

ABSTRACT
Germ line mutations of the gene encoding the tricarboxylic acid (TCA) cycle enzyme fumarate hydratase (FH) cause a hereditary cancer syndrome known as hereditary leiomyomatosis and renal cell cancer (HLRCC). HLRCC-associated tumors harbor biallelic FH inactivation that results in the accumulation of the TCA cycle metabolite fumarate. Although it is known that fumarate accumulation can alter cellular signaling, if and how fumarate confers a growth advantage remain unclear. Here we show that fumarate accumulation confers a chronic proliferative signal by disrupting cellular iron signaling. Specifically, fumarate covalently modifies cysteine residues on iron regulatory protein 2 (IRP2), rendering it unable to repress ferritin mRNA translation. Simultaneously, fumarate increases ferritin gene transcription by activating the NRF2 (nuclear factor [erythroid-derived 2]-like 2) transcription factor. In turn, increased ferritin protein levels promote the expression of the promitotic transcription factor FOXM1 (Forkhead box protein M1). Consistently, clinical HLRCC tissues showed increased expression levels of both FOXM1 and its proliferation-associated target genes. This finding demonstrates how FH inactivation can endow cells with a growth advantage.
KEYWORDS: ferritin, FH, FOXM1, fumarate, HLRCC, NRF2
INTRODUCTION
Hereditary leiomyomatosis and renal cell cancer (HLRCC) patients carry a germ line-inactivating mutation in one of the fumarate hydratase (FH) alleles and are prone to developing skin leiomyomas, uterine fibroids, and renal cell carcinoma of type 2 papillary morphology (1). HLRCC-associated tumors harbor a loss of heterozygosity at the FH locus, indicating biallelic FH inactivation as the tumor-initiating event (1). However, it remains unclear how FH inactivation drives carcinogenesis.
The most direct consequence of FH inactivation is intracellular fumarate accumulation. Accumulated fumarate can covalently modify cysteine residues of proteins in an uncatalyzed process termed succination and cause many alterations in cellular signaling (2). Succination in HLRCC cells was first discovered on Kelch-like ECH-associated protein 1 (KEAP1), a negative regulator of the nuclear factor (erythroid-derived 2)-like 2 (NRF2) transcription factor (3). Since HLRCC is driven by FH inactivation, chronic succination of KEAP1 results in constitutive NRF2 activation and increased expression of its target genes (3). Besides KEAP1, the Krebs cycle enzyme aconitase 2 (Aco2) was reported to be a succination target in Fh knockout mouse tissues, and succination inhibited its activity (2). Despite dramatic cellular changes induced by protein succination, it remains unclear whether succination contributes to FH inactivation-driven carcinogenesis.
The expansion of FH-inactivated cells into a tumor mass indicates that FH inactivation somehow endows the cells with a chronic proliferative signal, which is a fundamental hallmark of cancer (4). While the mechanisms by which cancer cells with oncogenic mutations in growth signaling genes acquire such a signal are clear, how the loss of a tricarboxylic acid (TCA) cycle enzyme induces proliferative signaling is enigmatic. Analogous to chronic exposure to an electrophilic carcinogen, FH-inactivated cells are chronically exposed to high levels of intracellular fumarate. Thus, understanding the mechanisms by which FH inactivation contributes to carcinogenesis has profound implications in cancer biology.
RESULTS
Fumarate accumulation confers a chronic proliferative signal in HLRCC cells.
To determine whether fumarate accumulation confers a chronic proliferative signal, we created a pair of isogenic FH-reconstituted (FHres) and control (FH−/−) HLRCC cell lines by stably transducing the well-characterized HLRCC cell line UOK262 (5) with functional Flag-tagged FH (UOK262-FHres) or with the corresponding empty vector control (UOK262-FH−/−). Since increased NRF2 protein levels in HLRCC cells are a direct consequence of KEAP1 succination, the NRF2 protein level and the upregulation of its target genes serve as good indicators of cellular protein succination in these cells (3). Expectedly, FH reconstitution resulted in decreased protein levels of NRF2 and of the NRF2 target gene AKR1B10, indicating that it drove a reduction in the intracellular fumarate concentration and therefore decreased protein succination (Fig. 1A). Using this isogenic pair of HLRCC cell lines in a cell proliferation assay, we found that FH-reconstituted UOK262-FHres cells had a significantly lower proliferation rate than did control UOK262-FH−/− cells (Fig. 1B). This reduced growth rate can be abrogated by the addition of a membrane-permeable form of fumarate, monomethyl fumarate (MMF) (Fig. 1C), supporting that fumarate promotes cell proliferation in FH-inactivated cells. Thus, we sought to determine the mechanism by which FH inactivation promotes cell proliferation.
FIG 1.

Fumarate accumulation confers chronic proliferation. (A) Western blots of NRF2 and its transcription target AKR1B10 after reintroduction of FH into the FH−/− HLRCC tumor cell line UOK262. β-Actin (ACTB) was used as a loading control. (B) Cell viability measured by formazan production in UOK262 cells (FH−/−) after reintroduction of FH (UOK262-FHres). Data are presented as means ± standard deviations of results from a representative experiment. Curves were statistically significantly different, as determined by two-way analysis of variance, for the FH genotype (P < 0.001) but not for time (P > 0.05). (C) Cell viability measured by formazan production in UOK262 cells (FH−/−) after reintroduction of FH (UOK262-FHres) and 80 μM MMF treatment. Data are presented as means ± standard deviations of results from a representative experiment. Curves were statistically significantly different, as determined by two-way analysis of variance, for MMF treatment (P < 0.001) but not for time (P > 0.05).
Fumarate accumulation increases intracellular ferritin levels.
We focused our investigation on protein succination, as it is one of the most prominent cellular changes induced by FH inactivation (6). To date, the succination of two proteins in the context of fumarate accumulation has been described in detail. These two proteins are KEAP1, which was identified in HLRCC, and Aco2, which was identified in Fh knockout mouse tissue. Succination of KEAP1 resulted in NRF2 activation, while succination of Aco2 inhibited its aconitase activity (2, 3). Aco2 belongs to a family of proteins that includes iron regulatory protein 1 (IRP1) (also known as aconitase 1 [ACO1]) and IRP2 (also known as ACO3) (7). Both IRP1 and IRP2 (collectively denoted IRPs) play central roles in cellular iron signaling, with IRP2 exerting a dominant effect (8). Interestingly, both NRF2 and IRPs interplay to regulate the expression of the ferritin light chain (FTL) and the ferritin heavy chain (FTH1) genes (9). These genes encode different subunits of ferritin, which is a cellular iron storage protein recently shown to promote cancer cell proliferation (10). Specifically, NRF2 promotes FTL and FTH1 transcription, while IRP2 represses the translation of the resulting transcripts by binding to a hairpin structure located in the 5′ untranslated region (UTR) known as an iron response element (IRE) (Fig. 2A). Thus, we hypothesized that fumarate accumulation in FH-inactivated cells drives a chronic growth signal by increasing ferritin expression in a concerted manner: it increases ferritin gene transcription through NRF2 activation and increases ferritin mRNA translation by inhibiting IRP2 (Fig. 2A).
FIG 2.

Fumarate accumulation increases ferritin levels. (A) Ferritin transcriptional and translational regulation. NRF2 can transcribe the FTL and FTH1 genes. IRP2 can bind FTL and FTH1 mRNA iron response element hairpins in the 5′ untranslated regions to repress translation. (B) Western blots of HK2 cells show relative increases in FTL and FTH1 protein levels upon siRNA-mediated FH knockdown. ACTB was used as a loading control. (C) Western blots of HEK293 cells show relative increases in FTL and FTH1 protein levels upon CRISPR-Cas9-mediated FH knockout. Increased NRF2 levels indicate a stable knockout. ACTB was used as a loading control. (D) Western blots of FH-rescued (FHres) and control (FH−/−) UOK262 cells show increased FTL and FTH1 levels under conditions of nonfunctional FH. ACTB was used as a loading control. (E) Addition of 80 μM MMF or 40 μM DMF to HEK293 cells increases FTL and FTH1 protein levels. ACTB was used as a loading control.
To determine the effects of FH inactivation on the intracellular ferritin level, we transiently knocked down FH in an immortalized human kidney epithelial cell line, HK2. Knockdown resulted in increased intracellular FTL and FTH1 protein levels (Fig. 2B), indicating that FH inactivation increases the cellular ferritin level. Since the effects of transient small interfering RNA (siRNA)-mediated knockdown could be different from those of biallelic FH inactivation, we further tested the observed effects by creating FH knockout HEK293 cells using clustered regularly interspaced short palindromic repeat (CRISPR) gene-editing technology. The knockout was confirmed by Sanger sequencing (see Fig. S1A and S1B in the supplemental material) and immunoblotting (Fig. S1C). Consistently, the knockout also resulted in higher FTL and FTH1 protein levels (Fig. 2C). Moreover, the reconstitution of functional FH in UOK262 HLRCC cells decreased FTL and FTH1 protein levels (Fig. 2D), supporting that FH inactivation increases the cellular ferritin level. Since the most direct consequence of FH inactivation is intracellular fumarate accumulation, we proceeded to simulate fumarate accumulation in HEK293 cells by treating them with two different membrane-permeable forms of fumarate: MMF and dimethyl fumarate (DMF). Treatments with either DMF or MMF led to increased FTL and FTH1 protein levels (Fig. 2E), supporting that fumarate accumulation increases the intracellular ferritin level.
IRP2 is a succination target.
To date, the only aconitase family member reported to be a succination target has been mouse Aco2 (2). Multiple-protein-sequence alignment of mouse Aco2 with human IRP1 and IRP2 revealed that the succination sites identified in mouse Aco2 are conserved in both human IRP1 and IRP2, suggesting that these proteins may be succination targets as well (Fig. 3A). We focused our investigation on IRP2 because it is the functionally dominant member (8). Using tandem mass spectrometry (MS/MS) analyses, we identified multiple succinated cysteine residues on ectopically expressed Flag-tagged IRP2 immunoprecipitated from DMF-treated HEK293 cells, supporting human IRP2 as a succination target (Fig. 3B; see also Table S1 in the supplemental material). However, peptides derived from the regions of IRP2 spanning residues C512 to C516 and C578 to C581 were not detected in this analysis, as the peptides generated by trypsin digestion were too large to be identified by using our proteomics platform. To more closely interrogate these regions, we generated an IRP2-A523R mutant, which introduced an additional tryptic digestion site. Tandem mass spectrometry analyses of IRP2-A523R revealed that C512 and C581 are succinated. These residues are conserved between human IRP2 and mouse Aco2 (Fig. 3C and Table S2). C512 was previously shown to be critical in maintaining the translational repression function of IRP2 (11). Thus, succination of IRP2 may partly contribute to the increased ferritin protein levels in FH-inactivated cells.
FIG 3.

IRP2 is a succination target. (A) Multiple-sequence alignment of mouse Aco2, human IRP2 (ACO3), and IRP1 (ACO1). Succination sites reported for Fh−/− mouse Aco2 are conserved in human IRP1 and IRP2. These sites are highlighted in red. (B) Adduct sites on Flag-tagged wild-type IRP2, as determined by LC-MS/MS. Black bars represent peptides detected by mass spectrometry analysis. (C) Adduct sites on Flag-tagged IRP2-A523R, as determined by LC-MS/MS. Black bars represent peptides detected by mass spectrometry analysis.
Fumarate inhibits IRP-mediated translational repression.
To quantify the effects of fumarate on IRP-mediated FTL and FTH1 translational repression, we developed constructs that allow the expression of a chimeric gene consisting of a firefly luciferase open reading frame carrying either the 5′ UTR of FTL (pECE-FTL-IRE-LUX) or the 5′ UTR of FTH1 (pECE-FTH1-IRE-LUX) (see Fig. S2 in the supplemental material). The transcription of this chimeric gene is controlled by a simian virus 40 (SV40) promoter, which allows constitutive transcription in transfected cells. Together with these vectors, we also created a transfection control vector, pECE-RL (Fig. S2). pECE-RL has the Renilla luciferase gene controlled by the same SV40 promoter as pECE-FTL-IRE-LUX and pECE-FTH1-IRE-LUX. The 5′ UTRs of the FTL and the FTH1 mRNAs each contain an IRE. By fusing them to the firefly luciferase gene, we enabled IRPs to control the translation of the firefly luciferase gene, allowing the quantification of IRP-IRE binding-mediated translational repression by luciferase reporter assays. Under conditions of high intracellular iron levels (supplemented with 200 μM iron citrate [Fe]), IRPs do not bind to IREs, resulting in higher firefly luciferase activity in HEK293 cells transfected with the constructs (Fig. 4A and B). Inversely, in the presence of an iron chelator (200 μM deferoxamine [DFO]), IRP represses the translation of firefly luciferase, resulting in lower activity (Fig. 4A and B). Expectedly, Western blot analysis showed that iron and DFO treatments increased FTL and FTH1 levels and decreased IRP2 levels accordingly (Fig. 4C). The cellular IRP2 level is regulated by the cellular iron sensor F-box and leucine-rich repeat protein 5 (FBXL5), which is stabilized under conditions of high iron levels (12, 13). FBXL5 mediates IRP2 ubiquitylation, marking it for proteasomal degradation. Hence, iron treatment increases the FBXL5 protein level, while DFO treatment decreases it (Fig. 4C). Using the luciferase reporter constructs, we found that DMF or MMF treatments, which simulate intracellular fumarate accumulation, can decrease the ferritin translational-repression activity of IRPs (Fig. 4D to G). Accordingly, Western blot analysis of DMF- and MMF-treated samples also showed increases in ferritin levels in the DMF- and MMF-treated groups compared to the vehicle control groups (Fig. 4H and I). DMF and MMF treatments also led to increased FBXL5 levels (Fig. 4H and I); however, treatment also resulted in concurrently increased IRP2, FTL, and FTH1 protein levels. Since in canonical iron signaling, IRP2 represses the translation of FTL and FTH1, the concurrent increases in FBXL5, FTL, FTH1, and IRP2 levels upon DMF and MMF treatments indicate that fumarate accumulation disrupts normal iron signaling. We also evaluated levels of iron signaling proteins in FH-inactivated cells. Consistently, UOK262-FH−/− cells had higher protein levels of FBXL5, FTL, FTH1, and IRP2 than did UOK262-FHres cells (Fig. 4J), indicating that FH inactivation can alter iron signaling.
FIG 4.
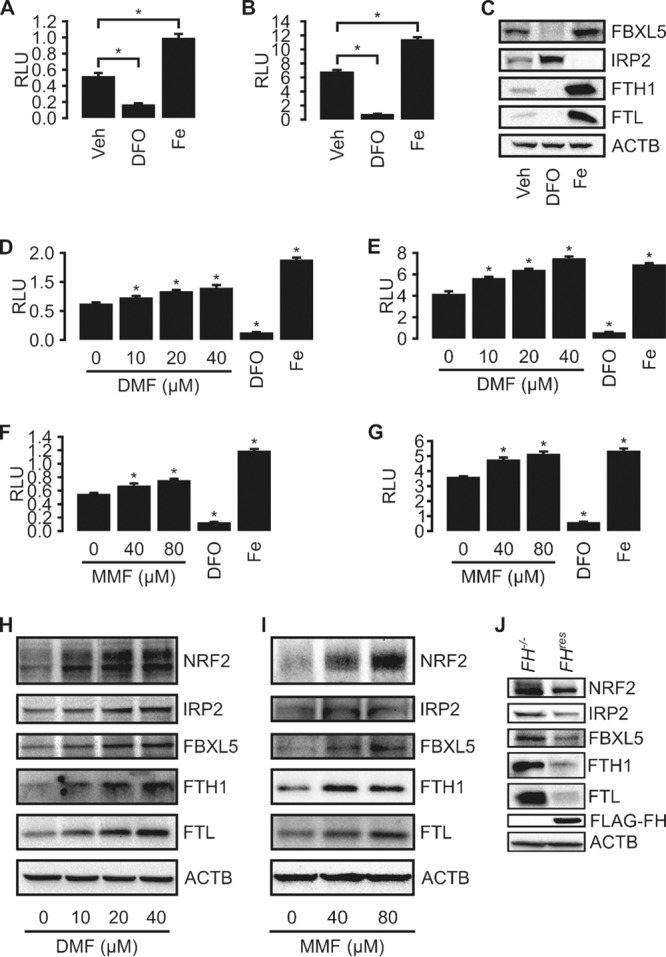
Fumarate inhibits IRP-mediated translational repression. (A and B) IRE reporter assays using the Dual-Glo luciferase assay (Promega) indicate appropriate responsiveness to iron levels for both pECE-FTL-IRE-LUX (A) and pECE-FTH1-IRE-LUX (B) cotransfected with pECE-RL into HEK293 cells. Cells were treated with ferric citrate (Fe; 200 micromolar) or an iron chelator (DFO; 200 micromolar) to modulate iron levels. Data are presented as means ± standard deviations. * indicates a P value of <0.05 relative to vehicle (Veh) treatment, as determined by Student's t test. RLU, relative light units. (C) Immunoblots of HEK293 cells treated with DFO show increased IRP2 and decreased FTL, FTH1, and FBXL5 protein levels. Treatment with Fe resulted in opposite effects. ACTB was used as a loading control. (D to G) IRE reporter assays using the Dual-Glo luciferase assay (Promega) with dimethyl fumarate and monomethyl fumarate treatments. HEK293 cells were cotransfected with pECE-RL and either pECE-FTL-IRE-LUX (D and F) or pECE-FTH1-IRE-LUX (E and G). Cells were treated with increasing concentrations of either dimethyl fumarate (D and E) or monomethyl fumarate (F and G). Data are presented as means ± standard deviations. * indicates a P value of <0.05 relative to vehicle treatment, as determined by Student's t test. (H and I) Western blots of HEK293 cells treated with increasing concentrations of DMF (H) or MMF (I) show dysregulated iron signaling, as indicated by simultaneously high protein levels of IRP2, FBXL5, FTL, and FTH1. ACTB was used as a loading control. (J) Western blots of UOK262-FH−/− control cells and UOK262-FHres cells. FH reconstitution leads to decreased NRF2, IRP2, FBXL5, FTH1, and FTL protein levels. ACTB was used as a loading control.
In canonical iron signaling, IRP2 is marked for ubiquitylation and subsequent proteasomal degradation by FBXL5. The paradoxical concurrent increases in FBXL5 and IRP2 levels following fumarate treatment or FH inactivation may be due to an impaired ubiquitylation of IRP2 by FBXL5, as evidenced by the decreased IRP2 ubiquitylation following MMF treatment (Fig. S3).
Sustained NRF2 activation partly contributes to increased intracellular ferritin levels.
Both FH inactivation and fumarate accumulation activate NRF2, and both FTL and FTH1 are transcription targets of NRF2. Thus, NRF2 activation may partly contribute to the observed increased ferritin gene expression levels in FH-inactivated cells by increasing their transcription. Quantitative PCR (qPCR) analyses revealed that the transcript levels of both FTL and FTH1 were higher in UOK262-FH−/− cells than in UOK262-FHres cells, suggesting that FH inactivation promotes the transcription of the FTL and FTH1 genes (Fig. 5A). To determine the contribution of NRF2 to promoting the transcription of the FTL and FTH1 genes in the context of fumarate accumulation, we generated NRF2 knockout HEK293 cells (HEK293-NRF2−/−) using CRISPR gene-editing technology. The knockout was confirmed by Sanger sequencing and immunoblotting (see Fig. S4 in the supplemental material). Using this cell line, we found that DMF treatment increased FTL and FTH1 transcript levels in HEK293-NRF2+/+ but not in HEK293-NRF2−/− cells, indicating that NRF2 mediates fumarate-induced FTL and FTH1 transcription (Fig. 5B). Moreover, HEK293 cells transfected with Myc-tagged NRF2 showed increased FTH1 and FTL protein levels compared to those in cells transfected with the empty vector, indicating that NRF2 plays an important role in modulating the expression of these genes (Fig. 5C). In Myc-NRF2-transfected cells, MMF treatment further increased FTL protein levels without further activating NRF2 (Fig. 5C). This effect may be attributed to the inhibition of IRP2 activity.
FIG 5.

NRF2 activation by fumarate contributes to ferritin accumulation. (A) Quantitative reverse transcription-PCR shows that FH reconstitution in UOK262 (UOK262-FHres) cells decreases relative FTL and FTH1 transcript levels. Changes in expression levels were normalized to values for UOK262-FHres cells. GCLM is an NRF2 target gene known to be upregulated in HLRCC. Error bars represent standard deviations. * indicates a P value of <0.05 as determined by Student's t test. (B) Quantitative reverse transcription-PCR of HEK293-NRF2−/− and HEK293-NRF2+/+ cells treated with 40 μM DMF or the vehicle (Veh). Changes in expression levels were normalized to values for vehicle treatment. Error bars represent standard deviations. * indicates a P value of <0.05 as determined by Student's t test. (C) Western blot of HEK293 cells transfected with pIRESpuro3 (vector) or NRF2-Myc and treated with the vehicle or 80 μM MMF. ACTB was used as a loading control. (D) Western blot of HEK293-NRF2−/− and HEK293-NRF2+/+ cells treated with the vehicle, 80 μM MMF, or 40 μM DMF. ACTB was used as a loading control. (E) Western blot of HEK293-NRF2−/− cells treated with increasing concentrations of DMF, 200 μM DFO, or 200 μM Fe.
To better understand how fumarate-mediated IRP2 inhibition contributes to increased ferritin protein levels independently of NRF2, we treated both HEK293-NRF2+/+ and HEK293-NRF2−/− cells with MMF and DMF. As expected, MMF and DMF treatments lead to increased FTL and FTH1 protein levels in HEK293-NRF2+/+ cells (Fig. 5D). However, in HEK293-NRF2−/− cells, MMF and DMF treatments increased only the FTL protein level (Fig. 5D). As anticipated, increasing concentrations of DMF in HEK293-NRF2−/− cells increased FTL but not FTH1 protein levels in a dose-responsive manner (Fig. 5E). These results indicate that in the absence of NRF2, fumarate can still mediate an increase in the FTL protein level; this effect is likely mediated by IRP2 inhibition and demonstrates that both increased transcription and translation cooperate to increase the FTL protein level, while the FTH1 protein level is more dependent on NRF2-mediated transcription.
UOK262 cells cannot be transfected efficiently by using routine DNA transfection strategies. Furthermore, UOK262-FH−/− and UOK262-FHres cells exhibit large differences in transduction and transfection efficiencies, making the direct evaluation of FTL and FTH1 translation using the dual-luciferase method impossible. Therefore, we quantified the increased FTH1 and FTL protein levels in UOK262-FH−/− by immunoblot densitometry analysis and normalized the values by their relative transcript levels (Fig. S5). Consistently, this analysis revealed that increases in FTL protein levels could not be accounted for by increased transcription alone, implying that both increased transcription and increased translation contributed to the fumarate-mediated increased FTL protein level, while the increase in the FTH1 protein level could be accounted for by increased transcription alone.
FH inactivation activates the FOXM1 transcription factor.
Ferritin confers a chronic growth signal by activating FOXM1 (Forkhead box protein M1) signaling (10). FOXM1 controls the expression of genes regulating progression through the G2/M phases of the cell cycle, and overexpression of FOXM1 is associated with cancer progression (14). Hence, we evaluated the activities of FOXM1 signaling in the context of FH inactivation and fumarate accumulation. Immunoblot analyses showed that UOK262-FH−/− cells have a higher level of FOXM1 than do UOK262-FHres cells, suggesting that FH inactivation activates FOXM1 signaling (Fig. 6A). qPCR analyses revealed increases in the levels of FOXM1 transcripts as well as the transcripts of the FOXM1 target genes AURKA, AURKB, and CDK1 in UOK262-FH−/− cells compared to UOK262-FHres cells, supporting that FOXM1 is activated in FH-inactivated cells (Fig. 6B). To determine if the observed activation of FOXM1 signaling was a result of fumarate accumulation, we simulated fumarate accumulation in UOK262-FHres cells by MMF treatment. Immunoblot analyses revealed that MMF-treated cells had higher levels of FOXM1 (Fig. 6C). To ascertain the role of ferritin in mediating the observed activation of FOXM1 signaling, we overexpressed FTL and FTH1 by cotransducing UOK262-FHres and HEK293 cells with lentiviruses carrying these genes. When ferritin was overexpressed, both cell lines showed increases in FOXM1 protein levels, indicating that ferritin increases the FOXM1 protein level (Fig. 6D). Consistently, sequential FTL and FTH1 knockdowns in UOK262 cells using short hairpin RNAs (shRNAs) also resulted in decreased FOXM1 levels, supporting that ferritin activates FOXM1 signaling in FH-inactivated cells (Fig. 6E). To evaluate if ferritin contributes to a progrowth phenotype in UOK262-FH−/− cells, we assessed proliferation following FTL or FTH1 knockdown. The knockdown of either FTL or FTH1 decreased the proliferation rate in UOK262 cells (Fig. 6F), indicating that ferritin can mediate growth in UOK262 cells.
FIG 6.

FH inactivation promotes FOXM1 signaling. (A) Western blotting of the FH-inactive UOK262-FH−/− cell line and the FH-reconstituted UOK262-FHres cell line. (B) Quantitative reverse transcription-PCR shows that FH reconstitution in UOK262 cells decreases the relative transcript levels of FOXM1 and its downstream targets AURKA, AURKB, and CDK1. Changes in expression levels were normalized to values for UOK262-FHres cells. GCLM is an NRF2 target gene known to be upregulated in HLRCC. Data are presented as means ± standard deviations of results from a representative experiment. * indicates a P value of <0.05 as determined by Student's t test. (C) Western blots of UOK262-FHres cells treated with the vehicle or 80 μM monomethyl fumarate show increased FOXM1 signaling with MMF. (D) Western blots of UOK262-FHres cells and HEK293 cells transduced with the pLKO-CMV empty vector (control) or pLKO-CMV-FTL and pLKO-CMV-FTH1 (ferritin) show that ferritin overexpression increases FOXM1 protein levels. (E) Western blots of UOK262 cells transduced with shRNAs targeting FTL (shRNA-FTL) or FTH1 (shRNA-FTH1) show decreased FOXM1 signaling relative to shRNA-Scr, a nontargeting shRNA control. (F) Cell viability measured by formazan production in UOK262 cells after transduction with shRNA-Scr, shRNA-FTL, and shRNA-FTH1. Data are presented as means ± standard deviations of results from a representative experiment. shRNA-FTL and shRNA-FTH1 knockdown curves were statistically significantly different from the shRNA-Scr curve for shRNA treatment (P < 0.001) but not for time (P > 0.05), as determined by two-way analysis of variance. (G) Relative mRNA levels of ferritin genes (FTL and FTH1), FOXM1, FOXM1 target genes (CDK1, AURKA, and AURKB), and nonferritin NRF2 targets (GCLM and AKR1B10) in HLRCC tumors and in normal kidney tissues. FDR, false discovery rate.
Previous reports indicated that FOXM1 was activated by ferritin through STAT3 signaling (10). Although UOK262-FH−/− cells had increased STAT3 phosphorylation relative to UOK262-FHres cells (see Fig. S6A in the supplemental material) and shRNA-mediated knockdown of either FTL or FTH1 decreased FOXM1 levels, only FTL knockdown reduced STAT3 phosphorylation (Fig. S6B). Additionally, ferritin overexpression increased FOXM1 levels (Fig. 6D) without altering STAT3 phosphorylation in UOK262-FHres cells, indicating that ferritin may activate FOXM1 through a different mechanism (Fig. S6C).
To evaluate the validity of the ferritin-FOXM1 signaling pathway in clinical samples, we reanalyzed previously reported gene expression microarray data derived from HLRCC tumors and normal kidney tissues (GEO accession no. GSE26574 and GSE20896) (3, 15). Concordant with our cell culture models, HLRCC tumors showed significantly increased expression levels of FTL, FTH1, and FOXM1 as well as the FOXM1 target genes AURKA, AURKB, and CDK1 compared to those in normal kidney tissues (Fig. 6G and Table S3).
DISCUSSION
Studies of hereditary cancers have led to the discovery and characterization of many physiologically and pathologically important tumor suppressors, including TP53 (16), RB (17), PTEN (18), and APC (19). Many of these classical tumor suppressors, such as the cell cycle's retinoblastoma protein, RB, mediate cell growth. Indeed, sustaining chronic proliferation is one of the most fundamental hallmarks of cancer (4). While it is clear how somatic mutations in genes involved in growth signaling mediate the chronic proliferation signal, mechanisms by which mutations of metabolic tumor suppressors such as FH confer such a signal were not known. In this study, we show that fumarate, which accumulates as a result of FH inactivation, functions as a mitogen to activate chronic proliferative signaling. Mechanistically, fumarate increases ferritin gene transcription through NRF2 activation and increases ferritin gene translation by inhibiting IRP2. In turn, ferritin activates the progrowth transcription factor FOXM1 (Fig. 7). Previous reports on ferritin-induced growth signaling described how glioblastoma cancer cells had high ferritin levels due to increased iron intake (10). In contrast, we demonstrate how cancer cells use an electrophilic oncometabolite to hijack ferritin regulation in an iron-independent mechanism to increase ferritin levels.
FIG 7.
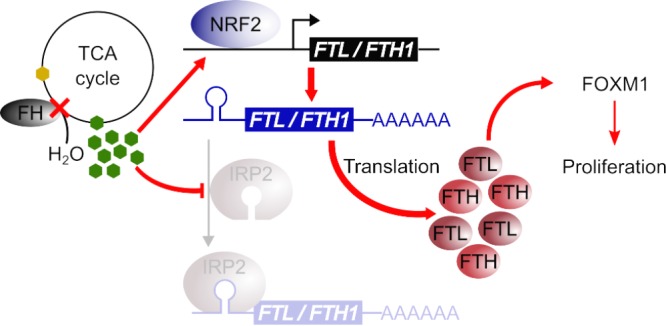
FH inactivation confers chronic proliferative signaling. The diagram shows how FH inactivation increases ferritin levels to promote proliferation. FH, a TCA cycle enzyme that catalyzes the hydration of fumarate (represented by green hexagons) to form malate (represented by yellow hexagons), is inactivated (represented by a red cross) in HLRCC. FH inactivation causes fumarate accumulation. The accumulated fumarate activates the NRF2 transcription factor, which transcribes the ferritin genes FTL and FTH1. Concurrently, the accumulated fumarate also inhibits IRP2, which normally represses the translation of FTL and FTH1, leading to a net increase in the ferritin level and subsequent FOXM1-dependent proliferative signaling.
The implications of increased ferritin levels in FH-deficient cells for signaling networks require further investigation. The established function of ferritin is to sequester free iron. Fumarate-mediated, iron-independent upregulation of ferritin could induce an intracellular iron deficiency, yet the concurrent accumulation of FBXL5 would indicate otherwise. These counterintuitive changes in IRP2, ferritin, and FBXL5 levels should be explored in the context of intracellular signaling; because iron is an important cofactor in heme- and iron-sulfur-containing enzymes, many carcinogenesis-associated pathways could be altered by iron-independent ferritin upregulation, including DNA synthesis (20, 21), DNA repair (22–24), metabolism (25, 26), and iron-dependent dioxygenase activity (27). Moreover, iron-independent functions of ferritin, such as its role in chemokine receptor-mediated cell migration or proinflammatory phosphatidylinositol 3-kinase (PI3K) signaling (28, 29), could be altered in FH-deficient cancers. Considering that we have shown that FOXM1 can be activated when ferritin is overexpressed, without changes to STAT3 signaling, as was previously reported, future inquiries into this pathway should evaluate if high ferritin levels activate FOXM1 through modulation of iron levels or through iron-independent signaling pathways.
Our results indicate that the thiol reactivity of fumarate underlies the mechanism by which it activates the observed chronic proliferative signaling, suggesting that other thiol-reactive compounds may also exert a similar effect. In addition to fumarate, fumarylacetoacetate (FAA) is another known cysteine-reactive oncometabolite (30). FAA accumulates following fumarylacetoacetate hydrolase (FAH) inactivation in hereditary tyrosinemia type 1 (HT1), a genetic disorder that can progress to hepatocellular carcinoma (HCC) (31, 32). Inhibition of FAA formation can significantly decrease cancer incidences, indicating that FAA accumulation drives HCC formation in HT1 patients (33). Our data imply that FAA may provide a chronic growth signal in HT1-associated HCC through a similar mechanism. Oncometabolites are not the only thiol-modifying compounds implicated in cancer. DMF, which is used clinically to treat psoriasis and multiple sclerosis, can induce kidney tumors akin to HLRCC at sufficiently high concentrations (200 and 400 mg/kg of body weight/day) (34).
Additionally, some environmental toxicants or their activated metabolites are also electrophiles capable of modifying cysteine residues: toxicants like acrolein can form cysteine adducts (35) and may also elicit growth modulation through concerted NRF2 activation and IRP2 inhibition. However, while NRF2 activation may be a common outcome of thiol-modifying compounds and Michael acceptors (36, 37), any generalizable inhibition of IRP2 repression by endogenous compounds and xenobiotics is premature. Indeed, in opposition to our description of fumarate-mediated decreases in IRP2-IRE binding activity, mice exposed to cigarette smoke showed increased IRP2-IRE binding activity, and a thiophene derivative has been shown to enhance IRP2 binding to the FTL IRE (38, 39). Furthermore, fumarate seems to differentially affect the translation of FTL and FTH1. Looking forward, a “cysteine code” may exist for IRP2, whereby increases or decreases to IRP2-IRE binding interactions are determined by which IRP2 cysteines are modified, what the structures of those modifications are, and how the adducts interact with IREs of diverse nucleotide compositions. Given the multitude of genes under IRE control, the alterations to iron signaling networks and downstream pathways may vary widely.
In conclusion, our results show how FH inactivation disrupts cellular iron signaling and induces a chronic proliferative phenotype, providing a mechanistic explanation for how the inactivation of FH can give rise to a fundamental cancer hallmark.
MATERIALS AND METHODS
Alignment of aconitase protein family members.
Amino acid sequences for mouse Aco2 (NCBI RefSeq accession no. NP_542364.1), human IRP2 (IREB2/ACO3) (accession no. NP_004127.1), and human IRP1 (IREB1/ACO1) (accession no. NP_001265281.1) were aligned by using Clustal Omega (40).
Reagents and chemicals.
Stock solutions of 50 mM DMF (catalog no. sc-239774; Santa Cruz Biotechnology, Dallas, TX) and 100 mM MMF (catalog no. 651419; Sigma, St. Louis, MO) were dissolved in dimethyl sulfoxide (DMSO) and serum-free medium (pH adjusted to 7.4 with HEPES buffer) (catalog no. 15630080-1M; Thermo Fisher, Waltham, MA), respectively. Stock solutions of 20 mM Fe (catalog no. F3388; Sigma) and 50 mM DFO (catalog no. D9533; Sigma) were dissolved in water. MG132 (catalog no. M7449; Sigma) was used as a proteasome inhibitor.
Cell culture conditions.
UOK262 cells were a generous gift from Marston Linehan (National Cancer Institute, NIH, Bethesda, MD) (5). HEK293 and HK2 cell lines were obtained from the ATCC (Manassas, VA). HEK293FT cells were purchased from Life Technologies (Thermo Fisher). UOK262, HK2, and derivative cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS). All FBS was heat inactivated for 30 min at 56°C. HEK293 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with high glucose (4.5 g/liter) (no pyruvate) supplemented with 10% FBS. HEK293FT cells were cultured in DMEM with no pyruvate and high glucose supplemented with 10% FBS and 500 μg/ml Geneticin. Cells were cultured at 37°C in atmospheric air enriched with 5% CO2. For both long-term culture and experiments, HEK293 and HEK293FT cells were cultured on vessels coated with poly-d-lysine.
Construction of Flag-tagged FH vectors.
Transient expression of FH in UOK262 cells was achieved by using a lentiviral construct based on the pLKO.5 vector (Sigma). The U6 promoter was swapped out for a cytomegalovirus (CMV) promoter to generate pLKO.5-CMV. The FH open reading frame was cloned into pLKO.5-CMV to give rise to pLKO-FH.
Construction of FTL, FTH1, and Flag-tagged IRP2 expression vectors.
Total RNA was extracted from HEK293 cells by using TRIzol reagent (Life Technologies, Thermo Scientific) according to the manufacturer's protocols. First-strand cDNA synthesis was performed by using SuperScript IV reverse transcriptase (Life Technologies), using FTL-, FTH1-, or IREB2-specific primers. Subsequent second-strand syntheses, amplifications, and the introduction of an N-terminal Flag tag to IREB2 were performed by using Phusion DNA polymerase (NEB, Ipswich, MA). The resulting IREB2 open reading frame PCR product was cloned into the pIRESpuro3 vector (Clontech, Mountain View, CA) to make the pIRESpuro3-N-Flag-IRP2 construct. Site-directed mutagenesis was performed with the Q5 site-directed mutagenesis kit (catalog no. E0552S; NEB) to generate the A523R mutant pIRESpuro3-N-Flag-IRP2. FTL and FTH1 open reading frame PCR products were cloned into pLKO.5-CMV to generate pLKO-CMV-FTL and pLKO-CMV-FTH1, respectively.
Construction of FTL and FTH1 5′-UTR luciferase vectors.
DNA fragments corresponding to chromosome 19, 48965309, through chromosome 19, 48965507 (human genome reference version GRCh38/hg38), and chromosome 11, 61967426 through chromosome 11, 61967660 (GRCh38/hg38), which correspond to the 5′ UTRs of the FTL and FTH1 genes, respectively, were amplified from human genomic DNA (gDNA) isolated from HEK293 cells. The firefly luciferase open reading frame was amplified from pGL4.21 (Promega, Madison, WI) by PCR using appropriate forward primers to allow splice by overlap extension PCR with the FTL and FTH1 5′ UTRs isolated previously. The resulting PCR products were cloned into the pECE vector (Addgene, Cambridge, MA), giving rise to pECE-FTL-IRE-LUX and pECE-FTH1-IRE-LUX, respectively. The Renilla luciferase open reading frame derived from pRL (Promega) was cloned into pECE to produce pECE-RL. This vector was used as a transfection control in dual-luciferase assays.
Construction of the Myc-NRF2 vector.
pCDNA3-Myc3-Nrf2 was made available from Addgene following its characterization (41). Myc-tagged NFE2L2 was isolated from pCDNA3-Myc3-Nrf2 and cloned into pIRESpuro3 to make NRF2-Myc. Cells were transfected with this plasmid by using Attractene transfection reagent (Qiagen, Valencia, CA).
shRNA-Scr, shRNA-FTL, and shRNA-FTH1 vectors.
The pLKO.5 vector (Sigma) was used to generate the following plasmids: shRNA-FTL (21-bp sequence of 5′-CTG GAG ACT CAC TTC CTA GAT-3′), shRNA-FTH1 (21-bp sequence of 5′-GCC GAA TCT TCC TTC AGG ATA-3′), and the shRNA-Scr scrambled control (21-bp sequence of 5′-GAC TAG AAG GCA CAG AGG GAT-3′). shRNA-Scr was used as a nontargeting control plasmid and targets no known human genes.
Lentivirus production and transient transduction.
Lentivirus for all pLKO plasmids was produced by using the ViraPower lentiviral expression system (Thermo Fisher) according to the manufacturer's protocols. For transient lentiviral transduction, cells were used for experiments 48 h after transduction. Transduction efficacy was verified via Western blotting.
Stable cell line production for UOK262-FHres and UOK262-FH−/− cells.
UOK262 parental cells were transduced with pLKO.5-CMV and pLKO.5-FH to generate UOK262-FH−/− and UOK262-FHres cells, respectively. Cells with FH stably incorporated into their genome were selected by using 2 μg/ml puromycin. Long-term cultures of the resulting stably transfected cells were maintained in puromycin medium, while experiments using these cells were conducted without puromycin.
Generation of NRF2 and FH knockout HEK293 lines.
NRF2 was knocked out in cells by using a commercial CRISPR-Cas9 NRF2 knockout system (catalog no. sc-400017; Santa Cruz) according to the manufacturer's recommendations. The knockout was verified by using Western blotting and Sanger sequencing. FH was knocked out in cells by using methods described previously (42). Two sets of oligonucleotides were used to generate single-guide RNAs (sgRNAs) to target Cas9 to FH gene loci: set A primers SiteAfor (5′-CAC CGG GAG GCA CTG CTG TTG GTA C-3′) and SiteArev (5′-AAA CGT ACC AAC AGC AGT GCC TCC C-3′) and set B primers SiteBfor (5′-CAC CGG AGC TCA TAG ATT CTT GGC A-3′) and SiteBrev (5′-AAA CTG CCA AGA ATC TAT GAG CTC C-3′). sgRNAs were cloned into pSpCas9(BB)-2A-GFP (Addgene) to generate pSpCas9-GFP-A and pSpCas9-GFP-B. To generate a homology-directed repair (HDR) template plasmid, we isolated the puromycin resistance cassette from pGL4.21 (Promega) using BamHI and SalI restriction sites and cloned the cassette into pUC19 (Addgene) to generate pUC19-Puro; DNA sequences flanking the sgRNA sites were added to the 5′ and 3′ ends of the puromycin cassette to generate homology-directed repair (HDR) arms. To generate a 5′-HDR arm, a DNA fragment corresponding to chromosome 1, 241668363, through chromosome 1, 241669362 (human genome reference version GRCh37/hg19), was isolated from HEK293 gDNA. To generate a 3′-HDR arm, a DNA fragment corresponding to chromosome 1, 241669413, through chromosome 1, 241670412 (GRCh37/hg19), was similarly isolated. Isolated 5′-HDR arm and 3′-HDR arm PCR products were subsequently cloned into pUC19-Puro to generate pUC19-Puro-FHHDR. HEK293 cells were transfected with pSpCas9-GFP-A, pSpCas9-GFP-B, and pUC19-Puro-FHHDR. Cells with FH stably knocked out were selected by using 2 μg/ml puromycin, and single colonies were screened for FH knockout by using immunoblotting and PCR amplification of the FH gene from HEK293-FH−/− cell gDNA. Primers used for gDNA amplification were 5′-CTG GTA GAT TTT AAT GGC ATG CTG-3′ and 5′-AAC CCT CAT CCT TCC CTA TAC TTT G-3′. Long-term cultures of the resulting stably transfected cells were maintained in puromycin medium, while experiments using these cells were conducted without puromycin.
MMF and DMF treatments.
For MMF and DMF treatments, cells were treated with the specified concentrations of MMF or DMF and were refreshed every 24 h for at least 48 h before being harvested for analyses.
UOK262-FH−/−, UOK262-FHres, and UOK262-shRNA cell proliferation assays.
Early-passage UOK262-FH−/− and UOK262-FHres cells were seeded into 96-well plates at 3,000 cells/well in medium containing 80 μM MMF or vehicle controls. Cell proliferation was measured by using the CellTiter 96 AQueous One Solution cell proliferation assay [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS)] (Promega) according to the manufacturer's recommended protocols. For all groups, media were refreshed every 24 h. For shRNA proliferation assays, parental UOK262 cells were transduced with specified shRNA viruses on a 10-cm dish. The following day, medium was refreshed. After an additional 24 h, cells were seeded into 96-well plates at 3,000 cells/well, and cell proliferation was measured daily.
Immunoblotting.
Primary antibodies to β-actin (ACTB) (1:10,000 in milk) (catalog no. A1978; Sigma), AKR1B10 (1:1,000 in bovine serum albumin [BSA]) (catalog no. sc-100501; Santa Cruz), FBXL5 (1:2,500 in milk) (catalog no. 672602; BioLegend, San Diego, CA), FH (1:1,000 in BSA) (catalog no. 4567; Cell Signaling, Danvers, MA), Flag (1:1,000 in milk) (catalog no. 8146; Cell Signaling), FOXM1 (1:500 in milk) (catalog no. sc-502; Santa Cruz), IRP2 (1:1,000 in BSA) (catalog no. sc-33682; Santa Cruz), FTH1 (1:1,000 in BSA) (catalog no. sc-25617; Santa Cruz), FTL (1:1,000 in BSA) (catalog no. sc-74513; Santa Cruz), Myc (1:1,000 in milk) (catalog no. 2276; Cell Signaling), NQO1 (1:1,000 in milk) (catalog no. sc-32793; Santa Cruz), NRF2 (1:1,000 in milk) (catalog no. sc-13032; Santa Cruz), phospho-STAT3 (1:1,000 in milk) (catalog no. 9145; Cell Signaling), STAT3 (1:1,000 in milk) (catalog no. 9139; Cell Signaling), and ubiquitin (1:1,000 in milk) (catalog no. 3936; Cell Signaling) were used for immunoblotting experiments. Band densitometry was quantified by using Image Lab software (Bio-Rad).
Immunoprecipitation and mass spectrometry analysis of Flag-tagged IRP2.
The Flag-tagged IRP2 protein was overexpressed in HEK293 cells by transiently transfecting the cells with the pIRESpuro3-N-Flag-IRP2 vector. On days 2 and 3 posttransfection, cells were cotreated with 40 μM dimethyl fumarate and 200 μM deferoxamine. Cells were harvested for immunoprecipitation on day 4 posttransfection. Immunoprecipitation experiments were performed by using a magnetic DYKDDDDK immunoprecipitation kit (catalog no. 635696; Clontech) according to the manufacturer's recommendations. Samples were eluted in 1× Laemmli sample buffer with a protease inhibitor and β-mercaptoethanol and then resolved on 7% SDS-PAGE gels. A band of approximately 105 kDa, as visualized by Coomassie blue staining, was excised for liquid chromatography (LC)-MS/MS analysis.
Excised bands were reduced, alkylated, and digested in gel by using trypsin as previously described (43). In-gel digests were then desalted, fractionated online by reversed-phase chromatography using a Thermo Fisher easy-nLC 1000 liquid chromatography system, and analyzed by tandem mass spectrometry on a Thermo Fisher Q-Exactive mass spectrometer (44, 45). Database searching was performed by using the MSGF+ search algorithm and a human protein database and considered differential modification of cysteine residues by carbamidomethylation (+57.021464 atomic mass units [amu]), 2-succination (+116.010959 amu), 2-monomethyl-succination (+130.026609 amu), and 2-dimethyl-succination (+144.042259 amu) (46). Peptide spectral matches were filtered by using a percolator-derived q value of 0.01 (47).
Ubiquitylation of IRP2.
HEK293 cells were transiently transfected with the pIRESpuro3-N-Flag-IRP2 vector. On days 2 and 3 posttransfection, cells were treated with 80 μM MMF. On day 4, cells were cotreated with 10 μM MG132, 80 μM MMF, or 200 μM Fe, as indicated, for 4 h. Whole-cell extracts were harvested, the protein level was normalized by a bicinchoninic acid (BCA) protein assay, and extracts were utilized for downstream immunoprecipitation and SDS-PAGE analyses as described above.
Luciferase assays.
HEK293 cells were plated onto 10-cm dishes in DMEM supplemented with 10% FBS and transfected the same day with plasmids pECE-RL and either pECE-FTL-IRE-LUX or pECE-FTH1-IRE-LUX by using the Attractene transfection reagent (Qiagen). The following day, transfected cells were transferred into 6-well plates. After cells were allowed to adhere for 24 h, the cells were dosed with appropriate concentrations of compounds diluted in RPMI plus 10% FBS. Media and chemicals were refreshed every 24 h. Following 48 h of exposure, cells were lysed and used in the dual-luciferase reporter (DLR) system (Promega) according to the manufacturer's recommendations.
RNA interference.
siRNA transfections were performed with Oligofectamine reagent (Thermo Fisher) according to the manufacturer's protocols. Two different species of FH-targeting siRNAs were used: siFH-1 (catalog no. FH HSS103687; Thermo Fisher) and siFH-2 (catalog no. FH HSS103688; Thermo Fisher). A nontargeting siRNA silencer negative control (catalog no. AM4637; Thermo Fisher-Ambion) was used as a nontargeting scrambled siRNA (siScr) transfection control. Cells were harvested for immunoblotting at 72 h posttransfection.
qPCR.
Total RNA was extracted from adherent cells by using TRIzol reagent according to the manufacturer's recommended protocols. cDNA was synthesized by using a High-Capacity cDNA reverse transcription kit (catalog no. 4368814; Thermo Fisher/Applied Biosystems), and qPCR was performed by using TaqMan Fast Advanced master mix (Applied Biosystems, Foster City, CA) with an appropriate TaqMan probe. TaqMan probes (Thermo Fisher/Applied Biosystems) targeting ACTB (catalog no. 4352935), AURKA (catalog no. Hs01582072_m1), AURKB (catalog no. Hs00945855_g1), CDK1 (catalog no. Hs00938777_m1), FOXM1 (catalog no. Hs01073586_m1), FTH1 (catalog no. Hs01000476_g1), FTL (catalog no. Hs00830226_gH), and GCLM (catalog no. Hs00157694_m1) were used in this study. Results were analyzed by using the 2−ΔΔCT method (48).
Statistical and microarray analyses.
All statistical analyses were performed in the R statistical environment (49). Gene expression microarray data were processed by using a robust multichip averaging algorithm (50). Differentially expressed genes between tumors and normal kidney tissues were determined by using the linear model for microarray analysis algorithm (51).
Supplementary Material
ACKNOWLEDGMENTS
We declare no conflicts of interest.
This material is based upon work supported by the National Science Foundation Graduate Research Fellowship Program under grant no. DGE-1143953 (M.J.K.). The project is partly funded by start-up funds from the University of Arizona College of Pharmacy and University of Arizona Health Sciences grant 214565 (A.O.).
Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/MCB.00079-17.
REFERENCES
- 1.Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, Leigh I, Gorman P, Lamlum H, Rahman S, Roylance RR, Olpin S, Bevan S, Barker K, Hearle N, Houlston RS, Kiuru M, Lehtonen R, Karhu A, Vilkki S, Laiho P, Eklund C, Vierimaa O, Aittomaki K, Hietala M, Sistonen P, Paetau A, Salovaara R, Herva R, Launonen V, Aaltonen LA, Multiple Leiomyoma Consortium . 2002. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 30:406–410. doi: 10.1038/ng849. [DOI] [PubMed] [Google Scholar]
- 2.Ternette N, Yang M, Laroyia M, Kitagawa M, O'Flaherty L, Wolhulter K, Igarashi K, Saito K, Kato K, Fischer R, Berquand A, Kessler BM, Lappin T, Frizzell N, Soga T, Adam J, Pollard PJ. 2013. Inhibition of mitochondrial aconitase by succination in fumarate hydratase deficiency. Cell Rep 3:689–700. doi: 10.1016/j.celrep.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ooi A, Wong JC, Petillo D, Roossien D, Perrier-Trudova V, Whitten D, Min BW, Tan MH, Zhang Z, Yang XJ, Zhou M, Gardie B, Molinie V, Richard S, Tan PH, Teh BT, Furge KA. 2011. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell 20:511–523. doi: 10.1016/j.ccr.2011.08.024. [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Yang Y, Valera VA, Padilla-Nash HM, Sourbier C, Vocke CD, Vira MA, Abu-Asab MS, Bratslavsky G, Tsokos M, Merino MJ, Pinto PA, Srinivasan R, Ried T, Neckers L, Linehan WM. 2010. UOK 262 cell line, fumarate hydratase deficient (FH−/FH−) hereditary leiomyomatosis renal cell carcinoma: in vitro and in vivo model of an aberrant energy metabolic pathway in human cancer. Cancer Genet Cytogenet 196:45–55. doi: 10.1016/j.cancergencyto.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang M, Ternette N, Su H, Dabiri R, Kessler BM, Adam J, Teh BT, Pollard PJ. 2014. The succinated proteome of FH-mutant tumours. Metabolites 4:640–654. doi: 10.3390/metabo4030640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gruer MJ, Artymiuk PJ, Guest JR. 1997. The aconitase family: three structural variations on a common theme. Trends Biochem Sci 22:3–6. doi: 10.1016/S0968-0004(96)10069-4. [DOI] [PubMed] [Google Scholar]
- 8.Meyron-Holtz EG, Ghosh MC, Iwai K, LaVaute T, Brazzolotto X, Berger UV, Land W, Ollivierre-Wilson H, Grinberg A, Love P, Rouault TA. 2004. Genetic ablations of iron regulatory proteins 1 and 2 reveal why iron regulatory protein 2 dominates iron homeostasis. EMBO J 23:386–395. doi: 10.1038/sj.emboj.7600041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. 2002. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res 62:5196–5203. [PubMed] [Google Scholar]
- 10.Schonberg DL, Miller TE, Wu Q, Flavahan WA, Das NK, Hale JS, Hubert CG, Mack SC, Jarrar AM, Karl RT, Rosager AM, Nixon AM, Tesar PJ, Hamerlik P, Kristensen BW, Horbinski C, Connor JR, Fox PL, Lathia JD, Rich JN. 2015. Preferential iron trafficking characterizes glioblastoma stem-like cells. Cancer Cell 28:441–455. doi: 10.1016/j.ccell.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zumbrennen KB, Wallander ML, Romney SJ, Leibold EA. 2009. Cysteine oxidation regulates the RNA-binding activity of iron regulatory protein 2. Mol Cell Biol 29:2219–2229. doi: 10.1128/MCB.00004-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vashisht AA, Zumbrennen KB, Huang X, Powers DN, Durazo A, Sun D, Bhaskaran N, Persson A, Uhlen M, Sangfelt O, Spruck C, Leibold EA, Wohlschlegel JA. 2009. Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science 326:718–721. doi: 10.1126/science.1176333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salahudeen AA, Thompson JW, Ruiz JC, Ma HW, Kinch LN, Li Q, Grishin NV, Bruick RK. 2009. An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science 326:722–726. doi: 10.1126/science.1176326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koo CY, Muir KW, Lam EW. 2012. FOXM1: from cancer initiation to progression and treatment. Biochim Biophys Acta 1819:28–37. doi: 10.1016/j.bbagrm.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 15.Ashrafian H, O'Flaherty L, Adam J, Steeples V, Chung YL, East P, Vanharanta S, Lehtonen H, Nye E, Hatipoglu E, Miranda M, Howarth K, Shukla D, Troy H, Griffiths J, Spencer-Dene B, Yusuf M, Volpi E, Maxwell PH, Stamp G, Poulsom R, Pugh CW, Costa B, Bardella C, Di Renzo MF, Kotlikoff MI, Launonen V, Aaltonen L, El-Bahrawy M, Tomlinson I, Pollard PJ. 2010. Expression profiling in progressive stages of fumarate-hydratase deficiency: the contribution of metabolic changes to tumorigenesis. Cancer Res 70:9153–9165. doi: 10.1158/0008-5472.CAN-10-1949. [DOI] [PubMed] [Google Scholar]
- 16.Malkin D, Li FP, Strong LC, Fraumeni JF Jr, Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA, Friend SH. 1990. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250:1233–1238. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- 17.Murphree AL, Benedict WF. 1984. Retinoblastoma: clues to human oncogenesis. Science 223:1028–1033. doi: 10.1126/science.6320372. [DOI] [PubMed] [Google Scholar]
- 18.Marsh DJ, Kum JB, Lunetta KL, Bennett MJ, Gorlin RJ, Ahmed SF, Bodurtha J, Crowe C, Curtis MA, Dasouki M, Dunn T, Feit H, Geraghty MT, Graham JM Jr, Hodgson SV, Hunter A, Korf BR, Manchester D, Miesfeldt S, Murday VA, Nathanson KL, Parisi M, Pober B, Romano C, Tolmie JL, Trembath R, Winter RM, Zackai EH, Zori RT, Weng L-P, Dahia PLM, Eng C. 1999. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet 8:1461–1472. doi: 10.1093/hmg/8.8.1461. [DOI] [PubMed] [Google Scholar]
- 19.Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, Horii A, Koyama K, Utsunomiya J, Baba S, Hedge P. 1991. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 253:665–669. doi: 10.1126/science.1651563. [DOI] [PubMed] [Google Scholar]
- 20.Hoffbrand AV, Ganeshaguru K, Hooton JW, Tattersall MH. 1976. Effect of iron deficiency and desferrioxamine on DNA synthesis in human cells. Br J Haematol 33:517–526. doi: 10.1111/j.1365-2141.1976.tb03570.x. [DOI] [PubMed] [Google Scholar]
- 21.Netz DJ, Stith CM, Stumpfig M, Kopf G, Vogel D, Genau HM, Stodola JL, Lill R, Burgers PM, Pierik AJ. 2011. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nat Chem Biol 8:125–132. doi: 10.1038/nchembio.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rudolf J, Makrantoni V, Ingledew WJ, Stark MJ, White MF. 2006. The DNA repair helicases XPD and FancJ have essential iron-sulfur domains. Mol Cell 23:801–808. doi: 10.1016/j.molcel.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 23.Aravind L, Koonin EV. 2001. The DNA-repair protein AlkB, EGL-9, and leprecan define new families of 2-oxoglutarate-and iron-dependent dioxygenases. Genome Biol 2:research0007. doi: 10.1186/gb-2001-2-3-research0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lukianova OA, David SS. 2005. A role for iron-sulfur clusters in DNA repair. Curr Opin Chem Biol 9:145–151. doi: 10.1016/j.cbpa.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 25.Oexle H, Gnaiger E, Weiss G. 1999. Iron-dependent changes in cellular energy metabolism: influence on citric acid cycle and oxidative phosphorylation. Biochim Biophys Acta 1413:99–107. doi: 10.1016/S0005-2728(99)00088-2. [DOI] [PubMed] [Google Scholar]
- 26.de Ungria M, Rao R, Wobken JD, Luciana M, Nelson CA, Georgieff MK. 2000. Perinatal iron deficiency decreases cytochrome c oxidase (CytOx) activity in selected regions of neonatal rat brain. Pediatr Res 48:169–176. doi: 10.1203/00006450-200008000-00009. [DOI] [PubMed] [Google Scholar]
- 27.Salminen A, Kauppinen A, Kaarniranta K. 2015. 2-Oxoglutarate-dependent dioxygenases are sensors of energy metabolism, oxygen availability, and iron homeostasis: potential role in the regulation of aging process. Cell Mol Life Sci 72:3897–3914. doi: 10.1007/s00018-015-1978-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruddell RG, Hoang-Le D, Barwood JM, Rutherford PS, Piva TJ, Watters DJ, Santambrogio P, Arosio P, Ramm GA. 2009. Ferritin functions as a proinflammatory cytokine via iron-independent protein kinase C zeta/nuclear factor kappaB-regulated signaling in rat hepatic stellate cells. Hepatology 49:887–900. doi: 10.1002/hep.22716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li R, Luo C, Mines M, Zhang J, Fan G-H. 2006. Chemokine CXCL12 induces binding of ferritin heavy chain to the chemokine receptor CXCR4, alters CXCR4 signaling, and induces phosphorylation and nuclear translocation of ferritin heavy chain. J Biol Chem 281:37616–37627. doi: 10.1074/jbc.M607266200. [DOI] [PubMed] [Google Scholar]
- 30.Lloyd AJ, Gray RG, Green A. 1995. Tyrosinaemia type 1 and glutathione synthetase deficiency: two disorders with reduced hepatic thiol group concentrations and a liver 4-fumarylacetoacetate hydrolase deficiency. J Inherit Metab Dis 18:48–55. doi: 10.1007/BF00711372. [DOI] [PubMed] [Google Scholar]
- 31.Jorquera R, Tanguay RM. 1997. The mutagenicity of the tyrosine metabolite, fumarylacetoacetate, is enhanced by glutathione depletion. Biochem Biophys Res Commun 232:42–48. doi: 10.1006/bbrc.1997.6220. [DOI] [PubMed] [Google Scholar]
- 32.Kubo S, Sun M, Miyahara M, Umeyama K, Urakami K, Yamamoto T, Jakobs C, Matsuda I, Endo F. 1998. Hepatocyte injury in tyrosinemia type 1 is induced by fumarylacetoacetate and is inhibited by caspase inhibitors. Proc Natl Acad Sci U S A 95:9552–9557. doi: 10.1073/pnas.95.16.9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lindstedt S, Holme E, Lock EA, Hjalmarson O, Strandvik B. 1992. Treatment of hereditary tyrosinaemia type I by inhibition of 4-hydroxyphenylpyruvate dioxygenase. Lancet 340:813–817. doi: 10.1016/0140-6736(92)92685-9. [DOI] [PubMed] [Google Scholar]
- 34.Temple R. 2014. NDA approval letter: Tecfidera (dimethyl fumarate NDA 204063). US Food and Drug Administration, Washington, DC. [Google Scholar]
- 35.Cai J, Bhatnagar A, Pierce WM Jr. 2009. Protein modification by acrolein: formation and stability of cysteine adducts. Chem Res Toxicol 22:708–716. doi: 10.1021/tx800465m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dinkova-Kostova AT. 2012. The role of sulfhydryl reactivity of small molecules for the activation of the KEAP1/NRF2 pathway and the heat shock response. Scientifica (Cairo) 2012:606104. doi: 10.6064/2012/606104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Talalay P, De Long MJ, Prochaska HJ. 1988. Identification of a common chemical signal regulating the induction of enzymes that protect against chemical carcinogenesis. Proc Natl Acad Sci U S A 85:8261–8265. doi: 10.1073/pnas.85.21.8261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zimmer M, Ebert BL, Neil C, Brenner K, Papaioannou I, Melas A, Tolliday N, Lamb J, Pantopoulos K, Golub T, Iliopoulos O. 2008. Small-molecule inhibitors of HIF-2a translation link its 5′UTR iron-responsive element to oxygen sensing. Mol Cell 32:838–848. doi: 10.1016/j.molcel.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cloonan SM, Glass K, Laucho-Contreras ME, Bhashyam AR, Cervo M, Pabon MA, Konrad C, Polverino F, Siempos II, Perez E, Mizumura K, Ghosh MC, Parameswaran H, Williams NC, Rooney KT, Chen ZH, Goldklang MP, Yuan GC, Moore SC, Demeo DL, Rouault TA, D'Armiento JM, Schon EA, Manfredi G, Quackenbush J, Mahmood A, Silverman EK, Owen CA, Choi AM. 2016. Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat Med 22:163–174. doi: 10.1038/nm.4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Furukawa M, Xiong Y. 2005. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol Cell Biol 25:162–171. doi: 10.1128/MCB.25.1.162-171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. 2013. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaiser P, Wohlschlegel J. 2005. Identification of ubiquitination sites and determination of ubiquitin-chain architectures by mass spectrometry. Methods Enzymol 399:266–277. doi: 10.1016/S0076-6879(05)99018-6. [DOI] [PubMed] [Google Scholar]
- 44.Kelstrup CD, Young C, Lavallee R, Nielsen ML, Olsen JV. 2012. Optimized fast and sensitive acquisition methods for shotgun proteomics on a quadrupole Orbitrap mass spectrometer. J Proteome Res 11:3487–3497. doi: 10.1021/pr3000249. [DOI] [PubMed] [Google Scholar]
- 45.Shimogawa MM, Saada EA, Vashisht AA, Barshop WD, Wohlschlegel JA, Hill KL. 2015. Cell surface proteomics provides insight into stage-specific remodeling of the host-parasite interface in Trypanosoma brucei. Mol Cell Proteomics 14:1977–1988. doi: 10.1074/mcp.M114.045146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim S, Pevzner PA. 2014. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat Commun 5:5277. doi: 10.1038/ncomms6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kall L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. 2007. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods 4:923–925. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- 48.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 49.R Core Team. 2013. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 50.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. 2003. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.