

Abstract
Goodpasture's syndrome (GPS) remains a very rare disease entity in the pediatric population characterized by the presence of pulmonary hemorrhage and rapidly evolving glomerulonephritis. We hereby describe the case of a 2-year-old girl who presented with renal failure and was diagnosed with GPS. A brief review of the literature in regard to data on demographics, pathogenesis, clinical features, diagnosis, treatment, and prognosis for renal recovery is also provided.
1. Introduction
Goodpasture's syndrome (GPS) is a rare and life threatening autoimmune condition with autoantibodies directed against the glomerular basement membrane (GBM) antigen. The term GPS refers to the triad of pulmonary hemorrhage, glomerulonephritis, and anti-GBM antibodies while Goodpasture's disease (GD) is the preferred terminology in the absence of pulmonary hemorrhage [1, 2]. The term antiglomerular basement membrane antibody disease (aGD) describes a patient with serum antibodies against the basement membrane and includes both Goodpasture's syndrome and disease.
2. Case Report
A 2-year, 11-month-old Hispanic female presented to her primary care physician's office with swelling of the hands and face following one week of fever, sore throat, and malaise. A screening urine analysis (U/A) revealed 3+ protein and blood with numerous red blood cells per high power field. Further work-up also demonstrated anemia (Hb of 8.5 g/dl) and several electrolyte imbalances with azotemia (BUN 116 mg/dl and Cr 7.3 g/dl) prompting immediate transfer to our Children's Hospital for further evaluation and management.
On admission, examination revealed a pale child with bilateral mild pedal edema and blood pressure of 125/71 mm Hg. She was afebrile, mildly tachycardic, and saturating at 100% in room air and parents denied any h/o joint pain/swelling or skin rash. Her urine output was noted to be darker and less frequent over the past few days.
Past medical history was significant for an admission about 7 months back with respiratory distress and presumed pneumonia. Labs at that time were significant for severe anemia (Hb 6.9 g/dl) and iron deficiency. Initial chest X-ray showed bilateral diffuse peribronchial cuffing and nodular opacities with concerns for severe bronchiolitis/bronchopneumonia (Figure 1). Patient was started on empiric antibiotic coverage but respiratory distress worsened to the point of requiring ventilator support. A work-up at that time showed elevated erythrocyte sedimentation rate (113 mm/hr) and negative serology for antinuclear antibody. A respiratory viral pathogen array came back positive for rhino virus. Patient's clinical condition continued to deteriorate and patient was placed on extracorporeal membrane oxygenation (ECMO). During this time patient was also started on high dose methylprednisolone with presumptive exaggerated inflammatory response to her viral pneumonia in a bid to reduce inflammation. Patient had a dramatic response to steroids and was off ECMO in 2 days and off ventilator support within a week time. A U/A done during this past hospital stay showed 1+ blood and no protein and a chemistry panel showed normal renal function with a serum creatinine of 0.41 mg/dl.
Figure 1.

Chest X-ray on initial presentation.
Work-up during this current admission confirmed anemia (Hb 7.8 g/dl) with a slightly elevated white blood cell count (10,400/mm3) and platelet count (234,000/mm3). Serum chemistry panel was abnormal for hyperkalemia (6 mmol/L), metabolic acidosis (Hco3 of 8 mmol/L), hypocalcemia (5.6 mg/dl), hyperphosphatemia (8.9 mg/dl), and renal failure (BUN 120 mg/dl and Cr 7.01 mg/dl). Further work-up involved evaluation as to identify the cause of glomerulonephritis and showed normal complement levels, normal coagulation profile, negative serology for viral etiology, lupus, and ANCA titers. Parathyroid hormone levels were elevated indicating a state of chronic kidney damage. ESR was elevated at 24 mm/hr but lesser than the prior admission value of 113 mm/hr and a CRP was not checked during the current admission. Urine protein to creatinine ratio was in the nephrotic range and antiglomerular basement membrane (GBM) titers were sent. Urine output recorded was between 1.5 and 2 ml/kg/hr during the initial few days but progressively got oliguric (0.3 to 0.5 ml/kg/hr) from the first week onwards. Renal ultrasound showed normal sized kidneys (right and left kidney around 7.2 cm) with increased cortical echogenicity bilaterally. A comparison of lab values during prior hospital stay and current admission is provided in Table 1.
Table 1.
Comparison of lab values between prior and current admission.
| Labs | 10/2015 | 5/2016 |
|---|---|---|
| Sodium (mmol/L) | 141 | 137 |
| Potassium (mmol/L) | 4.2 | 6.0 |
| Chloride (mmol/L) | 110 | 113 |
| Carbon dioxide (mmol/L) | 21 | 8 |
| Glucose (mg/dL) | 109 | 97 |
| BUN (mg/dL) | 5 | 120 |
| Creatinine (mg/dL) | 0.41 | 7.01 |
| Albumin (g/dL) | 2.9 | 2.5 |
| Calcium (mg/dL) | 9.3 | 5.6 |
| Phosphorus (mg/dL) | 8.9 | |
| White blood cell (×103/mcL) | 15.84 | 10.4 |
| Hemoglobin (g/dL) | 6.9 | 7.8 |
| Hematocrit (%) | 22.6 | 23.7 |
| Platelets (×103/mcL) | 616 | 234 |
| Ferritin (ng/mL) | 269 | 267 |
| Iron (mcg/dL) | 6 | 55 |
| Transferrin (mcg/dL) | 130 | 100 |
| TIBC (mcg/dL) | Not done | 125 |
| % saturation | Not done | 44 |
| Parathyroid hormone (pg/mL) | Not done | 1031 |
| C3 (mg/dL) | 166 | 114 |
| C4 (mg/dL) | 30 | 48 |
| ESR (mm/hr) | 113 | 12.5 |
| CRP (mg/dl) | 12.5 | Not done |
Patient underwent emergent hemodialysis to correct electrolyte imbalances. We proceeded with a renal biopsy to ascertain a tissue diagnosis for the glomerulonephritis. 12 glomeruli were available for light microscopic examination. 9/12 glomeruli showed global sclerosis (Figure 2). Remaining glomeruli showed cellular to fibrocellular crescents (Figure 3). The interstitium is involved by a dense inflammatory infiltrate composed of lymphocytes, plasma cells, and scattered eosinophils. No definite granulomas were identified. Immunofluorescence showed intense linear glomerular capillary staining with IgG, Kappa, and lambda chains (Figure 4). The renal biopsy findings were consistent with anti-GBM mediated crescentic glomerulonephritis.
Figure 2.
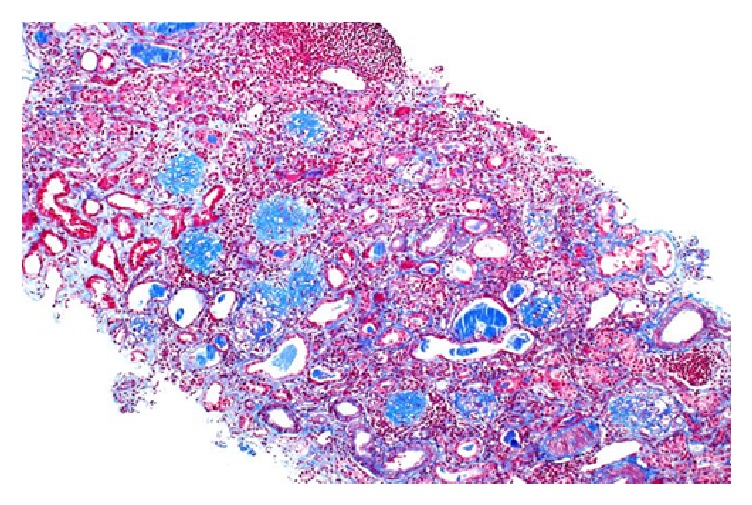
Trichrome stain showing global glomerulosclerosis.
Figure 3.

Glomerulus showing intraglomerular sclerosis.
Figure 4.

Immunofluorescence showing linear IgG deposits.
Patient was started on high dose methylprednisolone and plasmapheresis once the biopsy results were consistent with anti-GBM disease. Anti-GBM titers (IgG antibody) also came back elevated at 1.1 units (Normal < 1) confirming the diagnosis. The subtle elevation in anti-GBM titers could be secondary to the possibility of a serological remission though chronic damage to the kidneys has already happened as documented by the amount of fibrosis on renal biopsy. Unfortunately anti-GBM titers were not checked during the initial pneumonia like presentation 7 months back which likely represented the initial acute episode. With every other day plasmapheresis, anti-GBM titers started trending down but renal function did not recover. With the extent of global sclerosis noted in renal biopsy and with the very high PTH levels, chances for renal recovery remained slim. Rituximab was used as an alternate immunosuppressive agent instead of cyclophosphamide taking into consideration the amount of chronic damage noted on renal biopsy in an attempt to reduce infectious risk. Patient received a total of 5 sessions of plasmapheresis with no improvement in renal function and was transitioned to peritoneal dialysis. During the inpatient stay she suffered a hypertensive crisis with seizures and the control of blood pressure required multiple antihypertensive agents. Anti-GBM titers were periodically monitored by lab work on a monthly basis and remained negative on maintenance immunosuppression with mycophenolate and low dose prednisone. Patient received a diseased donor kidney transplant, 2 months back, and is currently doing well with normal renal function.
3. Discussion
3.1. Epidemiology
GPS is a rare condition occurring in approximately 0.5 to 1 per million per year in adults and even more rare in children [3]. According to the United States Renal Data Registry, incidence of pediatric end stage renal disease (ESRD) due to this rare entity is only 11-12 per year, accounting for 0.5% of pediatric ESRD in 2009–2013 [4]. It typically has a bimodal distribution with the first peak predominantly affecting males in their teens and twenties. The second peak which happens in older population (>60 years of age) affects male and female equally. GPS is rare in children, with only about 30 cases being reported in the pediatric literature, with the youngest reported child being 11 months of age. The previously reported cases of pediatric Goodpasture's syndrome over the past 25 years are detailed in Table 2 [5–13].
Table 2.
Prior reported cases of pediatric Goodpasture's syndrome.
| Age in years | Sex | Anti-GBM titers | Initial clinical presentation | Renal biopsy | Renal outcome | Pulmonary outcome | Treatment | Final outcome | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 4 | F | Positive | Pallor, fatigue oliguria, proteinuria, and microscopic hematuria with dominant renal involvement | End stage glomerulonephritis with crescent formation; linear deposition of IgG along basement membrane | No improvement | Stable | Prednisone, azathioprine, and cyclophosphamide | Died | [5] |
| 10 | F | Positive | Gross hematuria, oliguria, and uremia with dominant renal involvement Preceding infection with strep throat | Endocapillary and extracapillary proliferative GN with 80% crescents Immunofluorescence could not be done | Dialysis dependent with no improvement | Stable | Prednisolone, azathioprine, and plasmapheresis | Remained dialysis dependent | [5] |
| 7 | F | Positive | Diarrhea, vomiting, oliguria, and pallor with dominant renal involvement | Crescentic nephritis with linear IgG deposition | Initial improvement in urine output and GFR with subsequent decline and dialysis dependence | Stable | Plasmapheresis, prednisolone, and cyclophosphamide | Dialysis dependent | [5] |
| 6 | M | Positive | Dominant renal involvement | Diagnostic with crescentic nephritis | Improved | Stable | Steroid, plasmapheresis, and immunosuppression | Regained renal function | [6] |
| 10 | M | Positive | Cough, right lower lobe infiltrate, vomiting, and oliguria with dominant pulmonary involvement and pulmonary hemorrhage | Crescentic nephritis with extensive necrosis | Deterioration in renal function with dialysis dependence | Improved | Steroid, plasmapheresis, and immunosuppression | Dialysis dependent | [7] |
| 2.5 | F | Positive | Fever, anorexia with E. coli UTI as initial presentation with worsening renal function and oliguria | Extensive crescentic necrotizing nephritis with linear IgG deposits | No improvement | Stable | Steroid, plasmapheresis, and immunosuppression | Dialysis dependent | [8] |
| 11 months | F | Positive | Dominant renal involvement | Diagnostic with crescentic nephritis | No improvement | Stable | Steroid, plasmapheresis, and immunosuppression | Renal transplant | [9] |
| 5.6 | F | Positive | Fever, malaise, and gross hematuria with rapid decline in renal function | Diffuse cellular crescentic nephritis with linear IgG deposits | Recovery of renal function | Stable | Plasma exchange, solumedrol, and Cytoxan | CKD with stable renal function | [10] |
| 9 | M | Positive | Malaise, anorexia, and oligoanuria with pulmonary hemorrhage | Not done | Not improved | Pulmonary status improved | Plasma exchange, solumedrol, and Cytoxan | Dialysis dependent | [11] |
| 8 | F | Positive | Asymptomatic with persistent nephrotic range proteinuria and microhematuria | No crescents but with linear deposits of IgG | Improvement in proteinuria with stable renal function | Stable | Plasma exchange, prednisone, and oral Cytoxan | Asymptomatic | [12] |
| 19 months | M | Positive | Gross hematuria, proteinuria with rapid decline in renal function | Crescentic GN with weak global linear staining of IgG | Improvement in proteinuria and renal function | Stable | Plasma exchange, solumedrol, and Cytoxan | Asymptomatic | [13] |
3.2. Pathogenesis
The type IV collagen which provides the backbone for GBM formation is the target for autoantibody formation and damage in GPS. The type IV collagen has six genetically discrete chains (α1 to α6) which are arranged into triple helical protomers (α1α1α2, α3α4α5, and α5α5α6) of varying composition. The protomer has a 7S domain at the N-terminal, a collagenous part in the middle, and a noncollagenous (NC1) domain at the C-terminal [14]. The final collagen IV network in the GBM is a polymerized mesh such that the 7S domain forms a tetramer and the NC1 domain forms a hexamer providing the tensile strength to the basement membrane. α1α1α2-α1α1α2 is the predominant collagen prototype in embryonic GBM and a developmental switch happens to the final adult form of α3α4α5-α3α4α5 anywhere between 3 months and 3 years of age [15].
The specific target for autoantibody formation in GPS is the NC1 domain of the α3 subunit in the C-terminal. The NC1 domain also acts as the main promoter for collagen polymerization. The common presence of α3 collagen in the basement membrane of both kidneys and lungs explains the predominant organ involvement in this condition. A triggering event (upper respiratory infection, smoking, hydrocarbon exposure, and influenza) in a genetically susceptible individual causes exposure of the α3 NC1 domain and subsequent antibody formation [16, 17]. Strong HLA association with presence of HLA-DR15 and DR 4 allele in about 80% of affected individuals confirms a genetic predisposition as is the case in the majority of autoimmune diseases [18]. The absence of α3 subunit in younger children (before the developmental switch) could be attributed to the lesser incidence of aGD in younger children.
3.3. Clinical Features
Initial presentation of GPS can be nonspecific and often consists of symptoms such as malaise, weight loss, fever, and arthralgia [19]. Kidney disease may occur independently or with pulmonary disease. Renal manifestations vary widely and can range from hematuria and proteinuria to rapidly progressing renal failure with oliguria, fluid overload, and severe hypertension. Pulmonary symptoms may precede renal symptoms by weeks to months with hemoptysis being the most common pulmonary manifestation. Pulmonary bleeding can be occult leading to anemia and iron deficiency but the usual presentation is with profound pulmonary hemorrhage causing respiratory failure and death in a matter of hours. Other organ system involvement is very rare though cerebral vasculitis with confusion, aphasia, and seizures has been reported in the literature [20].
3.4. Pathology
The diagnosis of antiglomerular basement membrane disease is reliant on detection of anti-GBM antibodies either in circulation or in the tissue by means of renal or pulmonary biopsies. Serological testing for anti-GBM antibody titers (IgG1 subclass) usually employs ELISA methodology. The sensitivity of available commercial kits can vary from 63% to 100% underlying the possibility of missed diagnosis if solely reliant on serological testing [21, 22]. Renal biopsy can help confirm the diagnosis of GPS and also provides important clues on the amount of chronicity/activity helping to guide treatment. Light microscopy usually shows crescentic glomerulonephritis but the characteristic linear IgG deposition along the capillary wall is noted in immunofluorescence microscopy clinching the diagnosis. Lung biopsy also shows the linear IgG deposits but this finding is not as constant as in kidney [23].
3.5. Treatment
Early diagnosis is important in terms of ability to recover renal function. Treatment of choice initially is plasmapheresis to remove circulating antibodies. The preferred immunosuppressive therapy includes corticosteroids and cyclophosphamide to reduce antibody production. Alternate immunosuppressive therapy including rituximab has been tried in resistant cases [24]. Anti-GBM antibodies are monitored weekly until two negatives are achieved, at which time levels are monitored monthly for up to 6 months. Low dose prednisone, azathioprine, or mycophenolate may be used for maintenance immunosuppression once remission is established with cessation of antibody production. If the antibody titer levels remain positive, the immunosuppression therapy should be continued [25].
3.6. Prognosis
Unfortunately, many patients die secondary to pulmonary hemorrhage or renal failure before plasmapheresis and immunosuppression can be initiated. Currently the mortality rate is 20% in adults and 30% in children. Prognosis for renal recovery is worse in the presence of oliguria, presenting creatinine >6.8 or renal biopsy showing >50% crescent formation within glomeruli at time of diagnosis [26]. Evidences of chronic damage as documented by moderate or severe interstitial fibrosis and global glomerulosclerosis always carry a worse prognosis. Renal outcome is dependent on timing of diagnosis with improved outcomes if treatment is initiated within 4 weeks of renal involvement [27]. Despite the potential seriousness of lung hemorrhage with increased fatality, no residual pulmonary deficit or fibrosis is noted once patient recovers from the acute presentation. Though rare, recurrence of disease may occur years after initial presentation.
GPS as a cause of pulmonary renal syndrome in childhood remains extremely rare. A review of pediatric cases in the literature (Table 2) shows a female preponderance in children in comparison to majority male involvement in adults. Anti-GBM titers were positive in almost all of the reported cases. Majority of cases also showed dominant renal involvement with gross hematuria and oligoanuria being the most common presentation. Treatment strategies involved using a combination of steroids, plasma exchange, and cyclophosphamide in most of the patients. Renal recovery was noted only in 3/11 patients among whom one had presentation with nephrotic range proteinuria but with normal renal function [8]. The prognosis for renal recovery seems to be better in the absence of interstitial fibrosis and early treatment initiation as is noted in the adult literature.
4. Conclusion
In conclusion, GPS is a rare autoimmune condition presenting with significant mortality and morbidity in children. We report a case of 2-year, 11-month-old child who presented with this condition and the difficulties involved in coming to an accurate diagnosis. In hindsight, her initial presentation with pneumonia was likely an occult pulmonary hemorrhage as documented by the severe anemia and iron deficiency. Her response to steroids during the initial admission likely constituted a partial treatment. GPS though rare should be considered in the differential diagnosis of clinical presentation with lung and kidney involvement and early diagnosis and intervention are essential for a favorable outcome.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- 1.Gallagher H., Kwan J. T. C., Jayne D. R. W. Pulmonary renal syndrome: a 4-year, single-center experience. American Journal of Kidney Diseases. 2002;39(1):42–47. doi: 10.1053/ajkd.2002.29876. [DOI] [PubMed] [Google Scholar]
- 2.Lee R. W., D'Cruz D. P. Pulmonary renal vasculitis syndromes. Autoimmunity Reviews. 2010;9(10):657–660. doi: 10.1016/j.autrev.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 3.Fischer E. G., Lager D. J. Anti-glomerular basement membrane glomerulonephritis: a morphologic study of 80 cases. American Journal of Clinical Pathology. 2006;125(3):445–450. doi: 10.1309/NPTP-4UKV-7JU3-ELMQ. [DOI] [PubMed] [Google Scholar]
- 4.USRDS U.S. Renal Data System 2015 Annual Data Report: pediatric ESRD. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Disease, Bethesda. 2015.
- 5.Levin M., Rigden S. P. A., Pincott J. R., Lockwood C. M., Barratt T. M., Dillon M. J. Goodpasture's syndrome: treatment with plasmapheresis, immunosuppression, and anticoagulation. Archives of Disease in Childhood. 1983;58(9):697–702. doi: 10.1136/adc.58.9.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilvarry J., Doyle G. F., Gill D. G. Good outcome in anti-glomerular basement membrane nephritis. Pediatric Nephrology. 1992;6(3):244–246. doi: 10.1007/BF00878358. [DOI] [PubMed] [Google Scholar]
- 7.McCarthy L. J., Cotton J., Danielson C., Graves V., Bergstein J. Goodpasture's syndrome in childhood: treatment with plasmapheresis and immunosuppression. Journal of Clinical Apheresis. 1994;9(2):116–119. doi: 10.1002/jca.2920090204. [DOI] [PubMed] [Google Scholar]
- 8.Boven K., Miljoen H. P. J., Van Hoeck K. J., Van Marck E. A., Van Acker K. J. Anti-glomerular basement membrane glomerulopathy in a young child. Pediatric Nephrology. 1996;10(6):745–747. doi: 10.1007/s004670050207. [DOI] [PubMed] [Google Scholar]
- 9.Bigler S. A., Parry W. M., Fitzwater D. S., Baliga R. An 11-month-old with anti-glomerular basement membrane disease. American Journal of Kidney Diseases. 1997;30(5):710–712. doi: 10.1016/S0272-6386(97)90497-2. [DOI] [PubMed] [Google Scholar]
- 10.Bakkaloglu S. A., Kasapkara C. S., Soylemezoglu O., et al. Successful management of anti-GBM disease in a 5 1/2-year-old girl. Nephrology Dialysis Transplantation. 2006;21(10):2979–2981. doi: 10.1093/ndt/gfl232. [DOI] [PubMed] [Google Scholar]
- 11.Poddar B., Singhal S., Azim A., Gulati S., Baronia A. Goodpasture's syndrome in children. Saudi Journal of Kidney Diseases and Transplantation. 2010;21(5):935–939. [PubMed] [Google Scholar]
- 12.Nagano C., Goto Y., Kasahara K., Kuroyanagi Y. Case report: anti-glomerular basement membrane antibody disease with normal renal function. BMC Nephrology. 2015;16:p. 185. doi: 10.1186/s12882-015-0179-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bjerre A., Hogåsen K., Grotta J., Scott H., Tangeraas T., Dörje C. Rescue of kidney function in a toddler with anti-GBM nephritis. Clinical Kidney Journal. 2012;5(6):584–586. doi: 10.1093/ckj/sfs146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thorner P. S., Baumal R., Eddy A., Marrano P. Characterization of the NC1 domain of collagen type IV in glomerular basement membranes (GBM) and of antibodies to GBM in a patient with anti-GBM nephritis. Clinical Nephrology. 1989;31(3):160–168. [PubMed] [Google Scholar]
- 15.Hudson B. G., Reeders S. T., Tryggvason K. Type IV collagen: structure, gene organization, and role in human diseases. Molecular basis of goodpasture and alport syndromes and diffuse leiomyomatosis. Journal of Biological Chemistry. 1993;268(35):26033–26036. [PubMed] [Google Scholar]
- 16.Goodpasture E. W. Landmark publication from The American Journal of the Medical Sciences: the significance of certain pulmonary lesions in relation to the etiology of influenza. The American Journal of the Medical Sciences. 2009;338(2):148–151. doi: 10.1097/MAJ.0b013e31818fff94. [DOI] [PubMed] [Google Scholar]
- 17.Donaghy M., Rees A. J. Cigarette smoking and lung haemorrhage in glomerulonephritis caused by autoantibodies to glomerular basement membrane. The Lancet. 1983;322(8364):1390–1393. doi: 10.1016/s0140-6736(83)90923-6. [DOI] [PubMed] [Google Scholar]
- 18.Fisher M., Pusey C. D., Vaughan R. W., Rees A. J. Susceptibility to anti-glomerular basement membrane disease is strongly associated with HLA-DRB1 genes. Kidney International. 1997;51(1):222–229. doi: 10.1038/ki.1997.27. [DOI] [PubMed] [Google Scholar]
- 19.Dammacco F., Battaglia S., Gesualdo L., Racanelli V. Goodpasture's disease: a report of ten cases and a review of the literature. Autoimmunity Reviews. 2013;12(11):1101–1108. doi: 10.1016/j.autrev.2013.06.014. [DOI] [PubMed] [Google Scholar]
- 20.Gittins N., Basu A., Eyre J., Gholkar A., Moghal N. Cerebral vasculitis in a teenager with Goodpasture's syndrome. Nephrology Dialysis Transplantation. 2004;19(12):3168–3171. doi: 10.1093/ndt/gfh448. [DOI] [PubMed] [Google Scholar]
- 21.Salama A. D., Dougan T., Levy J. B., et al. Goodpasture's disease in the absence of circulating anti-glomerular basement membrane antibodies as detected by standard techniques. The American Journal of Kidney Diseases. 2002;39(6):1162–1167. doi: 10.1053/ajkd.2002.33385. [DOI] [PubMed] [Google Scholar]
- 22.Litwin C. M., Mouritsen C. L., Wilfahrt P. A., Schroder M. C., Hill H. R. Anti-glomerular basement membrane disease: Role of enzyme-linked immunosorbent assays in diagnosis. Biochemical and Molecular Medicine. 1996;59(1):52–56. doi: 10.1006/bmme.1996.0064. [DOI] [PubMed] [Google Scholar]
- 23.Briggs W. A., Johnson J. P., Teichman S., Yeager H. C., Wilson C. B. Antiglomerular basement membrane antibody-mediated glomerulonephritis and goodpasture's syndrome. Medicine (United States) 1979;58(5):348–361. doi: 10.1097/00005792-197909000-00002. [DOI] [PubMed] [Google Scholar]
- 24.Shah M. K., Hugghins S. Y. Characteristics and outcomes of patients with Goodpasture's syndrome. Southern Medical Journal. 2002;95(12):1411–1418. doi: 10.1097/00007611-200295120-00012. [DOI] [PubMed] [Google Scholar]
- 25.Kluth D. C., Rees A. J. Anti-glomerular basement membrane disease. Journal of the American Society of Nephrology. 1999;10(11):2446–2453. doi: 10.1681/ASN.V10112446. [DOI] [PubMed] [Google Scholar]
- 26.Merkel F., Pullig O., Marx M., Netzer K. O., Weber M. Course and prognosis of anti-basement membrane antibody (anti-BM-Ab)-mediated disease: report of 35 cases. Nephrology Dialysis Transplantation. 1994;9(4):372–376. [PubMed] [Google Scholar]
- 27.Alchi B., Griffiths M., Sivalingam M., Jayne D., Farrington K. Predictors of renal and patient outcomes in anti-GBM disease: clinicopathologic analysis of a two-centre cohort. Nephrology Dialysis Transplantation. 2015;30(5):814–821. doi: 10.1093/ndt/gfu399. [DOI] [PubMed] [Google Scholar]