

Abstract
Background: Tumor Necrosis Factor α (TNFα) upregulation during acute inflammatory response has been associated with numerous cardiac effects including modulating Connexin43 and vascular permeability. This may in turn alter cardiac gap junctional (GJ) coupling and extracellular volume (ephaptic coupling) respectively. We hypothesized that acute exposure to pathophysiological TNFα levels can modulate conduction velocity (CV) in the heart by altering electrical coupling: GJ and ephaptic.
Methods and Results: Hearts were optically mapped to determine CV from control, TNFα and TNFα + high calcium (2.5 vs. 1.25 mM) treated guinea pig hearts over 90 mins. Transmission electron microscopy was performed to measure changes in intercellular separation in the gap junction-adjacent extracellular nanodomain—perinexus (WP). Cx43 expression and phosphorylation were determined by Western blotting and Cx43 distribution by confocal immunofluorescence. At 90 mins, longitudinal and transverse CV (CVL and CVT, respectively) increased with control Tyrode perfusion but TNFα slowed CVT alone relative to control and anisotropy of conduction increased, but not significantly. TNFα increased WP relative to control at 90 mins, without significantly changing GJ coupling. Increasing extracellular calcium after 30 mins of just TNFα exposure increased CVT within 15 mins. TNFα + high calcium also restored CVT at 90 mins and reduced WP to control values. Interestingly, TNFα + high calcium also improved GJ coupling at 90 mins, which along with reduced WP may have contributed to increasing CV.
Conclusions: Elevating extracellular calcium during acute TNFα exposure reduces perinexal expansion, increases ephaptic, and GJ coupling, improves CV and may be a novel method for preventing inflammation induced CV slowing.
Keywords: TNFα, conduction, calcium, ephaptic coupling, connexin43
Introduction
Myocardial inflammation is associated with many cardiac diseases (Marchant et al., 2012; De Jesus et al., 2015) and modulates several determinants of cardiac function, both mechanical and electrical. Mechanically, myocardial inflammation can cause cardiac dysfunction and reduced ejection fraction (Lurz et al., 2012; Banka et al., 2015). Myocardial inflammation can also alter electrical impulse propagation by modulating gap junctional coupling (GJC) (Zhu et al., 2000; Xu H. F. et al., 2012), ionic currents (Tang et al., 2007; De Jesus et al., 2015), and tissue hydration state (Logstrup et al., 2015).
Inflammation is a complex process associated with the modulation of several physiologic factors, including the up- and downregulation of many cytokines, which are cell signaling molecules (Zhang and An, 2007). Cytokines modulate numerous cellular processes, some pro-inflammatory and others anti-inflammatory. Tumor Necrosis Factor α (TNFα) is a pro-inflammatory cytokine whose upregulation is a key marker of the acute inflammatory phase in several pathophysiologic states including ischemia, myocarditis, and cardiomyopathies (Matsumori et al., 1994; Intiso et al., 2004). TNFα upregulation modulates the expression of other cytokines and has a cascading effect on the inflammatory process. The effects of TNFα on various cellular functions have been extensively studied in cardiac and other tissue types. For example, some studies (Celes et al., 2007; Kimura and Nishida, 2010) demonstrated that exposure to TNFα reduces Connexin43 (Cx43) functional expression, the principle gap junctional protein in cardiac ventricles, while others reported no change (Sawaya et al., 2007). Other studies determined that TNFα can modulate Cx43 phosphorylation states in anterior pituitary cells (Meilleur et al., 2007). Studies have also demonstrated a temporal change in the regulation of Cx43 expression by TNFα where an increase in Cx43 mRNA and protein expression was reported at 6 h of TNFα exposure and a decrease at longer durations up to 48 h (Liu et al., 2012). However, the electrophysiologic effects of acute TNFα exposure in cardiac tissue are not fully understood.
In addition to its effect on GJ coupling, TNFα can also modulate vascular permeability which can alter tissue hydration state (Hansen et al., 1994). However, it is not known how this translates to the level of intercellular separation at nanodomains within the intercalated disc, such as the gap junction adjacent perinexus (Rhett et al., 2011). Additionally, TNFα has also been reported to reduce the expression of structural proteins along the intercalated disc (ID) and cause ID dehiscence (Celes et al., 2007). Both factors could cause perinexal widening which is associated with CV slowing possibly due to weaker ephaptic coupling (EpC) between myocytes (George et al., 2015, 2016; Veeraraghavan et al., 2015). Therefore, TNFα alone can modulate various determinants of CV similar to previously reported models of myocardial inflammation. In this study, we use pathophysiologic TNFα exposure as a model for myocardial inflammation and focus on the acute effects of TNFα on ventricular conduction. We hypothesized, that TNFα modulates CV by reducing electrical coupling in the heart—both EpC and GJC.
Here, we determined the ventricular conduction velocity response to TNFα exposure (100 pg/ml) over 90 mins. Our results suggest that CV slows with TNFα exposure relative to control. CV slowing is associated with reduced EpC but no significant modulation of GJC. Elevating extracellular calcium ion concentration ([Ca2+]o) improved both forms of electrical coupling in the presence of TNFα, which could have contributed to restoring CV to control values.
Methods
All experimental protocols have been approved by the Institutional Animal Care and Use Committee at Virginia Polytechnic Institute and State University and are in accordance with the NIH Guide for Care and Usage of Laboratory Animals.
Landgendorff preparation
Adult male Hartley Guinea Pigs (1,000–1,300 g) were anesthetized by exposure to isoflurane and hearts were excised following thoracotomy as previously described (Veeraraghavan et al., 2015). The heart was then attached to a Langendorff perfusion system and perfused with a solution containing, in mM, 1.25 CaCl2, 140 NaCl, 5.5 NaOH, 4.5 KCl, 5.5 Dextrose, 0.7 MgCl2, 9.9 HEPES, pH 7.4 at 37°C. The atria were removed and the heart was suspended in a bath containing the same perfusate at 37°C. Pressure was maintained at ~50 mmHg.
Optical mapping
After a 30 min stabilization period, hearts were perfused with 7.5 μM Di-4-ANEPPS for ~10 mins after which excess dye was washed out, which is t = 0 mins for all experiments. The electromechanical uncoupler, 2,3-butanedionemonoxime was added to the perfusate to reduce motion. A silver pacing wire was placed on the anterior ventricular surface of the heart in the center of the mapping field and a reference wire was introduced in the back of the bath. Hearts were stimulated at 1 V for 1 ms stimuli at a BCL of 300 ms. The dye was then excited by light at 510 nm and the emitted light was filtered by a 610 nm filter and captured by a Micam Ultima L-type CMOS camera as previously described (George et al., 2015; Entz et al., 2016).
Optical data were analyzed to measure CV—both longitudinal (CVL) and transverse (CVT), anisotropic ratio (AR = CVL/CVT), action potential duration (APD), and rise time (RT). Briefly, activation times were assigned at the maximum rate of rise of the action potential and were fitted to a parabolic surface to determine CV vectors. APD was defined as the time interval between activation time and 90% repolarization. RT was calculated as the time interval between 20 and 80% of the upstroke of the action potential.
Hearts were subjected to one of three interventions over 90 mins and optical recordings were obtained at 15 min intervals (n = 6 hearts for each of three intervention). In the first group (control) hearts were continuously perfused with control Tyrode for the entire 90 mins. In the second group (TNFα) hearts were perfused with control Tyrode + TNFα at 100 pg/ml for the 90 min duration. Finally, in the third group (TNFα + high calcium) hearts were perfused with control Tyrode + TNFα for the first 30 mins, followed by calcium elevation to 2.5 mM still in the presence of TNFα from t = 31 to 90 mins.
Electrocardiography
Volume conducted ECGs were recorded by silver chloride electrodes placed in the bath. The signals were recorded using the PowerLab 4/35 data acquisition system and LabChart Pro software. Signals were sampled at 1,000 Hz and filtered (0.1 and 50 Hz low and high cut off frequencies) to remove noise. Paced QRS duration and QT intervals were measured every 15 mins.
Transmission electron microscopy
Anterior epicardial tissue from the left ventricle (n = 3 hearts × 3 intervention × 15 images) was collected from hearts at t = 0 mins and after 90 mins during the three interventions—control, TNFα, and TNFα + high calcium, sliced into 1 mm3 sections, fixed in 2.5% glutaraldehyde overnight at 4°C and then washed and stored in PBS also at 4°C. Samples were then processed for TEM as previously described (George et al., 2015) and imaged using a JEM JEOL1400 Electron Microscope at X150,000 magnification. Fifteen images were acquired per sample, which were then analyzed using ImageJ to measure perinexal width. The average of six intermembrane distances between 30 and 105 nm away from the edge of the GJ plaque, 15 nm apart, is reported as WP. Data are reported at mean ± standard error.
Western blotting
Samples (n = 3 hearts × 3 conditions × 3 runs) were snap frozen at t = 0 or after 90 mins of control, TNFα or TNFα + high calcium treatment and immunoblotting was performed as previously described (Smyth et al., 2010) to determine Cx43 and pCx43—Ser368 expression. Briefly, samples were homogenized in RIPA lysis buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, 2 mM NaF, 200 μM Na3VO4) supplemented with HALT protease and phosphatase inhibitors (ThermoFisher Scientific) and electrophoresis was performed to separate proteins which were then transferred onto a PVDF membrane. This was then blocked with 5% BSA for 1 h at room temperature, followed by incubation with pCx43-Ser368 primary antibody (1:1,000, #3511, Cell Signaling Technologies) overnight at 4°C and, after washing, secondary antibody (1:5,000, Goat Anti-Rabbit HRP, abcam) for 1 h at room temperature. Protein expression was then quantified by ECL assay using a BioRad Chemidoc MP system. The membranes were then stripped with ReBlot Plus Strong (EMD Millipore) as per manufacturer's instructions and blocked with 5% milk for 1 h at room temperature. Membranes were then incubated with primary anitbodies against Cx43 (1:3,000, C2619 rabbit, Sigma Aldrich) and GAPDH (1:3,000, T6199 mouse, Sigma Aldrich) overnight at 4°C, followed by the corresponding secondary anitbodies (both 1:1,000, goat anti-mouse AlexaFluor555 and goat anti-rabbit AlexaFluor647) for 1 h at room temperature. Finally, total Cx43 and GAPDH protein expression was quantified using the BioRad Chemidoc MP system. Total Cx43 was normalized to GAPDH and pCx43 was normalized to total Cx43.
Confocal immunofluorescence
Ventricular sections from control (n = 3), TNFα (n = 6), and TNFα + high calcium (n = 3) hearts at t = 0 mins and after 90 mins were snap frozen in OCT. Samples were sectioned at 5 μm thickness onto glass slides and fixed with 2% paraformaldehyde for 5 mins on a rotator. Slides were then washed and samples were blocked with a solution containing 1% BSA and 1% Triton X-100 in PBS for 1 h at room temperature. Samples were then incubated with primary antibody against Cx43 (1:4,000, C2619 rabbit, Sigma Aldrich) and N-Cadherin (1:100, 610920, mouse, BD Biosciences) overnight at 4°C. Slides were then washed and samples were incubated with the corresponding secondary antibodies (1:4,000, Goat Anti-Rabbit AlexaFluor 488 and Goat Anti-Mouse AlexaFluor 633) for 2 h at room temperature. Prolong Gold Antifade (Life Technologies) was then applied to the slides and slide covers were applied. Slides were cured for ~48 h. Cx43 and N-Cadherin distribution were imaged using a Leica TCS SP8 laser scanning confocal microscope using a X63 oil immersion lens. Images acquired (3 per heart) were then analyzed similar to previously described methods. (Smyth et al., 2014; Yan et al., 2014) Briefly, images were converted to a binary format after thresholding, and the percent of Cx43 colocalized with N-Cadherin and normalized to total Cx43 was quantified to estimate Cx43 localization at the intercalated disc.
Statistical analysis
Single factor or two way ANOVA tests were performed to detect significant differences in the data and Student's t-test was applied as a post-hoc analysis. Bonferroni correction was applied as necessary with multiple comparisons. All data are reported as mean ± standard deviation unless stated otherwise. p < 0.05 was reported as significant.
Results
Conduction velocity-control vs. TNFα
Hearts were optically mapped during perfusion of control Tyrode's solution over a 90 min period, and representative isochrones maps are illustrated in Figure 1A, Upper Panel. CVL and CVT were calculated and are reported in Figure 1B.
Figure 1.

Conduction velocity quantified by optical mapping increases with time. (A) Representative isochrone maps from control Tyrode's solution perfused hearts at 0, 90 and 90, 180 mins. (B) Summary CVL and CVT measured up to 180 mins. Black curves indicate experiments where dye was perfused before t = 0 mins and gray curves indicates experiments where dye was perfused at 75 mins. (C) Percent increase in CVT and CVL over 90 min durations in the two sets of experiments. Black and gray *Indicates p < 0.05 relative to t = 0 or 90 mins respectively by paired comparison. Black # indicates p < 0.1 comparing between black and gray data point at 90 mins. Gray # indicates p < 0.1 comparing gray data point 90 and 180 mins.
In hearts perfused with control Tyrode's solution, both CVL and CVT isotropically increased over time. We hypothesized that the gradual CV increase over time was a result of either dye washout or degradation. Therefore, in order to compare CV at t = 0 and 90 mins without the effect of the dye, we delayed Di-4-ANEPPS perfusion for 75 mins in another set of experiments. Importantly, Figure 1 demonstrates that CV in the delayed dye perfusion experiments was not statistically different from CV in the original and early dye perfusion experiments. Further, CV still increased in the delayed dye perfusion experiment over the additional 90 mins (Figure 1). The percent increase in CVL and CVT over the first and second 90 min durations was similar (Figure 1C). Taken together, these data suggest that the observed rise in CV is related to dye washout or degradation.
However, in TNFα perfused hearts, CVL alone increased over time with no change in CVT as seen in representative isochrones (Figure 2A) and summary data (Figure 2B), and as a result an increasing trend in AR was also observed (p = 0.055, Figure 2B). Additionally, CVT was significantly slower in TNFα perfused hearts relative to control at t = 90 mins. Taking into consideration the temporal effects of dye on our measurements as demonstrated above, the data suggest that TNFα slows CVT over 90 mins. We therefore conclude that the presence of TNFα in the perfusate slows CV, preferentially in the transverse direction, and increases conduction anisotropy.
Figure 2.

TNFα slows conduction. (A) Representative isochrones maps from control and TNFα perfused hearts at t = 0 and 90 mins. (B) Summary of CVL, CVT, and AR values calculated from the optical recordings are graphed. Black and gray *Indicates p < 0.05 between t = 0 and 90 mins in control and TNFα perfused hearts respectively by paired comparison. #Indicates p < 0.05 between control and TNFα (unpaired).
Conduction velocity-control vs. TNFα + high calcium
Elevating extracellular calcium has been demonstrated to acutely increase CV possibly by improving EpC (George et al., 2015, 2016). We next perfused TNFα treated hearts with a physiologically high calcium solution (2.5 mM) introduced 30 mins into the experiment to determine if elevating extracellular calcium can attenuate TNFα induced CV slowing. Percent changes in CVL, CVT, and AR are reported in Figure 3 to compare the effects of TNFα + high calcium to control Tyrode perfused hearts. At t = 90 mins, CVL significantly increased relative to t = 0 mins and was similar to control at t = 90 mins. This suggests that neither TNFα nor high calcium have a significant impact on CVL, though the change in CVL may be below our ability to detect. On the other hand, CVT, as illustrated in Figure 3B, began to separate from control after the initial 30 mins of just TNFα perfusion, suggesting that TNFα can acutely slow CVT. Interestingly, elevating calcium in the presence of TNFα acutely increased CVT within 15 mins and restored CVT back to control values at 90 mins. Additionally, CVT was significantly greater at t = 90 mins relative to 0 mins during TNFα + high calcium. Finally, AR was not significantly different in control or TNFα + high calcium perfused hearts at t = 90 mins. Taken together, these data demonstrate that high calcium restored CV in TNFα treated hearts to control values.
Figure 3.

Conduction rescue by high calcium. Percent change in CVL (A), CVT (B), and AR (C) over time induced by control Tyrode perfusion and hearts treated with TNFα + high calcium at t > 30 mins. Black and gray *Indicates p < 0.05 between t = 0 and 90 mins in control and TNFα perfused hearts respectively by paired comparison.
Action potential
TNFα can modulate several ionic currents in the heart (Grandy and Fiset, 2009; Guillouet et al., 2011). We therefore quantified action potential parameters such as rise time (RT) and APD to determine the effects our interventions had on cardiac electrophysiology. RT was not significantly different between t = 0 and 90 mins during control or TNFα perfusion. However, RT increased at t = 90 mins with TNFα + high calcium. This can be seen in Figure 4A, and summary data is presented in Figure 4B, left panel.
Figure 4.

Action potential parameters (A) Representative action potentials recorded from the anterior epicardium of control, TNFα, and TNFα + high calcium perfused hearts. Solid long lines indicate peak of AP relative to the time aligned beginning of the action potential. Solid and dashed short lines indicate fiducials in upper and lower panels respectively (t = 0 and 90 mins) and (B) Summary of RT (left) and APD (right) calculated from these hearts at t = 0 and 90 mins. (C) APD quadrant analysis for hearts perfused with control Tyrode's, TNFα and TNFα + high calcium. *Indicates p < 0.05 relative to t = 0 mins by paired comparison. + indicates p < 0.05 and #Indicates p = 0.05 relative to zero.
APD, on the other hand, is significantly prolonged over time during control Tyrode perfusion but this effect was not as pronounced in the presence of TNFα or TNFα + high calcium (Figure 4B, Right Panel) suggesting that TNFα may be modulating ionic currents that determine APD. The optically mapped region on the anterior epicardial surface was then divided into 4 quadrants corresponding to the right ventricular (RV) base and apex, and the LV base and apex. APD was compared to determine if the effects of TNFα and TNFα + high calcium were homogenous (Figure 4C) across the epicardial surface. Although, APD was significantly prolonged only in 3 of 4 quadrants in control Tyrode's perfused hearts, there were no significant APD changes in hearts perfused with TNFα or TNFα + high calcium hearts. Furthermore, no significant changes in APD were observed between quadrants with any intervention suggesting homogenous APD modulation.
ECG
Optical maps were obtained from the anterior epicardial surface of the heart from a region that was ~16 × 16 mm. The CV, RT, and APD parameters reported above are based on changes in this specific field of view. However, several conditions can result in a heterogeneous modulation of CV which then increases risk for arrhythmias (Gutstein et al., 2001; Poelzing and Rosenbaum, 2004). Therefore, the ECG was assessed during pacing to determine QRS duration, which would indicate if the effect of TNFα on CV was also observed at the whole heart level. Representative ECG traces recorded from control, TNFα, and TNFα + high calcium treated hearts are presented in Figure 5A, and QRS durations are reported in Figure 5B. Changes in QRS duration over time (t = 0 to 90 mins) were not significantly different with any of the interventions. However, QRS duration at t = 90 mins was significantly prolonged in TNFα perfused hearts relative to control, consistent with CV slowing observed with TNFα relative to control reported above. Also, QRS duration was similar between control and TNFα + high calcium treated hearts at t = 90 mins further suggesting that elevating calcium improves conduction in the presence of TNFα.
Figure 5.
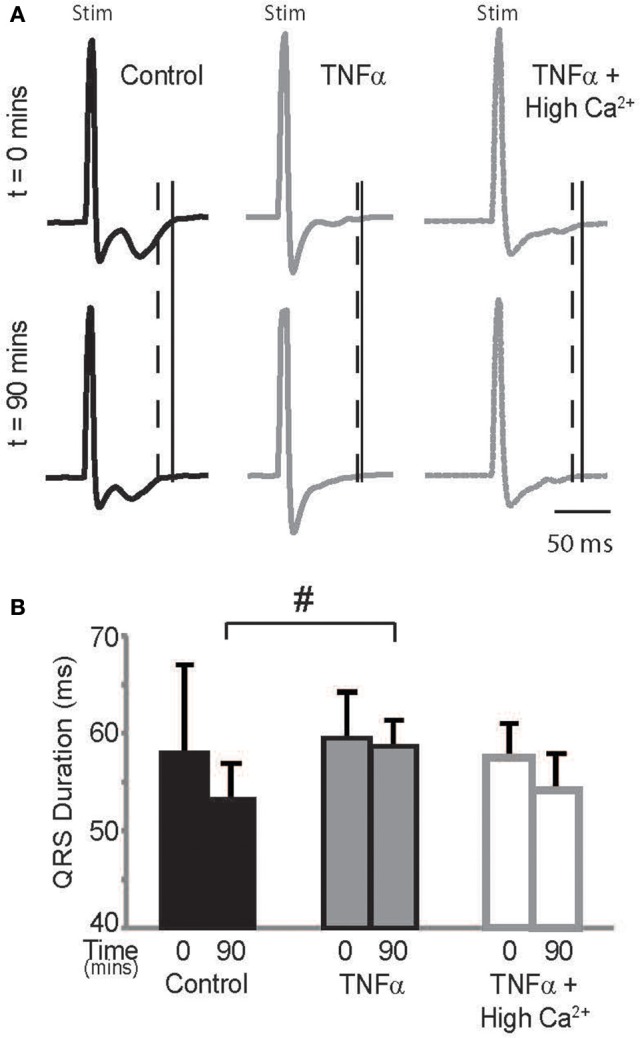
ECG (A) Paced QRS complexes from volume-conducted ECG traces recorded from control, TNFα and TNFα + high calcium perfused hearts. Solid vertical lines indicate end of QRS of ECGs from t = 0 mins and dashed vertical lines indicate end of QRS and T waves of ECGs from t = 90 mins. “Stim” indicates stimulus artifacts. (B) Summary of QRS duration. #indicates p < 0.05 relative to control (unpaired).
Perinexus
Next, the effect of TNFα on proposed modulators of ephaptic coupling like the perinexus was determined, since we previously demonstrated that elevating calcium within the physiologic range can decrease perinexal width in mouse ventricular myocardium (George et al., 2015, 2016). Perinexal width (WP) was not significantly different at t = 0 or 90 mins in control Tyrode perfused hearts, but TNFα significantly increased WP over the same time course (Figure 6). Elevating calcium in the presence of TNFα reduced WP back to control values. This is consistent with our previous mouse study, where we demonstrated that increasing extracellular calcium decreases perinexal width. These data suggest that WP correlates with observed CV changes induced by TNFα or TNFα + high calcium, and are consistent with predictions of EpC.
Figure 6.

TNFα modulates perinexal width (A) Electron micrographs of representative perinexi (highlighted in yellow) from hearts treated with control, TNFα or TNFα + high calcium over 90 mins. (B) Average perinexal width. *Indicates p < 0.05 relative to t = 0 (unpaired comparison).
Connexin43 expression, phosphorylation, and distribution
Finally, we sought to determine if TNFα alters Cx43 protein expression, phosphorylation or distribution. Representative western blots in Figure 7A and summary data in Figure 7B demonstrate that total Cx43 and the ratio of pCx43/Cx43 was not significantly altered over 90 mins with either control Tyrode or TNFα perfusion. Interestingly, elevating extracellular calcium with TNFα significantly increased total Cx43 expression relative to TNFα alone, but the ratio of pCx43/Cx43 did not change. Therefore, though Cx43 modulation may not contribute to CV slowing by TNFα, improving GJ coupling may be a mechanism that contributes to CV preservation with high calcium at 90 mins.
Figure 7.

Cx43 expression and phosphorylation modulation by TNFα (A) Representative images of membranes blotted for total Cx43, Cx43 phosphorylated at Ser368 and GAPDH as a loading control. (B) Protein expression in hearts treated with control, TNFα and TNFα + high calcium at t = 0 and 90 mins. *Indicates p < 0.05 by unpaired comparison.
Next, the distribution of Cx43 was also quantified to determine if TNFα causes Cx43 remodeling over 90 mins (Figure 8). Although Cx43 expression was not significantly altered over time with control Tyrode or TNFα, co-localization of Cx43 with the intercalated disc protein N-Cadherin was significantly reduced for both interventions. This suggests that Cx43 redistributes around the myocyte over 90 mins in these isolated heart preparations. Finally, TNFα + high calcium did not alter total Cx43 expression as mentioned above, and Cx43 distribution around the myocyte at t = 90 mins was preserved similar to control at t = 0 mins.
Figure 8.

Cx43 distribution modulation by TNFα (A) Representative samples stained for Cx43 (green) and N-Cadherin (red, to mark the intercalated disc). The colocalization of the two signals is indicated in yellow. Samples from control, TNFα, and TNFα + high calcium were compared and 1 h of no flow ischemia was used as a positive control. (B) Summary data of percent Cx43 colocalized with N-Cadherin. *Indicates p < 0.05 relative to t = 0 mins by unpaired comparison.
Lastly, as a positive control, hearts were exposed to 1 h of no flow ischemia. In these hearts, Cx43 co-localization with N-Cadherin was significantly reduced relative to control suggesting redistribution from the intercalated disc, as expected from previous publications (Smith et al., 1991; Huang et al., 1999; Jain et al., 2003).
Discussion
Conduction is a multifactorial phenomenon that can be modulated by several factors including tissue architecture, intercellular coupling and excitability of the tissue. Tissue architecture includes cellular dimensions and extracellular space, both extracellular volume and composition (fibrosis). Two modes of intercellular electrical coupling have currently been suggested in cardiac tissue, electrotonic coupling mediated by gap junctions and electric field coupling at ephapses. While modulating the gap junctional proteins, such as Cx43, can alter gap junctional coupling, altering the width of the perinexus, and/or its ionic composition has been suggested to alter ephaptic coupling. Finally, excitability of the tissue can be modulated by altering ion channel functional expression. In this study, we investigated the role of several of these determinants of cardiac conduction in modulating CV during acute TNFα exposure.
Hearts were exposed to 100 pg/ml TNFα, similar to concentrations previously reported in cardiac tissue during diseases like end-stage dilated cardiomyopathy and ischemic heart disease. (Giroir et al., 1992; Torre-Amione et al., 1996) We sought to determine whether acute TNFα exposure over 90 mins was associated with changes in CV modulators like ephaptic and/or GJC. Briefly, CV slowing was observed in TNFα perfused hearts relative to control and this was associated with perinexal expansion without a concomitant change in Cx43 expression or phosphorylation. Additionally, Cx43 cellular localization was altered by TNFα. Elevating extracellular calcium within guinea pig physiologic limits in the presence of TNFα rescued CV at 90 mins by restoring WP to control values and improving GJ expression, phosphorylation, and distribution.
Another novel finding of this study was that after Di-4-ANEPPS perfusion and excess dye washout, CV increased over time up to 90 min. Addition of more Di-4-ANEPPS at 90 min reversed this increase and restored CV to initial values. However, a similar increase in CV over time was observed after an additional 90 min. This can be interpreted as acute CV slowing due to Di-4-ANEPPS perfusion and restoration of CV over time due to either washout or degradation of the dye. It was previously suggested that Di-4-ANEPPS slows CV due to inhibition the sodium-potassium ATPase (Fedosova et al., 1995; Larsen et al., 2012). Restoration of CV, as in the current study, may be an effect of reversal of the inhibition of the sodium-potassium ATPase. However, this requires further investigation.
TNFα and ephaptic coupling
TNFα is one marker of acute inflammation and is important to many physiological processes. One important effect of TNFα is its ability to modulate vascular leakiness as evidenced by studies demonstrating that elevated TNFα concentrations increase vascular permeability (Hansen et al., 1994). Increased vascular permeability can then lead to extracellular edema formation in tissue (Logstrup et al., 2015). In the heart, extracellular edema has been demonstrated to slow CV and increase arrhythmogenesis possibly due to reduced ephaptic coupling between myocytes (George and Poelzing, 2015; George et al., 2015; Veeraraghavan et al., 2015; George et al., 2016).
In addition to gross extracellular edema, the results of this study indicate that TNFα can increase extracellular volumes in restricted nanodomains within intercalated discs, like the perinexus. Fluid retention in the bulk extracellular space could be one causative factor of TNFα-induced perinexal edema. Another explanation for perinexal expansion could be the effect of TNFα on structural junction proteins along the intercalated disc. For example, TNFα can reduce N-Cadherin (Celes et al., 2007) and plakoglobin (Asimaki et al., 2011) expression in the heart, which are essential components of the structural junctions that hold the two adjacent membranes together. Structurally uncoupling these junctions could also increase intercellular separation at the perinexus and thereby cause perinexal edema.
Finally, structural proteins like N-Cadherin, desmoglein, and desmocollin have calcium sensitive domains that determine binding affinity (Chitaev and Troyanovsky, 1997; Vleminckx and Kemler, 1999). Hypocalcemia and calcium-free solutions have been demonstrated to cause intercalated disc dehiscence by reduced binding affinity of these proteins (Chitaev and Troyanovsky, 1997; Vleminckx and Kemler, 1999). This can result in perinexal widening. Additionally, we recently demonstrated that increasing extracellular calcium can reduce perinexal width and maintain the structural integrity of the ephapse possibly by similarly modulating the structural protein binding affinity (George et al., 2016). In this study, elevating extracellular calcium could enhance adhesion at these junctions during TNFα exposure, thereby restoring WP to control values.
TNFα and gap junctional coupling
While the effects of TNFα on Cx43 expression have been extensively studied, some groups report that TNFα reduces (Celes et al., 2007; Sawaya et al., 2007) or increases (Liu et al., 2012) Cx43 expression. Some factors that can explain these diverse results are the type of tissue studied, concentration of TNFα, period of exposure or other yet to be explored experimental differences (Celes et al., 2007; Sawaya et al., 2007; Kimura and Nishida, 2010; Liu et al., 2012). Additionally, TNFα has also been demonstrated to reduce Cx43 phosphorylation at serine 368 in anterior pituitary cells (Meilleur et al., 2007), which is important in modulating Cx43 GJ channel conductance. Cx43 remodeling and lateralization has also been reported in the atria of TNFα overexpressing mice (Sawaya et al., 2007).
In this study, we explored the effect of TNFα on Cx43 expression at a time scale (90 mins) significantly shorter than previous studies. Total Cx43 expression and the ratio of phosphorylated to total Cx43 was not significantly different in the presence of TNFα relative to control at 90 mins. Interestingly, the distribution of Cx43 around the myocyte was heterogeneously altered even within anterior left ventricular (LV) tissue samples analyzed here. This finding is similar to that observed in the atria of TNFα overexpressing mice where Cx43 expression was not altered but Cx43 was redistributed around the myocyte (Sawaya et al., 2007).
Interestingly, elevating extracellular calcium to 2.5 mM in the presence of TNFα improved GJ coupling. Previous studies have demonstrated that increasing intracellular calcium can decrease GJ coupling between cells (Maurer and Weingart, 1987; Kurebayashi et al., 2008) by dephosphorylating Cx43 by a Ca2+/Calmodulin pathway (Xu Q. et al., 2012). However, calcium concentrations used in other previous studies were significantly greater than the concentration used in this study. Our previous study in mice demonstrated that modulating calcium in the range used here did not affect Cx43 expression, phosphorylation or function over 30 mins (George et al., 2016). However, here we report that in the presence of TNFα, GJ coupling in guinea pig hearts improved when calcium was increased to 2.5 mM for 60 mins.
Calcium and TNFα have been identified as key cell signaling regulators of protein transcription including Cx43. For example, elevated calcium can activate MAPK–dependent pathways that have been reported to either increase (Squecco et al., 2006; Stanbouly et al., 2008) or decrease (Petrich et al., 2002) Cx43 expression and phosphorylation. In this study, the specific mechanisms underlying enhanced GJ coupling in guinea pig hearts in the presence of TNFα + high calcium is not fully understood and may involve the activation of one or more of these cell signaling pathways.
TNFα and ionic currents
Ventricular heterogeneities of membrane proteins that form ion channels in the myocardium is well-established (Di Diego et al., 1996; Veeraraghavan and Poelzing, 2008), and TNFα modulates a variety of these ionic currents (Grandy and Fiset, 2009; Guillouet et al., 2011). In this study, APD prolongation was observed over 90 mins in control hearts but not in the presence TNFα with or without high calcium. APD prolongation in the control hearts could be a result of the phototoxic effects of Di-4-ANEPPS over time. (Schaffer et al., 1994; Hardy et al., 2009) In the presence of TNFα, several studies have reported that repolarizing potassium currents are reduced (Grandy and Fiset, 2009) which should theoretically increase APD. However, other studies have also demonstrated that inflammation is associated with APD shortening due to reduced L-type calcium current (Zhong et al., 1997; Greensmith and Nirmalan, 2013). In this study, the lack of APD prolongation with TNFα could be due to similar modulation of ionic currents.
Lastly, elevating calcium was also associated with increased action potential RT, suggesting that calcium decreased membrane excitability despite increasing conduction velocity. Increased RT could have been the effect of (1) decreasing sodium currents which would manifest as changes in the maximum rate of rise of the action potential (dV/dtmax) or (2) modulating diastolic membrane potential during the initial phase of excitation which can indirectly affect sodium channel availability. Calcium can modulate sodium channel phosphorylation and gating by a calcium/calmodulin kinase II dependent pathway (Wagner et al., 2006) and a five-fold increase in intracellular calcium has been reported to increase late sodium current (Wagner et al., 2006). However, the effects of physiologically elevating extracellular calcium, as we did in this study, on fast sodium current during cellular depolarization are unknown. The complex result that elevated calcium can increase RT while increasing CV requires further investigation. Finally, it is also possible that the increased GJ coupling in these hearts, with TNFα + high calcium, provides a greater sink to the excitatory current, thereby increasing RT. However, this theory also requires additional investigation.
Limitations
Most of the analysis described above involves only the anterior epicardial region of guinea pig hearts and TNFα could be having different effects on cellular functioning in different regions of the heart. Transmural and interventricular differences in several parameters like protein expression and APD have been previously described (Grandy and Fiset, 2009; Guillouet et al., 2011) and amplification of these differences by factors like TNFα needs to be better understood in order to identify therapeutic options to treat cardiac conduction slowing caused by inflammation. Nonetheless, this study is the first to highlight that acute TNFα exposure detrimentally affects CV in ventricles and identifies perinexal and gap junction remodeling as potential underlying mechanisms.
Finally, TNFα is one of the many cytokines that are involved in the inflammatory process. Several others like IL-6, IFNγ, and IL-8 have all been identified as physiologic modulators during inflammation. In this study, we focused on understanding the effects of individually modulating only TNFα. This is an important step prior to identifying the cumulative effects of inflammatory factors that occur during the complex process of myocardial inflammation. Furthermore, TNFα inhibition has also developed as a therapy for diseases associated with inflammation (Moe et al., 2004; Buyukakilli et al., 2012), which also increases the significance of understanding how TNFα and its inhibitors may affect cardiac functioning.
Conclusions
TNFα upregulation during inflammation can have significant effects on cardiac electrophysiology which includes anisotropic conduction slowing. Altering the perfusate calcium composition has been identified as a means to conceal the effects of TNFα on cardiac conduction acutely. Importantly, increasing extracellular calcium concentration in guinea pig hearts improves both proposed forms of electrical coupling between cardiac myocytes—Ephaptic and GJC and preserves cardiac conduction during acute TNFα exposure.
Author contributions
SG: experimental design, data acquisition, analysis and interpretation, drafting manuscript, and approval. PC: data acquisition, analysis and interpretation, manuscript editing, and approval. RG: data interpretation, manuscript editing, and approval. JS: Data interpretation, manuscript editing, and approval. SP: experimental design, data interpretration, manuscript editing, and approval.
Funding
This work was supported by an R01-HL102298 awarded to SP and R01-HL056728 awarded to RG, a VTCRI Medical Research Scholar Award, an American Heart Association Pre-doctoral fellowship, and the David W. Francis and Lillian Francis Scholarship Fund awarded to SG.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Asimaki A., Tandri H., Duffy E. R., Winterfield J. R., Mackey-Bojack S., Saffitz J. E., et al. (2011). Altered desmosomal proteins in granulomatous myocarditis and potential pathogenic links to arrhythmogenic right ventricular cardiomyopathy. Circ. Arrhythm. Electrophysiol. 4, 743–752. 10.1161/CIRCEP.111.964890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banka P., Robinson J. D., Uppu S. C., Harris M. A., Hasbani K., Powell A. J., et al. (2015). Cardiovascular magnetic resonance techniques and findings in children with myocarditis: a multicenter retrospective study. J. Cardiovasc. Magn. Reson. 17, 96. 10.1186/s12968-015-0201-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buyukakilli B., Atici A., Ozkan A., Balli E., Gunes S., Turhan A., et al. (2012). The effect of tumor necrosis factor-α inhibitor soon after hypoxia-ischemia on heart in neonatal rats. Life Sci. 90, 838–845. 10.1016/j.lfs.2012.03.036 [DOI] [PubMed] [Google Scholar]
- Celes M. R., Torres-Duenas D., Alves-Filho J. C., Duarte D. B., Cunha F. Q., Rossi M. A. (2007). Reduction of gap and adherens junction proteins and intercalated disc structural remodeling in the hearts of mice submitted to severe cecal ligation and puncture sepsis. Crit. Care Med. 35, 2176–2185. 10.1097/01.CCM.0000281454.97901.01 [DOI] [PubMed] [Google Scholar]
- Chitaev N. A., Troyanovsky S. M. (1997). Direct Ca2+-dependent heterophilic interaction between desmosomal cadherins, desmoglein and desmocollin, contributes to cell-cell adhesion. J. Cell Biol. 138, 193–201. 10.1083/jcb.138.1.193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jesus N. M., Wang L., Herren A. W., Wang J., Shenasa F., Ripplinger C. M., et al. (2015). Atherosclerosis exacerbates arrhythmia following myocardial infarction: role of myocardial inflammation. Heart Rhythm 12, 169–178. 10.1016/j.hrthm.2014.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Diego J. M., Sun Z. Q., Antzelevitch C. (1996). I(to) and action potential notch are smaller in left vs. right canine ventricular epicardium. Am. J. Physiol. 271(2 Pt 2), H548–H561. [DOI] [PubMed] [Google Scholar]
- Entz M., II., George S. A., Zeitz M. J., Raisch T., Smyth J. W., Poelzing S. (2016). Heart rate and extracellular sodium and potassium modulation of gap junction mediated conduction in guinea pigs. Front. Physiol. 7:16. 10.3389/fphys.2016.00016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedosova N. U., Cornelius F., Klodos I. (1995). Fluorescent styryl dyes as probes for Na,K-ATPase reaction mechanism: significance of the charge of the hydrophilic moiety of RH dyes. Biochemistry 34, 16806–16814. 10.1021/bi00051a031 [DOI] [PubMed] [Google Scholar]
- George S. A., Bonakdar M., Zeitz M., Davalos R., Smyth J. W., Poelzing S. (2016). Extracellular sodium dependence of the conduction velocity-calcium relationship: evidence of ephaptic self-attenuation. Am. J. Physiol. Heart Circ. Physiol. 310, H1129–H1139. 10.1152/ajpheart.00857.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George S. A., Poelzing S. (2015). Cardiac conduction in isolated hearts of genetically modified mice - Connexin43 and salts. Prog. Biophys. Mol. Biol. 120, 189–198. 10.1016/j.pbiomolbio.2015.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George S. A., Sciuto K. J., Lin J., Salama M. E., Keener J. P., Poelzing S., et al. (2015). Extracellular sodium and potassium levels modulate cardiac conduction in mice heterozygous null for the Connexin43 gene. Pflugers Arch. 467, 2287–2297. 10.1007/s00424-015-1698-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giroir B. P., Johnson J. H., Brown T., Allen G. L., Beutler B. (1992). The tissue distribution of tumor necrosis factor biosynthesis during endotoxemia. J. Clin. Invest. 90, 693–698. 10.1172/JCI115939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandy S. A., Fiset C. (2009). Ventricular K+ currents are reduced in mice with elevated levels of serum TNFα. J. Mol. Cell. Cardiol. 47, 238–246. 10.1016/j.yjmcc.2009.02.025 [DOI] [PubMed] [Google Scholar]
- Greensmith D. J., Nirmalan M. (2013). The effects of tumor necrosis factor-α on systolic and diastolic function in rat ventricular myocytes. Physiol. Rep. 1:e00093. 10.1002/phy2.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillouet M., Gueret G., Rannou F., Giroux-Metges M. A., Gioux M., Pennec J. P., et al. (2011). Tumor necrosis factor-α downregulates sodium current in skeletal muscle by protein kinase C activation: involvement in critical illness polyneuromyopathy. Am. J. Physiol. Cell Physiol. 301, C1057–C1063. 10.1152/ajpcell.00097.2011 [DOI] [PubMed] [Google Scholar]
- Gutstein D. E., Morley G. E., Vaidya D., Liu F., Chen F. L., Fishman G. I., et al. (2001). Heterogeneous expression of Gap junction channels in the heart leads to conduction defects and ventricular dysfunction. Circulation 104, 1194–1199. 10.1161/hc3601.093990 [DOI] [PubMed] [Google Scholar]
- Hansen P. R., Svendsen J. H., Hoyer S., Kharazmi A., Bendtzen K., Haunso S. (1994). Tumor necrosis factor-α increases myocardial microvascular transport in vivo. Am. J. Physiol. 266 (1 Pt 2), H60–H67. [DOI] [PubMed] [Google Scholar]
- Hardy M. E., Pollard C. E., Small B. G., Bridgland-Taylor M., Woods A. J., Abi-Gerges N., et al. (2009). Validation of a voltage-sensitive dye (di-4-ANEPPS)-based method for assessing drug-induced delayed repolarisation in beagle dog left ventricular midmyocardial myocytes. J. Pharmacol. Toxicol. Methods 60, 94–106. 10.1016/j.vascn.2009.03.005 [DOI] [PubMed] [Google Scholar]
- Huang X. D., Sandusky G. E., Zipes D. P. (1999). Heterogeneous loss of connexin43 protein in ischemic dog hearts. J. Cardiovasc. Electrophysiol. 10, 79–91. 10.1111/j.1540-8167.1999.tb00645.x [DOI] [PubMed] [Google Scholar]
- Intiso D., Zarrelli M. M., Lagioia G., Di Rienzo F., Checchia De Ambrosio C., Cioffi Dagger R. P., et al. (2004). Tumor necrosis factor α serum levels and inflammatory response in acute ischemic stroke patients. Neurol. Sci. 24, 390–396. 10.1007/s10072-003-0194-z [DOI] [PubMed] [Google Scholar]
- Jain S. K., Schuessler R. B., Saffitz J. E. (2003). Mechanisms of delayed electrical uncoupling induced by ischemic preconditioning. Circ. Res. 92, 1138–1144. 10.1161/01.RES.0000074883.66422.C5 [DOI] [PubMed] [Google Scholar]
- Kimura K., Nishida T. (2010). Role of the ubiquitin-proteasome pathway in downregulation of the gap-junction protein Connexin43 by TNF-α in human corneal fibroblasts. Invest. Ophthalmol. Vis. Sci. 51, 1943–1947. 10.1167/iovs.09-3573 [DOI] [PubMed] [Google Scholar]
- Kurebayashi N., Nishizawa H., Nakazato Y., Kurihara H., Matsushita S., Daida H., et al. (2008). Aberrant cell-to-cell coupling in Ca2+-overloaded guinea pig ventricular muscles. Am. J. Physiol. Cell Physiol. 294, C1419–C1429. 10.1152/ajpcell.00413.2007 [DOI] [PubMed] [Google Scholar]
- Larsen A. P., Sciuto K. J., Moreno A. P., Poelzing S. (2012). The voltage-sensitive dye di-4-ANEPPS slows conduction velocity in isolated guinea pig hearts. Heart Rhythm 9, 1493–1500. 10.1016/j.hrthm.2012.04.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Gao Z., Zhang L., Su L., Dong G., Yu H., et al. (2012). Temporal dynamic changes of connexin 43 expression in C6 cells following lipopolysaccharide stimulation. Neural Regen. Res. 7, 1947–1953. 10.3969/j.issn.1673-5374.2012.25.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logstrup B. B., Nielsen J. M., Kim W. Y., Poulsen S. H. (2015). Myocardial oedema in acute myocarditis detected by echocardiographic 2D myocardial deformation analysis. Eur. Heart. J. Cardiovasc. Imaging 17, 1018–1026. 10.1093/ehjci/jev302 [DOI] [PubMed] [Google Scholar]
- Lurz P., Eitel I., Adam J., Steiner J., Grothoff M., Desch S., et al. (2012). Diagnostic performance of CMR imaging compared with EMB in patients with suspected myocarditis. JACC Cardiovasc. Imaging 5, 513–524. 10.1016/j.jcmg.2011.11.022 [DOI] [PubMed] [Google Scholar]
- Marchant D. J., Boyd J. H., Lin D. C., Granville D. J., Garmaroudi F. S., McManus B. M. (2012). Inflammation in myocardial diseases. Circ. Res. 110, 126–144. 10.1161/CIRCRESAHA.111.243170 [DOI] [PubMed] [Google Scholar]
- Matsumori A., Yamada T., Suzuki H., Matoba Y., Sasayama S. (1994). Increased circulating cytokines in patients with myocarditis and cardiomyopathy. Br. Heart J. 72, 561–566. 10.1136/hrt.72.6.561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer P., Weingart R. (1987). Cell pairs isolated from adult guinea pig and rat hearts: effects of [Ca2+]i on nexal membrane resistance. Pflugers Arch. 409, 394–402. 10.1007/BF00583793 [DOI] [PubMed] [Google Scholar]
- Meilleur M. A., Akpovi C. D., Pelletier R. M., Vitale M. L. (2007). Tumor necrosis factor-α-induced anterior pituitary folliculostellate TtT/GF cell uncoupling is mediated by connexin 43 dephosphorylation. Endocrinology 148, 5913–5924. 10.1210/en.2007-0767 [DOI] [PubMed] [Google Scholar]
- Moe G. W., Marin-Garcia J., Konig A., Goldenthal M., Lu X., Feng Q. (2004). In vivo TNF-α inhibition ameliorates cardiac mitochondrial dysfunction, oxidative stress, and apoptosis in experimental heart failure. Am. J. Physiol. Heart Circ. Physiol. 287, H1813–H1820. 10.1152/ajpheart.00036.2004 [DOI] [PubMed] [Google Scholar]
- Petrich B. G., Gong X., Lerner D. L., Wang X., Brown J. H., Wang Y., et al. (2002). c-Jun N-terminal kinase activation mediates downregulation of connexin43 in cardiomyocytes. Circ. Res. 91, 640–647. 10.1161/01.RES.0000035854.11082.01 [DOI] [PubMed] [Google Scholar]
- Poelzing S., Rosenbaum D. S. (2004). Altered connexin43 expression produces arrhythmia substrate in heart failure. Am. J. Physiol. Heart Circ. Physiol. 287, H1762–H1770. 10.1152/ajpheart.00346.2004 [DOI] [PubMed] [Google Scholar]
- Rhett J. M., Jourdan J., Gourdie R. G. (2011). Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Mol. Biol. Cell 22, 1516–1528. 10.1091/mbc.E10-06-0548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya S. E., Rajawat Y. S., Rami T. G., Szalai G., Price R. L., Khoury D. S., et al. (2007). Downregulation of connexin40 and increased prevalence of atrial arrhythmias in transgenic mice with cardiac-restricted overexpression of tumor necrosis factor. Am. J. Physiol. Heart Circ. Physiol. 292, H1561–H1567. 10.1152/ajpheart.00285.2006 [DOI] [PubMed] [Google Scholar]
- Schaffer P., Ahammer H., Muller W., Koidl B., Windisch H. (1994). Di-4-ANEPPS causes photodynamic damage to isolated cardiomyocytes. Pflugers Arch. 426, 548–551. 10.1007/BF00378533 [DOI] [PubMed] [Google Scholar]
- Smith J. H., Green C. R., Peters N. S., Rothery S., Severs N. J. (1991). Altered patterns of gap junction distribution in ischemic heart disease. An immunohistochemical study of human myocardium using laser scanning confocal microscopy. Am. J. Pathol. 139, 801–821. [PMC free article] [PubMed] [Google Scholar]
- Smyth J. W., Hong T. T., Gao D., Vogan J. M., Jensen B. C., Shaw R. M., et al. (2010). Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. J. Clin. Invest. 120, 266–279. 10.1172/JCI39740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth J. W., Zhang S. S., Sanchez J. M., Lamouille S., Vogan J. M., Shaw R. M., et al. (2014). A 14-3-3 mode-1 binding motif initiates gap junction internalization during acute cardiac ischemia. Traffic 15, 684–699. 10.1111/tra.12169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squecco R., Sassoli C., Nuti F., Martinesi M., Chellini F., Nosi D., et al. (2006). Sphingosine 1-phosphate induces myoblast differentiation through Cx43 protein expression: a role for a gap junction-dependent and -independent function. Mol. Biol. Cell 17, 4896–4910. 10.1091/mbc.E06-03-0243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanbouly S., Kirshenbaum L. A., Jones D. L., Karmazyn M. (2008). Sodium hydrogen exchange 1 (NHE-1) regulates connexin 43 expression in cardiomyocytes via reverse mode sodium calcium exchange and c-Jun NH2-terminal kinase-dependent pathways. J. Pharmacol. Exp. Ther. 327, 105–113. 10.1124/jpet.108.140228 [DOI] [PubMed] [Google Scholar]
- Tang Q., Huang J., Qian H., Chen L., Wang T., Wang H., et al. (2007). Antiarrhythmic effect of atorvastatin on autoimmune myocarditis is mediated by improving myocardial repolarization. Life Sci. 80, 601–608. 10.1016/j.lfs.2006.11.045 [DOI] [PubMed] [Google Scholar]
- Torre-Amione G., Kapadia S., Lee J., Durand J. B., Bies R. D., Mann D. L., et al. (1996). Tumor necrosis factor-α and tumor necrosis factor receptors in the failing human heart. Circulation 93, 704–711. 10.1161/01.CIR.93.4.704 [DOI] [PubMed] [Google Scholar]
- Veeraraghavan R., Lin J., Hoeker G. S., Keener J. P., Gourdie R. G., Poelzing S. (2015). Sodium channels in the Cx43 gap junction perinexus may constitute a cardiac ephapse: an experimental and modeling study. Pflugers Arch. 467, 2093–2105. 10.1007/s00424-014-1675-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeraraghavan R., Poelzing S. (2008). Mechanisms underlying increased right ventricular conduction sensitivity to flecainide challenge. Cardiovasc. Res. 77, 749–756. 10.1093/cvr/cvm090 [DOI] [PubMed] [Google Scholar]
- Vleminckx K., Kemler R. (1999). Cadherins and tissue formation: integrating adhesion and signaling. Bioessays 21, 211–220. [DOI] [PubMed] [Google Scholar]
- Wagner S., Dybkova N., Rasenack E. C., Jacobshagen C., Fabritz L., Maier L. S., et al. (2006). Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Invest. 116, 3127–3138. 10.1172/JCI26620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H. F., Ding Y. J., Shen Y. W., Xue A. M., Xu H. M., Zhao Z. Q., et al. (2012). MicroRNA- 1 represses Cx43 expression in viral myocarditis. Mol. Cell. Biochem. 362, 141–148. 10.1007/s11010-011-1136-3 [DOI] [PubMed] [Google Scholar]
- Xu Q., Kopp R. F., Chen Y., Yang J. J., Roe M. W., Veenstra R. D. (2012). Gating of connexin 43 gap junctions by a cytoplasmic loop calmodulin binding domain. Am. J. Physiol. Cell Physiol. 302, C1548–C1556. 10.1152/ajpcell.00319.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J., Thomson J. K., Wu X., Zhao W., Pollard A. E., Ai X. (2014). Novel methods of automated quantification of gap junction distribution and interstitial collagen quantity from animal and human atrial tissue sections. PLoS ONE 9:e104357. 10.1371/journal.pone.0104357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. M., An J. (2007). Cytokines, inflammation, and pain. Int. Anesthesiol. Clin. 45, 27–37. 10.1097/AIA.0b013e318034194e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J., Hwang T. C., Adams H. R., Rubin L. J. (1997). Reduced L-type calcium current in ventricular myocytes from endotoxemic guinea pigs. Am. J. Physiol. 273(5 Pt 2), H2312–H2324. [DOI] [PubMed] [Google Scholar]
- Zhu Y., Mao Z., Lou D., Zhang H. (2000). The expression of connexin 43 and desmin in viral myocarditis. Zhonghua Bing Li Xue Za Zhi 29, 288–290. [PubMed] [Google Scholar]