

Abstract
The poliovirus (PV)-induced cytopathic effect (CPE) was blocked in neural cells but not in HeLa cells by the addition of monoclonal antibody (MAb) against PV or the human PV receptor (CD155) 2 h postinfection (hpi). Since each MAb has the ability to block viral infection, no CPE in PV-infected neural cells appeared to result from the blockade of multiple rounds of viral replication. Pulse-labeling experiments revealed that virus-specific protein synthesis proceeded 5 hpi with or without MAbs. However, in contrast to the results obtained without MAbs, virus-specific protein synthesis with MAbs was not detected 7 hpi. Shutoff of host translation was also not observed in the presence of MAbs. Western blot analysis showed that 2Apro, the viral protein which mediates the cleavage of eukaryotic translation initiation factor eIF4G, was still present 11 hpi. However, intact eIF4G appeared 11 hpi. An immunocytochemical study indicated that 2Apro was detected only in the nucleus 11 hpi. These results suggest that neural cells possess protective response mechanisms against PV infection as follows: (i) upon PV infection, neural cells produce a factor(s) to suppress PV internal ribosome entry site activity by 7 hpi, (ii) a factor which supports cap-dependent translation for eIF4G may exist in infected cells when no intact eIF4G is detected, and (iii) the remaining 2Apro is not effective in cleaving eIF4G because it is imported into the nucleus by 11 hpi.
Poliovirus (PV), the causative agent of poliomyelitis, is an enterovirus that belongs to the Picornaviridae. The genome of PV is a single-stranded, positive-sense RNA of approximately 7.5 kb and functions as mRNA after entry into the host cell cytoplasm. PV mRNA is uncapped and its translation is initiated by binding of the ribosome to viral mRNA downstream of the 5′ end, an RNA structure termed the internal ribosome entry site (IRES) (24, 29). The mRNA has only one long open reading frame encoding the viral polyprotein, which consists of the capsid precursor (P1) and the noncapsid precursors (P2 and P3) (38). The polyprotein is cotranslationally processed by virus-specific proteases 2Apro and 3Cpro (3CDpro) to generate mature viral proteins (17, 33). 2Apro cleaves a Tyr-Gly bond (13, 19, 28, 36), and 3Cpro (3CDpro) cleaves a Gln-Gly bond.
Upon infection, PV induces the shutoff of almost all host cell translation and induces a severe cytopathic effect (CPE) in infected cells. Both phenomena are thought to be induced mainly by 2Apro expression (3). The shutoff of host cell translation has been thought to result from the cleavage of eukaryotic translation initiation factor eIF4G (9, 18, 41) and poly(A) binding protein (34). eIF4G is a subunit of eIF4F, which also contains eIF4E, the cap binding protein, and eIF4A, the RNA helicase (4, 26). eIF4G itself serves as a scaffold protein which interacts with eIF4E and eIF4A, and its association with eIF3 has been suggested to promote attachment of the small ribosomal subunit at the 5′ ends of mRNAs (4, 26, 30). The cleavage of eIF4G blocks the formation of the cap-dependent translation complex, leading to the shutoff of host translation (10, 16, 20). Contrary to cap-dependent translation, the C-terminal cleavage product of eIF4G is sufficient to carry out IRES-dependent translation, and the synthesis of viral polyprotein continues after eIF4G cleavage (4, 8, 10, 12, 18, 41).
PV-infected cells show typical signs of the CPE, such as rounding up, accumulation of membranous vesicles (5, 7), condensation of chromatin (6), and detachment from the basal surface of culture dishes. Furthermore, a number of host nuclear proteins are redistributed from the nucleus to the cytoplasm during PV infection (23, 25, 32, 37). In addition, some components of the nuclear pore complex are degraded during PV infection (11). However, little is known about the molecular mechanisms responsible for CPE expression due to PV infection.
Tolskaya et al. (35) reported that the CPE in human neuroblastoma cells infected with virulent PV is suppressed by the addition of anti-PV hyperimmune serum shortly after the infection. They argued that the antibodies penetrate the cells, interact with assembled viral particles, and inhibit an unknown reaction responsible for cell death. Here we describe a similar observation obtained with an anti-human PV receptor (hPVR; CD155) monoclonal antibody (MAb) as well as an anti-PV MAb. Both MAbs have the ability to block PV infection. Thus, a new concept that elucidates the mechanisms responsible for this phenomenon is desirable. Our biochemical and immunocytochemical studies suggest the existence of specific mechanisms that produce a protective response against PV infection in neural cells.
MATERIALS AND METHODS
Cells and viruses.
Human neuroblastoma cell lines SK-N-SH and IMR-32 were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal calf serum. African green monkey kidney (AGMK) cells and HeLa S3 monolayer cells were maintained in DMEM supplemented with 5% newborn calf serum. AGMK cells were used for transfection and virus titration experiments. The Mahoney strain of PV type 1 (PV1/Mahoney) and a mutant PV (2A-HA virus) expressing hemagglutinin (HA)-tagged 2Apro for intact 2Apro were produced in AGMK cells transfected with the corresponding RNAs transcribed in vitro from infectious cDNA clones of pOM1 (31) and p2A-HA (an infectious cDNA clone of 2A-HA virus; see below), respectively. Cells transfected with RNA transcribed from pOM1 or p2A-HA were incubated in DMEM containing 5% newborn calf serum at 37°C for up to 3 days to recover the corresponding virus.
DNA procedures.
Infectious cDNA clone pOM1 was used as a template for the construction of p2A-HA, in which the nucleotide sequence for intact 2Apro in pOM1 was replaced by sequences encoding 2Apro, the HA tag, and an artificial 3Cpro cleavage site, in that order. Silent mutations were introduced into the nucleotide sequence of the artificial 3Cpro cleavage site to avoid recombination with the authentic nucleotide sequence at the 3′ terminus of the 2Apro coding sequence. See Fig. 7 for the genome structure of 2A-HA virus.
FIG. 7.

Structure of the 2A-HA virus genome. VPg represents a viral protein genome located at the 5′ terminus, AAA(n) represents poly(A) located at the 3′ terminus, P1 is a structural (capsid) protein precursor, and P2 and P3 are nonstructural (noncapsid) precursors. Nucleotide and amino acid sequences at the junction between regions 2A and 2B are indicated at the bottom of the figure.
To construct a plasmid which expressed only HA-tagged 2Apro driven by the PV IRES in transfected cells, nucleotides 108 to 742 of pOM1 and nucleotide sequences encoding methionine, the 2Apro cleavage site DLTTY*G (the asterisk denotes a scissile bond), 2Apro, and the HA tag were joined in frame in that order and inserted into mammalian expression vector pCI-neo (Promega). The resultant expression vector was designated pI108-2A-HA. DOTAP liposomal transfection reagent (Roche) was used for transfection of cells with this expression vector.
Antibodies.
Anti-PV1/Mahoney MAb 7m008 and anti-PV1/Sabin MAb 8a034 were kindly provided by the Japan Poliomyelitis Research Institute. MAb 7m008 can neutralize both PV1/Mahoney and PV1/Sabin, and MAb 8a034 is a neutralizing antibody specific for PV1/Sabin but not for PV1/Mahoney. Rabbit anti-PV1/Mahoney hyperimmune serum was prepared by immunizing rabbits with purified PV1/Mahoney. Rabbit polyclonal antibodies against 2Apro were prepared by using the C-terminal 14 amino acids of 2Apro (IRDLYAYEEEAMEQ) as an antigen. Those against eIF4G were elicited against a peptide of 17 residues (FYSWESSKDPAEQQGKG) corresponding to amino acid residues 37 to 21 of the C terminus of eIF4G. The anti-hPVR MAb (p286) used in this study was able to bind domain 1 of hPVR and block PV infection.
PV infection.
SK-N-SH and HeLa cells were infected with PV1/Mahoney or PV1/Sabin at a multiplicity of infection (MOI) of 10. After incubation at 37°C for 30 min, the infected cells were washed with serum-free medium and then incubated in fresh medium at 37°C for 1.5 h. Next, the medium was replaced with fresh medium containing MAb 7m008 (1:10 dilution), MAb 8a034 (1:10 dilution), or MAb p286 (1:3 dilution). Cell morphology was observed 24 hours postinfection (hpi) by light microscopy (Olympus IX70).
To study growth kinetics in a single cycle of infection, PV1/Mahoney-infected cell cultures were subjected to freezing-thawing three times at various times. The virus titers of the supernatants were measured by plaque assays after the removal of cell debris by centrifugation.
Pulse-labeling.
Infected cells were washed twice with methionine- and cysteine-free DMEM and incubated in the same medium at 37°C for 30 min. The cells then were supplemented with 100 μCi of [35S]methionine and [35S]cysteine per ml at various times and incubated at 37°C for 30 min. Next, the cells were washed three times with phosphate-buffered saline (PBS; 8 g of NaCl, 0.2 g of KCl, 1.15 g of Na2HPO4, and 0.2 g of KH2PO4 per liter) and lysed in TSA solution (10 mM Tris-HCl [pH 8.0], 140 mM NaCl, 0.025% NaN3, 1 mM phenylmethylsulfonyl fluoride, 10 μg of leupeptin/ml, 10 μg of aprotinin/ml) containing 1% Nonidet P-40 (NP-40). After the removal of cell debris by centrifugation, the lysates were separated by 12% polyacrylamide gel electrophoresis (PAGE) in a buffer containing 0.1% sodium dodecyl sulfate. The gels were dried, and the protein bands were visualized by autoradiography.
Northern blot analysis.
Infected SK-N-SH cells were treated with MAb p286 2 hpi and collected 7 hpi. They were homogenized in a Dounce homogenizer with PBS containing 1% NP-40 and 0.1% bovine serum albumin (BSA, fraction V; Sigma). After centrifugation to remove cell debris, the supernatants were applied to a 15 to 30% sucrose density gradient in PBS containing 1% NP-40 and 0.1% BSA. Centrifugation was performed at 41,000 rpm for 1 h at 4°C in a Beckman SW55Ti rotor. RNA was extracted from each fraction, treated at 65°C for 15 min in MOPS buffer [20 mM 3-(N-morpholino)propanesulfonic acid (MOPS) (pH 7.0), 5 mM CH3COONa, 1 mM EDTA] containing formaldehyde and formamide, and separated by 1% agarose gel electrophoresis in MOPS buffer. Northern blot analysis was carried out with AlkPhos Direct (Amersham Pharmacia Biotech) in accordance with the manufacturer's instructions. The probe used was negative-strand RNA complementary to nucleotides 1 to 742 of pOM1 to detect only positive-strand PV RNA. An AmpliScribe T3 transcription kit (Epicentre Technologies) was used for in vitro transcription.
Western blot analysis.
Infected cells were lysed in TSA solution containing 1% NP-40 at various times. After centrifugation to remove cell debris, the lysates were separated by 15% PAGE to detect 2Apro and by 6% PAGE to detect eIF4G in a buffer containing 0.1% sodium dodecyl sulfate. The proteins were transferred to an Immobilon transfer membrane (Millipore), probed with rabbit anti-2Apro antibodies or rabbit anti-eIF4G antibodies, and treated with goat anti-rabbit immunoglobulins conjugated with peroxidase. Protein bands were visualized by using enhanced chemiluminescence detection reagents (Amersham).
Immunofluorescence study.
Infected cells were washed once with PBS, fixed with 2% paraformaldehyde at room temperature for 10 min, and washed with PBS. After treatment with PBS containing 100 mM glycine at room temperature for 20 min, the cells were subjected to permeation with PBS containing 0.5% Triton X-100 at 4°C for 5 min and then were washed with PBS. Nonspecific staining was blocked by treatment with 3% BSA and 0.02% NaN3 in PBS at 37°C for 30 min. The cells were treated with rabbit hyperimmune serum against PV at 37°C for 2 h and then treated with goat anti-rabbit immunoglobulin G conjugated with Alexa Fluor 488 (Molecular Probes) at 37°C for 2 h. Nucleic acids were stained with 4′,6′-diamidino-2-phenylindole. Next, the cells were mounted with 80% (vol/vol) glycerol and analyzed with an inverted microscope (DM IRE2; Leica Microsystems) equipped with a confocal imaging spectrophotometer (TCS SP2; Leica Microsystems).
To detect HA-tagged 2Apro, a rat anti-HA MAb (Roche) and goat anti-rat immunoglobulin G conjugated with Alexa Fluor 488 were used as primary and secondary antibodies, respectively.
RESULTS
Effect of MAb against PV or the hPVR on the PV-induced CPE.
The PV-induced CPE in neural cells is blocked by the addition of rabbit hyperimmune serum against PV 2 hpi (35). To gain insight into the mechanisms responsible for this effect, MAb against PV or the hPVR was used for rabbit hyperimmune serum. SK-N-SH cells infected with PV1/Mahoney at an MOI of 10 showed typical signs of the CPE by 24 hpi without MAb, as expected (Fig. 1B). However, CPE expression was blocked when MAb against PV type 1 (7m008) was added 2 hpi (Fig. 1C). This blockade was not successful with MAb 8a034, a neutralizing MAb specific for PV1/Sabin, an oral poliovaccine strain derived from parental virulent PV1/Mahoney (Fig. 1E). CPE progression in neural cells infected with PV1/Sabin was inhibited by the addition of MAb 7m008 or 8a034 (data not shown). Thus, only neutralizing antibodies were effective in inhibiting CPE expression in neural cells. Neutralizing antibodies may inhibit PV infection before eclipse of the virus, and eclipse of PV by HeLa cells occurs much earlier than 2 hpi, but SK-N-SH cells may differ in that regard (14). This situation may result in efficient blockade of the CPE in PV-infected SK-N-SH cells by the addition of neutralizing antibodies 2 hpi. However, the eclipse scenario is not likely, because the PV generation time in SK-N-SH cells is very similar to that in HeLa cells (Fig. 2).
FIG. 1.

Inhibition of the PV-induced CPE in neural cells by MAb against PV or hPVR. Neural cells (A, B, C, D, and E) or HeLa cells (F, G, H, I, and J) were infected with PV1/Mahoney at an MOI of 10 (mock infection in panels A and F). At 2 hpi, the cells were washed three times. Then, medium not supplemented with MAbs (A, B, F, and G) or medium supplemented with MAbs against both PV1/Mahoney and PV1/Sabin (C and H), against hPVR (D and I), or against PV1/Sabin (E and J) was added to the culture. PV-infected cells were observed 24 hpi by microscopy.
FIG. 2.

PV replication in a single cycle of infection with or without MAbs. Virus titers in PV-infected cells at the indicated times were measured as described in Materials and Methods and plotted. SK-N-SH cells (A) and HeLa cells (B) were infected with PV1/Mahoney. The cells were treated with MAb against hPVR (□) or not treated with MAb (•) 2 hpi.
Similar inhibition was observed with the addition of MAb against hPVR (p286) (Fig. 1D). MAb p286 blocks PV infection by binding to domain 1 of hPVR, which is the PV binding site. This MAb also blocked the CPE induced by PV1/Sabin (data not shown). These results suggest that the inhibition of CPE expression in neural cells is due to the blockade of multiple rounds of viral replication. In fact, efficient CPE blockade by the addition of MAbs disappeared when the MAbs were added later than 5 hpi, the time by which some progeny virions had already been released into the culture medium, as in PV-infected HeLa cells (data not shown). HeLa cells, however, displayed a severe CPE regardless of whether the cells were treated with MAbs or not (Fig. 1G to J).
Virus yield with or without MAb.
It is possible that virus yield resulting from the lytic replication of PV determines whether or not CPE expression occurs. To test this hypothesis, PV yields in HeLa and SK-N-SH cells were examined in the presence or absence of MAb p286. The time profiles are shown in Fig. 2. The growth rate and final yield for PV1/Mahoney in HeLa and SK-N-SH cells with MAb p286 were almost identical to those without the MAb. Thus, viral replication efficiency is not affected by the addition of MAb in HeLa or SK-N-SH cells. These data indicate that virus yield is not a determinant of CPE expression in this situation. A similar experiment involving MAb 7m008 was not successful due to its neutralizing activity against PV.
Clearance of PV antigens in neural cells.
PV-infected neural cells treated with MAbs 2 hpi can be maintained by further passaging. Thus, infected cells are eventually cured of viral infection. It is possible that, after a single cycle of viral replication (Fig. 2), the clearance of PV occurs in infected neural cells. To examine the amounts of PV antigens present in neural cells, PV-infected neural cells in the absence or presence of MAb 7m008 or p286 were examined in an immunofluorescence study 11 and 24 hpi. As shown in Fig. 3, in the absence of MAb, neural cells showed a CPE by 11 hpi (Fig. 3A) and were destroyed by 24 hpi (Fig. 3D). They also retained significant amounts of PV antigens. In the presence of MAb, however, the amounts of PV antigens observed 11 hpi (Fig. 3B and C) dramatically decreased by 24 hpi (Fig. 3E and F). These data suggest that neural cells possess a mechanism for eliminating PV antigens with the aid of MAbs.
FIG. 3.

PV antigens in PV-infected neural cells with or without MAbs. PV-infected cells were examined in an immunofluorescence study 11 hpi (A, B, and C) and 24 hpi (D, E, and F). At 2 hpi the cells were not treated with MAb (A and D) or were treated with MAb against PV (B and E) or against hPVR (C and F). Red indicates nucleic acids, and green indicates PV antigens.
Protein synthesis in PV-infected neural cells.
Pulse-labeling experiments were performed as described in Materials and Methods to examine host and viral protein synthesis in neural cells (Fig. 4A) and HeLa cells (Fig. 4B) at various times after PV infection. In neural cells without MAb, viral protein synthesis was detected by 5 hpi and continued until at least 11 hpi, when the shutoff of host translation was observed (Fig. 4A) and the cells displayed a CPE (Fig. 3A). In the presence of MAb 7m008 or p286, however, PV-specific protein synthesis was detected by 5 hpi but not at 7 hpi (Fig. 4A), and host translation continued to occur (Fig. 4A). In HeLa cells (Fig. 4B), the shutoff of host translation was evident by 5 hpi, and PV-specific protein synthesis continued until at least 7 hpi regardless of whether the cells were treated with MAbs or not. These data also indicate that neural cells possess a mechanism that provides protection against PV infection.
FIG. 4.

Protein synthesis in PV-infected cells. Protein synthesis in PV-infected cells was investigated by a pulse-labeling assay with [35S]methionine. M (lane 1), mock infection. Neural cells (A) or HeLa cells (B) were pulse-labeled for 30 min beginning at the indicated times. Neural cells were not treated 2 hpi with MAb (lanes 2 to 5) or were treated 2 hpi with MAb against PV (lanes 6 to 9) or against hPVR (lanes 10 to 13). HeLa cells were not treated 2 hpi with MAb (lanes 2 to 5) or were treated 2 hpi with MAb against PV (lanes 6 to 8) or against hPVR (lanes 9 to 11).
It is possible that the inhibition of PV-specific protein synthesis was due to the degradation of PV mRNA in neural cells between 5 and 7 hpi. To exclude this possibility, a PV-infected neural cell culture was treated with MAb p286 2 hpi and harvested 7 hpi, and the cell extract was subjected to sucrose density gradient centrifugation to separate PV mRNA and PV virion particles. Northern blot analysis was carried out to detect the positive-sense PV RNA in each fraction (Fig. 5). The results indicate that a significant amount of free positive-sense PV RNA (PV mRNA) was present in the cell extract. These data suggest that PV-specific protein synthesis was inhibited at a stage of translation, that is, PV IRES activity was inhibited, although it is not known at present whether the antibody treatment also affected PV-specific RNA synthesis. It is possible that neural cells produce a factor(s) that inhibits PV IRES activity in response to PV infection in the presence of MAb.
FIG. 5.
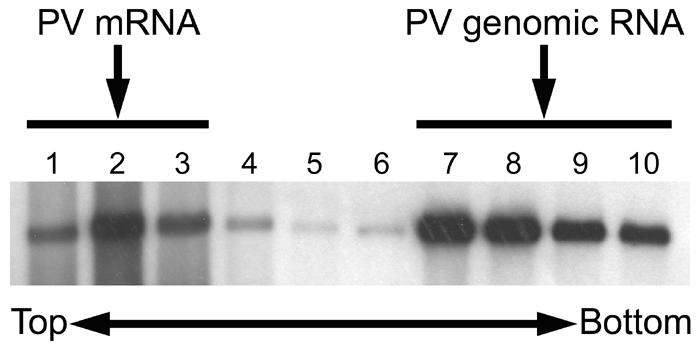
Detection of PV mRNA. PV-infected neural cells treated with MAb against hPVR 2 hpi were collected 7 hpi and homogenized. The supernatants of the homogenates were analyzed by sucrose density gradient centrifugation as described in Materials and Methods. Total RNA was extracted from each fraction and subjected to Northern blot analysis.
Existence of 2Apro in PV-infected neural cells.
Among PV-specific proteins, 2Apro is thought to be the most important viral molecule for inducing the CPE and host translation shutoff in infected cells. Accordingly, the existence of 2Apro in PV-infected neural cells was examined by Western blot analysis. As shown in Fig. 6A, the maximum amount of 2Apro was detected 5 hpi with or without MAb. The amount of 2Apro detected 11 hpi was similar to that detected 5 hpi in the absence of MAb. However, the amount of 2Apro decreased by 11 hpi in the presence of MAb. This result was expected because PV-specific protein synthesis was inhibited by 7 hpi (Fig. 4A). The turnover of 2Apro was probably not complete by 11 hpi but appeared to be complete by 24 hpi. In the absence of MAb, only a small amount of 2Apro was detected 24 hpi because many cells had been destroyed or were detached (Fig. 3D). In any event, a considerable amount of 2Apro still existed 11 hpi even in the presence of MAb.
FIG. 6.

Western blotting of 2Apro and elF4G. 2Apro expression (A) or eIF4G cleavage (B) in PV-infected neural cells was detected by Western blot analysis. M (lane 1), mock infection. Neural cells were not treated 2 hpi with MAb (lanes 2 to 6) or were treated 2 hpi with MAb against PV (lanes 7 to 10) or against hPVR (lanes 11 to 14).
PV-infected neural cells in the presence of MAb did not exhibit a CPE 11 hpi (Fig. 3B and C), although a significant amount of 2Apro was present in the cells 11 hpi. Another 2Apro-mediated phenomenon, the cleavage of eIF4G, was examined by Western blot analysis with anti-eIF4G antibodies (Fig. 6B). Most eIF4G was cleaved by 3.5 hpi, and no intact eIF4G was detected 5 hpi with or without MAb. In the presence of MAb, intact eIF4G began to appear 11 hpi, when a considerable amount of 2Apro was present (Fig. 6A); only intact eIF4G was detected 24 hpi, whereas, in the absence of MAb, no intact eIF4G was detected 5 hpi or later.
These results indicate that two 2Apro-mediated phenomena, CPE expression and eIF4G cleavage, were suppressed in PV-infected neural cells in the presence of MAb. 2Apro-mediated CPE expression is discussed below. As for eIF4G cleavage, it is possible that 2Apro activity was inhibited by cellular factors or by the compartmentalization of 2Apro from eIF4G in neural cells.
Localization of 2Apro.
To determine the localization of 2Apro in neural cells infected with PV, 2A-HA virus was constructed as described in Materials and Methods. The genomic structure of this virus is shown in Fig. 7. The virus had phenotypes similar to those of PV1/Mahoney (data not shown), such as induction of CPE progression in neural cells with or without MAb (Fig. 1), growth profile (Fig. 2), plaque size phenotype, pulse-labeling pattern (Fig. 4A), 2Apro expression (Fig. 6A), and eIF4G cleavage profile (Fig. 6B).
SK-N-SH cells were infected with 2A-HA virus at an MOI of 10, and the localization of 2Apro was investigated 5 and 11 hpi as described in Materials and Methods (Fig. 8). HA-tagged 2Apro was present in the nucleus and cytoplasm 5 hpi with or without MAb (Fig. 8A to C). In the presence of MAb, HA-tagged 2Apro was detected only in the nucleus 11 hpi (Fig. 8E and F), and the cells did not show signs of the CPE (Fig. 8H and I). In the absence of MAb, however, HA-tagged 2Apro was localized both in the nucleus and in the cytoplasm 11 hpi, and the cells displayed typical signs of the CPE by 11 hpi (Fig. 8D and G). These data indicate that 2Apro in PV-infected neural cells was imported into the nucleus. Thus, the compartmentation of 2Apro from eIF4G in neural cells may result in the appearance of intact eIF4G 11 hpi in the presence of MAb. Similar experiments were carried out with HeLa cells; the results obtained with MAb and without MAb were similar to those obtained for neural cells without MAb (data not shown).
FIG. 8.

Localization of HA-tagged 2Apro in PV-infected neural cells. Neural cells infected with 2A-HA virus were cultured as described in Materials and Methods. The cells were not treated 2 hpi with MAb (A, D, and G) or were treated 2 hpi with MAb against PV (B, E, and H) or against hPVR (C, F, and I). The cells were fixed 5 hpi (A, B, and C) or 11 hpi (D, E, F, G, H, and I) and subjected to an immunofluorescence study. Red indicates nucleic acids, and green indicates HA-tagged 2Apro.
Expression of HA-tagged 2Apro alone.
To investigate the abilities of 2Apro to induce the CPE and to be transported to the nucleus, HA-tagged 2Apro alone was expressed in SK-N-SH and HeLa cells by using a mammalian expression vector. The cells were fixed 2 days after transfection and stained as described in Materials and Methods. As shown in Fig. 9, only HeLa cells exhibited the CPE (Fig. 9E), and neural cells appeared not to be damaged (Fig. 9A). HA-tagged 2Apro was detected only in the nucleus in neural cells, but it was found both in the nucleus and in the cytoplasm in HeLa cells. HA-tagged 2Apro may be synthesized at higher levels in HeLa cells than in SK-N-SH cells. These data indicate that the harmful effect of 2Apro observed in HeLa cells is suppressed in neural cells and that HA-tagged 2Apro itself has the ability to be distributed to the nucleus.
FIG. 9.

Transient expression of HA-tagged 2Apro. Mammalian expression vector pCI-neo encoding HA-tagged 2Apro was transfected into cells, which were subjected to an immunofluorescence study. Neural cells (A, B, C, and D) and HeLa cells (E, F, G, and H) were fixed 2 days posttransfection. Red indicates nucleic acids, and green indicates HA-tagged 2Apro.
DISCUSSION
Cultured cells, such as HeLa cells, infected with PV die of a severe CPE. This phenomenon is thought to be attributed mainly to the expression of viral 2Apro, which is also responsible for eIF4G cleavage and host cell translation shutoff. An exception was reported by Tolskaya et al. (35). Neural cells infected with PV were eventually cured when they were treated with anti-PV hyperimmune serum shortly after infection. This report is a continuation of this observation and indicates the existence of protective mechanisms against PV infection in neural cells.
Since Tolskaya et al. (35) assumed that antibodies penetrate infected cells, we examined the possibility of penetration of MAbs by using HeLa cells and SK-N-SH cells. The results indicated that both cell lines take up MAbs (data not shown). However, it is not likely that MAbs inhibit PV replication inside cells, because the penetration of MAbs into HeLa cells occurred more efficiently than that into SK-N-SH cells (data not shown). Furthermore, the argument by Tolskaya et al. (35) does not correlate with the observation that an anti-hPVR MAb also blocked the PV-induced CPE in neural cells.
Our data strongly suggest that antibody treatment prevents multiple rounds of viral replication and that subsequent rounds of particle propagation inside cells result in CPE expression in neural cells. To confirm this notion, neural cells were infected with defective interfering particles of PV, which were not able to produce progeny virions because of the lack of a capsid protein coding sequence in the genome. Our preliminary results showed that defective interfering particles did not induce a CPE in neural cells but did induce a severe CPE in HeLa cells. However, it is not clear which process in the subsequent infections is necessary to induce the CPE. Indeed, virus yields were not different between infected cell cultures in the presence and in the absence of MAbs. It is known that virus yield is not altered when the efficiency of viral protein synthesis is slightly lowered artificially. This means that PV-specific protein synthesis in infected cells is present in surplus compared with PV-specific RNA synthesis. Thus, CPE expression may depend on the amounts of PV-specific proteins expressed in infected cells. It should be noted that the level of 2Apro expression in the absence of MAb was always higher than that in the presence of MAb 11 hpi, when virus production was already complete (Fig. 6A). The expression of 2Apro alone in neural cells (Fig. 9) may not be over the threshold needed to induce the CPE under the conditions used in this study. It is also possible that neural and HeLa cells differ in their sensitivities to harmful PV-specific proteins.
PV-specific protein synthesis was inhibited by 7 hpi (Fig. 4A), and virus production was completed by 8 hpi (Fig. 2). Therefore, the amounts of PV antigens were reduced until 24 hpi (Fig. 3). It is possible that PV-specific proteins are degraded by turnover mechanisms in cells. However, the reasons for the rapid disappearance of PV particles are not clear, considering the high stability of PV particles. They may be released into the culture medium. Alternatively, neural cells may possess mechanisms for destroying PV particles.
In the absence of MAb, host cell protein synthesis in infected neural cells continued until at least 9 hpi (data not shown), whereas eIF4G appeared to be completely cleaved by 5 hpi (Fig. 6B). These data suggest the existence of a factor(s) other than eIF4G that compensates for the lack of intact eIF4G in neural cells, although a small amount of residual intact eIF4G may function to continue host cell protein synthesis. It is possible that there are multiple eIF4G-related molecules in neural cells. Molecule p97/NAT1/DAP5 is one example, and it is similar to the C-terminal half of eIF4G (15, 21, 39). Such a factor(s) may be specifically expressed in neural cells and function in place of eIF4G. The factor(s) may be fairly resistant to PV infection, because translation shutoff in neural cells is delayed until 11 hpi.
HA-tagged 2Apro is imported into the nucleus. Since the HA tag itself does not show such activity (data not shown), the 2Apro moiety of HA-tagged 2Apro must carry a region(s) like a nuclear localization signal. Indeed, there are basic amino acid-rich regions in the N-terminal half of 2Apro. The function(s) of 2Apro in the nucleus is not known at present. However, the components of the nuclear pore complex are degraded in PV-infected cells, a process which might disturb nuclear import and export systems and therefore inhibit the cell signaling pathways which induce the onset of an antiviral response (11). According to our preliminary results, the degradation of components of the nuclear pore complex is not due to 2Apro (data not shown). In addition, our data suggest that 2Apro imported into the nucleus of neural cells is silent in the induction of host translation shutoff and CPE progression. Thus, 2Apro may be active only in the cytoplasm. Furthermore, it was reported that 2Apro may be involved in PV replication through mechanisms independent of its protease activity (22, 27, 40).
Besides 2Apro, 2BC is cytotoxic in HeLa cells, although its cytotoxicity is lower than that of 2Apro (1, 2). Since the PV-infected neural cells used in this study did not show a CPE when MAb to PV or the hPVR was added to the culture 2 hpi, it is possible that the MAb inhibits the cytotoxicity of 2BC in neural cells. Alternatively, 2BC may not have any toxic effect on neural cells.
The mechanism responsible for the inhibition of PV IRES activity in neural cells is unknown at present. Upon PV infection, neural cells may respond by producing a factor(s) that inhibits PV IRES activity. A search for such a factor(s) is essential to understanding the neurovirulence of PV and is presently being conducted. Such a putative factor(s) may be useful as an anti-PV agent.
Our hypothesis is summarized in Fig. 10. In the presence of MAb against PV or the hPVR, multiple rounds of viral replication are blocked. Thus, the viral infection cycle induced by the primary infection can be observed. As shown in Fig. 2, viral replication in neural cells is not affected by the addition of MAb 2 hpi. eIF4G is then cleaved by 2Apro in the cytoplasm by 5 hpi. Host translation continues without detectable intact eIF4G, probably because a neural cell factor(s) compensates for its absence. By 7 hpi, PV-specific protein synthesis ceases due to unknown mechanisms that may involve the induction of a factor(s) to prevent PV IRES activity. This effect results in inhibition of the accumulation of PV-specific proteins. Existing 2Apro in neural cells is imported into the nucleus by 11 hpi, and intact eIF4G begins to appear because of 2Apro localization in the nucleus. The resurrection of intact eIF4G supports host translation. As a result, PV-infected neural cells do not show a CPE and are eventually cured of PV infection.
FIG. 10.

Schematic explanation of the process responsible for inhibition of the PV-induced CPE in neural cells by MAb against PV or against hPVR. See the legend to Fig. 7 for definitions of abbreviations.
Neural cells in the central nervous system (CNS) may possess a specific immune system response that differs from that of other tissues. It should be noted that the curing of PV-infected neural cells in the CNS of monkeys has been suggested (6). Microglia derived from monocytes are mainly responsible for immune system activity in the CNS. Besides the utilization of microglia, it is possible that neural cells themselves prepare specific intracellular immune system features, such as double-stranded RNA-dependent protein kinase and RNA interference. The protective response against PV infection reported here may be an intracellular immune system response specific to neural cells. Alternatively, this kind of intracellular immune system may also be present in HeLa cells but may be masked. Much work should be done to elucidate the mechanisms of the protective response observed only in neuroblastoma cells.
Acknowledgments
We thank Etsuko Suzuki and Yuri Matsushita for help in preparing the manuscript.
This work was supported by a Grant-in-Aid for Specially Promoted Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
REFERENCES
- 1.Aldabe, R., A. Barco, and L. Carrasco. 1996. Membrane permeabilization by poliovirus proteins 2B and 2BC. J. Biol. Chem. 271:23134-23137. [DOI] [PubMed] [Google Scholar]
- 2.Aldabe, R., A. Irurzun, and L. Carrasco. 1997. Poliovirus protein 2BC increases cytosolic free calcium concentrations. J. Virol. 71:6214-6217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barco, A., E. Feduchi, and L. Carrasco. 2000. A stable HeLa cell line that inducibly expresses poliovirus 2Apro: effects on cellular and viral gene expression. J. Virol. 74:2383-2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belsham, G. J., and N. Sonenberg. 1996. RNA-protein interactions in regulation of picornavirus RNA translation. Microbiol. Rev. 60:499-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bienz, K., D. Egger., Y. Rasser., and W. Bossart. 1980. Kinetics and location of poliovirus macromolecular synthesis in correlation to virus-induced cytopathology. Virology 100:390-399. [DOI] [PubMed] [Google Scholar]
- 6.Bodian, D. 1949. Histopathlogic basis of clinical finding in poliomyelitis. Am. J. Med. 6:563-578. [DOI] [PubMed] [Google Scholar]
- 7.Dales, S., H. J. Eggers., I. Tam., and G. E. Palade. 1965. Electron microscopic study of the formation of poliovirus. Virology 379-389. [DOI] [PubMed]
- 8.Ehrenfeld, E. 1996. Initiation of translation by picornavirus RNAs, p. 549-573. In J. W. B. Hershey, M. B. Mathews, and N. Sonenberg (ed.), Translational control. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 9.Ehrenfeld, E. 1982. Poliovirus-induced inhibition of host-cell protein synthesis. Cell 28:435-436. [DOI] [PubMed] [Google Scholar]
- 10.Etchison, D., S. C. Milburn, I. Edery, N. Sonenberg, and J. W. Hershey. 1982. Inhibition of HeLa cell protein synthesis following poliovirus infection correlates with the proteolysis of a 220,000-dalton polypeptide associated with eucaryotic initiation factor 3 and a cap binding protein complex. J. Biol. Chem. 257:14806-14810. [PubMed] [Google Scholar]
- 11.Gustin, K. E., and P. Sarnow. 2001. Effects of poliovirus infection on nucleo-cytoplasmic trafficking and nuclear pore complex composition. EMBO J. 20:240-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hambidge, S. J., and P. Sarnow. 1992. Translational enhancement of the poliovirus 59 noncoding region mediated by virus-encoded polypeptide 2A. Proc. Natl. Acad. Sci. USA 89:10272-10276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanecak, R., B. L. Semler, C. W. Anderson, and E. Wimmer. 1982. Proteolytic processing of poliovirus polypeptides: antibodies to polypeptide P3-7c inhibit cleavage at glutamine-glycine pairs. Proc. Natl. Acad. Sci. USA 79:3973-3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holland, J. J. 1962. Irreversible eclipse of poliovirus by HeLa cells. Virology 16:163-176. [DOI] [PubMed] [Google Scholar]
- 15.Imataka, H., H. S. Olsen, and N. Sonenberg. 1997. A new translational regulator with homology to eukaryotic translation initiation factor 4G. EMBO J. 16:817-825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krausslich, H. G., M. J. Nicklin, H. Toyoda, D. Etchison, and E. Wimmer. 1987. Poliovirus proteinase 2A induces cleavage of eucaryotic initiation factor 4F polypeptide p220. J. Virol. 61:2711-2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krausslich, H. G., and E. Wimmer. 1988. Viral proteinases. Annu. Rev. Biochem. 57:701-754. [DOI] [PubMed] [Google Scholar]
- 18.Lamphear, B. J., R. Kirchweger., T. Skern., and R. E. Rhoads. 1995. Mapping of functional domains in eukaryotic protein synthesis initiation factor 4G (eIF4G) with picornaviral proteases-implications for cap-dependent and cap-independent translational initiation. J. Biol. Chem. 270:21975-21983. [DOI] [PubMed] [Google Scholar]
- 19.Lee, C. K., and E. Wimmer. 1988. Proteolytic processing of poliovirus polyprotein: elimination of 2Apro-mediated, alternative cleavage of polypeptide 3CD by in vitro mutagenesis. Virology 166:405-414. [DOI] [PubMed] [Google Scholar]
- 20.Leibowitz, R., and S. Penman. 1971. Regulation of protein synthesis in HeLa cells. 3. Inhibition during poliovirus infection. J. Virol. 8:661-668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levy-Strumpf, N., L. P. Deiss, H. Berissi, and A. Kimchi. 1997. DAP-5, a novel homolog of eukaryotic translation initiation factor 4G isolated as a putative modulator of gamma interferon-induced programmed cell death. Mol. Cell. Biol. 17:1615-1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu, H. H., X. Y. Li, A. Cuconati, and E. Wimmer. 1995. Analysis of picornavirus 2Apro proteins: separation of proteinase from translation and replication functions. J. Virol. 69:7445-7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McBride, A. E., A. Schlegel, and K. Kirkegaard. 1996. Human protein Sam68 relocalization and interaction with poliovirus RNA polymerase in infected cells. Proc. Natl. Acad. Sci. USA 93:2296-2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meerovitch, K., J. Pelletier, and N. Sonenberg. 1989. A cellular protein that binds to the 5′-noncoding region of poliovirus RNA: implications for internal translation initiation. Genes Dev. 3:1026-1034. [DOI] [PubMed] [Google Scholar]
- 25.Meerovitch, K., Y. V. Svitkin, H. S. Lee, F. Lejbkowicz, D. J. Kenan, E. K. Chan, V. I. Agol, J. D. Keene, and N. Sonenberg. 1993. La autoantigen enhances and corrects aberrant translation of poliovirus RNA in reticulocyte lysate. J. Virol. 67:3798-3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Merrick, W. C., and J. W. B. Hershey. 1996. The pathway and mechanism of eukaryotic protein synthesis, p. 31-69. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 27.Molla, A., A. V. Paul., M. Schmid., S. K. Jang., and E. Wimmer. 1993. Studies on dicistronic polioviruses implicate viral proteinase 2Apro in RNA replication. Virology 196:739-747. [DOI] [PubMed] [Google Scholar]
- 28.Pallansch, M. A., O. M. Kew, B. L. Semler, D. R. Omilianowski, C. W. Anderson, E. Wimmer, and R. R. Rueckert. 1984. Protein processing map of poliovirus. J. Virol. 49:873-880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pelletier, J., and N. Sonenberg. 1988. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature 334:320-325. [DOI] [PubMed] [Google Scholar]
- 30.Sachs, A. B., P. Sarnow., and M. W. Hentze. 1997. Starting at the beginning, middle, and end: translation initiation in eukaryotes. Cell 89:831-838. [DOI] [PubMed] [Google Scholar]
- 31.Shiroki, K., T. Ishii, T. Aoki, M. Kobashi, S. Ohka, and A. Nomoto. 1995. A new cis-acting element for RNA replication within the 5′ noncoding region of poliovirus type 1 RNA. J. Virol. 69:6825-6832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shiroki, K., T. Isoyama, S. Kuge, T. Ishii, S. Ohmi, S. Hata, K. Suzuki, Y. Takasaki, and A. Nomoto. 1999. Intracellular redistribution of truncated La protein produced by poliovirus 3Cpro-mediated cleavage. J. Virol. 73:2193-2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skern, T., and H. D. Liebig. 1994. Picornains 2A and 3C. Methods Enzymol. 244:583-595. [DOI] [PubMed] [Google Scholar]
- 34.Sonenberg, N. 1990. Poliovirus translation. Curr. Top. Microbiol. Immunol. 161:23-47. [DOI] [PubMed] [Google Scholar]
- 35.Tolskaya, E. A., T. A. Ivannikova, M. S. Kolesnikova, S. G. Drozdov, and V. I. Agol. 1992. Postinfection treatment with antiviral serum results in survival of neural cells productively infected with virulent poliovirus. J. Virol. 66:5152-5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toyoda, H., M. J. Nicklin, M. G. Murray, C. W. Anderson, J. J. Dunn, F. W. Studier, and E. Wimmer. 1986. A second virus-encoded proteinase involved in proteolytic processing of poliovirus polyprotein. Cell 45:761-770. [DOI] [PubMed] [Google Scholar]
- 37.Waggoner, S., and P. Sarnow. 1998. Viral ribonucleoprotein complex formation and nucleolar-cytoplasmic relocalization of nucleolin in poliovirus-infected cells. J. Virol. 72:6699-6709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wimmer, E., C. U. Hellen, and X. Cao. 1993. Genetics of poliovirus. Annu. Rev. Genet. 27:353-436. [DOI] [PubMed] [Google Scholar]
- 39.Yamanaka, S., K. S. Poksay, K. S. Arnold, and T. L. Innerarity. 1997. A novel translational repressor mRNA is edited extensively in livers containing tumors caused by the transgene expression of the apoB mRNA-editing enzyme. Genes Dev. 11:321-333. [DOI] [PubMed] [Google Scholar]
- 40.Yu, S. F., P. Benton, M. Bovee, J. Sessions, and R. E. Lloyd. 1995. Defective RNA replication by poliovirus mutants deficient in 2A protease cleavage activity. J. Virol. 69:247-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ziegler, E., A. M. Borman., F. G. Deliat., H. D. Liebig., D. Jugovic., K. M. Kean., T. Skern., and E. Kuechler. 1995. Picornavirus 2A proteinase-mediated stimulation of internal initiation of translation is dependent on enzymatic activity and the cleavage products of cellular proteins. Virology 213:549-557. [DOI] [PubMed] [Google Scholar]