

Abstract
Protein–protein interactions are essential for life. Yet, our understanding of the general principles governing binding is not complete. In the present study, we show that the interface between proteins is built in a modular fashion; each module is comprised of a number of closely interacting residues, with few interactions between the modules. The boundaries between modules are defined by clustering the contact map of the interface. We show that mutations in one module do not affect residues located in a neighboring module. As a result, the structural and energetic consequences of the deletion of entire modules are surprisingly small. To the contrary, within their module, mutations cause complex energetic and structural consequences. Experimentally, this phenomenon is shown on the interaction between TEM1-βlactamase and β-lactamase inhibitor protein (BLIP) by using multiple-mutant analysis and x-ray crystallography. Replacing an entire module of five interface residues with Ala created a large cavity in the interface, with no effect on the detailed structure of the remaining interface. The modular architecture of binding sites, which resembles human engineering design, greatly simplifies the design of new protein interactions and provides a feasible view of how these interactions evolved.
Keywords: cluster analysis, binding energy, protein–protein interaction, structure
The evolution of protein–protein binding sites is a result of optimization with three boundary conditions: the definition of the complex structure, optimization of the binding affinity for a given task, and the generation of specificity. Thermodynamic, kinetic, and structural studies of protein–protein interactions have taught us much of how proteins interact rapidly, tightly, and in a specific manner. These results have been translated into the development of in silico tools to calculate binding free energies, to weigh the energetic consequence of mutations, and for protein design (1–4). However, from the modest degree of success of the aforementioned methods, it is clear that our understanding of protein–protein interactions is still lacking (5).
The most popular approach for studying the energetic contribution of an amino acid to the free energy of binding (or stability) of proteins is to introduce a mutation and then measure the subsequent change in free energy (6). However, accurate interpretation of the results of such experiments is limited by the fact that the properties of a protein are a function of the entire system, and not necessarily the sum of its parts (7). As a result, the energetic impact of any given residue on binding is not a simple additive quantity (8). A more elaborate approach to study the contributions of amino acids to binding and stability involves the use of double and higher-order mutant cycles, where interactions between amino acids are treated within their native contexts (7, 9). Such cycles reveal whether the contributions from a pair of residues are additive, or whether the effects of mutations are coupled (10, 11).
Chakrabarti and Janin (12) have shown that small recognition sites between proteins (<2,000 Å2) are comprised of a single continuous patch, whereas large interfaces may be divided into several patches (with a distance threshold of >13 Å between patches). In the present study we further divide the interface based on the biochemical interactions made between all residues, as derived from the structure of the complex. We show that protein–protein binding sites have a modular architecture made up of clusters of residues with both strong intracluster connections and weak intercluster connections. The network of non-covalent interactions within or between proteins was defined based on an atomic distance threshold and the chemical properties of the involved groups. The networks obtained resemble “small world”-like features, which have been used to identify key residues in proteins, as well as the native conformations from nonnative decoys (13–16). As a model system, we use the interface between TEM1-β-lactamase (TEM1) and its protein inhibitor, β-lactamase inhibitor protein (BLIP). The structures of the complex (17) and of the unbound proteins (18, 19) have been determined to high resolution. The affinity of this interaction is in the nM range, with an association rate constant of ≈3 × 105 M-1·s-1 and a dissociation rate constant of 3 × 10-4 s-1, values which are commonly found for many protein–protein interactions (20). By means of cluster analysis, we divide the TEM1–BLIP interface into five clusters. Extensive multiple-mutant analysis of two of these clusters indicated that residue clusters are energetically independent of each other but have a high degree of cooperativity within each cluster. We further demonstrated that the deletion of a whole cluster of residues has no impact on the structure of the interface, whereas single mutations within a given cluster may lead to structural rearrangement of their cluster.
Materials and Methods
Protein Expression and Purification. Mutagenesis, expression and purification of TEM1 and BLIP were undertaken as described (20).
Kinetic Measurements. Kinetic constants were evaluated by surface plasmon resonance (SPR) detection by using a BIAcore 3000 (Uppsala) and a Laboratory SPR System (Proteoptics, Haifa, Israel). The Proteon F is a prototype biosensor that measures protein–protein interactions in a 6 × 6 format in real time yielding results of a quality similar to the BIAcore. Measurements were done in HBS (10 mM Hepes/3.4 mM EDTA/150 mM NaCl/0.05% surfactant P20, pH 7.4) at 25°C (20). For all measurements, TEM1 was immobilized to the sensor chip, and BLIP was the analyte, applied at six different protein concentrations. Binding curves were evaluated by using a simple one-to-one kinetic model. The change in free energy (ΔΔGKA) upon mutation was calculated from 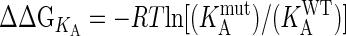 , with KA values being determined in two ways, the first (for interactions of kon < 5 × 106 M-1·s-1 and koff < 0.2 s-1) being the ratio of the kinetic constants (KA = koff/kon), and the (KA second [termed KA(ma)], by following the change in the refractive index (RU) at the equilibrium-binding signal and then fitting the data to the mass action expression RU = [C·KA(ma)·Rmax]/[C·KA(ma) + 1], where C represents the protein concentration. Values of ΔΔGKA (determined from koff/kon) and ΔΔGKA(ma) (determined by mass action) are highly correlated (with a correlation coefficient of 0.98; data not shown).
, with KA values being determined in two ways, the first (for interactions of kon < 5 × 106 M-1·s-1 and koff < 0.2 s-1) being the ratio of the kinetic constants (KA = koff/kon), and the (KA second [termed KA(ma)], by following the change in the refractive index (RU) at the equilibrium-binding signal and then fitting the data to the mass action expression RU = [C·KA(ma)·Rmax]/[C·KA(ma) + 1], where C represents the protein concentration. Values of ΔΔGKA (determined from koff/kon) and ΔΔGKA(ma) (determined by mass action) are highly correlated (with a correlation coefficient of 0.98; data not shown).
Crystallization and Structure Determination. Single crystals of both complexes were obtained by the microbatch method under oil by using the IMPAX 1–5 robot (Douglas Instruments, East Garston, Hungerford, Berkshire, U.K.). The proteins were crystallized at a concentration of 12 mg/ml. Crystals of the TEM-WT,BLIP(F142A) complex were grown from a precipitating solution of 100 mM Hepes (pH, 7.5), 30% wt/vol polyethylene glycol (PEG) 8000 and 2% vol/vol dioxane. Crystals formed in space group P212121, with cell constants a = 45.88 Å, b = 125.51 Å, and c = 158.76 Å, and contain one monomer in the asymmetric unit diffracting to 2.3 Å resolution. Crystals of BLIP(K74A,F142A,Y143A)–TEM1(E104A,Y105A) (KFYEY) multiple mutant complex, were grown from a precipitating solution of 50 mM sodium acetate (pH, 5), 10% PEG 6000, and 0.1 M LiCl. Crystals formed in space group P212121, with cell constants a = 45.71 Å, b = 124.47 Å, and c = 156.95 Å, and contain one monomer in the asymmetric unit diffracting to 1.9 Å resolution. Details of the data collection and analysis are described in Table 2, which is published as supporting information on the PNAS web site. The structures were solved by molecular replacement using the program molrep, by using the 1.73-Å refined structure of the complex (1jtg in the Brookhaven Data Bank, Protein Data Bank) as a model. Atomic refinement was carried out with the program cns (21). The atomic coordinates of TEM-WT,BLIP (F142A) and of KFYEY multiple mutant complex have been deposited in the Protein Data Bank (PDB ID codes 1S0W and 1XXM, respectively).
Clustering. Interactions between residues are defined by using the csu software package (22) (http://bip.weizmann.ac.il/oca-bin/lpccsu). Given a specific protein's structure as input, the software produces a list of interatomic interactions and their distance. The csu program further divides the atoms involved into categories, based on their biochemical properties. We considered only those interactions that fall within a predefined threshold and chemistry (Table 3, which is published as supporting information on the PNAS web site). Different thresholds gave a less clear yet similar picture of clusters. Two residues are defined as interacting if their atoms are interacting. Only sequence-specific interactions are counted; backbone–backbone interactions were not included, because they are not sequence-specific. We assigned a weight of 1 to atomic backbone–side chain interactions, and a weight of 2 for sidechain–sidechain interactions. The weight of any given interaction is defined by the sum of the (weighted) atomic interactions. We can thus define the network, or weighted graph, of interactions as G = (V,E,W), where V, the set of vertices (or nodes) is defined as the residues in the interface; E, the edges, constitute the set of interactions (both inter- and intrachain interactions); and the weight function W gives each edge its weight, as defined above.
For clustering, we used the superparamagnetic clustering tool, spc 3.3.13 (23, 24) which was previously used to cluster gene expression networks. The input to spc is a distance matrix representation of the network. The order of separation of the nodes is summarized in a dendogram. spc further calculates the stability parameter for each possible residue set, represented by its branch length (Fig. 1B).
Fig. 1.

Cluster analysis of the TEM1–BLIP interface. The interactions between residues located within the interface were extracted by using the csu package (22) [for parameters, see Table 3; clustered with the spc 3.3.13 tool (23)]. The interface was divided into five clusters of interactions, shown in A as a connectivity map, with the dendogram given in B, where the final nodes are the residues. A minimum of three residues is needed to form a cluster. The black lines indicate two residue interactions. (C) The location of the clusters is marked on the protein surfaces. An enlarged view of the two clusters (C1 and C2) is shown in D and includes the four water molecules separating the two clusters. The same color-coding is preserved throughout Fig. 1. In A, red squares mark BLIP residues, and blue circles mark TEM1 residues. In D, blue residues are for TEM1 and yellow for BLIP.
Results
The TEM1–BLIP Interface as a Cluster Aggregate. Traditionally, the interface of protein–protein interactions is described and analyzed as a network of interactions between residues of proteins A and B. In the present study, we used standard clustering techniques to separate the network into binding units or clusters. The bond definitions, along with the clustering tool, were specified in Materials and Methods. We define a cluster as including at least three residues that form a continuous network. The clusters are semiindependent of one another, having a maximum of a single weak edge connecting them. The contact map of the interface, indicating the five clusters, is shown in Fig. 1 A. The width of each edge relates to the interaction weight whereas the length is arbitrary. Clusters were extracted according to the stability parameters, calculated by the spc (Fig. 1B). Using the same color scheme, the five clusters are superimposed on the binding surfaces of TEM1 and BLIP (Fig. 1C). A magnification of the two clusters, which were experimentally analyzed, is shown in Fig. 1D.
Free Energy Relationships Within and Between Clusters. To determine inter and intracluster relations of mutations on the free energy of binding, most combinations of residues located in clusters C1 and C2 were analyzed. C1 includes BLIP D49 and the four TEM1 residues S130, S235, R243, and K234. C2 includes three BLIP residues (K74, F142, and Y143) and four TEM1 residues (Y105, E104, and N170) (Gly and Ala are ignored because their side chains cannot be deleted by mutation). The two clusters are adjacent to one another, with four water molecules being located between them (Fig. 1D). Mutant proteins (to Ala) were prepared to include the single mutations, as well as the double and multiple mutations of all residues located on each of the two clusters. The effects of mutations on the free energy of binding, from a single mutation located on one of the proteins and up to the deletion of whole clusters, were measured by means of surface plasmon resonance. ΔΔGKA(ma) values for all combinations of TEM1–BLIP mutants of C2, and of mutations of residues located both in C1 and C2, are given in Table 1. ΔΔGKA values for mutations involving only C1 are taken from Albeck et al. (8).
Table 1. Changes in binding free energies for TEM1 and BLIP upon mutation.
| TEM1 mutants
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| BLIP mutants | WT | E104A* | Y105A* | E104A, Y105A* | R243A† | K234A† | S130A† | SSR† | KSSR† |
| WT | 0.0 | 6.5 | –0.7 | 4.3 | 5.3 | 4.3 | 1.4 | 7.3 | 10.2 |
| D49A† | 7.5 | 4.6 | 6.7 | 1.5 | 3.7 | 7.1 | |||
| K74A* | 14.9 | 6.6 | 13.9 | 5.8 | 27.7 | 27.4 | 13.3 | 21.0 | 22.9 |
| F142A* | 8.8 | 11.5 | 2.9 | 6.3 | 14.2 | 14.2 | 11.9 | 14.8 | 17.9 |
| Y143A* | 1.6 | 7.8 | 4.5 | 8.6 | 9.5 | 10.7 | 7.5 | 11.1 | 15.4 |
| K74,F142A* | 20.3 | 13.1 | 20.0 | 13.1 | 23 | 24.4 | 21.6 | 24.3 | 23.7 |
| K74A,Y143A* | 12.8 | 4.4 | 14.2 | 7.4 | 18.3 | 20.4 | 15.6 | 18.4 | 27.7 |
| F142A,Y143A* | 11.9 | 12.1 | 12.5 | 11.9 | 19.0 | 19.6 | 17.4 | 20.0 | 20.4 |
| K74A,F142A,Y143A* | 16.3 | 10.0 | 17.7 | 10.1 | 22.0 | 22.9 | 21.2 | 23.2 | 22.9 |
Binding energies (in kJ/mol) were measured with TEM1 proteins immobilized to the sensor chip. The average standard deviation in the value of ΔΔGKA(ma) is ± 0.5 kJ/mol. KA(ma) is the equilibrium affinity constant derived by action mass analysis (see Materials and Methods). The change in free energy upon mutation, ΔΔGKA is calculated from RT In (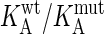 ) in kJ/mol. SSR protein, TEM1 mutated to S130A, S235A, and R243A. KSSR protein, TEM1 mutated to S130A, S235A, R243A, and K234A. Underlined in bold are the two multiple mutants where entire clusters were removed.
) in kJ/mol. SSR protein, TEM1 mutated to S130A, S235A, and R243A. KSSR protein, TEM1 mutated to S130A, S235A, R243A, and K234A. Underlined in bold are the two multiple mutants where entire clusters were removed.
Mutations located in C2
Mutations located in C1
Interactions Within a Cluster Are Nonadditive. The additivity of ΔΔG of multiple mutations was determined by plotting the experimentally determined changes in free energy (ΔΔG) versus the sum of ΔΔG values of the individual mutations. Analyzing the additivity of mutations of residues located within one cluster on both TEM1 and BLIP (Fig. 2A) shows that most combinations of mutations are subadditive. In other words, the sum of ΔΔG values of the individual mutations is much larger than the value measured for the multiple mutant. This effect was demonstrated at its extreme when an entire cluster was mutated to Ala. Summing up the loss of free energy of binding of the five single mutants of C2 [BLIP(K74A,F142A,Y143A) and TEM1(E104A,Y105A)] yields a value of 31.1 kJ/mol. This number is composed of an additive loss of 25.3 kJ/mol for the three BLIP mutants, and 5.8 kJ/mol for the two TEM1 mutants. (The mutation N170A on TEM1 had no effect on binding in all combinations tested, and therefore was ignored). The ΔΔG of the triple BLIP mutant was found to be 16.3 kJ/mol, and that of the double-TEM1 mutant, 4.3 kJ/mol. Removing all five residues simultaneously (by mutation to Ala) resulted in a loss of only 10.1 kJ/mol of binding free energy. Even the single BLIP mutation K74A destabilized the complex to a greater extent than the multiple C2 mutant KFYEY (Table 1). The same phenomenon was detected for C1. Here, the additive loss in binding energy was found to be 28 kJ/mol, of which 7.5 kJ/mol came from the BLIP (D49A) mutant, and 20.5 kJ/mol, from the sum of the four single TEM1 mutants. Mutating the whole C1 cluster to Ala reduced the free energy of binding by only 7.1 kJ/mol.
Fig. 2.

Additivity of free energy of binding between mutations on TEM1 and BLIP. Additive ΔΔG is defined as ΔΔGmut1 + ΔΔGmut2, plotted versus the experimentally determined values of the same mutations applied simultaneously (ΔΔGmut1,mut2). (A) Additivity of mutations located on TEM1 and BLIP and in the same cluster (either C1 or C2). (B) Additivity of mutations located on both proteins, and on different clusters (C1 in one protein, and C2 in the second). (C) Additivity of mutations located on the same protein and in the same cluster.
The Binding Energies of C1 and C2 Are Additive with Respect to One Another. What is the energy relationship between residues located on two independent clusters that are structurally adjacent? Are they independent binding units, in which case mutations will be additive, or is the interface a continuum? To answer this question, we determined the additivity of intercluster mutants (Fig. 2B). In stark contrast to the intracluster mutations, all 23 tested combinations of intercluster mutations were additive. These measurements were between five different TEM1 mutant proteins in C1 [the three single mutations S130A, K234A, and R243A; and the two multiple mutations (K243A-S130A,S235A,R243A) and (R243A,S130A,S235A)] and the seven BLIP mutants in C2 [the three single mutants K74A, F142A, and Y143A; and four multiple mutants (K74A,F142A), (F142A,Y143A), (K74A,Y143A), and (K74A,F142A,Y143A)]. Although the clusters are in close structural proximity, they are energetically independent (Figs. 1 C and D and 2B). In other words, mutations in C1 do not affect residue in C2, and vice versa.
In a third set of experiments, the additivity of multiple mutations located on one protein and within the same cluster was measured and compared with the sum of the individual mutations (Fig. 2C). In this case, the degree of additivity obtained is between those shown in Fig. 2 A and B. In other words, mutations located on the same protein and within the same cluster are more additive than mutations mapped to one cluster but located on both proteins forming the complex, and less additive than mutations located on two separate clusters.
Effects of Mutations on the Structure of the Binding Site. To better understand the complex energetic relationships between mutations located within the same cluster, relative to the simple additivity of mutations located on different clusters, we determined two mutant structures: that of the single BLIP mutant F142A (in C2) in complex with WT TEM, and that of the multiple KFYEY mutant where, in essence, cluster 2 is completely eliminated. Fig. 3A shows the TEM1–BLIP binding interface of the KFYEY mutant structure, overlaid on the WT complex interface. The two structures are basically identical, except for the deletion of the five mutated side-chains. [All-atom rms deviation (rmsd) between the interfaces of the WT versus the KFYEY structures is 0.37 Å.] The deletion of C2 results in the creation of a large hole within the interface (Fig. 3B). Interestingly, this hole is not filled with structural water, and the residues surrounding the hole maintain the same structure as in the wild type.
Fig. 3.

TEM1–BLIP mutant structures, as determined by x-ray crystallography. (A and B) The structure of the multiple mutant BLIP(K74A,F142A,Y143A) in complex with TEM1(E104A,Y105A) (PDB ID code 1XXM). (A) An overlay of the interface residues (green is the WT, and blue is of the mutant structure), with the mutated residues marked in red. (B) A surface representation of the KFYEY mutant complex, with the hole in the interface field by the wild-type residues superimposed in green. (C) BLIP(F142A) in complex with WT TEM1 (green; PDB ID code 1SOW) in comparison with the WT structure of the complex (magenta; PDB ID code 1JTG) and the two protein structures as determined in their unbound state (purple; PDB ID code 1BTL for TEM1; see ref. 18 for BLIP). T marks residues of TEM1 and B of BLIP. The distance between TEM Glu-104 and BLIP Lys-74 is marked for the WT and BLIP(F142A) complex structures.
The structure of the single mutant F142A of BLIP (located at the center of C2) reveals a more complex picture (Fig. 3C). This structure shows a number of local perturbations around the mutated F142A residue. For example, TEM1 Y105 flips away from its place in C2 and takes on a new role as part of C4. Secondly, small but significant movements of both K74(BLIP) and E104(TEM1) result in an unraveling of this important salt bridge. The distance between these two residues is increased from 2.68 Å in the WT proteins to 4.45 Å in the F142A mutant (Fig. 3C). Overall, the whole organization of C2 seems to be affected by the F142A mutation. Analyzing this mutant structure in comparison with the unbound TEM1 and BLIP structures (18, 19) provides interesting insights into the cause of the observed structural movements. The conformation of Y105 of TEM1 in the mutant complex is similar to that found in unbound protein (Fig. 3C). Moreover, both E104 of TEM1 and K74 of BLIP move outwards in the unbound structures, similar to what is seen in the F142A mutant structure. Thus, the F142A mutant structure seems to suggest that the energetic importance of F142 in stabilizing the complex is related to its role in structuring C2. The structure of the complex of TEM-WT with BLIP (F142A) provides a logical explanation for the chaotic energetic picture of this mutant, which is very much dependent on its environment (Table 1). For example, ΔΔG of F142A alone is 8.8 kJ/mol, but ΔΔG of the double mutant BLIP(F142A),TEM(Y105A) is only 2.9 kJ/mol. Furthermore, the ΔΔG of the triple BLIP mutant (F142A,E104A,Y105A) binding TEM-WT is only 6.3 kJ/mol (in comparison with an additive value of 14.7 kJ/mol for the three mutations independently).
Discussion
In the present study we demonstrate that a protein–protein interface is built in a modular fashion. Each cluster of residues represents a module that can be removed or changed with retention of the quaternary structure, and with a relatively small loss in affinity. An interface cluster is herein defined as a group of amino acids that is mainly interconnected (i.e., very few connections are made with residues outside the cluster). The cluster map is produced by clustering the bond information obtained from interatomic contacts, as defined by distance and chemical composition.
Five such clusters were identified for the TEM1–BLIP complex. Of these, two proximate clusters, C1 and C2, were thoroughly tested for inter- and intracluster relationships. Our results clearly showed intracluster cooperativity, as well as intercluster additivity. Subadditivity for intracluster mutations was found to be more pronounced when the probed residues are located on different proteins, and weaker (although significant) when they are on the same protein (compare Fig. 2 A with C). Yet, with respect to binding, the existence of partial nonadditivity between residues on the same protein strongly suggests that amino acids within a cluster are organized in a cooperative manner.
The method most commonly used to analyze the contribution of residues toward the stability of a protein–protein complex involves evaluating the loss in free energy of binding upon mutation (6). However, this method is not without problems, because the loss in the measured free energy of binding caused by single mutations in C1 and C2 equals, or even exceeds, that of removing the entire cluster. The sum of the loss in free energy of all of the single mutations within a cluster exceeds by far (up to 4-fold) the loss in free energy generated when all of the residues of the cluster are mutated simultaneously. Comparing the structure of the BLIP(F142A) mutant binding TEM1-WT with the structure of the C2 deletion mutant KFYEY provides some interesting insights into this phenomenon. The single mutation causes a structural relaxation within its cluster whereas, in the KFYEY mutant structure, a large hole is created in place of the original residues, but no underlying structural changes are noted. It seems, therefore, that the energetic effect of many single mutations is larger than their net contribution, due to a penalty paid by leaving the rest of the cluster behind. Once the entire cluster is mutated, the change in binding free energy is related to the simple lack of those interactions.
Based on our results, can any generalizations be made concerning the cluster composition of a binding site? The cluster composition of four additional protein complexes with diverse affinities [from 2 μM for the CheA–Chey complex (25) to nanomolar for both the human growth hormone–receptor and Ran–importin-β complexes (26, 27) and 0.01 pM affinity measured for barnase/barstar (28)] is presented in Fig. 4. The cluster composition of the two nM affinity binding complexes resembles that of TEM1–BLIP (Fig. 1), with the binding site being composed of a number of medium-sized clusters. The weak interaction between CheA and Chey is characterized by relatively few contacts between the two proteins, with only one medium-sized developed cluster. On the other side of the spectrum of affinities, we have barnase-barstar. Although its interface is composed of only two clusters, it is obvious that at least the larger of the two is a highly developed cluster that forms a very elaborate network of interactions. Thus, the degree of connectivity is much higher in this interface, compared with those of the other four complexes analyzed. This result is intriguing and suggests that the extent of connectivity, rather than the size of the interface, is the driving force behind tight binding. A protein–protein binding site is not just a conglomerate of proximate residues forming interactions; rather, it has a high degree of internal organization. This organization may be necessary if the two proteins are to bind to each other in an aqueous solution. Indeed, computerized docking simulations have shown multiple binding solutions that bury at least as much surface compared with that measured for the real complex, but don't generate binding (29–33). Thus, binding seems to be a result of higher organization of the binding sites, and not just of surface complementarity.
Fig. 4.

Cluster analysis of four protein–protein interactions with diverse affinities (μM for CheA/CheY; nanomolar for human growth hormone–receptor and Ran–importin-β; and 0.01 pM for barnase/barstar).
So why aren't all interfaces organized as tightly as barnase and barstar? The answer may be found in the inherent difficulty of maintaining this high level of organization, which requires a constant, strong driving force. The case of barnase-barstar is indeed unique, as the penalty for not having a tight binding barstar inhibitor is instant death of the bacteria (resulting from the RNase activity of barnase) (34). With such strong pressure, a very highly ordered binding site can be maintained. On the other hand, the construction of a binding site from a number of well-organized clusters is a much easier solution to provide cooperativity on one hand, but without the need to simultaneously organize the structure of the entire interface. An interface architecture that is modular in nature facilitates the adaptation of proteins to evolutionary pressure, because only small units rather than whole binding sites have to change, to modulate specificity or binding affinity. A modular architecture provides the underlying flexibility necessary if different protein partners are to be bound to one another with high specificity at overlapping binding sites, as is indeed observed.
The loss in free energy of binding caused by deleting C1 and C2 is 7.1 and 10.1 kJ/mol, respectively, which constitutes 36% of the free energy of the entire TEM1–BLIP complex (47.5 kJ/mol). With the energetics of clusters being additive, it will be interesting to see to what extent the other three interface clusters contribute to TEM1–BLIP binding. These data will provide corroboration of the entropic cost of the complex formation, which is estimated from theoretical considerations to be ≈60 kJ/mol (35). Thus, the cluster view not only explains the evolution of high affinity and specificity but also may be used to simplify energy calculations and to create interface design.
Supplementary Material
Acknowledgments
We thank Dr. Kay Gottschalk and Dr. Tal Peleg-Shulman for their critical comments and help, Renne Abramovich for her assistance in the purification of some of the proteins, the Israel Structural Proteomics Center (ISPC) for doing the crystallization and solving the structures, and the staff of Proteoptics for their collaboration. This research was funded by Minerva Grant 8444.
Author contributions: G.S. designed research; D.R., O.R., S.A., R.M., and O.D. performed research; D.R., O.R., O.D., and G.S. analyzed data; and G.S. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: KFYEY, BLIP(K74A,F142A,Y143A)–TEM1(E104A,Y105A) multiple mutant complex; TEM1, TEM1-β-lactamase; BLIP, β-lactamase inhibitor protein; C1, cluster 1; C2, cluster 2.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 1S0W and 1XXM).
References
- 1.Selzer, T., Albeck, S. & Schreiber, G. (2000) Nat. Struct. Biol. 7, 537-541. [DOI] [PubMed] [Google Scholar]
- 2.Kortemme, T., Joachimiak, L. A., Bullock, A. N., Schuler, A. D., Stoddard, B. L. & Baker, D. (2004) Nat. Struct. Mol. Biol. 11, 371-379. [DOI] [PubMed] [Google Scholar]
- 3.Looger, L. L., Dwyer, M. A., Smith, J. J. & Hellinga, H. W. (2003) Nature 423, 185-190. [DOI] [PubMed] [Google Scholar]
- 4.Guerois, R., Nielsen, J. E. & Serrano, L. (2002) J. Mol. Biol. 320, 369-387. [DOI] [PubMed] [Google Scholar]
- 5.Bosshard, H. R., Marti, D. N. & Jelesarov, I. (2004) J. Mol. Recognit. 17, 1-16. [DOI] [PubMed] [Google Scholar]
- 6.Clackson, T. & Wells, J. A. (1995) Science 267, 383-386. [DOI] [PubMed] [Google Scholar]
- 7.Horovitz, A. (1996) Folding Des. 1, 121-126. [DOI] [PubMed] [Google Scholar]
- 8.Albeck, S., Unger, R. & Schreiber, G. (2000) J. Mol. Biol. 298, 503-520. [DOI] [PubMed] [Google Scholar]
- 9.Carter, P. J., Winter, G., Wilkinson, A. J. & Fersht, A. R. (1984) Cell 38, 835-840. [DOI] [PubMed] [Google Scholar]
- 10.Hidalgo, P. & MacKinnon, R. (1995) Science 268, 307-310. [DOI] [PubMed] [Google Scholar]
- 11.Roisman, L. C., Piehler, J., Trosset, J. Y., Scheraga, H. A. & Schreiber, G. (2001) Proc. Natl. Acad. Sci. USA 98, 13231-13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chakrabarti, P. & Janin, J. (2002) Proteins 47, 334-343. [DOI] [PubMed] [Google Scholar]
- 13.Kannan, N. & Vishveshwara, S. (1999) J. Mol. Biol. 292, 441-464. [DOI] [PubMed] [Google Scholar]
- 14.Vendruscolo, M., Dokholyan, N. V., Paci, E. & Karplus, M. (2002) Phys. Rev. E. Stat. Nonlin. Soft Matter Phys. 65, 061910. [DOI] [PubMed] [Google Scholar]
- 15.Samudrala, R. & Moult, J. (1998) J. Mol. Biol. 279, 287-302. [DOI] [PubMed] [Google Scholar]
- 16.Dokholyan, N. V., Li, L., Ding, F. & Shakhnovich, E. I. (2002) Proc. Natl. Acad. Sci. USA 99, 8637-8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strynadka, N. C., Jensen, S. E., Alzari, P. M. & James, M. N. (1996) Nat. Struct. Biol. 3, 290-297. [DOI] [PubMed] [Google Scholar]
- 18.Strynadka, N. C., Jensen, S. E., Johns, K., Blanchard, H., Page, M., Matagne, A., Frere, J. M. & James, M. N. (1994) Nature 368, 657-660. [DOI] [PubMed] [Google Scholar]
- 19.Jelsch, C., Mourey, L., Masson, J.-M. & Samama, J.-P. (1993) Proteins Struc. Funct. Genet. 16, 364-383. [DOI] [PubMed] [Google Scholar]
- 20.Albeck, S. & Schreiber, G. (1999) Biochemistry 38, 11-21. [DOI] [PubMed] [Google Scholar]
- 21.Brunger, A. T., Adams, P. D., Clore, G., M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D Biol. Crystallogr. 54, 905-921. [DOI] [PubMed] [Google Scholar]
- 22.Sobolev, V., Sorokine, A., Prilusky, J., Abola, E. E. & Edelman, M. (1999) Bioinformatics 15, 327-332. [DOI] [PubMed] [Google Scholar]
- 23.Blatt, M., Wiseman, S. & Domany, E. (1996) Phys. Rev. Lett. 76, 3251-3254. [DOI] [PubMed] [Google Scholar]
- 24.Blatt, M., Wiseman, S. & Domany, E. (1997) Neural Comput. 9, 1805-1842. [Google Scholar]
- 25.Li, J., Swanson, R. V., Simon, M. I. & Weis, R. M. (1995) Biochemistry 34, 14626-14636. [DOI] [PubMed] [Google Scholar]
- 26.Cunningham, B. C. & Wells, J. A. (1993) J. Mol. Biol. 234, 554-563. [DOI] [PubMed] [Google Scholar]
- 27.Villa Braslavsky, C. I., Nowak, C., Gorlich, D., Wittinghofer, A. & Kuhlmann, J. (2000) Biochemistry 39, 11629-11639. [DOI] [PubMed] [Google Scholar]
- 28.Schreiber, G. & Fersht, A. R. (1993) Biochemistry 32, 5145-5150. [DOI] [PubMed] [Google Scholar]
- 29.Vakser, I. A., Matar, O. G. & Lam, C. F. (1999) Proc. Natl. Acad. Sci. USA 96, 8477-8482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang, C., Chen, J. & DeLisi, C. (1999) Proteins 34, 255-267. [PubMed] [Google Scholar]
- 31.Camacho, C. J., Gatchell, D. W., Kimura, S. R. & Vajda, S. (2000) Proteins 40, 525-537. [DOI] [PubMed] [Google Scholar]
- 32.Fernandez-Recio, J., Totrov, M. & Abagyan, R. (2004) J. Mol. Biol. 335, 843-865. [DOI] [PubMed] [Google Scholar]
- 33.Gray, J. J., Moughon, S., Wang, C., Schueler-Furman, O., Kuhlman, B., Rohl, C. A. & Baker, D. (2003) J. Mol. Biol. 331, 281-299. [DOI] [PubMed] [Google Scholar]
- 34.Hartley, R. W. (1989) Trends Biochem. Sci. 14, 450-454. [DOI] [PubMed] [Google Scholar]
- 35.Janin, J. (1997) Proteins 28, 153-161. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.