

We have witnessed significant advances in the management of pulmonary arterial hypertension (PAH) (1), a rare and severe condition characterized by intense pulmonary vascular remodeling, vasoconstriction, endothelial dysfunction, inflammation, and in situ thrombosis (2). More than 10 drugs targeting the endothelin, nitric oxide, and prostacyclin pathways (3) have been developed and commercialized. At present, available therapies have been shown to decrease patients’ risk of short-term mortality (4) and clinical worsening (5). However, despite these advances with modern treatment modalities, most patients continue to experience poor quality of life (6, 7), and long-term prognosis remains poor (8–10). Intriguingly, despite the publication of numerous preclinical studies proposing new therapeutic targets for PAH (11), few have reached clinical trial phases (12) and none of these innovative therapies are currently approved for clinical use.
The journey from the discovery of a potential therapeutic target to its commercialization is long, complex, and costly. The series of clinical trials can cost hundreds of millions of dollars. Moreover, patient time commitment for current and upcoming PAH clinical trials is in the range of months to years, thus limiting the number of patients with PAH to be enrolled in trials testing for other therapies. In addition, even when animal studies show promising results, more than 80% of these therapies ultimately fail when tested in humans (13). Potential reasons for unpredicted clinical response in humans and delayed drug development may include the inherent limitations of the currently available animal and in vitro models that mimic (part of) the spectrum of the human disease, as well as bias in the analysis or reporting of findings and limited data reproducibility (14). Importantly, this is not unique to PAH, because translating research into improved patient care exists across diseases (13, 15). Nonetheless, given the limited financial resources, the persistent medical need for improved therapy in PAH, and the restricted study population available for clinical trials, there is a need to reduce the number of false positive signals in preclinical studies and to optimize the development of innovative therapeutic targets through performance of clinical trials based on more robust experimental data. The current review discusses the challenges, pitfalls, and opportunities in preclinical research to foster drug development in PAH.
Exploratory versus Confirmatory Preclinical Research: Two Different and Complementary Modes of Investigation
Exploratory and confirmatory PAH preclinical studies are two important components of translational research. In exploratory research, investigators seek to provide a better understanding of the pathophysiological processes and to identify key cellular and molecular signaling pathways/targets involved in PAH development and/or progression. In confirmatory investigation, researchers seek to provide strong, reproducible, and detailed information on the dosage and toxicity of potential drug candidates to decide whether the drugs can be tested in PAH clinical trials. In this case, more stringent study design is recommended (Table 1). Taken together, it is clear that the prerequisites for these two modes of investigations differ and that the two different, albeit complementary, experimental strategies must be used.
Table 1.
Key Recommendations for Optimal Methods and More Stringent Study Design for Confirmatory Preclinical Studies in Pulmonary Arterial Hypertension
| General methodological considerations |
| • Predetermined power calculation to define adequate sample size yielding to relevant results |
| • Unbiased approaches using randomization and blinded analyses |
| • Registration of preclinical studies to be considered to minimize publication/reporting bias |
| • Confirmation of results by independent investigators |
| In vitro studies |
| • Validation of the in vitro findings in situ and within various animal models |
| • Limitation of the number of passages and characterization and purity assessment of the primary cell cultures |
| • Careful selection of controls that most likely represent healthy cells isolated and cultured according to the same protocols |
| • Replication of the results in multiple cell lines |
| • Better reproduction of the cellular natural microenvironment (e.g., three-dimensional cell culture models in which cells are grown within extracellular matrix gels, pathological hypoxia, shear stress) |
| In vivo experiments |
| • Comprehensive phenotypical characterization of animal models |
| • Standardization of experimental conditions (e.g., route of administration and dose of Sugen, duration of hypoxia, timing of the intervention) |
| • Confirmation of in vivo findings in >1 animal model, taking into account genetic diversity, age, and comorbidities |
| • Appropriate splitting of age-/sex-matched littermates between control and treatment groups |
| • Realization of pharmacodynamic studies (e.g., tolerability, duration of the effect) |
| • Use of meaningful end points (e.g., survival, comprehensive hemodynamics) |
| • Comparison of the efficacy of the tested molecule alone or in combination with current clinical therapies |
| • Confirmation of drug efficacy in larger animal models to be considered |
Human Samples in PAH Preclinical Studies
Despite the importance of scientific results obtained from animal models, most of these studies have been hampered by the fact that the models do not entirely encompass the typical features of human PAH. Human samples of high quality from patients with PAH and appropriate control subjects are thus invaluable resources for improving our understanding of PAH and validating emerging hypotheses. Human tissues may serve for exploratory studies aimed at demonstrating candidate cellular and molecular processes, genetic or proteomic markers (16) associated with PAH, critical histological and phenotypical characterization, as well as isolation and culture of pulmonary vascular cells (Figure 1) (17). In addition to characterizing human PAH, these samples are useful to rigorously support our assumption that animal models of PAH that share a similar end-stage pathology with the human disease are actually based on similar pathogenic mechanisms and further validate the translation potential of animal models before undertaking clinical studies.
Figure 1.
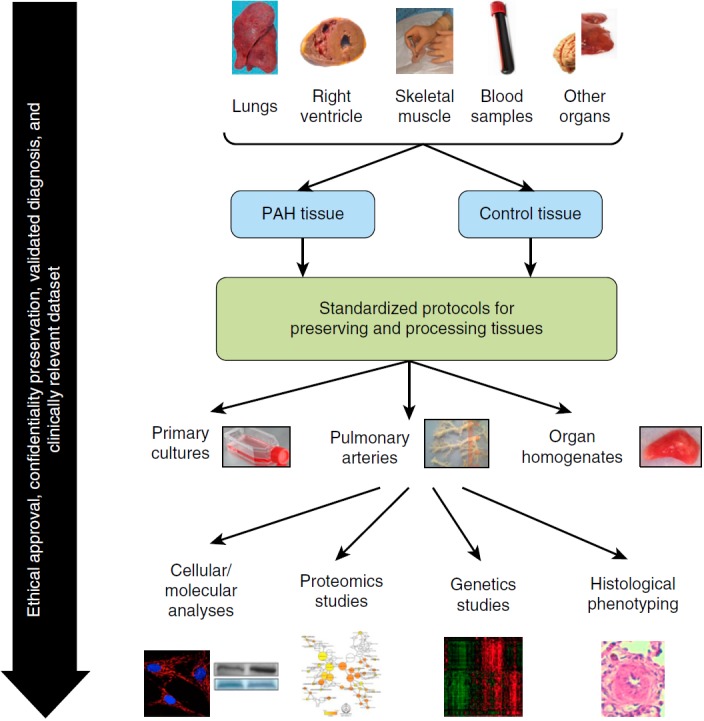
Pulmonary arterial hypertension (PAH) preclinical studies using human specimens. The access to human samples represents an invaluable resource for improving our understanding of PAH. Throughout this process, ethical approval, informed consent, and preservation of participants’ confidentiality are crucial prerequisites. The availability of a clinically relevant data set to confirm the suitability of the samples is also essential. Control samples should be matched for age, sex, and localization. In addition, careful selection of control tissues that most likely represent healthy tissues is crucial. The elaboration of preestablished and standardized operating procedures to ensure optimal techniques for acquiring, preserving, processing, and eventually destroying tissue samples is required. Finally, the creation of a structured PAH biobank and network facilitating access to human tissue samples is essential.
Human PAH lungs (tissues and cells) have been most commonly obtained at the time of lung transplantation. Preestablished protocols are mandatory to ensure the use of optimal techniques for acquiring, preserving, and processing lung tissue samples, such as those described by the Pulmonary Hypertension Breakthrough Initiative (www.ipahresearch.org/PHBI-Research-Project.html). More rarely, human PAH tissues have been obtained at the time of “warm”/early autopsy. Because PAH is associated with systemic metabolic, mitochondrial, inflammatory, and angiogenic disturbances (18–20), early autopsy has the advantage of allowing access to a wide variety of organs of interest. Nonetheless, extreme care is required to ensure that tissues are obtained before cellular and protein degradation occurs. In both cases, however, tissue samples are obtained from patients with end-stage PAH, when proteins and signaling pathways may be influenced by the patients’ treatment or merely represent a consequence of long-standing disease rather than mechanisms accounting for PAH development or progression. However, it is virtually impossible to obtain human PAH lungs from untreated patients with early disease or during longitudinal assessments of disease evolution. The development of specialized endoarterial biopsy catheters may soon overcome this limitation, and future clinical trials must demonstrate the safety of this approach in patients (21).
Although we believe that the confirmation of new pathogenic features in human PAH significantly strengthens the conclusions of preclinical experiments, the use of human tissue samples is also associated with important responsibilities and challenges. Ethical approval, patient/family informed consent, and preservation of participants’ confidentiality are crucial prerequisites for such procedures. Although increasing the external validity of new findings, interindividual variability among patients with PAH is expected to be more common, when compared with experimental animal models, potentially requiring larger sample size. Given the effort required to harvest and prepare human tissues, the diagnosis of PAH should be fully validated according to current guidelines (1). The availability of clinical data is thus mandatory to confirm the suitability of the samples used for experiments.
The choice of cells and tissues with which PAH samples are compared is also crucially important. Control samples should be age and sex matched. Ideally, control subjects should also be matched for their underlying disease (e.g., scleroderma patients without PAH vs. scleroderma-associated PAH). Although this is desirable for samples obtained from living patients, it is virtually impossible to obtain true control samples for lung tissues. In many cases, researchers have relied on resected lung tissue for cancer. However, special attention is required to obtain tissues sufficiently distal from the tumor, which may significantly influence the phenotype and genotype of neighboring cells (22). More rarely, lungs from unused organ donors or at the time of early autopsy after the sudden death of patients with no lung disease have been used. In all cases, equivalent tissue specimens should be collected from the same organ areas. This is particularly important within the lungs, as distal versus more proximal pulmonary arteries (PAs) may significantly differ phenotypically. In addition, the same handling and processing must be used. Taken together, careful selection of control tissues that most likely represent healthy lungs/tissues is crucial.
Finally, few lung transplantations are performed for PAH worldwide yearly (23). Therefore, every effort should be made not to miss opportunities to collect specimens. The creation of structured PAH network and biobank facilities dedicated to harvesting and preserving explanted lung tissues, facilitating access to human tissue and ensuring homogeneity in tissue processing, is thus warranted because human tissues are currently underexploited in PAH experimental research. Despite the present ethical concerns and complex national and local regulations, such samples should ideally be placed in a public/network repository with governance rules allowing others to have access, based on scientific rationale, to such invaluable material to promote pathophysiological and therapeutic target discoveries.
Lessons from Conventional Two-Dimensional Culture Systems to Organs-on-Chip Technologies
The extensive structural and functional remodeling of the vasculature in PAH lungs involves changes in all three layers of the vessel wall (intima, media, and adventitia) and are the consequence of abnormal cellular hypertrophy, hyperplasia, inflammation, apoptosis, migration, and accumulation of extracellular matrix (ECM) (24–26). Many different cell types contribute to the dynamic nature of this process, including—among others—pulmonary artery endothelial cells (PAECs) and pulmonary artery smooth muscle cells (PASMCs), fibroblasts/myofibroblasts, and pericytes, as well as various inflammatory and immune cells (Figure 2).
Figure 2.
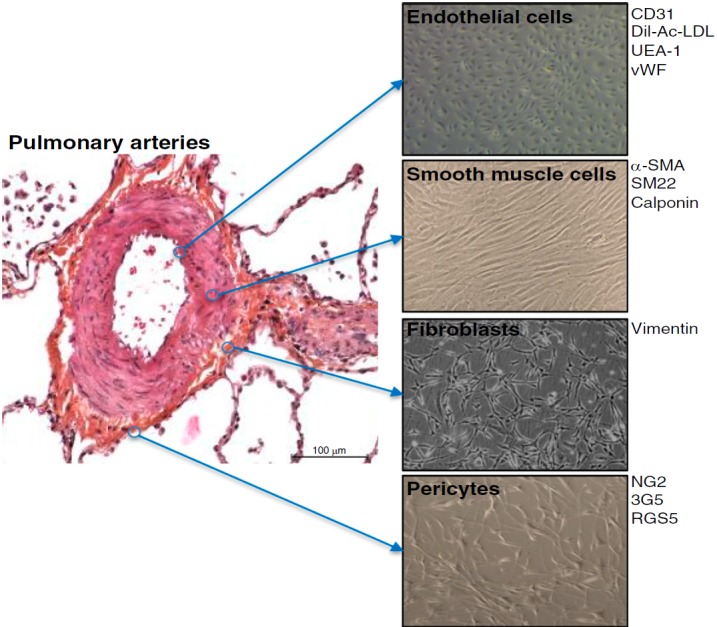
Key cell types constituting the vascular wall of small pulmonary arteries and contributing to pulmonary vascular remodeling. 3G5 = 3G5-reactive ganglioside antigen; α-SMA = α-smooth muscle actin; CD = clusters of differentiation; Dil-Ac-LDL = 3,3′-dioctadecyloxacarbocyanine-acetylated low-density lipoprotein; NG2 = nerve/glial antigen 2; RGS5 = regulator of G protein signaling 5; SM22 = smooth muscle protein 22-α; UEA-1 = Ulex europaeus agglutinin 1; vWF = von Willebrand factor.
Conventional two- and three-dimensional culture systems have made substantial contributions to PAH translational research and biomedical discovery and are being widely used (Table 2). Indeed, the conservation of an abnormal cellular phenotype outside their natural habitats has been demonstrated for primary cultures at early passages. In severe PAH, PAECs and PASMCs exhibit hyperproliferative and apoptosis-resistant phenotypes contributing to the extensive pulmonary vascular remodeling observed in PAH (27–32), as well as proinflammatory phenotypes (33, 34).
Table 2.
Phenotypic Signature of Primary Cultures of Pulmonary Vascular Cells Derived from Patients with Severe Pulmonary Arterial Hypertension
| Phenotypic Signature in Human Pulmonary Hypertension | References | |
|---|---|---|
| Pulmonary endothelial cells | Proproliferative and apoptosis-resistant phenotype | 27, 29, 70 |
| Proinflammatory phenotype | 33 | |
| Smooth muscle–like mesenchymal phenotype | 44, 71, 72 | |
| Decreased tube formation | 73 | |
| Increased migratory potential | 28 | |
| Changes in metabolic processes | 74, 75 | |
| Pulmonary arterial smooth muscle cells | Proproliferative and apoptosis-resistant phenotype | 28, 76–83 |
| Proinflammatory phenotype | 34 | |
| Increased migratory potential | 28 | |
| Senescent phenotype | 84, 85 | |
| Changes in metabolic processes | 41, 80, 86–90 | |
| Pulmonary fibroblasts | Proproliferative and apoptosis-resistant phenotype | 91–94 |
| Proinflammatory phenotype | 94, 95 | |
| Increased migratory potential | 91, 94 | |
| Changes in metabolic processes | 87 | |
| Pulmonary pericytes | Proproliferative phenotype | 96, 97 |
| Decreased migratory potential | 97 | |
| Decreased tube formation | 97 |
These cells are useful to unravel the pathophysiological mechanisms of this disease, to identify intrinsic abnormalities and to explore the panel of secreted factors contributing to aberrant cell–cell and cell–matrix communication. The cellular response can be tested in the presence of small-molecule agonists or antagonists under high-throughput conditions. Furthermore, they can also help in characterizing cell phenotypes from genetic studies and determining the activities of a promising therapeutic agent or even serve in cell-based assays for high-throughput screening. Although abnormal cell phenotypes are conserved over several passages in vitro, that does not necessarily imply that molecular expression patterns or distinct cellular responses will be identical to the in vivo situation. Moreover, culture conditions such as media or substrate stiffness may further add to phenotypic changes. It is thus crucial to study how these various pulmonary vascular cell types derived from human PAH lungs behave outside their pathological microenvironment.
Although there are clear limits to translate in vitro findings to the human disease, several measures should be adopted to increase the translational efficiency of the in vitro studies. Among the most important of these are to validate in situ the relevance of the in vitro findings. The research problem should also be defined as precisely as possible to choose the most relevant cell type and technique to use. Characterization and purity assessments of the primary cell cultures should also be assessed to ensure that they maintain phenotypic properties with passaging. As stated previously, the availability of a clinically relevant data set, appropriate selection of control cells, and the fact that the same protocols for the isolation and culture of “control cells” and “PAH cells” must be employed are essential.
Better in vitro models are also needed to more accurately mimic human disease. Indeed, the pulmonary vascular remodeling associated with PAH implies intense production of matrix structure. Indeed, cells interact physically and functionally with components of the ECM that actively contribute to the creation of a permissive pericellular/extracellular microenvironment for cell proliferation, differentiation, survival, and migration in PAH. In this context, several three-dimensional cell culture models in which cells are grown within ECM gels could be used to facilitate expression of differentiated characteristics that allow, for example, the study of tube formation and cell–matrix interactions. Pathological tissue hypoxia and hemodynamic forces (shear stress) are also two other key components of the cellular microenvironment in PAH that influence signaling pathways and cell function. To investigate the in vitro effects of chronic exposure to hypoxia, specially dedicated settings could be considered to lower oxygen to 0.1–1% as well as pharmacological inhibitors of prolyl hydroxylation such as cobalt chloride (CoCl2, a hypoxia mimetic), desferrioxamine, and dimethyloxalylglycine (a nonspecific inhibitor of prolyl hydroxylase domain–containing proteins) (35, 36). Similarly, in vitro steady shear stress flow patterns could be generated by various types of dedicated apparatus to deliver a specific, unidirectional, and constant flow rate across a cultured endothelial monolayer in a geometrically uniform flow chamber. Various levels of shear stress could be applied to pulmonary microvascular endothelial cell monolayers, ranging from physiological (2.5 dyn/cm2) to high shear (15 or ultimately 21 dyn/cm2), and maintained up to 72 hours or increased gradually with various flow profiles at intervals of 24 hours (37, 38). Finally, the availability of coculture technology could also be useful when answering specific biological questions and for studying both physiological and pathophysiological interactions between different cell populations (e.g., immune cells with PASMCs) (39). Similarly, a range of different coculture systems, two- and three-dimensional culture systems, as well as organs-on-chip technologies have also been developed for specific experimentation (40). Nevertheless, even with the best in vitro strategies, primary cell cultures may not always be appropriate or practical for some specific experimentation and do not fully reconstitute features of clinical PAH. Therefore, their strengths and weaknesses should be taken into account and most commonly be combined with in situ observations and animal models to validate in vitro observations and optimize adequate translational research.
In Vivo Preclinical Research in PAH
Animal models have played a crucial role in drug development in PAH. Multiple animal models have been developed to mimic as much as possible the abnormalities seen in the human PAH condition, to decipher the molecular mechanisms involved in the development and progression of the disease and to test the therapeutic potential of drugs. Although these models contribute to a better understanding of the pathology and therapeutic innovation, they display substantial limitations (Table 3). Among these, the monocrotaline exposure model (rats) and chronic exposure to hypoxia model (mice and rats) (the latter model now frequently combined with administration of the vascular endothelial cell growth factor receptor inhibitor SU5416 [Sugen]) are the most commonly investigated models. Fawn-hooded rats, which spontaneously develop PH in a serotonin-dependent fashion that replicates some PAH features, have also been used (41). More recently, the first models of pulmonary veno-occlusive disease induced by alkylating agents have been described (42, 43). Using advances in transgenic technologies, mutant mice and rats have been generated to mimic features of the most common genetic cause of PAH, namely BMPR2 loss of function (44). Because the generation and characterization of these new genetic models are expensive and time-consuming, they should ideally be shared through a public repository. It must also be kept in mind that although mice allow extensive genetic manipulations, they generally display modest vascular remodeling compared with rats. Importantly, few animal models have been developed and validated for other types of pulmonary hypertension, potentially explaining the limited therapeutic advances beyond group 1 disease. Developing such relevant animal models should thus be a priority for the scientific community.
Table 3.
Preclinical Models of Pulmonary Hypertension
| Species | Model (Ref) | Pros | Cons | Comments |
|---|---|---|---|---|
| Rat | Induced | |||
| Monocrotaline (98) |
|
|
To overcome the easy reversal of PH, some authors have attempted to combine the administration of MCT with other interventions, such as pneumonectomy or chronic hypoxia | |
| Chronic hypoxia (103) | Inexpensive and widely available | No obstructive intimal lesions | Assumed to be representative of group 3 (PH secondary to lung disease and/or hypoxia) rather than PAH | |
| Pulmonary vascular remodeling induced by chronic hypoxia exposure alone is reversible after cessation of hypoxia, contrasting with human PAH, which occurs under normoxic conditions and is associated with irreversible vascular lesions | ||||
| Severity of the phenotype differs among rat strains (Sprague-Dawley rats are highly susceptible, whereas Fischer rats are relatively resistant to hypoxia) | ||||
| Sugen–hypoxia (50) |
|
Sugen has been administered to various strains, including athymic nude rats, to decipher the role of regulatory T cells in the pathogenesis of PAH (106) | ||
| Mitomycin (42, 43) | May allow the understanding of pulmonary veno-occlusive disease, a largely ignored and misdiagnosed form of PAH | The first rodent model of pulmonary veno-occlusive disease | ||
| Pulmonary banding (107–109) | Allows study of chronic pressure-overloaded right heart hypertrophy and failure in the absence of pulmonary vascular remodeling | Only a few studies have characterized the PAB model | ||
| Spontaneous | ||||
| Fawn-hooded rats (41) | Spontaneously develop PH in a serotonin-dependent fashion that replicates some PAH features | Associated with developmental lung anomalies, including reduced number of alveoli (110) | ||
| More than 6 mo is required to observe moderate PH in this strain | ||||
| Available data are limited on therapeutic treatments that have been tested in this model | ||||
| Genetically modified | ||||
| BMPR2Δ140Exon1/+ (44) | Powerful tool to dissect the underlying pathophysiological mechanisms of PAH | Low penetrance and expressivity, precluding their use for drug testing | First rat model of BMPR2 heterozygosity generated by zinc finger nuclease–based gene editing of the first exon in the gene | |
| The identification of patient-relevant second triggers that would lead to high and consistent occurrence of PH would certainly facilitate their use in large-scale preclinical setting | ||||
| Mouse | Induced | |||
| Chronic hypoxia | Inexpensive and widely available | Reversal | ||
| Displays modest vascular remodeling after exposure to chronic hypoxia | ||||
| Sugen–hypoxia (111) | Allows the development of severe PH with more typical angioproliferative features of PAH compared with chronic hypoxia alone | Displays moderate vascular remodeling and PH only | ||
| Schistosomiasis mice (112) | May contribute to further understanding of underlying mechanisms such as the role of inflammation in PH associated with schistosomiasis | |||
| Genetically modified | ||||
| Bmpr2+/– (113) | Mice allow extensive genetic manipulations |
|
|
|
| SM22α-rtTA × TetO (7)-Bmpr2R899X (115) | Pulmonary vascular remodeling similar to plexogenic arteriopathy | Only one-third will develop elevated pulmonary pressures, vascular pruning, and muscularization of microvessels | Expressing a dominant-negative BMPR2 gene (with an arginine-to-termination mutation at amino acid 899) | |
| Tg(Alk1-Cre)-L1 × Bmpr2flox/flox (116) | Knockout mice developed mild PH, with only a subset of mice exhibiting RV hypertrophy and distal PA remodeling | |||
| Sirt3–/– (117) | Confirmed the implication of mitochondrial dysfunction | |||
| Ucp2–/– (118) | Confirmed the implication of mitochondrial dysfunction | |||
| ApoE–/– (119) | The fact that ApoE–/– mice spontaneously develop PH and PA remodeling suggested that insulin resistance and obesity contribute to the development of PH | |||
| S100A4/Mts-1 (overexpression) (120, 121) | Pulmonary vascular remodeling similar to plexogenic arteriopathy is seen with this model | Low penetrance (males remain essentially unaffected) | Overexpressing s100A4/mts1 gene under the control of the HMG-CoA reductase gene promoter | |
| May provide a possible explanation for sex differences, as seen in human idiopathic PAH | ||||
| Tg(SM22α-Cre) × Pparγflox/flox (122) | Confirmed the implication of PPARγ in the development of PAH | Mice with targeted deletion of PPARγ in PASMCs developed only mild PH and pulmonary vascular remodeling | ||
| SM22α–5-HTT+ (overexpression) (123) | Allowed the assessment of targeting the serotonin signaling pathway to reverse pulmonary vascular remodeling | 5-HTT–overexpressing mice exposed to chronic hypoxia developed RV hypertrophy and significant pulmonary vascular remodeling | ||
| CC10-IL6 (overexpression) (58) | IL-6–overexpressing mice exposed to chronic hypoxia developed elevated RV systolic pressure and hypertrophy, as well as significant obliterative pulmonary vasculopathic changes | May be specifically representative of PAH with marked inflammatory implication only | This model documented that IL-6 promotes the development and progression of PA remodeling | |
| Endothelin receptor-B knockout model (124) | Confirmed that the entire endothelin signaling system plays an important role in PH and contributed to the development of endothelin receptor antagonists as a treatment for PAH | |||
| Vip–/– (125) | VIP–/– mice spontaneously develop moderate to severe PH with pulmonary vascular remodeling and RV hypertrophy | The exact role of VIP in PH remains unknown | ||
| This model differs from human PH regarding hemodynamic severity and histology |
Definition of abbreviations: Alk1 = activin receptor-like kinase 1; ApoE = apolipoprotein E; BMPR2 = bone morphogenetic protein receptor 2; CC10 = club cell 10 protein; FiO2 = fraction of inspired oxygen; HMG-CoA = 3-hydroxy-3-methylglutaryl-coenzyme A reductase; 5-HTT = serotonin transporter; MCT = monocrotaline; PA = pulmonary artery; PAB = pulmonary artery banding; PAH = pulmonary arterial hypertension; PASMCs = pulmonary artery smooth muscle cells; PH = pulmonary hypertension; PPARγ = peroxisome proliferator activated receptor γ; rtTA = reverse tetracycline-controlled transactivator; RV = right ventricle; S100A4 (Mts1) = S100 calcium-binding protein A4; Sirt3 = sirtuin 3; SM22α = smooth muscle protein 22-α; TetO (7) = seven tetracycline operator (tetO) repeats; Tg = transgenic mice; Ucp2 = uncoupling protein 2; VIP = vasoactive intestinal peptide.
As observed for virtually all multifactorial disorders, current animal models are useful but not perfect, and they mimic only part of the PAH pathophysiological process. This may explain why animal models are frequently considered poor predictors of whether an experimental drug can become an effective treatment. For this reason, the rationale for choosing one model should be stated (45, 46), and performing studies using more than one rodent model is recommended, whenever possible. This approach is now often applied in PAH preclinical exploratory studies (47, 48). Sometimes, however, the real reason is that confirmatory preclinical studies were not rigorously designed (Table 1). The probability that a research finding is indeed true not only depends on the level of statistical significance, but also on the prior probability of it being true (before doing the experiments) and the statistical power of the study (14). For confirmatory studies, it is thus mandatory that hypotheses and analyses be planned a priori to minimize multiple testing. In addition, the research problem should be defined as precisely as possible with prespecified hypotheses and analyses to minimize multiple testing, and the rationale for choosing one model should be stated (45, 46). Predetermined power calculation to define adequate sample size yielding relevant results is also recommended. Although registration would pose a challenge for exploratory studies, some kind of registration for confirmatory experiments should also be considered to minimize publication/reporting bias. Moreover, confirmatory studies by independent investigators would provide a promising approach to increase robustness of and confidence in novel therapeutic targets. Such an approach would require a shift in the present attitude toward the value of such studies for funding and publication.
Another major problem remains the heterogeneity in the experimental design and execution, even within a same model. In the Sugen–hypoxia model, for example, there is considerable inconsistency in terms of choice of route of Sugen administration (49, 50), duration of hypoxia, and initiation and termination of treatment. Therefore, better standardization of the experimental design will certainly contribute to better reproducibility.
When studies are designed to confirm the curative potential of a molecule, researchers should also investigate what dose animals can tolerate, whether the drug reaches the relevant tissue at the required dose, and how the drug is metabolized. Moreover, treatment of the animals should be initiated after noninvasive confirmation (e.g., echocardiography) of the full establishment of PH, as it is critical to demonstrate whether a molecule improves PAH or only slows down its progression. In addition to short-term studies, the potential benefit of new drugs should be determined after longer periods of treatment through determination of key pulmonary and systemic hemodynamic parameters and standardized morphometric analyses of pulmonary vascular remodeling, such as PA wall thickness, the percentage of muscularized distal PA, and right ventricular hypertrophy. Cardiac output, systemic blood pressure, right ventricular function, exercise tolerance, long-term toxicity, and overall survival should also be monitored when possible (Figure 3). Moreover, confirmatory studies should ideally compare the efficacy of the tested molecule with current clinical therapies (47), because new drugs are likely to be given to patients with PAH who are already receiving standard PAH therapies. Finally, the use of preclinical larger-animal models more closely related to humans, such as pigs (51, 52) or nonhuman primates (53), could result in increased clinical success rates of drugs in development. However, working with larger animals is extremely expensive and fraught with ethical concerns.
Figure 3.

Example of a confirmatory study design using animal specimens. BMPR2 = bone morphogenetic protein receptor, type II; CO = cardiac output; FHR = fawn-hooded rat; MCT = monocrotaline; mPAP = mean pulmonary artery pressure; PA = pulmonary artery; PAH = pulmonary arterial hypertension; PVR = pulmonary vascular resistance; RA = right atrium; RVSP = right ventricular systolic pressure; RV = right ventricle.
Facing Challenges in Translating Confirmatory Preclinical Investigations into Clinical Trials
Although current treatments may delay disease progression, these therapies have not shown potential in reversing the vasculopathy that underlies PAH.
Novel potential targets of PAH drug development include vascular inflammation, metabolic derangements, and aberrant BMPR2 (bone morphogenetic protein receptor, type 2) signaling.
Among the first approaches to encounter these challenges are immunotherapy trials. The first multicenter blinded, randomized, placebo-controlled clinical trial (RCT) to evaluate an immunotherapy in PAH began enrolling patients in 2010 and is testing the efficacy of B-cell depletion with rituximab in systemic sclerosis–associated PAH. ASC01 is a National Institutes of Health–sponsored trial scheduled to complete enrolment this year (clinicaltrials.gov, NCT01086540). This trial includes a number of mechanistic studies including evaluating B-cell phenotype and clonality, multiplex cytokine assays, and regulatory T-cell properties. For this study, as for other emerging PAH therapies, there is anticipation that this careful phenotyping may provide biomarkers that may help predict which patients will benefit most from a given adjuvant therapy.
Although ASC01 has targeted one aspect of the adaptive immune response, two innate immune mediators are the focus of forthcoming clinical trials: leukotriene B4 (LTB4) and neutrophil elastase. Targeting these mediators has posed unique challenges to development. Macrophage-derived LTB4 was identified as a potentially important cause of endothelial injury that may also be involved in the genesis and maintenance of the occlusive arteriopathy (54). Ubenimex (Eiger Biosciences, Palo Alto, CA) is a leukotriene A4 hydrolase inhibitor, which inhibits LTB4 biosynthesis; ubenimex has been used as a chemotherapy adjuvant in Japan and required orphan drug designation for use in the forthcoming U.S. PAH trial (clinicaltrials.gov, NCT02664558). There is evidence to suggest that different forms of PAH may result from different types of immune injury (e.g., helper T-cell type 1 [Th1]/Th17 vs. Th2 immunity) (55), and it is not currently clear how various patients with PAH will respond; it may depend on whether their immunity is skewed toward one phenotype or serum levels of LTB4 may predict drug responsiveness. Another approach targeting innate immunity will use the drug elafin (Proteo Biotech Inc, Kiel, Germany) to inhibit neutrophil elastase activity, which is also strongly implicated in PAH pathogenesis and BMPR2 activation (56, 57). Because elafin has only previously been given to humans as a single intravenous injection in Europe, chronic subcutaneous therapy will require Good Laboratory Practices studies in at least one large-animal species as well as a phase 1 clinical trial before a multicenter, phase 2 RCT in patients with PAH takes place. IL-6, a molecule that bridges both innate and adaptive immunity, and that appears to have a causal role in PAH pathogenesis (58–60), is being tested in an open-label phase 2 safety trial of tocilizumab. As with the rituximab trial, a principal issue is whether the immune suppression caused by IL-6 modulation is safe in an already tenuous population.
Apoptosis signal-regulating kinase 1 (ASK1) is a ubiquitously expressed redox-sensitive serine/threonine protein kinase that activates the p38 mitogen-activated protein kinase and Jnk pathway. A phase 2, dose-ranging, randomized and placebo-controlled study of GS-4997 in patients with PAH has been initiated (clinicaltrials.gov, NCT02234141), which addresses safety, tolerability, and efficacy.
Dysfunctional BMPR2 signaling is implicated in the pathogenesis of PAH (61). A study screening 3,756 U.S. Food and Drug Administration–approved drugs identified FK506 (tacrolimus) as an effective up-regulator of BMP signaling, and low-dose FK506 was an effective treatment for experimental PAH (62). Preliminary data also suggested that compassionate use of low-dose FK506 in three patients with end-stage PAH might have been associated with clinical benefit (63). On the basis of these findings, a phase 2a RCT in PAH was initiated to evaluate the safety and tolerability of FK506 in stable patients with PAH (clinicaltrials.gov, NCT01647945).
As in cancer, vascular remodeling in PAH is a state of apoptosis resistance of the distal PA cells. Several studies have demonstrated that a switch from antiapoptotic glycolytic metabolism toward proapoptotic oxidative phosphorylation metabolism causes regression of vascular remodeling and PAH in several animal models (20, 64, 65). This has been achieved with the small-molecule metabolic modulator dichloroacetate, an inhibitor of the mitochondrial enzyme pyruvate dehydrogenase kinase. Dichloroacetate has been used in humans for more than 30 years, mostly in the treatment of inherited mitochondrial disorders, and is also currently being evaluated as a potential therapy in cancer (66–68). On the basis of these findings, a phase 1 RCT in subjects with advanced PAH was completed (clinicaltrials.gov, NCT01083524). Although results are still pending, a particular challenge faced for further developing this promising PAH therapy is securing financial backing for a clinical trial when using a repurposed and off-patent drug. In the apparent absence of a major return of financial investment, needed to lure venture capital support, it appears that exceptional dedication by committed investigators backed by governmental and foundational support may be required to advance this and other similarly promising PAH therapies.
We are entering an exciting era for new therapeutics in the treatment of PAH (69). Gene expression studies and high-throughput studies have emerged as powerful tools to identify novel therapeutic targets and biologically active compounds able to modulate dedicated pathways in PAH. This approach was employed for the identification of FK506 described previously, for example (62).
The use of inducible pluripotent stem cells (iPSCs) provides another exciting approach to drug discovery in PAH. Use of iPSCs to derive endothelial or smooth muscle cells from patients with PAH could certainly help us in further elucidating the genetic and molecular mechanisms of PAH and translating this into a platform for screening novel PAH-specific compounds. This approach could also be used to predict the pharmacological profile of a given compound in an individual patient, as these cells contain the patient’s unique genetic and epigenetic markers. This could predict how various patients might respond to a given compound and help us move PAH treatment into personalized medicine.
However, various challenges exist, reinforcing the need for more robust experimental data with reduced false positive signals, allowing the prioritization of the most promising compound. First, new approaches need to show benefit on top of optimized treatment with currently approved therapy; a barrier not faced in the original vasodilator trials. This makes the detection of any putative benefit potentially challenging, particularly if adverse interactions occur with existing therapies, or the benefit of the new compounds is best observable in treatment-naive patients. Second, complex regulatory agency approvals are required to target novel pathways for PAH treatment. For all emerging PAH therapies, it is essential that regulatory authorities grant orphan drug status before proceeding to clinical trials to help advance the evaluation and development of such products. Third, some new therapies carry novel risks not encountered with approved agents, such as hypersensitivity reactions and immunosuppression. Fourth, the pathway to development for these novel drugs can be highly complex and not receive standard industry support. Therefore, investigator-initiated trials must provide a sufficiently convincing rationale to entice investigators who are accustomed to working with well-funded corporations that offer potentially more attractive compensation for examining already-trusted vasodilation pathways. Given these problems, it is heartening to note that clinician-scientists appear to be rising to the challenge, and the next decade should offer patients with PAH considerable hope for better cures.
Conclusions
A better translation of preclinical research findings into sustainable improvements in patient outcomes is clearly needed, but remains a substantial obstacle for many human diseases, including PAH. At present, research on PAH is characterized by significant discordance between preclinical findings and clinical results. Bridging this translational gap is thus vital and can be achieved only by rigorous use and characterization of human PAH tissues, a better design of preclinical studies, significant governmental and foundational support and funding, and close collaboration and significant commitment from clinical trial sites.
Footnotes
Originally Published in Press as DOI: 10.1164/rccm.201607-1515PP on September 20, 2016
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, et al. Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS). 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2015;46:903–975. doi: 10.1183/13993003.01032-2015. [DOI] [PubMed] [Google Scholar]
- 2.Schermuly RT, Ghofrani HA, Wilkins MR, Grimminger F. Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol. 2011;8:443–455. doi: 10.1038/nrcardio.2011.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med. 2004;351:1425–1436. doi: 10.1056/NEJMra040291. [DOI] [PubMed] [Google Scholar]
- 4.Galiè N, Manes A, Negro L, Palazzini M, Bacchi-Reggiani ML, Branzi A. A meta-analysis of randomized controlled trials in pulmonary arterial hypertension. Eur Heart J. 2009;30:394–403. doi: 10.1093/eurheartj/ehp022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lajoie AC, Lauzière G, Lega JC, Lacasse Y, Martin S, Simard S, Bonnet S, Provencher S. Combination therapy versus monotherapy for pulmonary arterial hypertension: a meta-analysis. Lancet Respir Med. 2016;4:291–305. doi: 10.1016/S2213-2600(16)00027-8. [DOI] [PubMed] [Google Scholar]
- 6.Taichman DB, Shin J, Hud L, Archer-Chicko C, Kaplan S, Sager JS, Gallop R, Christie J, Hansen-Flaschen J, Palevsky H. Health-related quality of life in patients with pulmonary arterial hypertension. Respir Res. 2005;6:92. doi: 10.1186/1465-9921-6-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shafazand S, Goldstein MK, Doyle RL, Hlatky MA, Gould MK. Health-related quality of life in patients with pulmonary arterial hypertension. Chest. 2004;126:1452–1459. doi: 10.1378/chest.126.5.1452. [DOI] [PubMed] [Google Scholar]
- 8.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, Yaïci A, Weitzenblum E, Cordier JF, Chabot F, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122:156–163. doi: 10.1161/CIRCULATIONAHA.109.911818. [DOI] [PubMed] [Google Scholar]
- 9.Humbert M, Sitbon O, Yaici A, Montani D, O’Callaghan DS, Jais X, Parent F, Savale L, Natali D, Gunther S, et al. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur Respir J. 2010;36:549–555. doi: 10.1183/09031936.00057010. [DOI] [PubMed] [Google Scholar]
- 10.Benza RL, Miller DP, Barst RJ, Badesch DB, Frost AE, McGoon MD. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest. 2012;142:448–456. doi: 10.1378/chest.11-1460. [DOI] [PubMed] [Google Scholar]
- 11.Malenfant S, Margaillan G, Loehr JE, Bonnet S, Provencher S. The emergence of new therapeutic targets in pulmonary arterial hypertension: from now to the near future. Expert Rev Respir Med. 2013;7:43–55. doi: 10.1586/ers.12.83. [DOI] [PubMed] [Google Scholar]
- 12.Hoeper MM, Barst RJ, Bourge RC, Feldman J, Frost AE, Galié N, Gómez-Sánchez MA, Grimminger F, Grünig E, Hassoun PM, et al. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation. 2013;127:1128–1138. doi: 10.1161/CIRCULATIONAHA.112.000765. [DOI] [PubMed] [Google Scholar]
- 13.Perrin S. Preclinical research: make mouse studies work. Nature. 2014;507:423–425. doi: 10.1038/507423a. [DOI] [PubMed] [Google Scholar]
- 14.Ioannidis JP. Why most published research findings are false. PLoS Med. 2005;2:e124. doi: 10.1371/journal.pmed.0020124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moeller A, Ask K, Warburton D, Gauldie J, Kolb M. The bleomycin animal model: a useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int J Biochem Cell Biol. 2008;40:362–382. doi: 10.1016/j.biocel.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malenfant S, Potus F, Fournier F, Breuils-Bonnet S, Pflieger A, Bourassa S, Tremblay È, Nehmé B, Droit A, Bonnet S, et al. Skeletal muscle proteomic signature and metabolic impairment in pulmonary hypertension. J Mol Med. 2015;93:573–584. doi: 10.1007/s00109-014-1244-0. [DOI] [PubMed] [Google Scholar]
- 17.Potus F, Malenfant S, Graydon C, Mainguy V, Tremblay È, Breuils-Bonnet S, Ribeiro F, Porlier A, Maltais F, Bonnet S, et al. Impaired angiogenesis and peripheral muscle microcirculation loss contribute to exercise intolerance in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2014;190:318–328. doi: 10.1164/rccm.201402-0383OC. [DOI] [PubMed] [Google Scholar]
- 18.Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot-Keros L, Duroux P, Galanaud P, Simonneau G, Emilie D. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med. 1995;151:1628–1631. doi: 10.1164/ajrccm.151.5.7735624. [DOI] [PubMed] [Google Scholar]
- 19.Potus F, Ruffenach G, Dahou A, Thebault C, Breuils-Bonnet S, Tremblay È, Nadeau V, Paradis R, Graydon C, Wong R, et al. Downregulation of microRNA-126 contributes to the failing right ventricle in pulmonary arterial hypertension. Circulation. 2015;132:932–943. doi: 10.1161/CIRCULATIONAHA.115.016382. [DOI] [PubMed] [Google Scholar]
- 20.Paulin R, Michelakis ED. The metabolic theory of pulmonary arterial hypertension. Circ Res. 2014;115:148–164. doi: 10.1161/CIRCRESAHA.115.301130. [DOI] [PubMed] [Google Scholar]
- 21.Rothman A, Davidson S, Wiencek RG, Evans WN, Restrepo H, Sarukhanov V, Ruoslahti E, Williams R, Mann D. Vascular histomolecular analysis by sequential endoarterial biopsy in a shunt model of pulmonary hypertension. Pulm Circ. 2013;3:50–57. doi: 10.4103/2045-8932.109913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kadara H, Shen L, Fujimoto J, Saintigny P, Chow CW, Lang W, Chu Z, Garcia M, Kabbout M, Fan YH, et al. Characterizing the molecular spatial and temporal field of injury in early-stage smoker non–small cell lung cancer patients after definitive surgery by expression profiling. Cancer Prev Res (Phila) 2013;6:8–17. doi: 10.1158/1940-6207.CAPR-12-0290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yusen RD, Edwards LB, Kucheryavaya AY, Benden C, Dipchand AI, Dobbels F, Goldfarb SB, Levvey BJ, Lund LH, Meiser B, et al. International Society for Heart and Lung Transplantation. The registry of the International Society for Heart and Lung Transplantation: thirty-first adult lung and heart–lung transplant report—2014; focus theme: retransplantation. J Heart Lung Transplant. 2014;33:1009–1024. doi: 10.1016/j.healun.2014.08.004. [DOI] [PubMed] [Google Scholar]
- 24.Guignabert C, Dorfmuller P. Pathology and pathobiology of pulmonary hypertension. Semin Respir Crit Care Med. 2013;34:551–559. doi: 10.1055/s-0033-1356496. [DOI] [PubMed] [Google Scholar]
- 25.Guignabert C, Tu L, Girerd B, Ricard N, Huertas A, Montani D, Humbert M. New molecular targets of pulmonary vascular remodeling in pulmonary arterial hypertension: importance of endothelial communication. Chest. 2015;147:529–537. doi: 10.1378/chest.14-0862. [DOI] [PubMed] [Google Scholar]
- 26.Tuder RM, Archer SL, Dorfmüller P, Erzurum SC, Guignabert C, Michelakis E, Rabinovitch M, Schermuly R, Stenmark KR, Morrell NW. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25) Suppl:D4–D12. doi: 10.1016/j.jacc.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masri FA, Xu W, Comhair SA, Asosingh K, Koo M, Vasanji A, Drazba J, Anand-Apte B, Erzurum SC. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;293:L548–L554. doi: 10.1152/ajplung.00428.2006. [DOI] [PubMed] [Google Scholar]
- 28.Tu L, De Man FS, Girerd B, Huertas A, Chaumais MC, Lecerf F, François C, Perros F, Dorfmüller P, Fadel E, et al. A critical role for p130Cas in the progression of pulmonary hypertension in humans and rodents. Am J Respir Crit Care Med. 2012;186:666–676. doi: 10.1164/rccm.201202-0309OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tu L, Dewachter L, Gore B, Fadel E, Dartevelle P, Simonneau G, Humbert M, Eddahibi S, Guignabert C. Autocrine fibroblast growth factor-2 signaling contributes to altered endothelial phenotype in pulmonary hypertension. Am J Respir Cell Mol Biol. 2011;45:311–322. doi: 10.1165/rcmb.2010-0317OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meloche J, Potus F, Vaillancourt M, Bourgeois A, Johnson I, Deschamps L, Chabot S, Ruffenach G, Henry S, Breuils-Bonnet S, et al. Bromodomain-containing protein 4: the epigenetic origin of pulmonary arterial hypertension. Circ Res. 2015;117:525–535. doi: 10.1161/CIRCRESAHA.115.307004. [DOI] [PubMed] [Google Scholar]
- 31.Ruffenach G, Chabot S, Tanguay VF, Courboulin A, Boucherat O, Potus F, Meloche J, Pflieger A, Breuils-Bonnet S, Nadeau V, et al. Role for RUNX2 in proliferative and calcified vascular lesions in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2016;194:1273–1285. doi: 10.1164/rccm.201512-2380OC. [DOI] [PubMed] [Google Scholar]
- 32.Paulin R, Courboulin A, Barrier M, Bonnet S. From oncoproteins/tumor suppressors to microRNAs, the newest therapeutic targets for pulmonary arterial hypertension. J Mol Med. 2011;89:1089–1101. doi: 10.1007/s00109-011-0788-5. [DOI] [PubMed] [Google Scholar]
- 33.Le Hiress M, Tu L, Ricard N, Phan C, Thuillet R, Fadel E, Dorfmüller P, Montani D, de Man F, Humbert M, et al. Proinflammatory signature of the dysfunctional endothelium in pulmonary hypertension: role of the macrophage migration inhibitory factor/CD74 complex. Am J Respir Crit Care Med. 2015;192:983–997. doi: 10.1164/rccm.201402-0322OC. [DOI] [PubMed] [Google Scholar]
- 34.Savai R, Al-Tamari HM, Sedding D, Kojonazarov B, Muecke C, Teske R, Capecchi MR, Weissmann N, Grimminger F, Seeger W, et al. Pro-proliferative and inflammatory signaling converge on FoxO1 transcription factor in pulmonary hypertension. Nat Med. 2014;20:1289–1300. doi: 10.1038/nm.3695. [DOI] [PubMed] [Google Scholar]
- 35.Ahn YT, Kim YM, Adams E, Lyu SC, Alvira CM, Cornfield DN. Hypoxia-inducible factor-1α regulates KCNMB1 expression in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2012;302:L352–L359. doi: 10.1152/ajplung.00302.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huertas A, Tu L, Thuillet R, Le Hiress M, Phan C, Ricard N, Nadaud S, Fadel E, Humbert M, Guignabert C. Leptin signalling system as a target for pulmonary arterial hypertension therapy. Eur Respir J. 2015;45:1066–1080. doi: 10.1183/09031936.00193014. [DOI] [PubMed] [Google Scholar]
- 37.Bonnet S, Provencher S. Shear stress maladaptation in pulmonary arterial hypertension: an ageless concept. Am J Respir Crit Care Med. 2016;193:1331–1332. doi: 10.1164/rccm.201601-0113ED. [DOI] [PubMed] [Google Scholar]
- 38.Szulcek R, Happé CM, Rol N, Fontijn RD, Dickhoff C, Hartemink KJ, Grünberg K, Tu L, Timens W, Nossent GD, et al. Delayed microvascular shear adaptation in pulmonary arterial hypertension: role of platelet endothelial cell adhesion molecule-1 cleavage. Am J Respir Crit Care Med. 2016;193:1410–1420. doi: 10.1164/rccm.201506-1231OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bonnet S, Rochefort G, Sutendra G, Archer SL, Haromy A, Webster L, Hashimoto K, Bonnet SN, Michelakis ED. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc Natl Acad Sci USA. 2007;104:11418–11423. doi: 10.1073/pnas.0610467104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Hsin HY, Ingber DE. Reconstituting organ-level lung functions on a chip. Science. 2010;328:1662–1668. doi: 10.1126/science.1188302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thébaud B, Bonnet S, Haromy A, Harry G, Moudgil R, McMurtry MS, et al. An abnormal mitochondrial-hypoxia inducible factor-1α–Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation. 2006;113:2630–2641. doi: 10.1161/CIRCULATIONAHA.105.609008. [DOI] [PubMed] [Google Scholar]
- 42.Perros F, Günther S, Ranchoux B, Godinas L, Antigny F, Chaumais MC, Dorfmüller P, Hautefort A, Raymond N, Savale L, et al. Mitomycin-induced pulmonary veno-occlusive disease: evidence from human disease and animal models. Circulation. 2015;132:834–847. doi: 10.1161/CIRCULATIONAHA.115.014207. [DOI] [PubMed] [Google Scholar]
- 43.Ranchoux B, Günther S, Quarck R, Chaumais MC, Dorfmüller P, Antigny F, Dumas SJ, Raymond N, Lau E, Savale L, et al. Chemotherapy-induced pulmonary hypertension: role of alkylating agents. Am J Pathol. 2015;185:356–371. doi: 10.1016/j.ajpath.2014.10.021. [DOI] [PubMed] [Google Scholar]
- 44.Ranchoux B, Antigny F, Rucker-Martin C, Hautefort A, Péchoux C, Bogaard HJ, Dorfmüller P, Remy S, Lecerf F, Planté S, et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation. 2015;131:1006–1018. doi: 10.1161/CIRCULATIONAHA.114.008750. [DOI] [PubMed] [Google Scholar]
- 45.Mitani Y, Mutlu A, Russell JC, Brindley DN, DeAlmeida J, Rabinovitch M. Dexfenfluramine protects against pulmonary hypertension in rats. J Appl Physiol (1985) 2002;93:1770–1778. doi: 10.1152/japplphysiol.00500.2002. [DOI] [PubMed] [Google Scholar]
- 46.Pullamsetti SS, Berghausen EM, Dabral S, Tretyn A, Butrous E, Savai R, Butrous G, Dahal BK, Brandes RP, Ghofrani HA, et al. Role of Src tyrosine kinases in experimental pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2012;32:1354–1365. doi: 10.1161/ATVBAHA.112.248500. [DOI] [PubMed] [Google Scholar]
- 47.Meloche J, Pflieger A, Vaillancourt M, Paulin R, Potus F, Zervopoulos S, Graydon C, Courboulin A, Breuils-Bonnet S, Tremblay E, et al. Role for DNA damage signaling in pulmonary arterial hypertension. Circulation. 2014;129:786–797. doi: 10.1161/CIRCULATIONAHA.113.006167. [DOI] [PubMed] [Google Scholar]
- 48.Paulin R, Courboulin A, Meloche J, Mainguy V, Dumas de la Roque E, Saksouk N, Côté J, Provencher S, Sussman MA, Bonnet S. Signal transducers and activators of transcription-3/pim1 axis plays a critical role in the pathogenesis of human pulmonary arterial hypertension. Circulation. 2011;123:1205–1215. doi: 10.1161/CIRCULATIONAHA.110.963314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Long L, Ormiston ML, Yang X, Southwood M, Gräf S, Machado RD, Mueller M, Kinzel B, Yung LM, Wilkinson JM, et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat Med. 2015;21:777–785. doi: 10.1038/nm.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, Voelkel NF, Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death–dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001;15:427–438. doi: 10.1096/fj.00-0343com. [DOI] [PubMed] [Google Scholar]
- 51.Courboulin A, Kang C, Baillard O, Bonnet S, Bonnet P. Increasing pulmonary artery pulsatile flow improves hypoxic pulmonary hypertension in piglets. J Vis Exp. 2015;99:e52571. doi: 10.3791/52571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boulate D, Perros F, Dorfmuller P, Arthur-Ataam J, Guihaire J, Lamrani L, Decante B, Humbert M, Eddahibi S, Dartevelle P, et al. Pulmonary microvascular lesions regress in reperfused chronic thromboembolic pulmonary hypertension. J Heart Lung Transplant. 2015;34:457–467. doi: 10.1016/j.healun.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 53.George MP, Champion HC, Simon M, Guyach S, Tarantelli R, Kling HM, Brower A, Janssen C, Murphy J, Carney JP, et al. Physiologic changes in a nonhuman primate model of HIV-associated pulmonary arterial hypertension. Am J Respir Cell Mol Biol. 2013;48:374–381. doi: 10.1165/rcmb.2011-0434OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tian W, Jiang X, Tamosiuniene R, Sung YK, Qian J, Dhillon G, Gera L, Farkas L, Rabinovitch M, Zamanian RT, et al. Blocking macrophage leukotriene B4 prevents endothelial injury and reverses pulmonary hypertension. Sci Transl Med. 2013;5:200ra117. doi: 10.1126/scitranslmed.3006674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res. 2014;115:165–175. doi: 10.1161/CIRCRESAHA.113.301141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zaidi SH, You XM, Ciura S, Husain M, Rabinovitch M. Overexpression of the serine elastase inhibitor elafin protects transgenic mice from hypoxic pulmonary hypertension. Circulation. 2002;105:516–521. doi: 10.1161/hc0402.102866. [DOI] [PubMed] [Google Scholar]
- 57.Nickel NP, Spiekerkoetter E, Gu M, Li CG, Li H, Kaschwich M, Diebold I, Hennigs JK, Kim KY, Miyagawa K, et al. Elafin reverses pulmonary hypertension via caveolin-1–dependent bone morphogenetic protein signaling. Am J Respir Crit Care Med. 2015;191:1273–1286. doi: 10.1164/rccm.201412-2291OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res. 2009;104:236–244. doi: 10.1161/CIRCRESAHA.108.182014. , 28p following 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Savale L, Tu L, Rideau D, Izziki M, Maitre B, Adnot S, Eddahibi S. Impact of interleukin-6 on hypoxia-induced pulmonary hypertension and lung inflammation in mice. Respir Res. 2009;10:6. doi: 10.1186/1465-9921-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Meloche J, Renard S, Provencher S, Bonnet S. Anti-inflammatory and immunosuppressive agents in PAH. Handbook Exp Pharmacol. 2013;218:437–476. doi: 10.1007/978-3-642-38664-0_18. [DOI] [PubMed] [Google Scholar]
- 61.Soubrier F, Chung WK, Machado R, Grünig E, Aldred M, Geraci M, Loyd JE, Elliott CG, Trembath RC, Newman JH, et al. Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62(25) Suppl:D13–D21. doi: 10.1016/j.jacc.2013.10.035. [DOI] [PubMed] [Google Scholar]
- 62.Spiekerkoetter E, Tian X, Cai J, Hopper RK, Sudheendra D, Li CG, El-Bizri N, Sawada H, Haghighat R, Chan R, et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J Clin Invest. 2013;123:3600–3613. doi: 10.1172/JCI65592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Spiekerkoetter E, Sung YK, Sudheendra D, Bill M, Aldred MA, van de Veerdonk MC, Vonk Noordegraaf A, Long-Boyle J, Dash R, Yang PC, et al. Low-dose FK506 (tacrolimus) in end-stage pulmonary arterial hypertension. Am J Respir Crit Care Med. 2015;192:254–257. doi: 10.1164/rccm.201411-2061LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sutendra G, Bonnet S, Rochefort G, Haromy A, Folmes KD, Lopaschuk GD, Dyck JRB, Michelakis ED. Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci Transl Med. 2010;2:44ra58. doi: 10.1126/scitranslmed.3001327. [DOI] [PubMed] [Google Scholar]
- 65.Sutendra G, Michelakis ED. The metabolic basis of pulmonary arterial hypertension. Cell Metab. 2014;19:558–573. doi: 10.1016/j.cmet.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 66.Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, et al. A mitochondria–K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 67.Kinnaird A, Dromparis P, Saleme B, Gurtu V, Watson K, Paulin R, Zervopoulos S, Stenson T, Sutendra G, Pink DB, et al. Metabolic modulation of clear-cell renal cell carcinoma with dichloroacetate, an inhibitor of pyruvate dehydrogenase kinase. Eur Urol. 2016;69:734–744. doi: 10.1016/j.eururo.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 68.Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010;2:31ra34. doi: 10.1126/scitranslmed.3000677. [DOI] [PubMed] [Google Scholar]
- 69.Sung YK, Yuan K, de Jesus Perez VA. Novel approaches to pulmonary arterial hypertension drug discovery. Expert Opin Drug Discov. 2016;11:407–414. doi: 10.1517/17460441.2016.1153625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Patel M, Predescu D, Tandon R, Bardita C, Pogoriler J, Bhorade S, Wang M, Comhair S, Hemnes AR, Chen J, et al. A novel p38 mitogen–activated protein kinase/Elk-1 transcription factor–dependent molecular mechanism underlying abnormal endothelial cell proliferation in plexogenic pulmonary arterial hypertension. J Biol Chem. 2013;288:25701–25716. doi: 10.1074/jbc.M113.502674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hopper RK, Moonen JA, Diebold I, Cao A, Rhodes CJ, Tojais NF, Hennigs JK, Gu M, Wang L, Rabinovitch M. In pulmonary arterial hypertension, reduced BMPR2 promotes endothelial-to-mesenchymal transition via HMGA1 and its target slug. Circulation. 2016;133:1783–1794. doi: 10.1161/CIRCULATIONAHA.115.020617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stenmark KR, Frid M, Perros F. Endothelial-to-mesenchymal transition: an evolving paradigm and a promising therapeutic target in PAH. Circulation. 2016;133:1734–1737. doi: 10.1161/CIRCULATIONAHA.116.022479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.de Jesus Perez VA, Yuan K, Orcholski ME, Sawada H, Zhao M, Li CG, Tojais NF, Nickel N, Rajagopalan V, Spiekerkoetter E, et al. Loss of adenomatous polyposis coli–α3 integrin interaction promotes endothelial apoptosis in mice and humans. Circ Res. 2012;111:1551–1564. doi: 10.1161/CIRCRESAHA.112.267849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fijalkowska I, Xu W, Comhair SA, Janocha AJ, Mavrakis LA, Krishnamachary B, Zhen L, Mao T, Richter A, Erzurum SC, et al. Hypoxia inducible-factor1α regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am J Pathol. 2010;176:1130–1138. doi: 10.2353/ajpath.2010.090832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xu W, Koeck T, Lara AR, Neumann D, DiFilippo FP, Koo M, Janocha AJ, Masri FA, Arroliga AC, Jennings C, et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci USA. 2007;104:1342–1347. doi: 10.1073/pnas.0605080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Eddahibi S, Guignabert C, Barlier-Mur AM, Dewachter L, Fadel E, Dartevelle P, Humbert M, Simonneau G, Hanoun N, Saurini F, et al. Cross talk between endothelial and smooth muscle cells in pulmonary hypertension: critical role for serotonin-induced smooth muscle hyperplasia. Circulation. 2006;113:1857–1864. doi: 10.1161/CIRCULATIONAHA.105.591321. [DOI] [PubMed] [Google Scholar]
- 77.Guignabert C, Raffestin B, Benferhat R, Raoul W, Zadigue P, Rideau D, Hamon M, Adnot S, Eddahibi S. Serotonin transporter inhibition prevents and reverses monocrotaline-induced pulmonary hypertension in rats. Circulation. 2005;111:2812–2819. doi: 10.1161/CIRCULATIONAHA.104.524926. [DOI] [PubMed] [Google Scholar]
- 78.Izikki M, Guignabert C, Fadel E, Humbert M, Tu L, Zadigue P, Dartevelle P, Simonneau G, Adnot S, Maitre B, et al. Endothelial-derived FGF2 contributes to the progression of pulmonary hypertension in humans and rodents. J Clin Invest. 2009;119:512–523. doi: 10.1172/JCI35070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fernandez RA, Wan J, Song S, Smith KA, Gu Y, Tauseef M, Tang H, Makino A, Mehta D, Yuan JX. Upregulated expression of STIM2, TRPC6, and Orai2 contributes to the transition of pulmonary arterial smooth muscle cells from a contractile to proliferative phenotype. Am J Physiol Cell Physiol. 2015;308:C581–C593. doi: 10.1152/ajpcell.00202.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guignabert C, Tu L, Izikki M, Dewachter L, Zadigue P, Humbert M, Adnot S, Fadel E, Eddahibi S. Dichloroacetate treatment partially regresses established pulmonary hypertension in mice with SM22α-targeted overexpression of the serotonin transporter. FASEB J. 2009;23:4135–4147. doi: 10.1096/fj.09-131664. [DOI] [PubMed] [Google Scholar]
- 81.Eddahibi S, Humbert M, Fadel E, Raffestin B, Darmon M, Capron F, Simonneau G, Dartevelle P, Hamon M, Adnot S. Serotonin transporter overexpression is responsible for pulmonary artery smooth muscle hyperplasia in primary pulmonary hypertension. J Clin Invest. 2001;108:1141–1150. doi: 10.1172/JCI12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McMurtry MS, Archer SL, Altieri DC, Bonnet S, Haromy A, Harry G, Bonnet S, Puttagunta L, Michelakis ED. Gene therapy targeting survivin selectively induces pulmonary vascular apoptosis and reverses pulmonary arterial hypertension. J Clin Invest. 2005;115:1479–1491. doi: 10.1172/JCI23203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hameed AG, Arnold ND, Chamberlain J, Pickworth JA, Paiva C, Dawson S, Cross S, Long L, Zhao L, Morrell NW, et al. Inhibition of tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) reverses experimental pulmonary hypertension. J Exp Med. 2012;209:1919–1935. doi: 10.1084/jem.20112716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Noureddine H, Gary-Bobo G, Alifano M, Marcos E, Saker M, Vienney N, Amsellem V, Maitre B, Chaouat A, Chouaid C, et al. Pulmonary artery smooth muscle cell senescence is a pathogenic mechanism for pulmonary hypertension in chronic lung disease. Circ Res. 2011;109:543–553. doi: 10.1161/CIRCRESAHA.111.241299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mouraret N, Marcos E, Abid S, Gary-Bobo G, Saker M, Houssaini A, Dubois-Rande JL, Boyer L, Boczkowski J, Derumeaux G, et al. Activation of lung p53 by Nutlin-3a prevents and reverses experimental pulmonary hypertension. Circulation. 2013;127:1664–1676. doi: 10.1161/CIRCULATIONAHA.113.002434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McMurtry MS, Bonnet S, Wu X, Dyck JR, Haromy A, Hashimoto K, Michelakis ED. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res. 2004;95:830–840. doi: 10.1161/01.RES.0000145360.16770.9f. [DOI] [PubMed] [Google Scholar]
- 87.Zhao L, Chen CN, Hajji N, Oliver E, Cotroneo E, Wharton J, Wang D, Li M, McKinsey TA, Stenmark KR, et al. Histone deacetylation inhibition in pulmonary hypertension: therapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation. 2012;126:455–467. doi: 10.1161/CIRCULATIONAHA.112.103176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yuan XJ. Voltage-gated K+ currents regulate resting membrane potential and [Ca2+]i in pulmonary arterial myocytes. Circ Res. 1995;77:370–378. doi: 10.1161/01.res.77.2.370. [DOI] [PubMed] [Google Scholar]
- 89.Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JG, Weir EK. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS–HIF-1α–Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol. 2008;294:H570–H578. doi: 10.1152/ajpheart.01324.2007. [DOI] [PubMed] [Google Scholar]
- 90.Yu Y, Platoshyn O, Zhang J, Krick S, Zhao Y, Rubin LJ, Rothman A, Yuan JX. c-Jun decreases voltage-gated K+ channel activity in pulmonary artery smooth muscle cells. Circulation. 2001;104:1557–1563. doi: 10.1161/hc3801.095662. [DOI] [PubMed] [Google Scholar]
- 91.Anwar A, Li M, Frid MG, Kumar B, Gerasimovskaya EV, Riddle SR, McKeon BA, Thukaram R, Meyrick BO, Fini MA, et al. Osteopontin is an endogenous modulator of the constitutively activated phenotype of pulmonary adventitial fibroblasts in hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2012;303:L1–L11. doi: 10.1152/ajplung.00050.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Das M, Burns N, Wilson SJ, Zawada WM, Stenmark KR. Hypoxia exposure induces the emergence of fibroblasts lacking replication repressor signals of PKCζ in the pulmonary artery adventitia. Cardiovasc Res. 2008;78:440–448. doi: 10.1093/cvr/cvn014. [DOI] [PubMed] [Google Scholar]
- 93.Panzhinskiy E, Zawada WM, Stenmark KR, Das M. Hypoxia induces unique proliferative response in adventitial fibroblasts by activating PDGFβ receptor–JNK1 signalling. Cardiovasc Res. 2012;95:356–365. doi: 10.1093/cvr/cvs194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang D, Zhang H, Li M, Frid MG, Flockton AR, McKeon BA, Yeager ME, Fini MA, Morrell NW, Pullamsetti SS, et al. MicroRNA-124 controls the proliferative, migratory, and inflammatory phenotype of pulmonary vascular fibroblasts. Circ Res. 2014;114:67–78. doi: 10.1161/CIRCRESAHA.114.301633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li M, Riddle SR, Frid MG, El Kasmi KC, McKinsey TA, Sokol RJ, Strassheim D, Meyrick B, Yeager ME, Flockton AR, et al. Emergence of fibroblasts with a proinflammatory epigenetically altered phenotype in severe hypoxic pulmonary hypertension. J Immunol. 2011;187:2711–2722. doi: 10.4049/jimmunol.1100479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ricard N, Tu L, Le Hiress M, Huertas A, Phan C, Thuillet R, Sattler C, Fadel E, Seferian A, Montani D, et al. Increased pericyte coverage mediated by endothelial-derived fibroblast growth factor-2 and interleukin-6 is a source of smooth muscle–like cells in pulmonary hypertension. Circulation. 2014;129:1586–1597. doi: 10.1161/CIRCULATIONAHA.113.007469. [DOI] [PubMed] [Google Scholar]
- 97.Yuan K, Orcholski ME, Panaroni C, Shuffle EM, Huang NF, Jiang X, Tian W, Vladar EK, Wang L, Nicolls MR, et al. Activation of the Wnt/planar cell polarity pathway is required for pericyte recruitment during pulmonary angiogenesis. Am J Pathol. 2015;185:69–84. doi: 10.1016/j.ajpath.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kay JM, Harris P, Heath D. Pulmonary hypertension produced in rats by ingestion of Crotalaria spectabilis seeds. Thorax. 1967;22:176–179. doi: 10.1136/thx.22.2.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gomez-Arroyo JG, Farkas L, Alhussaini AA, Farkas D, Kraskauskas D, Voelkel NF, Bogaard HJ. The monocrotaline model of pulmonary hypertension in perspective. Am J Physiol Lung Cell Mol Physiol. 2012;302:L363–L369. doi: 10.1152/ajplung.00212.2011. [DOI] [PubMed] [Google Scholar]
- 100.Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1013–L1032. doi: 10.1152/ajplung.00217.2009. [DOI] [PubMed] [Google Scholar]
- 101.Montani D, Bergot E, Günther S, Savale L, Bergeron A, Bourdin A, Bouvaist H, Canuet M, Pison C, Macro M, et al. Pulmonary arterial hypertension in patients treated by dasatinib. Circulation. 2012;125:2128–2137. doi: 10.1161/CIRCULATIONAHA.111.079921. [DOI] [PubMed] [Google Scholar]
- 102.Montani D, Seferian A, Savale L, Simonneau G, Humbert M. Drug-induced pulmonary arterial hypertension: a recent outbreak. Eur Respir Rev. 2013;22:244–250. doi: 10.1183/09059180.00003313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hislop A, Reid L. New findings in pulmonary arteries of rats with hypoxia-induced pulmonary hypertension. Br J Exp Pathol. 1976;57:542–554. [PMC free article] [PubMed] [Google Scholar]
- 104.Kasahara Y, Tuder RM, Taraseviciene-Stewart L, Le Cras TD, Abman S, Hirth PK, Waltenberger J, Voelkel NF. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest. 2000;106:1311–1319. doi: 10.1172/JCI10259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jiang B, Deng Y, Suen C, Taha M, Chaudhary KR, Courtman DW, Stewart DJ. Marked strain-specific differences in the SU5416 rat model of severe pulmonary arterial hypertension. Am J Respir Cell Mol Biol. 2016;54:461–468. doi: 10.1165/rcmb.2014-0488OC. [DOI] [PubMed] [Google Scholar]
- 106.Taraseviciene-Stewart L, Nicolls MR, Kraskauskas D, Scerbavicius R, Burns N, Cool C, Wood K, Parr JE, Boackle SA, Voelkel NF. Absence of T cells confers increased pulmonary arterial hypertension and vascular remodeling. Am J Respir Crit Care Med. 2007;175:1280–1289. doi: 10.1164/rccm.200608-1189OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bogaard HJ, Mizuno S, Hussaini AA, Toldo S, Abbate A, Kraskauskas D, Kasper M, Natarajan R, Voelkel NF. Suppression of histone deacetylases worsens right ventricular dysfunction after pulmonary artery banding in rats. Am J Respir Crit Care Med. 2011;183:1402–1410. doi: 10.1164/rccm.201007-1106OC. [DOI] [PubMed] [Google Scholar]
- 108.Dias CA, Assad RS, Caneo LF, Abduch MC, Aiello VD, Dias AR, Marcial MB, Oliveira SA. Reversible pulmonary trunk banding. II. An experimental model for rapid pulmonary ventricular hypertrophy. J Thorac Cardiovasc Surg. 2002;124:999–1006. doi: 10.1067/mtc.2002.124234. [DOI] [PubMed] [Google Scholar]
- 109.Muller WH, Jr, Dammann JF., Jr The surgical significance of pulmonary hypertension. Ann Surg. 1952;136:495–509. doi: 10.1097/00000658-195209000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Le Cras TD, Kim DH, Gebb S, Markham NE, Shannon JM, Tuder RM, Abman SH. Abnormal lung growth and the development of pulmonary hypertension in the Fawn-Hooded rat. Am J Physiol. 1999;277:L709–L718. doi: 10.1152/ajplung.1999.277.4.L709. [DOI] [PubMed] [Google Scholar]
- 111.Ciuclan L, Bonneau O, Hussey M, Duggan N, Holmes AM, Good R, Stringer R, Jones P, Morrell NW, Jarai G, et al. A novel murine model of severe pulmonary arterial hypertension. Am J Respir Crit Care Med. 2011;184:1171–1182. doi: 10.1164/rccm.201103-0412OC. [DOI] [PubMed] [Google Scholar]
- 112.Crosby A, Jones FM, Southwood M, Stewart S, Schermuly R, Butrous G, Dunne DW, Morrell NW. Pulmonary vascular remodeling correlates with lung eggs and cytokines in murine schistosomiasis. Am J Respir Crit Care Med. 2010;181:279–288. doi: 10.1164/rccm.200903-0355OC. [DOI] [PubMed] [Google Scholar]
- 113.Beppu H, Ichinose F, Kawai N, Jones RC, Yu PB, Zapol WM, Miyazono K, Li E, Bloch KD. BMPR-II heterozygous mice have mild pulmonary hypertension and an impaired pulmonary vascular remodeling response to prolonged hypoxia. Am J Physiol Lung Cell Mol Physiol. 2004;287:L1241–L1247. doi: 10.1152/ajplung.00239.2004. [DOI] [PubMed] [Google Scholar]
- 114.Yasuda T, Tada Y, Tanabe N, Tatsumi K, West J. Rho-kinase inhibition alleviates pulmonary hypertension in transgenic mice expressing a dominant-negative type II bone morphogenetic protein receptor gene. Am J Physiol Lung Cell Mol Physiol. 2011;301:L667–L674. doi: 10.1152/ajplung.00423.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.West J, Harral J, Lane K, Deng Y, Ickes B, Crona D, Albu S, Stewart D, Fagan K. Mice expressing BMPR2R899X transgene in smooth muscle develop pulmonary vascular lesions. Am J Physiol Lung Cell Mol Physiol. 2008;295:L744–L755. doi: 10.1152/ajplung.90255.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hong KH, Lee YJ, Lee E, Park SO, Han C, Beppu H, Li E, Raizada MK, Bloch KD, Oh SP. Genetic ablation of the BMPR2 gene in pulmonary endothelium is sufficient to predispose to pulmonary arterial hypertension. Circulation. 2008;118:722–730. doi: 10.1161/CIRCULATIONAHA.107.736801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Paulin R, Dromparis P, Sutendra G, Gurtu V, Zervopoulos S, Bowers L, Haromy A, Webster L, Provencher S, Bonnet S, et al. Sirtuin 3 deficiency is associated with inhibited mitochondrial function and pulmonary arterial hypertension in rodents and humans. Cell Metab. 2014;20:827–839. doi: 10.1016/j.cmet.2014.08.011. [DOI] [PubMed] [Google Scholar]
- 118.Dromparis P, Paulin R, Sutendra G, Qi AC, Bonnet S, Michelakis ED. Uncoupling protein 2 deficiency mimics the effects of hypoxia and endoplasmic reticulum stress on mitochondria and triggers pseudohypoxic pulmonary vascular remodeling and pulmonary hypertension. Circ Res. 2013;113:126–136. doi: 10.1161/CIRCRESAHA.112.300699. [DOI] [PubMed] [Google Scholar]
- 119.Hansmann G, Wagner RA, Schellong S, Perez VA, Urashima T, Wang L, Sheikh AY, Suen RS, Stewart DJ, Rabinovitch M. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-γ activation. Circulation. 2007;115:1275–1284. doi: 10.1161/CIRCULATIONAHA.106.663120. [DOI] [PubMed] [Google Scholar]
- 120.Greenway S, van Suylen RJ, Du Marchie Sarvaas G, Kwan E, Ambartsumian N, Lukanidin E, Rabinovitch M. S100A4/Mts1 produces murine pulmonary artery changes resembling plexogenic arteriopathy and is increased in human plexogenic arteriopathy. Am J Pathol. 2004;164:253–262. doi: 10.1016/S0002-9440(10)63115-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ambartsumian N, Klingelhöfer J, Grigorian M, Karlstrøm O, Sidenius N, Georgiev G, Lukanidin E. Tissue-specific posttranscriptional downregulation of expression of the S100A4(mts1) gene in transgenic animals. Invasion Metastasis. 1998-1999;18:96–104. doi: 10.1159/000024502. [DOI] [PubMed] [Google Scholar]
- 122.Hansmann G, de Jesus Perez VA, Alastalo TP, Alvira CM, Guignabert C, Bekker JM, Schellong S, Urashima T, Wang L, Morrell NW, et al. An antiproliferative BMP-2/PPARγ/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J Clin Invest. 2008;118:1846–1857. doi: 10.1172/JCI32503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.MacLean MR, Deuchar GA, Hicks MN, Morecroft I, Shen S, Sheward J, Colston J, Loughlin L, Nilsen M, Dempsie Y, et al. Overexpression of the 5-hydroxytryptamine transporter gene: effect on pulmonary hemodynamics and hypoxia-induced pulmonary hypertension. Circulation. 2004;109:2150–2155. doi: 10.1161/01.CIR.0000127375.56172.92. [DOI] [PubMed] [Google Scholar]
- 124.Ivy D, McMurtry IF, Yanagisawa M, Gariepy CE, Le Cras TD, Gebb SA, Morris KG, Wiseman RC, Abman SH. Endothelin B receptor deficiency potentiates ET-1 and hypoxic pulmonary vasoconstriction. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1040–L1048. doi: 10.1152/ajplung.2001.280.5.L1040. [DOI] [PubMed] [Google Scholar]
- 125.Said SI, Hamidi SA, Dickman KG, Szema AM, Lyubsky S, Lin RZ, Jiang YP, Chen JJ, Waschek JA, Kort S. Moderate pulmonary arterial hypertension in male mice lacking the vasoactive intestinal peptide gene. Circulation. 2007;115:1260–1268. doi: 10.1161/CIRCULATIONAHA.106.681718. [DOI] [PubMed] [Google Scholar]