

Abstract
Mitochondria strongly contribute to the maintenance of cellular integrity through various mechanisms, including oxidative adenosine triphosphate production and calcium homeostasis regulation. Therefore, proper regulation of the abundance, distribution and activity of mitochondria is crucial for the maintenance of cellular homeostasis. Previous studies have shown that ionizing radiation (IR) alters mitochondrial functions, suggesting that mitochondria are likely to be an important target of IR. Though IR reportedly influences cellular mitochondrial abundance, the mechanism remains largely unknown. In this study, we examined how IR influences mitochondrial abundance in mouse fibroblasts. When mouse NIH/3T3 cells were exposed to X-rays, a time-dependent increase was observed in mitochondrial DNA (mtDNA) and mitochondrial mass, indicating radiation-induced upregulation of mitochondrial abundance. Meanwhile, not only did we not observe a significant change in autophagic activity after irradiation, but in addition, IR hardly influenced the expression of two mitochondrial proteins, cytochrome c oxidase subunit IV and cytochrome c, or the mRNA expression of Polg, a component of DNA polymerase γ. We also observed that the expression of transcription factors involved in mitochondrial biogenesis was only marginally affected by IR. These data imply that radiation-induced upregulation of mitochondrial abundance is an event independent of macroautophagy and mitochondrial biogenesis. Furthermore, we found evidence that IR induced long-term cell cycle arrest and cellular senescence, indicating that these events are involved in regulating mitochondrial abundance. Considering the growing significance of mitochondria in cellular radioresponses, we believe the present study provides novel insights into understanding the effects of IR on mitochondria.
Keywords: ionizing radiation, cellular radioresponse, mitochondria, mitochondrial biogenesis, cell cycle arrest, cellular senescence
INTRODUCTION
Mitochondria are double-membrane–enclosed eukaryotic organelles with a vital role in numerous cellular functions. Besides oxidative adenosine triphosphate (ATP) production, they are essential for other functions such as β-oxidation of free fatty acids, heme synthesis, and calcium homeostasis [1]. Since mitochondria contribute greatly towards the maintenance of cellular integrity, proper regulation of their abundance, distribution and activity is crucial for the maintenance of cellular homeostasis. Cellular mitochondrial abundance is governed by the balance between production and elimination [2, 3]. Mitochondrial biogenesis, the process whereby new mitochondria are produced, is a sophisticated multistep process that involves mitochondrial DNA (mtDNA) transcription and translation, translocation of nucleus-derived transcripts, and recruitment of newly synthesized proteins and lipids [4]. On the other hand, elimination of mitochondria is mainly achieved via mitochondrial autophagy or ‘mitophagy’, a process where severely damaged or superfluous mitochondria are degraded via macroautophagy [4]. Mitochondrial abundance within the cell is not constant, but rather fluctuates in response to various external stress stimuli such as cold exposure, energy deprivation, and physical exercise [2]. This regulation of mitochondrial abundance is intricate and requires precise coordination, where disruption or alterations in mitochondrial abundance have been associated with various pathologies. For example, increased mitochondrial abundance is a common denominator of several disorders, including neurodegenerative diseases and myopathies [5, 6]. Similar progressive mitochondrial accumulation is observed during aging in multiple cell types in a diversity of organisms [4]. Thus, deciphering the mechanisms that control mitochondrial abundance is important for fully understanding the growing significance of the role of mitochondria in human health.
Ionizing radiation (IR) elicits a variety of responses in eukaryotic cells, including DNA repair, reversible or irreversible cell cycle arrest, and the activation of stress signaling [7, 8]. These cellular responses ultimately influence the fate of irradiated cells. Radiation-induced cellular responses include the alteration of mitochondrial functions, such as mitochondrial respiration and reactive oxygen species (ROS) production [9–11]. We previously reported that IR affects various aspects of mitochondrial physiology, including mitochondrial respiration, ATP production, and mitochondrial dynamics [12, 13]. In addition, mitochondria are also suggested to be involved in radiation-induced intra- and intercellular signaling [14–16]. These findings indicate that mitochondria are likely to be an important target of IR and may play a critical role in cellular radioresponses.
Among various types of radioresponses, previous studies have indicated that IR influences cellular mitochondrial abundance [12, 17]. In support of these observations, Nugent et al. demonstrated that IR increases mitochondrial mass [10], and other groups observed increased mtDNA after irradiation [18, 19]. Similar results have also been reported in vivo [19, 20]. This suggests that a change in mitochondrial abundance after IR is a conserved radioresponse. However, the mechanism by which IR affects mitochondrial abundance remains unclear. In this study, we provide evidence that the radiation-induced increase of mitochondrial abundance is an event independent of macroautophagy and mitochondrial biogenesis.
MATERIALS AND METHODS
Reagents
MitoTracker Green FM was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Anti-LC3 antibody (PM036) was purchased from Medical and Biological Laboratories (Nagoya, Japan). Anti-cytochrome c (Cyt c) antibody was obtained from BD Biosciences (San Jose, CA, USA). Anti-cytochrome c oxidase IV (COX IV), anti-actin and horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The chemiluminescence detection kit, Western Lightning Plus-ECL, was purchased from Perkin-Elmer (Waltham, MA, USA).
Cell culture and X-irradiation
Mouse embryonic fibroblast NIH/3T3 cells were maintained in Dulbecco's Modified Eagle Medium (DMEM; Thermo Fisher Scientific) containing 10% (v/v) calf serum (Thermo Fisher Scientific) at 37°C in 5% CO2. SV40-immortalized mouse embryonic fibroblasts (MEFs) were maintained in DMEM containing 10% (v/v) fetal bovine serum (Biowest; Nuaille, France) at 37°C in 5% CO2. X-irradiation was performed at room temperature (RT) using a Shimadzu PANTAK HF-320 X-ray irradiator (Shimadzu; Kyoto, Japan) or an X-RAD iR-225 X-ray irradiator (Precision X-Ray; North Branford, CT, USA) with a dose rate of 2.54 Gy/min at 200 kVp, 20 mA with a 1.0-mm aluminum filter or 1.37 Gy/min at 200 kVp, 15 mA with a 1.0-mm aluminum filter, respectively. After irradiation, the culture medium was replaced with fresh growth medium and the cells were cultured for analysis.
Mitochondrial DNA copy number
Total genomic DNA containing mtDNA and nuclear DNA (nDNA) was extracted from cells using a Blood & Cell Culture DNA Mini Kit (QIAGEN; Hilden, Germany) according to the manufacturer's instructions. DNA was eluted with TE buffer and its quantity and purity was determined by spectrometric analysis. The relative copy number of mtDNA to nDNA was determined by real-time polymerase chain reaction (PCR) using primers specific to NADH dehydrogenase subunit 6 (ND6; mitochondrial) and beta-2 microglobulin (B2m; nuclear) genes (Table 1). The obtained DNA (5 ng for ND6; 20 ng for B2m) was subjected to real-time PCR analysis using a LightCycler Nano System (Roche Applied Science, Mannheim, Germany), with the samples being prepared using FastStart Essential DNA Green Master mix (Roche Applied Science). The following conditions were applied for PCR analysis: 95°C for 10 min, followed by 45 cycles at 95°C for 20 s, 60°C for 20 s and 72°C for 20 s. A melting curve analysis step was performed at the end of amplification, consisting of continuous heating from 60°C to 95°C with 0.1°C increments every 1 s. Serial dilutions of DNA from non-irradiated cells were analyzed to establish standard curves. The sizes of PCR products were verified by agarose gel electrophoresis (Supplementary Fig. 1A).
Table 1.
Sequences of primers used in the study
| Name | Symbol | Orientation | Sequence (5’–3’) | Size (bp) |
|---|---|---|---|---|
| NADH dehydrogenase subunit 6 | ND6 | Forward | ACCATCATTCAAGTAGCACAACTAT | 126 |
| (mtDNA) | Reverse | AGTTGGAGTAATTAATCTTGATGGT | ||
| beta-2 microglobulin | B2m | Forward | AGGAGTGGATCTCTGGAAAG | 169 |
| (nDNA) | Reverse | GCTTGATCACATGTCTCGAT | ||
| peptidylprolyl isomerase A | Ppia | Forward | GAGCTGTTTGCAGACAAAGTTC | 125 |
| Reverse | CCCTGGCACATGAATCCTGG | |||
| polymerase (DNA directed), gamma | Polg | Forward | GCAGATGTATGCAGTCACAA | 92 |
| Reverse | TGTCCACAGGAAGATTCAAC | |||
| peroxisome proliferator-activated receptor, gamma, coactivator 1 alpha | Ppargc1a | Forward | TCATTTGATGCACTGACAGA | 153 |
| Reverse | GTAGCTGAGCTGAGTGTTGG | |||
| transcription factor A, mitochondrial | Tfam | Forward | TCCCCTCGTCTATCAGTCTT | 98 |
| Reverse | TGTGGAAAATCGAAGGTATG | |||
| transcription factor B2, mitochondrial | Tfb2m | Forward | AGAGGAACATGGATGGAGAG | 230 |
| Reverse | GCAGGAGTACAGATCGAACA | |||
| nuclear respiratory factor 1 | Nrf1 | Forward | CGTACCATCACAGACCGTAG | 157 |
| Reverse | AGCCACAGCAGAGTAATTCA | |||
| GA repeat binding protein, beta 1 | Gabpb1 | Forward | TTTGGGGAAGAAGCTTTTAG | 107 |
| Reverse | GAGAAGTTCCCAACCAGTCT |
Mitochondrial mass
Mitochondrial mass was measured as previously described [12] with minor modification. Briefly, cells were incubated with serum-free DMEM containing 1 μM MitoTracker Green FM for 30 min at 37°C. Cells were then trypsinized and washed twice with phosphate-buffered saline (PBS), followed by resuspension in PBS. MitoTracker fluorescence was obtained using an EPICS XL flow cytometer (Beckman Coulter; Brea, CA, USA).
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis and western blotting
Cells were collected and lysed with lysis buffer [50 mM Tris-HCl (pH 7.5), 1% (v/v) Triton X-100, 5% (v/v) glycerol, 5 mM EDTA and 150 mM NaCl]. After centrifugation at 18 000 g for 15 min at 4°C, supernatant was collected and boiled for 3 min with 3-fold concentrated Laemmli sample buffer [0.1875 M Tris-HCl (pH 6.8), 15% (v/v) β-mercaptoethanol, 6% (w/v) sodium dodecyl sulfate (SDS), 30% (v/v) glycerol and 0.006% (w/v) bromophenol blue]. Proteins were separated by SDS-polyacrylamide gel electrophoresis and then transferred onto a nitrocellulose membrane (Advantec TOYO; Tokyo, Japan). The membrane was blocked with Tris-buffered saline with Tween 20 (TBST) [10 mM Tris-HCl (pH 7.4), 0.1 M NaCl and 0.1% Tween-20] containing 5% (w/v) non-fat skim milk and probed with specific antibodies diluted in TBST containing 5% (w/v) non-fat skim milk, overnight at 4°C. After probing with HRP-conjugated secondary antibodies, the bound antibodies were detected with Western Lightning Plus-ECL. Images were captured using a LAS 4000 mini image analyzer (Fujifilm; Tokyo, Japan) and analyzed with MultiGauge software (Fujifilm).
Quantitative reverse transcription–polymerase chain reaction
Quantitative reverse transcription (qRT)-PCR analysis was performed as previously described [21]. Total RNA was extracted using a Relia Prep™ RNA Cell Miniprep System (Promega Corporation, Madison, WI, USA). The RNA sample (2 μg) was reverse-transcribed using the ReverTra Ace® qPCR RT Master Mix (TOYOBO, Osaka, Japan). The obtained cDNA was subjected to real-time PCR analysis using a LightCycler Nano System, with the samples being prepared using FastStart Essential DNA Green Master mix. The sequences of the PCR primers used for qRT-PCR are shown in Table 1. The following conditions were applied for PCR analysis: initial denaturation at 95°C for 10 min, followed by 45 cycles at 95°C for 20 s, 60°C for 20 s and 72°C for 20 s. A melting curve analysis step was performed at the end of amplification, consisting of continuous heating from 60°C to 95°C with 0.1°C increments every 1 s. Serial dilutions of DNA from non-irradiated cells were analyzed to establish standard curves. The relative mRNA level of each gene was normalized to that of peptidylprolyl isomerase A, which was used as the internal control. The sizes of PCR products were verified by agarose gel electrophoresis (Supplementary Fig. 2).
Transmission electron microscopy
For electron microscopy, cultured cells were washed with PBS and fixed in 2.5% glutaraldehyde/0.1 M phosphate buffer for 4 h. Subsequently, cells were postfixed in 1% osmium tetroxide for 1.5 h. Fixed cells were collected by Cell Scrapers (Nalge Nunc International; Rochester, NY, USA) and centrifuged at 1000 rpm for 5 min. Collected cells were embedded in 3% agarose, dehydrated in a graded alcohol series, and embedded in Quetol 812 (Nisshin EM; Tokyo, Japan). Ultrathin sections (50 nm) were double-stained with uranyl acetate and lead citrate. All samples were photographically captured using a JOEL transmission electron microscope operated at 90 kV (JEM-1400 Plus, JEOL, Tokyo, Japan).
Cell cycle analysis
Cell cycle analysis was conducted as previously described [22]. Briefly, after cells were collected and washed with ice-cold PBS, they were fixed with ice-cold 70% (v/v) ethanol and kept at −20°C for 12 h. RNA was hydrolyzed with 100 μg/ml RNase A (NIPPON GENE; Tokyo, Japan) at 37°C for 30 min. The cells were stained with propidium iodide (Sigma-Aldrich) for 20 min. The DNA content of the cells was measured using an EPICS XL flow cytometer. Percentages of cells in each phase of the cell cycle were calculated by manually gating the histograms.
Senescence-associated β-gal staining
Cells were plated on 35-mm tissue culture dishes and incubated until exposure to 10 Gy X-rays. After irradiation, cells were stained with the Senescence β-Galactosidase Staining Kit (#9860) according to the manufacturer's instructions (Cell Signaling Technology, Danvers, MA, USA). Senescent cells were identified and scored under a light microscope.
Statistical analysis
All results are expressed as mean ± standard deviation (SD) of at least three separate experiments. Statistical analyses were performed by the Dunnett's test or Mann–Whitney U test. The minimum level of significance was set at P < 0.05.
RESULTS
Effect of X-irradiation on mitochondrial abundance
To determine whether IR influences the quantity of mitochondria, we first examined mtDNA content as a marker of mitochondrial abundance in NIH/3T3 cells exposed to X-irradiation. As shown in Fig. 1A, cellular mtDNA copy number was elevated after X-irradiation in a dose-dependent manner. Time-course analysis revealed that IR increased cellular mtDNA content progressively, up to 72 h post-irradiation (Fig. 1B). Similarly, X-irradiation resulted in a time-dependent increase in mitochondrial mass, measured by MitoTracker Green staining of mitochondria (Fig. 1C). These results indicate that X-irradiation stimulates an increase in mitochondrial abundance.
Fig. 1.

Effect of X-irradiation on mitochondrial abundance. (A) NIH/3T3 cells were exposed to X-rays at the indicated doses, followed by a 72-h incubation period. Total genomic DNA was extracted and the mtDNA/nDNA ratio was determined by real-time PCR. (B) NIH/3T3 cells were exposed to 10 Gy X-rays and incubated for the indicated times. Total genomic DNA was extracted and the mtDNA/nDNA ratio was determined by real-time PCR. (C) NIH/3T3 cells were exposed to 10 Gy X-rays and incubated for the indicated times. The cells were stained with MitoTracker Green and fluorescence intensity was evaluated by flow cytometry. Data are expressed as mean ± SD of three independent experiments. *P < 0.05, **P < 0.01 versus 0 Gy or 0 h (Dunnett's test).
Effect of X-irradiation on autophagic activity, mitochondrial protein expression and mitochondrial DNA replication
To examine whether macroautophagy was involved in the increase in mitochondrial abundance after irradiation, we utilized established biochemical measures of autophagic activity. Covalent lipidation of the ubiquitin-fold protein Atg8/microtubule–associated protein light chain 3 (LC3) is essential for autophagy induction [23]. The unconjugated LC3-I form is diffuse in the cytosol, whereas phosphatidylethanolamine-conjugated LC3-II is localized to autophagic vacuoles and exhibits greater mobility by SDS-PAGE [24]. After X-irradiation, LC3-II level was slightly diminished at 8 h post-irradiation, followed by a gradual increase toward 72 h post irradiation (Fig. 2A). Because the LC3-II amount is an index of autophagic activity [24], this result shows that autophagic activity initially drops after irradiation and then recovers to the basal level by 72 h after irradiation. This time-course is inconsistent with the time-course change of mitochondrial abundance after irradiation. Therefore, it suggests that radiation-induced upregulation of mitochondrial abundance is not due to the reduction of autophagic activity.
Fig. 2.

Effect of X-irradiation on autophagic activity, mitochondrial protein expression and mtDNA replication. NIH/3T3 cells were exposed to 10 Gy X-rays and incubated for the indicated times. (A) The expression levels of LC3-I and -II in whole-cell extracts were analyzed by western blotting. Actin was used as a loading control. Representative blots of three experiments are shown. (B) The expression levels of COX IV and Cyt c in whole-cell extract were analyzed by western blotting. Actin was used as a loading control. Representative blots of three experiments are shown. (C) Quantitative analysis of mitochondrial protein expression. Densitometric analysis was performed, and expression levels of COX IV and Cyt c were normalized to that of actin. n.s. = not significant (Dunnett's test). (D) The expression level of Polg mRNA was determined by qRT-PCR, using total RNA extracted from the cells. Data are expressed as mean ± SD of three independent experiments. *P < 0.05 versus 0 h (Dunnett's test).
We then explored whether IR affects mitochondrial protein expression and mtDNA replication, which are associated with the control of mitochondrial abundance [2]. To this end, we first tested the expression of two mitochondrial proteins, COX IV and Cyt c, in whole-cell extract obtained from NIH/3T3 cells exposed to 10 Gy X-rays. Contrary to our expectations, X-irradiation caused no significant impact on the expression of these mitochondrial proteins (Fig. 2B and C). To investigate whether the radiation-induced mtDNA upregulation was associated with an increase in mtDNA replication, we examined the mRNA expression of Polg, a component of DNA polymerase γ that regulates mtDNA replication [25]. As shown in Fig. 2D, X-irradiation did not cause an increase, but rather a slight decrease in Polg expression at 48 h post-irradiation. These results suggest that X-irradiation upregulates mitochondrial abundance without increasing mitochondrial protein expression and mtDNA replication.
Effect of X-irradiation on the expression of mitochondrial biogenesis-related genes
In general, increased mitochondrial abundance is induced via mitochondrial biogenesis, a process by which new mitochondria are produced via growth and division of pre-existing mitochondria [2, 4]. Since the transcriptional activation of genes related to mitochondrial biogenesis is generally regarded to be critical for this process, we examined whether IR influences it. After NIH/3T3 cells were irradiated with 10 Gy, total RNA was extracted and mRNA expression of the following genes was evaluated by qRT-PCR: peroxisome proliferator–activated receptor, gamma, coactivator 1 alpha (Ppargc1a); transcription factor A, mitochondrial (Tfam); transcription factor B2, mitochondrial (Tfb2m); nuclear respiratory factor 1 (Nrf1); and GA-repeat binding protein, beta 1 [Gabpb1, also known as nuclear respiratory factor 2 (Nrf2)]. While the expression of Ppargc1a, which is a co-transcriptional regulation factor that promotes mitochondrial biogenesis by activating various transcription factors, was significantly upregulated by X-irradiation up to 72 h post irradiation (Fig. 3A), the expression of other genes, which are all transcription factors involved in mitochondrial biogenesis, was only marginally affected by IR (Fig. 3B–E). Collectively, these data imply that, although X-irradiation upregulates cellular mitochondrial abundance, it is not associated with the activation of the conventional mitochondrial biogenesis process.
Fig. 3.

Effect of X-irradiation on the expression of mitochondrial biogenesis-related genes. NIH/3T3 cells were exposed to 10 Gy X-rays and incubated for the indicated times. Total RNA was extracted from the cells, and the mRNA levels of genes related to mitochondrial biogenesis were analyzed by qRT-PCR. The mRNA expression of the following genes was evaluated: Ppargc1a (A), Tfam (B), Tfb2m (C), Nrf1 (D) and Gabpb1 (E). Data are expressed as mean ± SD of three independent experiments. *P < 0.05 versus 0 h (Dunnett's test).
Ultrastructural analysis of mitochondria after X-irradiation
The above results prompted us to examine the number and the structure of mitochondria in the irradiated cells using transmission electron microscopy (TEM). Compared with non-irradiated cells (Fig. 4A and B), the irradiated cells (Fig. 4C–F) appeared to have higher numbers of mitochondria. In addition, it was noted that the mitochondria acquired a granular shape after irradiation, with the structure of cristae appearing blurred. These findings indicate that X-irradiation led to an increase in the number of mitochondria with an atypical cristae structure.
Fig. 4.
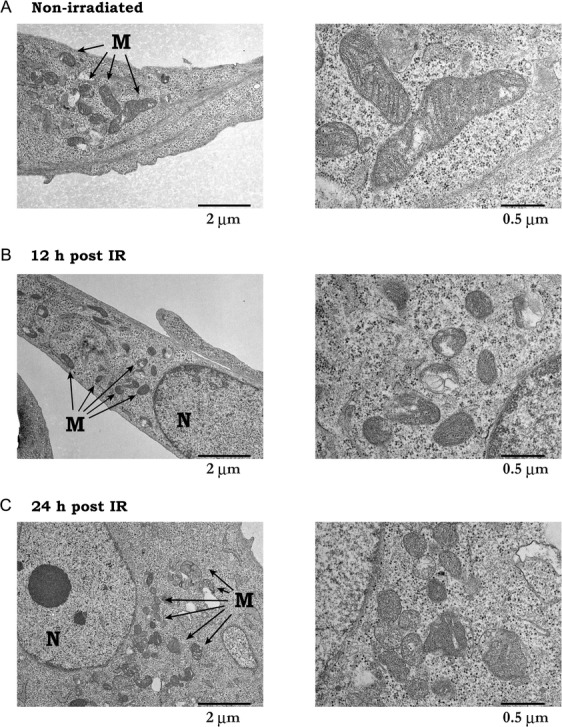
Ultrastructural analysis of mitochondria after X-irradiation. Mouse embryonic fibroblasts (MEFs) were exposed to 10 Gy X-rays, and ultrastructural morphology was visualized by TEM. (A and B) non-irradiated. (C and D) 12 h after irradiation. (E and F) 24 h after irradiation. B, D and F are magnified images of mitochondria in A, C and E, respectively. M = mitochondria, N = nucleus. Representative electron micrographs are shown.
Effect of X-irradiation on cell cycle distribution and cellular senescence
Because previous studies have shown that the cells in different phases of the cell cycle contain different amounts of mitochondria [26–28], we analyzed the cell cycle distribution after X-irradiation. As shown in Fig. 5A, irradiated cells displayed a significant increase of cells in the G2 phase until 24 h post irradiation, indicating a pronounced G2 arrest. The number of cells in S phase decreased at 8 to 24 h post irradiation compared with in the control cells, probably due to G1 arrest. Although the cell cycle distribution of the irradiated cells partially recovered at 48 and 72 h after the peak of G2 arrest at 24 h, the proportion of G2/M cells was still much higher at these time points than in the control, indicating the induction of long-term G2 arrest.
Fig. 5.

Effect of X-irradiation on cell cycle distribution and cellular senescence. (A) NIH/3T3 cells were exposed to 10 Gy X-rays and incubated for the indicated times. Cell cycle analysis was performed by propidium iodide staining and flow cytometry. Percentages of cells in each phase of the cell cycle were calculated, and data are expressed as mean ± SD of three independent experiments. (B) NIH/3T3 cells were left untreated [IR(-)] or exposed to 10 Gy X-rays and incubated for 72 h [IR(+)]. Cellular senescence was measured by senescence-associated (SA) β-gal assay. SA β-gal–positive cells were scored under a light microscope. Data are expressed as mean ± SD of three independent experiments. *P < 0.05 (Mann–Whitney U test).
Since it has been shown that cells under long-term G2 arrest undergo senescence and that mitochondria content is high in senescent cells [29, 30], we analyzed cellular senescence by senescence-associated (SA) β-gal assay in irradiated NIH/3T3 cells at 72 h post irradiation. As shown in Fig. 5B, the rates of SA β-gal–positive cells increased from 3.0 ± 0.3% in control cells to 9.2 ± 3.2% in irradiated cells. These results indicate that X-irradiation induces long-term G2 arrest and consequent cellular senescence, implying the involvement of these cellular responses in the upregulation of mitochondrial abundance after irradiation.
DISCUSSION
In this study, we demonstrated the time-dependent increase in mtDNA as well as mitochondrial mass after IR, indicating radiation-induced upregulation of mitochondrial abundance. While previous studies have reported a similar phenomenon, several conflicting results have also been presented [31, 32]. These suggest that, although IR normally results in the upregulation of mitochondrial abundance, differences in experimental settings (e.g. cell line, type of radiation, irradiation dose, etc.) could influence mitochondrial status after irradiation, leading to different outcomes. Further investigation is required for elucidating the mechanism behind this inconsistency.
The results from this study imply that the upregulation of mitochondrial abundance after IR is not due to activation of the conventional mitochondrial biogenesis process. In support of our results, Eaton and colleagues reported that exposure to IR results in an increase in mtDNA and mitochondrial mass without elevating Tfam protein expression in primary fibroblasts [33]. Our data also suggest that increased autophagy is unlikely to be responsible for the upregulation of mitochondrial abundance. Not only did we not observe the decrease in LC3-II level at the time-points when mitochondrial abundance increased (Fig. 2A), we did not find any trace of enhanced mitophagy after irradiation by TEM analysis. Additionally, the collapse of mitochondrial membrane potential, which is often concomitant with mitophagy, was not induced by X-irradiation (data not shown). These findings corroborate the idea that increased mitochondrial abundance by IR is independent of macroautophagy/mitophagy.
It has been demonstrated that mitochondrial abundance periodically fluctuates during cell cycle progression [26–28], indicating the association of cell cycle progression with the regulation of mitochondrial abundance. In this study, we found that, following the pronounced G2 arrest peak at 24 h after X-irradiation at 10 Gy, cell cycle distribution of the irradiated cells did not return to normal and the cells seemingly made a transition to long-term G2 arrest (Fig. 5A). Previous studies have shown that long-term G2 arrest after high-dose irradiation triggers cellular senescence, and cellular mitochondria content is high in senescent cells as well as in cells isolated from older individuals [29, 34]. In addition, the average number of mtDNA genomes per cell was reported to increase at late passages of various diploid human cells [35] and an increase in mitochondrial mass was reported during replicative senescence of MRC-5 human lung fibroblasts [36]. In the present study, we revealed that IR induced cellular senescence in NIH/3T3 fibroblasts. These lines of evidence imply that long-term cell cycle arrest and consequent cellular senescence contribute to the upregulation of mitochondrial abundance after IR.
At this point, it remains unclear how cellular senescence leads to the increase in mitochondria content. Lee and colleagues have suggested that stress-induced microtubule derangement is one of the molecular events involved in the increase in mitochondrial mass upon treatment with hydrogen peroxide [37]. Microtubules are the major components of cytoskeletal systems that are responsible for the regulation of the mitochondrial distribution in the cell. Because the appearance of senescent cells (enlarged and flattened) indicates the rearrangement (or derangement) of cytoskeletal systems, this seems to be a good follow-on point for future investigation. Meanwhile, as the senescent cells constitute only <10% of the whole population of the irradiated cells, it is uncertain that the increased mitochondrial abundance after IR can be explained solely by senescence induction. This should be elucidated by comparing the mitochondrial content in SA β-gal–positive and –negative cells after irradiation.
Additionally, Ppargc1a mRNA expression was significantly elevated after IR, unlike other mitochondrial biogenesis-related genes tested in this study (Fig. 3). Ppargc1a is a transcriptional co-activator and is widely recognized as the master regulator of mitochondrial biogenesis [38, 39]. However, since Ppargc1a was the only gene that responded to IR, it is suggested that Ppargc1a participates in biological processes other than mitochondrial biogenesis upon exposure to IR. Besides mitochondrial biogenesis, Ppargc1a has been associated with numerous signaling pathways and biological processes such as autophagy, cytokine production, and angiogenesis [39]. Since these processes are reportedly stimulated by IR, it would be worthwhile to investigate the role of Ppargc1a in these processes in future studies. Furthermore, the mechanism by which IR induces Ppargc1a mRNA expression is another area of research to be studied. Gene expression of Ppargc1a is stimulated by various external stimuli via the activation of transcription factors. Peroxisome proliferator–activated receptors (PPARs), cAMP response element-binding protein (CREB) and Yin Yang 1 (YY1) are well-known transcription factors regulating Ppargc1a expression, all of which are reported to be influenced by IR [40]. In particular, considering the involvement of CREB in the DNA damage response [41, 42], CREB could be responsible for the induction of Ppargc1a after IR.
The upregulation of mitochondrial abundance after exposure to IR may be consequently associated with increased cellular oxidative stress. Ljubicic et al. have shown that intracellular ROS levels are affected by the mitochondrial content of the cell [43]. In addition, we previously demonstrated that radiation-induced G2/M arrest led to a sustained increase in the number of cells with elevated mitochondrial content and those under the higher oxidative stress conditions [12]. Since oxidative stress is one of the main causes of late post-radiation effects [44], the upregulation of mitochondrial abundance after irradiation may influence the consequence of radiation exposure in mammalian cells.
TEM evaluation of the mitochondrial morphology in the irradiated cells revealed two distinct changes: increase in mitochondrial number and alteration in mitochondrial morphology. Mitochondria in the irradiated MEFs exhibited a more granular shape than those in the control MEFs. Similar morphological change in mitochondria was also found in irradiated NIH/3T3 cells when evaluated with mitochondria staining and laser confocal microscopy (Supplementary Fig. 3). This is a typical signature of mitochondrial fission. Since IR has been shown to stimulate mitochondrial fission [13, 45, 46], our TEM data may represent not only the increase in mitochondrial abundance, but also the induction of mitochondrial fission after irradiation. The other change noted was blurring of the cristae structure. The architecture of cristae is considered to be closely associated with mitochondrial integrity, and its disruption has been observed in various diseases [47]. Thus, the blurring of cristae may reflect a change in mitochondrial function, brought on by exposure to IR. These lines of evidence seem to support our view that the upregulation of mitochondrial abundance after IR is not due to an increase in intact mitochondria via the activation of mitochondrial biogenesis signaling.
In summary, we investigated the mechanism by which IR influences cellular mitochondrial abundance and found evidence suggesting that it is an event independent of macroautophagy and mitochondrial biogenesis. Furthermore, our data implied the involvement of long-term cell cycle arrest and consequent cellular senescence. Considering the growing significance of mitochondria in cellular radioresponses, we believe the present study provides novel insights to understand the effects of IR on mitochondria.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
SUPPLEMENTARY DATA
Supplementary data are available at the Journal of Radiation Research online.
CONFLICT OF INTEREST
The authors declare that there are no conflicts of interest.
FUNDING
This work was supported by the JSPS KAKENHI [Grant numbers 23780286 to T.Y., 25861045 to H.Y., and 24659551 to O.I.], the Takeda Science Foundation to T.Y., and a research grant for the Study of the Health Effects of Radiation Organized by Ministry of the Environment, Japan to O.I. and T.Y. The sponsors had no role in the study design, collection, analysis and interpretation of data, drafting of the manuscript, or the decision to submit the manuscript for publication.
REFERENCES
- 1. Duchen MR. Mitochondria in health and disease: perspectives on a new mitochondrial biology. Mol Aspects Med 2004;25:365–451. [DOI] [PubMed] [Google Scholar]
- 2. Michel S, Wanet A, De Pauw A et al. Crosstalk between mitochondrial (dys)function and mitochondrial abundance. J Cell Physiol 2012;227:2297–310. [DOI] [PubMed] [Google Scholar]
- 3. Zhu J, Wang KZQ, Chu CT.. After the banquet: mitochondrial biogenesis, mitophagy, and cell survival. Autophagy 2013;9:1663–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Palikaras K, Tavernarakis N.. Mitochondrial homeostasis: the interplay between mitophagy and mitochondrial biogenesis. Exp Gerontol 2014;56:182–8. [DOI] [PubMed] [Google Scholar]
- 5. Malpass K. Neurodegenerative disease: defective mitochondrial dynamics in the hot seat—a therapeutic target common to many neurological disorders. Nat Rev Neurol 2013;9:417. [DOI] [PubMed] [Google Scholar]
- 6. Wredenberg A, Wibom R, Wilhelmsson H et al. Increased mitochondrial mass in mitochondrial myopathy mice. Proc Natl Acad Sci U S A 2002;99:15066–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Eriksson D, Stigbrand T.. Radiation-induced cell death mechanisms. Tumour Biol 2010;31:363–72. [DOI] [PubMed] [Google Scholar]
- 8. Jackson SP, Bartek J.. The DNA-damage response in human biology and disease. Nature 2009;461:1071–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leach JK, Van Tuyle G, Lin PS et al. Ionizing radiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen. Cancer Res 2001;61:3894–901. [PubMed] [Google Scholar]
- 10. Nugent SME, Mothersill CE, Seymour C et al. Increased mitochondrial mass in cells with functionally compromised mitochondria after exposure to both direct gamma radiation and bystander factors. Radiat Res 2007;168:134–42. [DOI] [PubMed] [Google Scholar]
- 11. Ogura A, Oowada S, Kon Y et al. Redox regulation in radiation-induced cytochrome c release from mitochondria of human lung carcinoma A549 cells. Cancer Lett 2009;277:64–71. [DOI] [PubMed] [Google Scholar]
- 12. Yamamori T, Yasui H, Yamazumi M et al. Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radic Biol Med 2012;53:260–70. [DOI] [PubMed] [Google Scholar]
- 13. Yamamori T, Ike S, Bo T et al. Inhibition of the mitochondrial fission protein dynamin-related protein 1 (Drp1) impairs mitochondrial fission and mitotic catastrophe after x-irradiation. Mol Biol Cell 2015;26:4607–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gong B, Chen Q, Almasan A.. Ionizing radiation stimulates mitochondrial gene expression and activity. Radiat Res 1998;150:505–12. [PubMed] [Google Scholar]
- 15. Zhou H, Ivanov VN, Lien Y-C et al. Mitochondrial function and nuclear factor-κB-mediated signaling in radiation-induced bystander effects. Cancer Res 2008;68:2233–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rajendran S, Harrison SH, Thomas RA et al. The role of mitochondria in the radiation-induced bystander effect in human lymphoblastoid cells. Radiat Res 2011;175:159–71. [DOI] [PubMed] [Google Scholar]
- 17. Kam WW-Y, Banati RB. Effects of ionizing radiation on mitochondria. Free Radic Biol Med 2013;65:607–19. [DOI] [PubMed] [Google Scholar]
- 18. Nugent S, Mothersill CE, Seymour C et al. Altered mitochondrial function and genome frequency post exposure to gamma-radiation and bystander factors. Int J Radiat Biol 2010;86:829–41. [DOI] [PubMed] [Google Scholar]
- 19. Malakhova L, Bezlepkin VG, Antipova V et al. The increase in mitochondrial DNA copy number in the tissues of gamma-irradiated mice. Cell Mol Biol Lett 2005;10:721–32. [PubMed] [Google Scholar]
- 20. Gubina NE, Merekina OS, Ushakova TE.. Mitochondrial DNA transcription in mouse liver, skeletal muscle, and brain following lethal x-ray irradiation. Biochemistry 2010;75:777–83. [DOI] [PubMed] [Google Scholar]
- 21. Sakai Y, Yamamori T, Yasui H et al. Downregulation of the DNA repair enzyme apurinic/apyrimidinic endonuclease 1 stimulates transforming growth factor-β1 production and promotes actin rearrangement. Biochem Biophys Res Commun 2015;461:35–41. [DOI] [PubMed] [Google Scholar]
- 22. Suzuki M, Yamamori T, Yasui H et al. Effect of MPS1 inhibition on genotoxic stress responses in murine tumour cells. Anticancer Res 2016;36:2783–92. [PubMed] [Google Scholar]
- 23. Mizushima N, Ohsumi Y, Yoshimori T.. Autophagosome formation in mammalian cells. Cell Struct Funct 2002;27:421–9. [DOI] [PubMed] [Google Scholar]
- 24. Mizushima N, Yoshimori T.. How to interpret LC3 immunoblotting. Autophagy 2007;3:542–5. [DOI] [PubMed] [Google Scholar]
- 25. Holt IJ, Reyes A.. Human mitochondrial DNA replication. Cold Spring Harb Perspect Biol 2012;4, 10.1101/cshperspect.a012971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jahnke VE, Sabido O, Freyssenet D.. Control of mitochondrial biogenesis, ROS level, and cytosolic Ca2+ concentration during the cell cycle and the onset of differentiation in L6E9 myoblasts. Am J Physiol Cell Physiol 2009;296:C1185–94. [DOI] [PubMed] [Google Scholar]
- 27. Havens CG, Ho A, Yoshioka N et al. Regulation of late G1/S phase transition and APC Cdh1 by reactive oxygen species. Mol Cell Biol 2006;26:4701–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martínez-Diez M, Santamaría G, Ortega AD et al. Biogenesis and dynamics of mitochondria during the cell cycle: significance of 3’UTRs. PLoS One 2006;1:e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ye C, Zhang X, Wan J et al. Radiation-induced cellular senescence results from a slippage of long-term G2 arrested cells into G1 phase. Cell Cycle 2013;12:1424–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee H-C, Yin P-H, Chi C-W et al. Increase in mitochondrial mass in human fibroblasts under oxidative stress and during replicative cell senescence. J Biomed Sci 2002;9:517–26. [DOI] [PubMed] [Google Scholar]
- 31. Wang L, Kuwahara Y, Li L et al. Analysis of Common Deletion (CD) and a novel deletion of mitochondrial DNA induced by ionizing radiation. Int J Radiat Biol 2007;83:433–42. [DOI] [PubMed] [Google Scholar]
- 32. Zhou X, Li N, Wang Y et al. Effects of X-irradiation on mitochondrial DNA damage and its supercoiling formation change. Mitochondrion 2011;11:886–92. [DOI] [PubMed] [Google Scholar]
- 33. Eaton JS, Lin ZP, Sartorelli AC et al. Ataxia-telangiectasia mutated kinase regulates ribonucleotide reductase and mitochondrial homeostasis. J Clin Invest 2007;117:2723–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Toussaint O, Medrano EE, Von Zglinicki T. Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Exp Gerontol 2000;35:927–45. [DOI] [PubMed] [Google Scholar]
- 35. Shmookler Reis RJ, Goldstein S. Mitochondrial DNA in mortal and immortal human cells. Genome number, integrity, and methylation. J Biol Chem 1983;258:9078–85. [PubMed] [Google Scholar]
- 36. Hwang ES, Yoon G, Kang HT. A comparative analysis of the cell biology of senescence and aging. Cell Mol Life Sci 2009;66:2503–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee C-F, Liu C-Y, Hsieh R-H et al. Oxidative stress-induced depolymerization of microtubules and alteration of mitochondrial mass in human cells. Ann N Y Acad Sci 2005;1042:246–54. [DOI] [PubMed] [Google Scholar]
- 38. Wenz T. Regulation of mitochondrial biogenesis and PGC-1α under cellular stress. Mitochondrion 2013;13:134–42. [DOI] [PubMed] [Google Scholar]
- 39. Chan MC, Arany Z. The many roles of PGC-1α in muscle—recent developments. Metabolism 2014;63:441–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dominy JE, Puigserver P. Mitochondrial biogenesis through activation of nuclear signaling proteins. Cold Spring Harb Perspect Biol 2013;5, 10.1101/cshperspect.a015008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Amorino GP, Mikkelsen RB, Valerie K et al. Dominant-negative cAMP-responsive element-binding protein inhibits proliferating cell nuclear antigen and DNA repair, leading to increased cellular radiosensitivity. J Biol Chem 2003;278:29394–9. [DOI] [PubMed] [Google Scholar]
- 42. Shi Y, Venkataraman SL, Dodson GE et al. Direct regulation of CREB transcriptional activity by ATM in response to genotoxic stress. Proc Natl Acad Sci U S A 2004;101:5898–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ljubicic V, Hood DA. Kinase-specific responsiveness to incremental contractile activity in skeletal muscle with low and high mitochondrial content. Am J Physiol Endocrinol Metab 2008;295:E195–204. [DOI] [PubMed] [Google Scholar]
- 44. Szumiel I. Ionizing radiation-induced oxidative stress, epigenetic changes and genomic instability: the pivotal role of mitochondria. Int J Radiat Biol 2015;91:1–12. [DOI] [PubMed] [Google Scholar]
- 45. Zhang B, Davidson MM, Zhou H et al. Cytoplasmic irradiation results in mitochondrial dysfunction and DRP1-dependent mitochondrial fission. Cancer Res 2013;73:6700–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kobashigawa S, Suzuki K, Yamashita S. Ionizing radiation accelerates Drp1-dependent mitochondrial fission, which involves delayed mitochondrial reactive oxygen species production in normal human fibroblast-like cells. Biochem Biophys Res Commun 2011;414:795–800. [DOI] [PubMed] [Google Scholar]
- 47. Zick M, Rabl R, Reichert AS. Cristae formation-linking ultrastructure and function of mitochondria. Biochim Biophys Acta 2009;1793:5–19. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.