, Wang H, Tian J, Xie Z. Src-mediated inter-receptor cross-talk between the Na+/K+-ATPase and the epidermal growth factor receptor relays the signal from ouabain to mitogen-activated protein kinases. J Biol Chem. 2002;277:18694–18702. doi: 10.1074/jbc.M111357200. [DOI] [PubMed] [Google Scholar]
- 95.Reichold M, Zdebik AA, Lieberer E, Rapedius M, Schmidt K, Bandulik S, Sterner C, Tegtmeier I, Penton D, Baukrowitz T, Hulton SA, Witzgall R, Ben-Zeev B, Howie AJ, Kleta R, Bockenhauer D, Warth R. KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function. Proc Natl Acad Sci USA. 2010;107:14490–14495. doi: 10.1073/pnas.1003072107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Terker AS, Zhang C, McCormick JA, Lazelle RA, Zhang C, Meermeier NP, Siler DA, Park HJ, Fu Y, Cohen DM, Weinstein AM, Wang WH, Yang CL, Ellison DH. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab. 2015;21:39–50. doi: 10.1016/j.cmet.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]