

Abstract
Ionizing radiation causes biological damage that leads to severe health effects. However, the effects and subsequent health implications caused by exposure to low-dose radiation are unclear. The objective of this study was to determine phosphoprotein profiles in normal human fibroblast cell lines in response to low-dose and high-dose γ-radiation. We examined the cellular response in MRC-5 cells 0.5 h after exposure to 0.05 or 2 Gy. Using 1318 antibodies by antibody array, we observed ≥1.3-fold increases in a number of identified phosphoproteins in cells subjected to low-dose (0.05 Gy) and high-dose (2 Gy) radiation, suggesting that both radiation levels stimulate distinct signaling pathways. Low-dose radiation induced nucleic acid–binding transcription factor activity, developmental processes, and multicellular organismal processes. By contrast, high-dose radiation stimulated apoptotic processes, cell adhesion and regulation, and cellular organization and biogenesis. We found that phospho-BTK (Tyr550) and phospho-Gab2 (Tyr643) protein levels at 0.5 h after treatment were higher in cells subjected to low-dose radiation than in cells treated with high-dose radiation. We also determined that the phosphorylation of BTK and Gab2 in response to ionizing radiation was regulated in a dose-dependent manner in MRC-5 and NHDF cells. Our study provides new insights into the biological responses to low-dose γ-radiation and identifies potential candidate markers for monitoring exposure to low-dose ionizing radiation.
Keywords: MRC-5 cells, NHDF cells; explorer phospho antibody microarray; low-dose γ-radiation; phospho-BTK (Tyr550); phospho-Gab2 (Tyr643)
INTRODUCTION
Humans are continually exposed to low-dose background ionizing radiation (IR) from various natural sources [1]. Exposure to low doses (≤100 mGy) of man-made radiation is associated with certain occupations and medical diagnostic and therapeutic procedures [2–4]. However, there is a lack of suitable methods for directly assessing such low-dose IR exposure, and there are great uncertainties about its health risk [5]. Therefore, there has been a growing concern about the hazardous effects caused by low-dose IR.
Nevertheless, previous studies have shown that the biological effects of low-dose radiation differ from those observed with high doses, and are primarily unrelated to direct DNA damage [6–9]. Exposure to high-dose radiation ionizes the molecules in living cells, which causes DNA damage and promotes malignant tumor formation [10–12]. By contrast, low-dose radiation confers some beneficial effects, called hormesis, upon organisms [13]. The adaptive response has been studied primarily with low-dose radiation [14]. Previous studies suggest that DNA repair and cell cycle regulation are involved in the radiation-induced adaptive response [15]. Therefore, the LNT model of the health risk associated with low-dose radiation has been criticized on the basis of the different biological effects induced by low-dose versus high-dose radiation [16]. Cell behavior exposed to low-dose radiation has been well-documented as mentioned above, but the underlying regulatory mechanisms are still poorly understood.
The genomes of all living organisms constantly suffer deleterious attacks. DNA double-strand breaks (DSBs) are considered the most biologically damaging legions produced by IR and radiomimetic chemicals, or by endogenous agents, mainly reactive oxygen species [17]. If left unrepaired, DSBs can result in permanent cell cycle arrest, induction of apoptosis or mitotic cell death caused by loss of genomic material [18]. In order to cope with this lesion, cells activate a complex network of interacting pathways that lead to damage repair and resumption of the normal cellular life cycle or to programmed cell death. This network, called the DNA damage response (DDR), coordinates the activation of cell cycle checkpoint, the appropriate DNA repair pathways, and numerous other responses [19]. The detection and subsequent downstream signaling of DSBs requires the interplay of the actions of various proteins whose functions can be categorized as DNA damage sensors, transducers, mediators and effectors. Especially, the five most prominent DDR kinases—ataxia telangiectasia mutated (ATM), ATM and Rad3-related protein (ATR), DNA-dependent protein kinase (DNA-PKcs), Checkpoint kinase1 (Chk1), and Checkpoint kinase2 (Chk2)—have been extensively reviewed [20].
Activation of the p38 MAPK pathway by cytokines and receptor ligands normally leads to cell differentiation. Activation of p38 MAPK through environmental stress can mediate cell death. In response to DNA damage stimuli that induce DSBs (IR, UV, chemotherapeutic drugs) [21], activation of p38 MAPK can also lead to the induction of a G2/M cell cycle checkpoint through p53-dependent and -independent mechanisms [22–27]. Another member of the MAPK family, c-Jun kinase (JNK), is activated by IR [28], in an ATM-dependent manner [29, 30]. And also, p53 has been shown to be phosphorylated in vitro on both the N-terminal regulatory domains by a number of different kinases, including DNA-dependent protein kinase (DNA-PK) [31], ATM [32] and ATR [33, 34] at serine 15 (Ser15). Ser 15 has been studied particularly closely, as it is the site of p53 phosphorylation by the ATM kinase [32, 33], whose activity is required for p53 stabilization in response to IR and some other types of DNA damage [35, 36]. These phosphorylation events are involved in regulating p53 activity.
The relationship between mRNA and protein levels is not necessarily linear, but is determined by the activity of proteins that are directly responsible for maintaining the correct cellular signal function [37]. Therefore, phosphorylation events are likely to play important roles in rapid cellular response to radiation. As mentioned above, the early response of proteins to IR-induced DNA damage is well established. However, the complete profile of markers for biological responses to low-dose radiation (≤100 mGy) has not been elucidated to date. The ultimate goal is to identify a specific marker that can be applied to a non-invasively obtained biological sample to assist in a medical or policy risk–benefit analysis and decision-making processes in radiation protection or other radiation scenarios. The primary goals of our study were to determine early response proteins and phosphoprotein profiles that result from exposure to low-dose radiation in normal human fibroblast cell lines (MRC-5 and NHDF).
MATERIALS AND METHODS
Cell culture and radiation treatment
Normal human lung fibroblasts (MRC-5) and normal human dermal fibroblasts (NHDF) were acquired from the American Type Culture Collection (Mannassas, VA, USA). Fibroblast cells were cultured in MEM medium containing 10% fetal bovine serum, penicillin (100 U/ml), and streptomycin (100 U/ml) at 37°C under an atmosphere of 5% CO2. MRC-5 and NHDF cells at passage 8−10 were used for this study. NHDF and MRC-5 fibroblasts were seeded at a density of 5 × 105 cells in 100 mm dishes and irradiated with 0.05, 0.1, 1, 2 and 4 Gy using a 137Cs γ-irradiator (gammacell®40 Exactor, Best Theratronics, Ottawa, Ontario K2K 0E4, Canada), with a delivery rate of 1.03 Gy/min. The cells were allowed to exposure for 3, 6, 62, 125, and 249 s after irradiation, respectively. After irradiation, the cells were returned to the incubator, and western blotting or Explorer phosphoprotein microarray was performed. The γ-ray generator according to the manual for each set of radiation conditions was certified Gammacell 137Cs source irradiator calibrated by a physicist from the ACME Medical Inc.
Cell viability assay
Cell viability was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell proliferation assay (Sigma, St Louis, MO, USA) 48 h after irradiation. The yellow tetrazolium dye MTT is reduced to purple formazan in the mitochondria of living cells. MTT was added to the cells, and the cells were then incubated for another 3 h at 37°C. Then, the medium solution was removed, 100 μl of dimethyl sulfoxide (DMSO) was added to the cells in each well, and the cultures were mixed on a shaker for 15 min. The absorbance at 540 nm was measured using a spectrophotometer (Lab System, Helsinki, Finland). The MTT assay was repeated at least three times for each cell line in triplicate independent experiments, and then data were analyzed.
Western blotting
Cells were harvested, rinsed with ice-cold phosphate buffered saline, and lysed in homogenization buffer (50 mM Tris-Cl, pH 6.8) containing protease inhibitor and phosphatase inhibitor cocktail (Thermo Scientific, Waltham, MA), 10% sodium dodecyl sulfate (SDS), and 10% glycerol. Protein concentrations of whole-cell lysates were determined using bicinchoninic acid (BCA) protein assay (Thermo Scientific, Waltham, MA, USA). Blots were probed with primary antibodies against phospho-p53 (Ser15), phospho-ETK (Tyr40), ELK1 and ETK (Cell Signaling Technology, Beverly, MA, USA); γ-H2AX, phospho-Nibrin/Nbs1 (Ser343) and Nibrin (Upstate Biotechnology, Lake Placid, NY, USA); p21, p53 and β-actin (Santa Cruz Biotechnology, California, USA); phospho-Gab2 (Tyr643), Gab2, phospho-BTK (Tyr550) and BTK (Abcam, Cambridge, MA, USA); phospho-CamK4 (Thr196/200) and CamK4 (Aviva Systems Biology, San Diego, USA). Western blotting was performed using standard protocols, and membranes were visualized by enhanced chemiluminescence (ECL solution, Amersham Biosciences, Uppsala, Sweden).
Phosphoprotein profiling by the Phospho Explorer antibody microarray
The Phospho Explorer antibody microarray, which was designed and manufactured by Full Moon Biosystems, Inc. (Sunnyvale, CA), contains 1318 antibodies [38]. Each of the antibodies has two replicates that are printed on a coated glass microscope slide, along with multiple positive and negative controls. The antibody array experiment was performed according to manufacturer's protocol [39–42]. We performed both antibody array and analysis using the customized service from ebiogen (Ebiogen Inc., Seoul, Republic of Korea).
Antibody array data acquisition and analysis
Slide scanning was performed using the GenePix 4100A scanner (Molecular Devices Corporation, Sunnyvale, CA, USA). The slides were dried before scanning, and were scanned within 24−48 h. The slides were scanned at 10 μm resolution using optimal laser power and a photomultiplier tube. The scanned images were graded and quantified using GenePix 7.0 Software (Molecular Devices Corporation, Sunnyvale, CA, USA). The numerical data were analyzed using Genowiz 4.0TM (Ocimum Biosolutions, India). After these analyses, the protein information was annotated using the UniProt database. The phosphorylation ratio was calculated using the following formula:
Statistical analysis
Cell culture experiments were repeated at least three times. Values are shown as mean ± S.D. The comparison of quantitative data was analyzed by Student's t-test between two groups when data was normally distributed. (two-tailed; P-values < 0.05 were considered to be statistically significant). The statistical program used was Sigma Plot 12.0 (Systat Software Inc., Point Richmond CA). This normalization makes the average intensity of all samples numerically equivalent to the average intensity of the all genes. Differences between groups were evaluated using two-tailed unpaired or paired Student's t-tests for single comparison. Differentially expressed phosphoproteins were detected by analyzing with a t-test on each radiation group. The statistical program used was MeV 4.9.0 Software (TM4 Development Group, USA). The significantly differential proteomic features were determined by the MeV t-test (P < 0.05).
RESULTS
Low-dose radiation does not affect the cells’ sensitivity to the induction of DNA damage
To study the effect of low-dose radiation on cell viability, normal human fibroblast MRC-5 and NHDF cells were irradiated with 0.5 to 2 Gy of γ-radiation and compared with un-irradiated control cells. All cells were incubated for 48 h after irradiation. We used the MTT assay to compare cell proliferation after various doses of γ-radiation (Fig. 1A). As expected, MRC-5 cells treated with high-dose radiation had lower viability and proliferation than cells treated with low-dose radiation (0.05 Gy). The tumor suppressor p53 gene is a transcription factor that accumulates in cells in response to DNA damage [43, 44]. To examine whether high-dose radiation induces cellular stress in MRC-5 cells, we analyzed changes in DNA-damage proteins induced by low-dose and high-dose radiation. MRC-5 cells were irradiated with 0 (untreated control), 0.05 or 0.1 Gy (low dose), or 2 Gy (high dose). Cells were incubated for 8 h, and then whole-cell lysates were extracted and subjected to western blotting. The phospho-p53 (Ser15), p53 and p21 (CDKN1A) protein levels in MRC-5 cells were measured with specific antibodies. Consistent with the cell viability results shown in Fig. 1A, western blots showed that the expression of proteins involved in DNA-damage responses was higher in cells treated with 2 Gy of γ-radiation than in cells treated with 0.05 to 0.1 Gy. We also obtained similar results using human NHDF skin fibroblast cells (Fig. 1C and D).
Fig. 1.

Effect of γ-radiation in normal human fibroblast cells. (A) MRC-5 cells were incubated for 48 h after exposure to γ-radiation as indicated. Cell viability was determined by MTT assay, and data were expressed as means ± SD (*P < 0.05 and **P < 0.005 compared with untreated controls). (B) Cells were prepared at 8 h after exposure to γ-irradiation (0, 0.05 and 2 Gy), and expression levels of phospho-p53 (Ser15), p53 and p21 were determined by western blot analysis. β-actin was used as a loading control. (C) Cell viability was determined by MTT assay as shown Fig. 1A in NHDF cells. Data represent three independent experiments expressed as means ± SD (*P < 0.05 and **P < 0.005 compared with untreated controls). (D) Western blotting was performed as indicated in Fig. 1B in NHDF cells.
Low-dose and high-dose γ-radiation effects on phosphoprotein profiles in MRC-5 cells
The primary goal of our study was to identify changes in phosphoprotein profiles induced by exposure to low-dose γ-radiation in normal human fibroblast cells. Therefore, we determined specific phosphoproteins responding to low-dose and high-dose irradiation using the Explorer phospho antibody microarray and proteins extracted from MRC-5 cells treated with low-dose (0.05 Gy) and high-dose (2 Gy) γ-radiation. We used two different methods to compare changes in phosphoprotein profiles. First, we analyzed phosphoprotein expression levels in cells treated with 0.05 or 2 Gy of γ-radiation. Second, we compared the differences in protein class of phosphoproteins for early responses to 0.05 or 2 Gy of γ-radiation.
The phospho antibody microarray was used to identify differences in the phosphoprotein profiles in normal human fibroblast cell lines in response to low-dose (0.05 Gy) and high-dose (2 Gy) γ-radiation. Therefore, we identified differentially expressed phosphoproteins in irradiated MRC-5 cells. We screened 1318 proteins on the phospho antibody microarray slides, and obtained a total of 219 phosphoproteins that were upregulated more than 1.1-fold in response to 0.05 and 2 Gy of γ-radiation (Fig. 2A). The levels of 119 phosphoproteins increased after 0.5 h only in response to low-dose radiation (0.05 Gy). On the other hand, the levels of 73 phosphoproteins increased after 0.5 h in response to high-dose radiation (2 Gy). To analyze the statistical significance of the results, we chose proteins with a significance of P < 0.05. To examine the differences in genomic functional categories of proteins responding to different doses of irradiation, we extracted unique gene process categories that were activated early in response to either low-dose or high-dose treatment. We analyzed the expression profiles in MRC-5 cells 0.5 h after irradiation to identify expression signatures associated with molecular function.
Fig. 2.

Differences in phosphoprotein profiles in response to low-dose and high-dose radiation in MRC-5 cells. MRC-5 cells were treated with 0.05 and 2 Gy γ-radiation as indicated, and cell lysates were extracted from MRC-5 cells after 0.5 h. Early response phosphoproteins were identified based on ≥1.1-fold changes from three independent experiments. (A) Representative Venn diagram shows the numbers of phosphoproteins identified in antibody microarrays that were upregulated ≥1.1-fold in response to γ-radiation as indicated at the time and dose ranges. (B) Distribution of protein class of biological functions (GO processes) for upregulated phosphoproteins (≥1.1-fold) associated with the early response (0.5 h) to γ-radiation. In t-tests, all changes were significant (P < 0.05).
The representative graph shown in Fig. 2B indicates that the 0.05 Gy post-irradiation baseline signatures were enriched in transmembrane receptor regulatory/adaptor protein (2.1%), membrane traffic protein (1.4%), oxidoreductase (2.7%), nucleic acid binding (20.5%) and transcription factor (13%). By contrast, the 2 Gy post-irradiation baseline signatures were lower functional categories of transmembrane receptor regulator/adaptor protein (0%), membrane traffic protein (0.9%), oxidoreductase (0.9%), nucleic acid binding (14.8%) and transcription factor (9.3%), but were higher for the protein group of cytoskeletal protein (6.5%), transferase (22.2%), cell adhesion molecule (2.8%) and receptor (3.7%). Anything else group has similar effects (Fig. 2B). Tables 1 and 2 indicate that the proteins (P < 0.05) are phosphorylated exclusively in cells treated with low-dose radiation or high-dose radiation (≥1.3-fold), respectively.
Table 1.
Phosphoproteins responding to low-dose radiation (0.05 Gy) in human fibroblast cells
| Phosphoprotein name | Microarray ratio | P-value | Swiss prot. |
|---|---|---|---|
| Mean SD | Accession no. | ||
| Phospho-Gab2 (Tyr643) | (+) 1.573 ± 0.50 | 0.032 | Q9UQC2 |
| Phospho-P95/NBS (Ser343) | (+) 1.485 ± 0.22 | 0.007 | O60934 |
| Phospho-BTK (Tyr550) | (+) 1.355 ± 0.19 | 0.007 | Q06187 |
| Phospho-Elk1 (Ser383) | (+) 1.321 ± 0.30 | 0.017 | P19419 |
| Phospho-ETK (Tyr40) | (+) 1.312 ± 0.21 | 0.009 | P51813 |
| Phospho-CaMK4 (Thr196/200) | (+) 1.310 ± 0.47 | 0.040 | Q16566 |
Student's t-test indicated that average fold-change values were significant at the level of P < 0.05; (+) indicates increased protein expression in MRC-5 cells; SD indicates standard deviation of fold-change in triplicate independent experiments.
Table 2.
Phosphoproteins responding to high-dose radiation (2 Gy) in human fibroblast cells
| Phosphoprotein name | Microarray ratio | P-value | Swiss prot. |
|---|---|---|---|
| Mean ± SD | Accession no. | ||
| Phospho-MEK1 (Thr298) | (+) 1.570 ± 0.58 | 0.043 | Q02750 |
| Phospho-PLCG1 (Tyr1253) | (+) 1.558 ± 0.15 | 0.003 | P19174 |
| Phospho-IRS-1 (Ser612) | (+) 1.522 ± 0.32 | 0.015 | P35568 |
| Phospho-TFII-I (Tyr248) | (+) 1.519 ± 0.36 | 0.019 | P78347 |
| Phospho-IKK-alpha/beta (Ser176/177) | (+) 1.461 ± 0.53 | 0.042 | O15111 |
| Phospho-MEK1 (Thr286) | (+) 1.432 ± 0.48 | 0.036 | Q02750 |
| Phospho-Pyk2 (Tyr580) | (+) 1.405 ± 0.25 | 0.011 | Q14289 |
| Phospho-Keratin 8 (Ser431) | (+) 1.397 ± 0.53 | 0.045 | P05787 |
| Phospho-ERK3 (Ser189) | (+) 1.360 ± 0.31 | 0.017 | Q16659 |
| Phospho-Chk1 (Ser296) | (+) 1.322 ± 0.61 | 0.007 | O14757 |
| Phospho-CBL (Tyr700) | (+) 1.318 ± 0.02 | 0.000 | P22681 |
| Phospho-BTK (Tyr550) | (+) 1.316 ± 0.20 | 0.008 | Q06187 |
| Phospho-LIMK1/2 (Thr508/505) | (+) 1.302 ± 0.38 | 0.027 | P53667 |
The Student's t-test indicated that average fold-change values were significant at the level of P < 0.05; (+) indicates increased protein expression in MRC-5 cells; SD indicates standard deviation of fold-change in triplicate independent experiments.
Collectively, these results suggest that low-dose and high-dose γ-radiation treatments have different effects on early responsive phosphoproteins and gene ontology functional categories in human fibroblast cells.
Validation of candidate proteins by western blotting
We analyzed the phosphoproteins that displayed ≥1.3-fold increases (P < 0.05) in the Explorer phospho antibody microarray data and had a small standard deviation across triplicate independent experiments. To verify the accuracy of antibody array data for phosphorylated protein expression profiling, western blotting was performed for several phosphoproteins that displayed expression changes in MRC-5 and NHDF cells. In MRC-5 cells, the expression profiles of five phosphoproteins, including phospho-NBS1 (Ser343), phospho-Gab2 (Tyr643), phospho-BTK (Tyr550), phospho-ETK (Tyr40) and phospho-CamK4 (Thr196/200), were dramatically increased in response to low-dose radiation (0.05 Gy) compared with those under high-dose radiation (2 Gy). We verified that the expression level of phospho-BTK (Tyr550) was slightly increased in response to high-dose radiation (2 Gy) compared with that under low-dose radiation (0.05 Gy), although the level of phospho-BTK (Tyr550) expression in response to high-dose radiation was lower than that under low-dose radiation. On the other hand, phospho-Elk1 (Ser383) were slightly increased in MRC-5 cells after low-dose radiation (0.05 Gy). Collectively, this result is consistent with the antibody array data for MRC-5 cells (Fig. 3A).
Fig. 3.
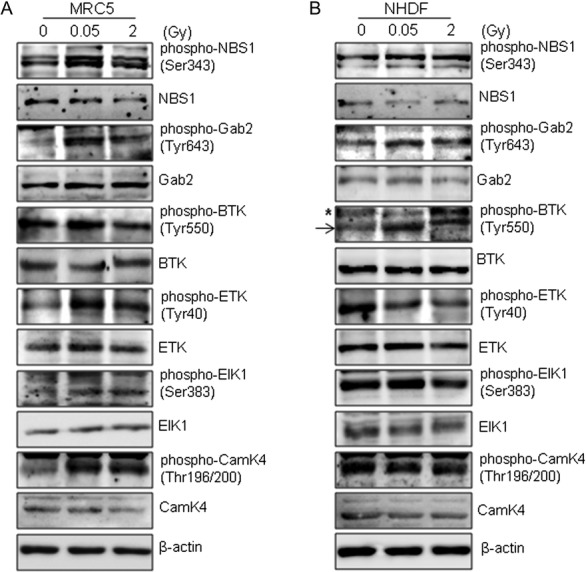
Antibody microarray analysis validates phosphoproteins involved in the early response to γ-radiation. (A and B) Normal human fibroblast cells were incubated for 0.5 h after exposure to 0.05 or 2 Gy of γ-radiation. Expression of levels of phospho-NBS1 (Ser343), NBS1, phospho-Gab2 (Tyr643), Gab2, phospho-BTK (Tyr550), BTK, phospho-ETK (Tyr40), ETK, phospho-Elk1 (Ser383), Elk1, phospho-CamK4 (Thr196/200) and CamK4 were determined by western blotting. (A, MRC-5 cells; B, NHDF cells). β-actin was used as a loading control. The asterisk (*) indicates nonspecific band.
To further confirm the induction of selected phosphoproteins, we performed western blotting to examine the levels of phosphoproteins in NHDF cells 0.5 h after exposure to low-dose (0.05 Gy) or high-dose (2 Gy) γ-radiation. Consistent with the results of western blotting for MRC-5 cells shown in Fig. 3A, the western blots showed that phospho-BTK (Tyr550) and phospho-Gab2 (Tyr643) expression levels in NHDF cells were higher after exposure to low-dose (0.05 Gy) radiation than after exposure to high-dose (2 Gy) radiation. However, the expression levels of phospho-NBS1 (Ser343), phospho-ETK (Tyr40) and phospho-CamK4 (Thr196/200) were not higher under low-dose radiation compared with those under high-dose radiation in NHDF cells. These data indicate that exposure to IR-induced dose-dependent changes in phospho-BTK (Tyr550) and phospho-Gab2 (Tyr643) expression levels, suggesting that specific responses to low-dose radiation regulate these phosphoproteins in normal cells.
Several proteins exhibit dose-dependent activation during early responses to γ-radiation
To evaluate DNA damage caused by γ-radiation, γ-H2AX protein levels were verified by western blotting. MRC-5 and NHDF cells were irradiated with 0 Gy (control), 0.05 and 0.1 Gy (low dose), and 2 and 4 Gy (high dose), and cells were incubated for 0.5 h after irradiation. Then, whole-cell lysates were extracted and subjected to western blotting. The γ-H2AX protein levels were detected with a specific antibody. The levels of γ-H2AX were essentially unchanged in MRC-5 and NHDF cells exposed to low-dose (0.05 and 0.1 Gy) radiation compared with the levels in untreated control cells (Fig. 4A). By contrast, exposure to high-dose radiation (2 and 4 Gy) significantly induced γ-H2AX accumulation. These results show that unrepairable DNA damage did not occur after low-dose irradiation (0.05 and 0.1 Gy), but high-dose radiation (2 and 4 Gy) induced the accumulation of γ-H2AX protein levels in response to DNA damage in MRC-5 and NHDF cells.
Fig. 4.
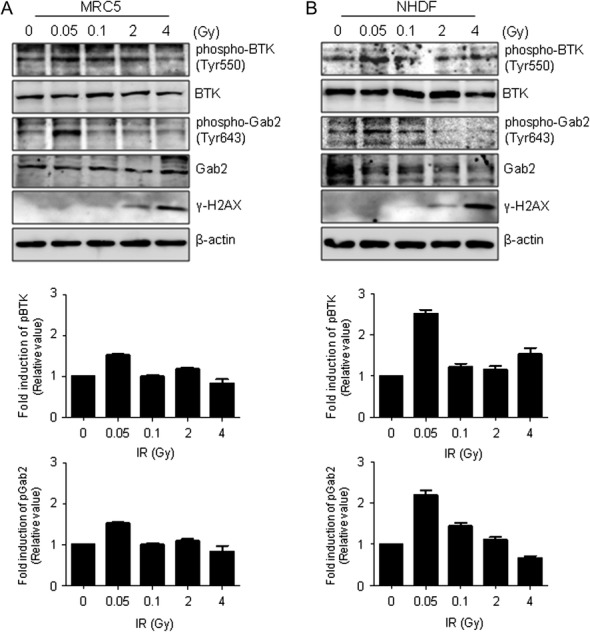
Effect of γ-radiation on phospho-BTK (-Tyr550) and phospho-Gab2 (Tyr643) levels in normal human fibroblast cells. MRC-5 and NHDF cells were exposed to γ-radiation at the indicated doses and then incubated for 0.5 h. (A) Phospho-BTK (Tyr550), BTK, phospho-Gab2 (Tyr643) and Gab2 protein expression levels in MRC-5 cells were determined by western blotting (top). The protein levels were calculated by densitometry and normalized with respect to unphosphorylated proteins. (B) Protein levels in NHDF cells were detected with the corresponding antibodies using MRC-5 cells (top). The consensus protein levels were analyzed by densitometry. γ-H2AX was used as a biochemical marker for DNA damage. β-actin was used as a loading control.
To clarify the effect of IR on phosphoprotein profiles in normal human fibroblast cells, we used western blotting to examine phospho-BTK (Tyr550) and phospho-Gab2 (Tyr643) levels during early responses to γ-radiation in MRC-5 and NHDF cells exposed to 0−4 Gy. We measured the phospho-BTK (Tyr550) and phospho-Gab2 (Tyr643) expression levels in MRC-5 and NHDF cells, 0.5 h after irradiation. The results showed dose-dependent responses of phospho-BTK (Tyr550) and phospho-Gab2 (Tyr643) in MRC-5 (Fig. 4A, Top) and NHDF (Fig. 4B, Top) cells, respectively. We analyzed these expression levels using Multi-Gauge ver. 3.0. The resulting graph represents the intensity of phospho-BTK (Tyr550) and phospho-Gab2 (Tyr643) compared with that of the control at the indicated radiation dose. The expression levels were normalized with respect to non-phosphorylated proteins, and expressed as the relative intensity at each radiation dose. In MRC-5 cells, the relative intensities for phospho-BTK (Tyr550) at 0, 0.05, 0.1, 2 and 4 Gy were 1.0, 1.5, 1.0, 1.2 and 0.8, respectively; the relative intensities for phospho-Gab2 (Tyr643) at the same radiation doses were 1.0, 2.2, 1.3, 1.3 and 0.6, respectively (Fig. 4A, Bottom). We also determined the relative intensity of activated phospho-BTK (Tyr550) and phospho-Gab2 (Tyr643) in NHDF cells treated with 0, 0.05, 0.1, 2 and 4 Gy of γ-radiation. The relative intensities for phospho-BTK (Tyr550) were 1.0, 2.5, 1.2, 1.2 and 1.5, respectively; the relative intensities for phospho-Gab2 (Tyr643) were 1.0, 2.2, 1.4, 1.1 and 0.7, respectively (Fig. 4B, Bottom). These combined results suggest that phospho-BTK (Tyr550) and phospho-Gab2 (Tyr643) are activated in response to low-dose γ-radiation (0.05 Gy), and are candidate markers with high sensitivity and specificity for low-dose radiation.
DISCUSSION
The LNT model of health risks associated with exposure to low-dose IR has been criticized on the basis of the different biological effects induced by low-dose versus high-dose radiation. The question of whether departure from linearity is skewed toward increased or decreased risk is a hot point of debate in the fields of radiation risk–benefit analysis and radiation protection. Understanding the risks of low-dose radiation has significant societal importance. Large-scale epidemiological studies are required in order to precisely determine the effects and quantify the risks of low-dose radiation on humans [45]. Therefore, we focused our study on the effects of low-dose radiation on normal human fibroblast cells, which are involved in determining the homeostasis of cellular stress responses.
The present study provides initial phosphoprotein profiles of normal human lung fibroblasts (MRC-5 cells) and normal human dermal fibroblasts (NHDF cells) subjected to low-dose γ-radiation, and explores potential differences between phosphorylated protein expression levels during early responses to low-dose and high-dose irradiation. The primary goal of our study was to determine whether there were quantitative differences between cellular responses to low-dose and high-dose radiation. First, we demonstrated the effects of low-dose and high-dose radiation on cellular stress pathways. DNA damage, especially double-strand breaks DSBs, induces a DNA damage response (DDR) [46] that leads to repair, apoptosis or permanent growth arrest (senescence). This response is characterized by activation of sensor kinase (ATM, ATR, DNA-PK), formation of DNA damage foci containing phosphorylated histone H2AX (γ-H2AX) and ultimately induction of checkpoint proteins such as p53 and the CDK inhibitor p21, which contribute to cell cycle arrest. p53 becomes phosphorylated on multiple sites in vivo in response to various types of stress, and many stress-activated kinases can phosphorylate p53 in vitro [47–51]. Treatment with low-dose radiation (0.05 Gy) did not significantly affect cell viability or induce DNA damage based on the expression levels of cell cycle regulatory proteins such as phospho-p53 (Ser15), p53 and p21 compared with the levels resulting from high-dose radiation (2 Gy) (Fig. 1). Therefore, we suggest that the late effects of high-dose radiation exposure on the function of fibroblast cells relate to unrepairable damage.
Low-dose short-term exposure to IR in the range of 0.01 to 0.5 Gy (1−50 rad) can induce cellular stimulatory effects, whereas high-dose radiation can induce damaging or lethal cellular effects [52]. The biological response of low dose radiation is known as radiation homeostasis, and is an adaptive response of biological organisms to low levels of stress or damage that results in a modest overcompensation of affected pathways and improved fitness [16]. During tissue homeostasis, there is an equilibrium between the net growth rate and the net rate of cell death [53]. Depending on the type of cellular stress and its severity, the cell's response can be manifold. Our study is the first to analyze changes in phosphoprotein profiles as the end point to investigate the effects of extremely low radiation doses on normal human fibroblast cells, and to compare these results with those of high radiation doses. Our data suggest that quantitative differences exist between phosphoprotein activation pathways induced by 0.05 and those induced by 2 Gy. The total number of phosphoproteins (≥1.1-fold) responding to low-dose radiation at all experimental time points was much more than that of phosphoproteins responding to high-dose radiation (Fig. 2A). We validated the results of phosphoprotein profiling using the Explorer phospho antibody array, which indicated that the activated protein changes range observed in this study were relatively smaller than high dose responding changes range observed in the arrays. And then, we acquired a good protein cohort from antibody array results. As predicted from the antibody microarray data, we demonstrated that gene ontology functional categories differed in cells exposed to low-dose and high-dose radiation (Fig. 2B). The term ‘functional genomics’ is often used broadly to refer to many possible approaches that can be used for understanding the properties and function of the entirety of an organism's genes and gene products. We suggest that it will be important to further explore the molecular mechanisms involved in low-dose radiation responses.
We utilized the Explorer antibody microarray and identified 119 phosphoproteins whose expression changed 0.5 h after treatment with 0.05 Gy of γ-radiation, and 73 phosphoproteins whose expression changed 0.5 h after treatment with 2 Gy of γ-radiation (≥1.1-fold). The number of 27 phosphoproteins increased in common. The representative Venn diagram was composed of validated proteins (P < 0.05) (Fig. 2A). Interestingly, we demonstrated that membrane receptor regulatory/adaptor proteins or traffic proteins were associated with activation by low-dose irradiation. In addition, cytoskeletal proteins or receptor and adhesion molecules were associated with activation by high-dose irradiation (Fig. 2B). Signaling pathway might be regulated by receptor binding of selective adaptor proteins in low-dose radiation exposure. Also, it could be action to cell fate from the activation of the various protein kinases (e.g., MAPK, JNK, and ATM) in the intracellular if the cells have been exposed to high-dose radiation. Table 1 lists the proteins (P < 0.05) that were phosphorylated only in cells subjected to low-dose radiation (≥1.3-fold increase in phosphorylated protein levels). In addition, Table 2 lists the phosphorylated proteins (P < 0.05) in cells subjected to high-dose radiation (≥1.3-fold increase in phosphorylated protein levels).
In essence, if the stress stimulus does not go beyond a certain threshold, the cell can cope with it by mounting an appropriate protective cellular response, which ensures the cell's survival. Conversely, the failure to activate or maintain a protective response, for example, if the stressful agent is too strong, results in activation of stress signaling cascades that eventually fuel into cell death pathways. Various stress-induced molecules, including NF-κB, p53, JNK and MAPK/ERK, have been implicated in propagating and modulating the cell death signal [54, 55]. From the validated high-dose response proteins listed in Table 2, we examined the connection between cellular stress signaling proteins and high-dose (2 Gy) radiation responding proteins. Subsequent activation of PI3K and phospholipase Cγ induces a Ca2+ efflux from the endoplasmic reticulum. Increased cytosolic [Ca2+] promotes exocytosis and mast cell degranulation [56]. An in vitro study using the human mast cell line HMC-1 revealed that ionizing radiation causes degranulation of mast cells [57]. ATM mediates IKK activation by DSBs [58]. RAFTK (also known as PYK2, CADTK) represents a stress-sensitive mediator of the p38 MAPK signaling pathway in response to certain cytotoxic agents [59]. During apoptosis, a cell undergoes dramatic changes in morphology due to a complete reorganization of its cytoplasmic and nuclear skeleton [60, 61]. K8 phosphorylation at Ser431 increased dramatically upon stimulation of cells with epidermal growth factor (EGF) or after mitotic arrest [62, 63]. The expression of LIMK2 has been reported to induce the formation of stress fibers and membrane blebs [64], suggesting that LIMK1/2 activation contributes to the apoptotic morphology. The phosphorylation of LIMK1 (Thr508) and LIMK (Thr505) is known to be involved in apoptosis [65]. DNA damage–induced CHK1 autophosphorylation at Ser 296 is regulated by an intramolecular mechanism [66]. BTK kinase stimulates phosphorylation of Tyr248 of TFII-1, which leads to transcriptional activity and nuclear localization. TFII-I and DBC1 mediate cellular mechanisms of cell-cycle regulation and DNA double strand damage repair [67].
Alternatively, p21-activated kinase (PAK) regulates adhesion-dependent phosphorylation of MEK1 on S298, and subsequent MEK1 (MAPK kinase 1) activation [68]. PAKs are activation loop kinases for ERK3/ERK4 [69]. In addition, Mek1 and Mek2 are regulated by their subcellular localization [70], which is determined in many cases by interaction with specific scaffold proteins. Serine 612 of IRS-1 is phosphorylated upon PKC activation in a human kidney fibroblasts cell line (293 cells) [71]. c-Cbl and Cbl-b are ubiqutin ligases [72] that Tyr700 of c-Cbl residue is efficiently phosphorylated by Syk and the Src-family kinases; Fyn, Yes, and Lyn involved in the phosphorylation, but not by Lck or ZAP-70 [73]. Therefore, high-dose–responding proteins result in either positive or negative regulation of the cellular stress response. Although the early responding proteins of high-dose (2 Gy) didn't directly induce DNA damage kinases such as ATM, ATR, or DNA-PK, there are associated with cellular stress inducible molecules as shown Table 2.
The primary goal of our study was to identify quantitative differences between cellular responses to low-dose and high-dose radiation. Two of the validated proteins in MRC-5 cells—phospho-ETK (Tyr40) and phospho-CamK4 (Thr196/200)—were not affected by radiation responses in NHDF cells (Fig. 3B). There may be hitherto unidentified features of MRC-5 and NHDF cells that result in divergent pathways in response to low-dose radiation. Although pBTK (Tyr550) protein was induced 0.5 h after both 0.05 and 2 Gy irradiation, expression was stronger after exposure to low-dose radiation than after exposure to high-dose radiation. Work done over the last ~20 years at The Gray Cancer Institute and other laboratories has identified a region of increased radio hypersensitivity to doses of ~0.5 Gy/fraction. Many cell lines exposed to doses in this range demonstrate an unexpectedly high cell kill per unit dose compared with the linear-quadratic prediction [74, 75]. On the basis of these studies, BTK might be related to the hypersensitivity in respond to low-dose radiation and the effect as a radiosensitizer, at least in tumor-targeted strategies. It is known that hyper-radiosensitivity is associated with both the adaptive response (classically thought of as the induction of some sort of protective mechanism, e.g. DNA repair) as well as with the inverse dose-rate effect. The requirement to exceed a threshold level of radiation injury for the full induction of repair processes has become known as the adaptive response [76, 77]. These responses temporarily upregulate defenses against, repair of, and removal of damages after a triggering event [9, 78]. Therefore, BTK might have the potential effect to regulate homeostasis, which is associated with both hyper-radiosensitivity as well as the adaptive response.
Previous research reveals that phosphorylation of the histone variant H2AX at the site of DNA DSBs results in the formation and accumulation of γ-H2AX foci in the cell nucleus within a few minutes of DNA damage [79]. The formation of γ-H2AX foci by DSBs induced after exposure to radiation doses as low as 1 mGy [80], and p-ATM (Ser1981) was detected after irradiation with 0.1 Gy for 15 min [81]. They showed the number of γ-H2AX foci formed 71 foci per cell for 2 Gy and 7.2 foci per cell for 0.2 Gy in MRC-5 cells. The analysis, however, was carried out by immunofluorescence detection, and western blotting may not provide the same sensitivity as measurement of foci formation. In addition, because of the weak signal of phospho-ATM (Ser1981) for 0.1 Gy at 15 min, ATM immunoprecipitation was performed for 0.1 Gy or constructs expressing Flag-tagged WT-ATM were transfected in cells for 0.5 Gy. Our studies showed the remaining a γ-H2AX signal at doses ≥2 Gy because we hard to see the γ-H2AX by western blotting at doses ≤0.1 Gy. Also, previous reports revealed the dose response of the number of phosphorylated ATM foci formation at doses of 1.2–200 mGy in MRC-5 cells 0.05 h after irradiation. In particular, the increase in the number of DSBs at the range of 1.2–5 mGy was largely due to radiation induced bystander effects [82]. The biological indicators of low dose radiation may help to evaluate the health risks involved in exposure to radiation, and to develop accurate instruments for measuring the exposure of personnel to low dose radiation. We found that IR inhibited the activation of phospho-BTK (Tyr550) and phospho-Gab2 (Tyr643) in a dose-dependent manner in MRC-5 and NHDF cells (Fig. 4). Therefore, we suggest that phospho-BTK (Tyr550) and phospho-Gab2 (Tyr643) demonstrate early responses to low-dose radiation, and that low-dose radiation (≤100 mGy) can activate these molecules without inducing cellular or genetic toxicity.
The interplay between cell survival and apoptotic signaling is regulated by cell surface receptors and cytoplasmic and nuclear regulatory molecules. These regulatory molecules may be simple unidirectional regulators or bidirectional regulators, such as BTK and Gab2. Previous reports demonstrated that BTK-deficient cells underwent apoptosis after stimulation with anti IgM [83], whereas wild-type cells survived and displayed increasing expression of BclxL. After B-cell receptor (BCR) engagement, BTK translocates to the plasma membrane, where it is phosphorylated at Y551 [84]. Conversely, engagement of the BCR with an antibody to immunoglobulin M (anti-IgM) triggered apoptosis in wild-type DT-40 cells [85]. By comparison, the lack of BTK results in increased sensitivity to TRAIL-induced apoptosis [86]. Thus, BTK could mediate protection from apoptosis via a different mechanism through interaction of a respective receptor with BTK when under exposure to γ-irradiation. Conventional Gab2 knockout (Gab2KO) mice don't show any obvious developmental defects, but display impaired allergic responses osteoclast defects, and abnormal hematopoiesis in adulthood. In Gab2-deficient mice, the number of mast cells was reduced markedly in the stomach and less severely in the skin. Gab2 adaptor proteins play important roles in mast cell degranulation [87–90]. Therefore, BTK activation might respond to specific stimuli such as reactive oxygen species, and protect the cell from death. In addition, Gab2 plays pivotal roles in the downstream signal transduction pathways of various growth factors, and therefore may have the potential to regulate homeostasis. Future work should examine the relationship between these biological markers and the potential health effects associated with them.
The skin is the first organ exposed to radiation in most radiological incidents and medical imaging procedures, and live samples can be obtained directly by minimally invasive procedures in order to estimate the biological damage in humans following radiation exposure. The present study illustrated a unique approach and provided potential indicators for better understanding of the biological responses of low-dose IR by analyzing protein phosphorylation levels. Reliable markers for exposure to low-dose IR may also provide a scientific basis for developing future radiation protection standards and for understanding the biological consequences of exposure to low-dose radiation. Thus, our study identified phospho-BTK (Tyr550) and phospho-Gab2 (Tyr643) as potential candidate markers for exposure to low-dose radiation and as indicators of low-dose radiation in normal human fibroblast cells.
CONFLICT OF INTEREST
The authors declare that there are no conflicts of interest.
FUNDING
This work was supported by the Ministry of Trade, Industry & Energy (MOTIE), Republic of Korea [grant number 20131610101840].
REFERENCES
- 1. Protection, NCoR, Measurements Exposure of the population in the United States and Canada from natural background radiation: recommendations of the National Council on Radiation Protection and Measurements: the Council, Report No. 45, 1987.
- 2. Brenner DJ, Hall EJ. Computed tomography—an increasing source of radiation exposure. N Engl J Med 2007;357:2277–84. [DOI] [PubMed] [Google Scholar]
- 3. Kleinerman RA. Cancer risks following diagnostic and therapeutic radiation exposure in children. Pediatr Radiol 2006;36(Suppl 2):121–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Smith-Bindman R, Lipson J, Marcus R, et al. Radiation dose associated with common computed tomography examinations and the associated lifetime attributable risk of cancer. Arch Intern Med 2009;169:2078–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mobbs SF, Muirhead CR, Harrison JD. Risks from ionising radiation: an HPA viewpoint paper for Safegrounds. J Radiol Prot 2011;31:289–307. [DOI] [PubMed] [Google Scholar]
- 6. Ray M, Yunis R, Chen X, et al. Comparison of low and high dose ionising radiation using topological analysis of gene coexpression networks. BMC Genomics 2012;13:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang F, Stenoien DL, Strittmatter EF, et al. Phosphoproteome profiling of human skin fibroblast cells in response to low- and high-dose irradiation. J Proteome Res 2006;5:1252–60. [DOI] [PubMed] [Google Scholar]
- 8. Blimkie MS, Fung LC, Petoukhov ES, et al. Repair of DNA double-strand breaks is not modulated by low-dose gamma radiation in C57BL/6J mice. Radiat Res 2014;181:548–59. [DOI] [PubMed] [Google Scholar]
- 9. Feinendegen LE. Evidence for beneficial low level radiation effects and radiation hormesis. Br J Radiol 2005;78:3–7. [DOI] [PubMed] [Google Scholar]
- 10. Park E, Ahn GN, Lee NH, et al. Radioprotective properties of eckol against ionizing radiation in mice. FEBS Lett 2008;582:925–30. [DOI] [PubMed] [Google Scholar]
- 11. Kodym E, Kodym R, Choy H, et al. Sustained metaphase arrest in response to ionizing radiation in a non-small cell lung cancer cell line. Radiat Res 2008;169:46–58. [DOI] [PubMed] [Google Scholar]
- 12. Wichmann A, Jaklevic B, Su TT. Ionizing radiation induces caspase-dependent but Chk2- and p53-independent cell death in Drosophila melanogaster. Proc Natl Acad Sci USA 2006;103:9952–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Calabrese EJ. Hormesis: a revolution in toxicology, risk assessment and medicine. EMBO Rep 2004;5:S37–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tapio S, Jacob V. Radioadaptive response revisited. Radiat Environ Biophys 2007;46:1–12. [DOI] [PubMed] [Google Scholar]
- 15. Wolff S. Failla Memorial Lecture. Is radiation all bad? The search for adaptation. Radiat Res 1992;131:117–23. [PubMed] [Google Scholar]
- 16.The 2007 Recommendations of the International Commission on Radiological Protection. ICRP publication 103. Annals of the ICRP 2007;37:1–332. [DOI] [PubMed]
- 17. Bensimon A, Aebersold R, Shiloh Y. Beyond ATM: the protein kinase landscape of the DNA damage response. FEBS Lett 2011;585:1625–39. [DOI] [PubMed] [Google Scholar]
- 18. Olive PL. The role of DNA single- and double-strand breaks in cell killing by ionizing radiation. Radiat Res 1998;150(5 Suppl):S42–51. [PubMed] [Google Scholar]
- 19. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010;40:179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev 2011;25:409–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res 2005;15:11–8. [DOI] [PubMed] [Google Scholar]
- 22. She QB, Chen N, Dong Z. ERKs and p38 kinase phosphorylate p53 protein at serine 15 in response to UV radiation. J Biol Chem 2000;275:20444–9. [DOI] [PubMed] [Google Scholar]
- 23. She QB, Bode AM, Ma WY, et al. Resveratrol-induced activation of p53 and apoptosis is mediated by extracellular-signal-regulated protein kinases and p38 kinase. Cancer Res 2001;61:1604–10. [PubMed] [Google Scholar]
- 24. Bulavin DV, Higashimoto Y, Popoff IJ, et al. Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature 2001;411:102–7. [DOI] [PubMed] [Google Scholar]
- 25. Mikhailov A, Shinohara M, Rieder CL. The p38-mediated stress-activated checkpoint. A rapid response system for delaying progression through antephase and entry into mitosis. Cell Cycle 2005;4:57–62. [DOI] [PubMed] [Google Scholar]
- 26. Kurosu T, Takahashi Y, Fukuda T, et al. p38 MAP kinase plays a role in G2 checkpoint activation and inhibits apoptosis of human B cell lymphoma cells treated with etoposide. Apoptosis 2005;10:1111–20. [DOI] [PubMed] [Google Scholar]
- 27. Thornton TM, Rincon M. Non-classical p38 map kinase functions: cell cycle checkpoints and survival. Int J Biol Sci 2009;5:44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kharbanda S, Saleem A, Shafman T, et al. Ionizing radiation stimulates a Grb2-mediated association of the stress-activated protein kinase with phosphatidylinositol 3-kinase. J Biol Chem 1995;270:18871–4. [DOI] [PubMed] [Google Scholar]
- 29. Lee SA, Dritschilo A, Jung M. Impaired ionizing radiation-induced activation of a nuclear signal essential for phosphorylation of c-Jun by dually phosphorylated c-Jun amino-terminal kinases in ataxia telangiectasia fibroblasts. J Biol Chem 1998;273:32889–94. [DOI] [PubMed] [Google Scholar]
- 30. Verheij M, Ruiter GA, Zerp SF, et al. The role of the stress-activated protein kinase (SAPK/JNK) signaling pathway in radiation-induced apoptosis. Radiother Oncol 1998;47:225–32. [DOI] [PubMed] [Google Scholar]
- 31. Lees-Miller SP, Chen YR, Anderson CW. Human cells contain a DNA-activated protein kinase that phosphorylates simian virus 40 T antigen, mouse p53, and the human Ku autoantigen. Mol Cell Biol 1990;10:6472–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Banin S, Moyal L, Shieh S, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998;281:1674–7. [DOI] [PubMed] [Google Scholar]
- 33. Canman CE, Lim DS, Cimprich KA, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998;281:1677–9. [DOI] [PubMed] [Google Scholar]
- 34. Lakin ND, Hann BC, Jackson SP. The ataxia-telangiectasia related protein ATR mediates DNA-dependent phosphorylation of p53. Oncogene 1999;18:3989–95. [DOI] [PubMed] [Google Scholar]
- 35. Kastan MB, Zhan Q, el-Deiry WS, et al. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 1992;71:587–97. [DOI] [PubMed] [Google Scholar]
- 36. Khanna KK, Beamish H, Yan J, et al. Nature of G1/S cell cycle checkpoint defect in ataxia-telangiectasia. Oncogene 1995;11:609–18. [PubMed] [Google Scholar]
- 37. Gygi SP, Rochon Y, Franza BR, et al. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol 1999;19:1720–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mitchell SJ, Martin-Montalvo A, Mercken EM, et al. The SIRT1 activator SRT1720 extends lifespan and improves health of mice fed a standard diet. Cell Rep 2014;6:836–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Havel LS, Kline ER, Salgueiro AM, et al. Vimentin regulates lung cancer cell adhesion through a VAV2-Rac1 pathway to control focal adhesion kinase activity. Oncogene 2015;34:1979–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pulito C, Mori F, Sacconi A, et al. Cynara scolymus affects malignant pleural mesothelioma by promoting apoptosis and restraining invasion. Oncotarget 2015;6:18134–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jiang HL, Sun HF, Gao SP, et al. SSBP1 suppresses TGFβ-driven epithelial-to-mesenchymal transition and metastasis in triple-negative breast cancer by regulating mitochondrial retrograde signaling. Cancer Res 2016;76:952–64. [DOI] [PubMed] [Google Scholar]
- 42. Xu J, Zhou L, Ji L, et al. The REGγ-proteasome forms a regulatory circuit with IκBε and NFκB in experimental colitis. Nat Commun 2016;7:10761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bhana S, Lloyd DR. The role of p53 in DNA damage-mediated cytotoxicity overrides its ability to regulate nucleotide excision repair in human fibroblasts. Mutagenesis 2008;23:43–50. [DOI] [PubMed] [Google Scholar]
- 44. Levine AJ, Hu W, Feng Z. The P53 pathway: what questions remain to be explored. Cell Death Differ 2006;13:1027–36. [DOI] [PubMed] [Google Scholar]
- 45. Brenner DJ, Doll R, Goodhead DT, et al. Cancer risks attributable to low doses of ionizing radiation: assessing what we really know. Proc Natl Acad Sci USA 2003;100:13761–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. van Attikum H, Gasser SM. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol 2009;19:207–17. [DOI] [PubMed] [Google Scholar]
- 47. Prives C, Hall PA. The p53 pathway. J Pathol 1999;187:112–26. [DOI] [PubMed] [Google Scholar]
- 48. Fuchs SY, Fried VA, Ronai Z. Stress-activated kinases regulate protein stability. Oncogene 1998;17:1483–90. [DOI] [PubMed] [Google Scholar]
- 49. Meek DW. Multisite phosphorylation and the integration of stress signals at p53. Cell Signal 1998;10:159–66. [DOI] [PubMed] [Google Scholar]
- 50. Giaccia AJ, Kastan MB. The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev 1998;12:2973–83. [DOI] [PubMed] [Google Scholar]
- 51. Jayaraman L, Prives C. Covalent and noncovalent modifiers of the p53 protein. Cell Mol Life Sci 1999;55:76–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cuttler JM, Pollycove M. Can cancer be treated with low doses of radiation. J Am Phys Surg 2003;8:108. [Google Scholar]
- 53. Lockshin RA, Zakeri Z. Cell death in health and disease. J Cell Mol Med 2007;11:1214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Cell Biol 2007;19:142–9. [DOI] [PubMed] [Google Scholar]
- 55. Perkins ND, Gilmore TD. Good cop, bad cop: the different faces of NF-kappaB. Cell Death Differ 2006;13:759–72. [DOI] [PubMed] [Google Scholar]
- 56. Sagi-Eisenberg R. The mast cell: where endocytosis and regulated exocytosis meet. Immunol Rev 2007;217:292–303. [DOI] [PubMed] [Google Scholar]
- 57. Albrecht M, Muller K, Kohn FM, et al. Ionizing radiation induces degranulation of human mast cells and release of tryptase. Int J Radiat Biol 2007;83:535–41. [DOI] [PubMed] [Google Scholar]
- 58. Li N, Banin S, Ouyang H, et al. ATM is required for IκB kinase (IKKk) activation in response to DNA double strand breaks. J Biol Chem 2001;276:8898–903. [DOI] [PubMed] [Google Scholar]
- 59. Pandey P, Avraham S, Kumar S, et al. Activation of p38 mitogen-activated protein kinase by PYK2/related adhesion focal tyrosine kinase-dependent mechanism. J Biol Chem 1999;274:10140–4. [DOI] [PubMed] [Google Scholar]
- 60. van Engeland M, Kuijpers HJ, Ramaekers FC, et al. Plasma membrane alterations and cytoskeletal changes in apoptosis. Exp Cell Res 1997;235:421–30. [DOI] [PubMed] [Google Scholar]
- 61. Oberhammer FA, Hochegger K, Froschl G, et al. Chromatin condensation during apoptosis is accompanied by degradation of lamin A+B, without enhanced activation of cdc2 kinase. J Cell Biol 1994;126:827–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schutte B, Henfling M, Kolgen W, et al. Keratin 8/18 breakdown and reorganization during apoptosis. Exp Cell Res 2004;297:11–26. [DOI] [PubMed] [Google Scholar]
- 63. Ku NO, Omary MB. Phosphorylation of human keratin 8 in vivo at conserved head domain serine 23 and at epidermal growth factor-stimulated tail domain serine 431. J Biol Chem 1997;272:7556–64. [DOI] [PubMed] [Google Scholar]
- 64. Amano T, Tanabe K, Eto T, et al. LIM-kinase 2 induces formation of stress fibres, focal adhesions and membrane blebs, dependent on its activation by Rho-associated kinase-catalysed phosphorylation at threonine-505. Biochem J 2001;354(Pt 1):149–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Croft DR, Coleman ML, Li S, et al. Actin-myosin–based contraction is responsible for apoptotic nuclear disintegration. J Cell Biol 2005;168:245–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Okita N, Minato S, Ohmi E, et al. DNA damage-induced CHK1 autophosphorylation at Ser296 is regulated by an intramolecular mechanism. FEBS Lett 2012;586:3974–9. [DOI] [PubMed] [Google Scholar]
- 67. Tanikawa M, Wada-Hiraike O, Yoshizawa-Sugata N, et al. Role of multifunctional transcription factor TFII-I and putative tumour suppressor DBC1 in cell cycle and DNA double strand damage repair. Br J Cancer 2013;109:3042–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Slack-Davis JK, Eblen ST, Zecevic M, et al. PAK1 phosphorylation of MEK1 regulates fibronectin-stimulated MAPK activation. J Cell Biol 2003;162:281–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Deleris P, Trost M, Topisirovic I, et al. Activation loop phosphorylation of ERK3/ERK4 by group I p21-activated kinases (PAKs) defines a novel PAK-ERK3/4-MAPK-activated protein kinase 5 signaling pathway. J Biol Chem 2011;286:6470–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pouysségur J, Lenormand P. Fidelity and spatio-temporal control in MAP kinase (ERKs) signalling. Eur J Biochem 2003;270:3291–9. [DOI] [PubMed] [Google Scholar]
- 71. Nawaratne R, Gray A, Jorgensen CH, et al. Regulation of insulin receptor substrate 1 pleckstrin homology domain by protein kinase C: role of serine 24 phosphorylation. Mol Endocrinol 2006;20:1838–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Thien CB, Langdon WY. c-Cbl and Cbl-b ubiquitin ligases: substrate diversity and the negative regulation of signalling responses. Biochem J 2005;391(Pt 2):153–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tsygankov AY, Mahajan S, Fincke JE, et al. Specific association of tyrosine-phosphorylated c-Cbl with Fyn tyrosine kinase in T cells. J Biol Chem 1996;271: 27130–7. [DOI] [PubMed] [Google Scholar]
- 74. Joiner MC, Marples B, Johns H. The response of tissues to very low doses per fraction: a reflection of induced repair. Recent Results Cancer Res 1993;130:27–40. [DOI] [PubMed] [Google Scholar]
- 75. Wouters BG, Skarsgard LD. Low-dose radiation sensitivity and induced radioresistance to cell killing in HT-29 cells is distinct from the “adaptive response” and cannot be explained by a subpopulation of sensitive cells. Radiat Res 1997;148:435–42. [PubMed] [Google Scholar]
- 76. Bonner WM. Phenomena leading to cell survival values which deviate from linear–quadratic models. Mutat Res 2004;568:33–9. [DOI] [PubMed] [Google Scholar]
- 77. Marples B. Is low-dose hyper-radiosensitivity a measure of G2-phase cell radiosensitivity. Cancer Metastasis Rev 2004;23:197–207. [DOI] [PubMed] [Google Scholar]
- 78. Feinendegen LE, Neumann RD. The issue of risk in complex adaptive systems: the case of low-dose radiation induced cancer. Hum Exp Toxicol 2006;25:11–7. [DOI] [PubMed] [Google Scholar]
- 79. Bonner WM. Low-dose radiation: thresholds, bystander effects, and adaptive responses. Proc Natl Acad Sci USA 2003;100:4973–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Rothkamm K, Löbrich M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc Natl Acad Sci USA 2003;100:5057–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003;421:499–506. [DOI] [PubMed] [Google Scholar]
- 82. Ojima M, Ban N, Kai M.. DNA double-strand breaks induced by very low X-ray doses are largely due to bystander effects. Radiat Res 2008;170:365–71. [DOI] [PubMed] [Google Scholar]
- 83. Anderson JS, Teutsch M, Dong Z, et al. An essential role for Bruton's [corrected] tyrosine kinase in the regulation of B-cell apoptosis. Proc Natl Acad Sci USA 1996;93:10966–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Mohamed AJ, Yu L, Backesjo CM, et al. Bruton's tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol Rev 2009;228:58–73. [DOI] [PubMed] [Google Scholar]
- 85. Uckun FM, Waddick KG, Mahajan S, et al. BTK as a mediator of radiation-induced apoptosis in DT-40 lymphoma B cells. Science 1996;273:1096–100. [DOI] [PubMed] [Google Scholar]
- 86. Schmidt U, van den Akker E, Parren-van Amelsvoort M, et al. Btk is required for an efficient response to erythropoietin and for SCF-controlled protection against TRAIL in erythroid progenitors. J Exp Med 2004;199:785–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Gu H, Saito K, Klaman LD, et al. Essential role for Gab2 in the allergic response. Nature 2001;412:186–90. [DOI] [PubMed] [Google Scholar]
- 88. Nishida K, Wang L, Morii E, et al. Requirement of Gab2 for mast cell development and KitL/c-Kit signaling. Blood 2002;99:1866–9. [DOI] [PubMed] [Google Scholar]
- 89. Wada T, Nakashima T, Oliveira-dos-Santos AJ, et al. The molecular scaffold Gab2 is a crucial component of RANK signaling and osteoclastogenesis. Nat Med 2005;11:394–9. [DOI] [PubMed] [Google Scholar]
- 90. Nishida K, Yamasaki S, Ito Y, et al. FcεRI-mediated mast cell degranulation requires calcium-independent microtubule-dependent translocation of granules to the plasma membrane. J Cell Biol 2005;170:115–26. [DOI] [PMC free article] [PubMed] [Google Scholar]