

Abstract
Our previous research showed that traumatic brain injury (TBI) induced by controlled cortical impact (CCI) causes not only massive cell death, but also results extensive dendrite degeneration in those spared neurons in the cortex. Cell death and dendrite degeneration in the cortex may contribute to persistent cognitive, sensory, and motor dysfunction. There is still no approach available to prevent cells from death and dendrites from degeneration following TBI. When we treated the animals with a small molecule, 7,8- Dihydroxyflavone (DHF) that mimics the function of BDNF through provoking TrkB activation, reduced dendrite swellings in the cortex. DHF treatment also prevented dendritic spine loss after TBI. Functional analysis showed that DHF improved rotarod performance on the third day after surgery. These results suggest that although DHF treatment did not significantly reduced neuron death, it prevented dendrites from degenerating, and protected dendritic spines against TBI insult. Consequently, DHF can partially improve the behavior outcomes after TBI.
Keywords: Traumatic brain injury; dendrite; degeneration; brain-derived neurotrophic factor; 7,8-dihydroxyflavone
Introduction
Traumatic brain injury (TBI) is a serious public health problem in the United States (US). According to statistics from the Centers for Disease Control and Prevention (CDC), TBI is a contributing factor to a third (30.5%) of all injury-related deaths in the US. Every year, at least 1.7 million TBIs occur either as an isolated injury or along with other injuries [1]. In 2010, the total of direct and indirect medical costs was an estimated $80 billion [2]. Traumatic brain injury is a significant problem because it induces not only direct damage like tissue lesion, but also indirect damage including extensive cell death and degeneration. Besides cell death, degeneration is usually seen in spared neurons after injury, suggesting that neurons may have survived from injury, but may have sustained damage that is not visible.
Dendrite degeneration is an aspect of the damage. Dendrites are the branched projections of a neuron that provide a mass membrane surface for forming dendritic spines and act to conduct the electrochemical stimulation received from other neural cells to the cell body. Our recent research revealed that mild TBI causes extensive dendritic degeneration of the cortex [3], and that moderate TBI even causes acute dendrite degeneration and synaptic loss in spared neurons of the hippocampus [4]. Dendrites play a critical role in integrating these synaptic inputs and in determining the extent to which action potentials are produced by the neurons. Therefore, dendrite degeneration may lead to persistent cognitive, sensory, and motor dysfunction [5].
Brain derived neurotrophic factor (BDNF) is a member of neurotrophin family and it plays an important role in neuron survival, differentiation, and synaptical plasticity during cell development [6,7]. Previous research showed that upregulation of BDNF is associated with therapeutic improvement after traumatic brain injury [8–14]. Conditionally knocking out BDNF in the early stage of neurons in the dendate gyrus decreased dendrite development [15] and reduced newborn neuron survival after TBI; whereas, infusion of BDNF in the dentate gyrus prevented immature neuron death after TBI [16]. However, application of BDNF in clinical treatment still has problems. Intravenous injection of BDNF is safer but BDNF cannot penetrate the blood-brain barrier (BBB). Furthermore, direct intracerebral injection of BDNF is too invasive.
A small molecule alternative that can mimic BDNF function with the capacity to cross the BBB is needed. Recent studies demonstrated that 7,8-dihydroxyflavone (DHF) may be the suitable candidate. 7,8-dihydroxyflavone belongs to the Flavonoids family, a diverse group of plant secondary metabolites present in fruits and vegetables. Flavonoids, exert diverse biological effects, including acting as antioxidants and cancer-preventing agents [17]. Flavonoids may improve cognitive performance by protecting vulnerable neurons, enhancing existing neuronal function, and stimulating neuronal regeneration [18]. DHF is a small molecule with a moelcular weight of 254 dolton, thus it can cross the BBB and binds to TrkB to mimic BDNF functions [19]. It has been shown that DHF is neuroprotective in mutiple disease models [19–30] including Alzheimer’s disease [28,31] and traumatic brain injury [21]. In this study we assessed the role of DHF on neurons survive, dendrite degeneration and the functional outcomes after TBI.
Materials and Methods
Animals
Mice (C57/BL6) were housed with a 12/12 light/dark cycle and had free access to food and water ad libitum according to the principles outlined in “Guidelines for Care and Use of Experimental Animals”. They were used in experiments at an age of 8–10 weeks. All procedures were approved by the Indiana University Institutional Care and Use Committee (IACUC).
Controlled Cortical Impact Traumatic Brain Injury
Male mice (n=96) at 8–10 weeks old were subjected to moderate controlled cortical impact (CCI) injury (n=72) or SHAM treatment (n=24), as we previously described [33–35], with the following exceptions: the amount of deformation was set at 1.0 mm and the piston velocity controlled at 3.0 m/sec. These modifications resulted in a moderate level of injury using an electromagnetic model [36] (Impact One TM Stereotaxic Impactor for CCI, Leica Microsystem, and Illinois USA). Briefly, the mice were anesthetized with avertin and placed in a stereotaxic frame (Kopf Instruments,Tujunga, CA) prior to TBI. Using sterile procedures, the skin was retracted and a 4-mm craniotomy centered between the lambda and bregma sutures was performed. A point was identified midway between the lambda and bregma sutures and midway between the central suture and the temporalis muscle laterally. The skullcap was carefully removed without disruption of the underlying dura. Prior to the injury, the impacting piston was angled so that the impacting tip (3 mm in diameter) was perpendicular to the exposed cortical surface. Then the mice received the impact with setup parameters. SHAM (non-injured) animals received the craniotomy, but no CCI injury.
7,8-dihydroxyflavone (DHF) Treatment
After surgery, the CCI-injured mice (n=72) were randomized for placement into PBS control, vehicle control and DHF treatment groups. The mice then received intraperitoneal (i.p.) injections of PBS, DMSO (17%), or DHF (5 mg/kg in 17% DMSO) four times at 1, 24, 48, and 72 hours after surgery. The sham-surgery mice (n=24) also received i.p. injections of PBS four times at 1, 24, 48, and 72 hours after sham treatments. Four hours after the final injection, 8 mice from each group were sacrificed for assessing cortical tissue lesion (n=4) with cresyl violet staining or dendrite degeneration (n=4) with Golgi staining. The rest of animals (n=16 in each group) were allowed to survive for another 3 weeks for behavioral analysis with rotarod test and Morris water-maze.
Tissue processing
Animals were deeply anesthetized with an overdose of avertin and then perfused transcardially with 0.9% saline, followed by an ice-cold fixative containing 4% PFA in PBS. The brains were removed and postfixed in 4% PFA overnight, then cryoprotected with 30% sucrose for 48 hr. Serial coronal sections (30 μm thick) were cut using a cryostat (Microm HM 500 M) and stored at −20°C. The evaluators of histology were blinded to treatment conditions.
Cresyl Violet staining
Frozen sections were transferred to the free-floating glass slides and stained using cresyl violet solution for 20 minutes.Afterward, they were rinsed in double-distilled water quickly. Sections were immersed in differentiation solution (95% Ethanol) for two minutes. Then sections were dehydrated in 100% Ethanol, and cleared in 100% Xylene. Finally, sections were dried and mounted for imaging.
Cortical cavity volume measurement
By 72 hours after traumatic brain injury, the cortical tissue shows loss in the impacted area after fixation and cryoprotection. However, the absolute volume of the spare tissue was reduced due to the tissue processing. Therefore, we used a described percent cortex to quantify the extent of the contusion [3]. One-in-six series of 30μm-thick brain sections (180μm apart) were stained with crysel violet staining to show the spare cortex. The boundary contours of the contralateral and ipsilateral spare cortex were drawn with a Zeiss microscope attached to a Neurolucida system (Microbrightfield Inc., Colchester, VT). The the enclosed volume with the contours was measured. The percent cortex of the cavity was calculated with the following formula: percentage of the cortical cavity = (contralateral cortex volume – ipsilateral spare cortex volume) / contralateral cortex volume × 100%.
Golgi Staining
We used the FD Rapid Golgi Stain kit (FD NeuroTechnologies) to perform Golgi staining following the vendor’s protocol. The freshly dissected brains were immersed in impregnation solution (made by mixing equal volumes of Solutions A and B) and stored at room temperature for at least 2 weeks in the dark. The brains were then transferred into solution C and kept for at least 48 hrs at 4°C in the dark. Afterward, they were sliced using a horizon sliding slicer (SM2010R; Leica, Nussloch, Germany) at a thickness of 200 μm and stained using standard staining procedures supplied by FD.
Dendrite Morphology Analysis
Pyramidal neurons were analyzed using the following selection criteria: all Golgi stained neurons located in layer II/III with apical dendrites extending toward the surface. For each selected neuron, all branches of the dendritic tree were reconstructed at 20x magnification using a motorized microscope (Zeiss Imager M2) with Neurolucida software (Microbrightfield, VT). A 3D analysis of the reconstructed neurons was performed using NeuroExplorer software (Microbrightfield). More than 10 neurons were studied for each of the 4 animals in different experimental groups. The branch data of neurons from the same animal were averaged. Several aspects of dendritic morphology were examined. To assess overall changes, total length of dendrite trees, average length of dendrites, and number of dendrite branches were compared across groups using one-way ANOVA test. The complexity of dendrite trees was assessed with the Sholl analysis [38,39]. The number of intersections of dendrites was calculated with concentric spheres positioned at radial intervals of 20 μm. To quantify the dendritic swellings, we counted the number of beadings on the dendrites. All of the pyramidal neurons matched the criteria in layer II/III within the 2-mm area from edge of cavity were imaged and beadings on those dendrites were counted. Dendritic beading were defined as a rounded appearance that extending beyond the diameter of the parent dendrite and separated by “interbead ” segment [40].
Spine Quantification
For analysis of dendritic spines, high magnification images (63x oil immersion objective) were captured using a camera (Zeiss Axio observer) attached to a Zeiss inverted microscope. The neurons meeting the criteria used for dendrite analysis were selected for imaging. The images containing a Z-stack maximum projection of a dendrite from a single neuron were assembled in Photoshop to reconstruct the cell structure. The spines located at the main apical dendrite were assessed (>2000 spines for each mouse were analyzed). To assess changes in spine morphology, spines in the selected dendrite were classified into mushroom, stubby, and filopodia categories. The proportion of spines in each category was compared using one-way ANOVA.
Behavior Test
Rotarod test
For motor function analysis, the rotarod test was used. Sixty-four mice were used for the test; 16 mice for each group. During the test, an animal was allowed to remain stationary for 10 seconds at 0 rpm. The speed was slowly increased to 3 rpm for 10 seconds and was steadily increased by 3 rpm in 10-second intervals until the maximum of 30 rpm was reached. There were 4 trials each day for each mouse with a 20-minute interval between trials. The first rotarod test was performed before surgery to obtain a baseline. After surgery, the rotarod test was administered 3 times, at 3 days, 7 days, and 14 days after surgery. Latency on the stage was recorded and analyzed using repeated measures. We then used one-way ANOVA to analyze specific time points between the 4 groups.
Morris Water Maze
Two weeks after TBI, the Morris water maze (MWM) test was used to assess the function of mouse spatial learning and memory. The test lasted 6 days, including a 5-day trial of learning and a 1-day trial of memory. During the learning days, mice underwent 4 trials from different start positions. Mice were released in cold water at 18°C. Each mouse had 1 minute to find the platform hidden 1 cm underwater. Once a mouse reached the platform, it was allowed to remain 15 seconds on the platform to build memories. If a mouse didn’t find the platform within 1 minute, then it was placed on the platform and allowed to remain for 15 seconds. During a 1-minute rest between trials, mice stayed in a dry cage with a towel and a warm light. On the sixth day, the platform was removed and each mouse started from a new position. Only 1 trial was performed on the last day. Latency of the learning trails and duration in the platform quadrant were recorded to analyze the function of learning and memory. The persons who evaluated the behaviors were blinded to injury and treatment conditions.
Statistical Analysis
All data are presented as mean ± standard error (SE), with the number of repetitive experiments or mice indicated. Mean values were statistically compared using one-way ANOVA followed by Tukey's post hoc testing.
Results
Post-injury treatment with DHF did not significantly decreased TBI-induced cortical lesion
To examine the effect of DHF on tissue lesion in the cortex, the animal received intraperitoneal (i.p.) injection of either DHF once (5 mg/kg in 17% DMSO) or vehicle (17% DMSO) at 1, 24, 48, and 72 hours hour after surgery. Four hours after surgery, the brains were removed to assess cortical lesion. The area of cortical cavity was assessed after staining of the sections with crysel violet staining. The normalized percentages of the cortical cavity was 15.9±4.5% in the TBI-injureed mice treated with vechicle (Fig. 1A and C). The normalized percentages of the cortical cavity in the mice treated with DHF was 14.9±1.9% (Fig. 1B and C). The result showed there is a slightly reduction of cortical cavity in the DHF treated mice following TBI, but this difference is not stastistically significant (p>0.05).
Figure 1. Post-injury treatment with DHF did not significantly decreased cortical lesion following moderate traumatic brain injury (TBI).
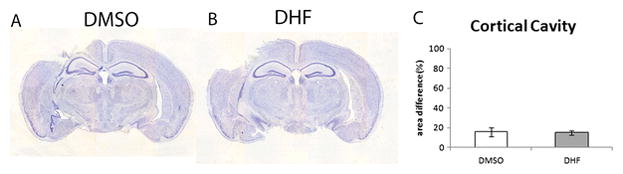
Crysel violet staining was performed to assess the cortical cavity in the brain received 7,8- Dihydroxyflavone (DHF) or vehicle treatment following TBI. (A) Cortical cavity of vehicle treatment mice after TBI. (B) Cortical cavity of DHF treatment mice after TBI. (C) Measurement of cavity (n=4, *, p<0.05).
DHF treatment protected dendritic degeneration after moderate TBI
As shown in previous studies [41,42], moderate TBI induces a cavity and cell death in the cortex (Fig. 2A and B). Although neurons near the cavity can survive after TBI, their survival does not mean they have not been injured. To assess whether TBI caused injury to those spared neurons around the lesion area in the cortex, we used Golgi staining to assess their dendritic morphology. Three sections at the epicenter of each mouse brain were selected for evaluation. All pyramidal neurons in layer II/III that were stained by Golgi and that matched the criteria were included in the analysis. The dendrites from the neurons in the SHAM-treated mice (n>30) were smooth with a high density of spines (Fig. 2E, F, and M). However, the dendrites of spared pyramidal neurons in the TBI mice were deformed and showed serious swelling (Fig. 1G, H, and N). Dendritic beading is a hallmark of postinjury degeneration, indicating the dendrite injury in the spared neurons around the lesion area in the cortex following moderate TBI. When the TBI-injured mice were treated with DHF for 3 days, dendrites were smoother with less swelling than dendrites in the TBI group (Fig. 2I, J, and O). The dendrites in the DMSO-treated (Vehicle) group were swelling similar to the dendrites in the TBI group (Fig. 2K, L and P). These data suggest that DHF treatment protected dendrites of spared neurons in the perilesion area from degeneration.
Figure 2. DHF treatment reduced dendritic swelling after moderate traumatic brain injury.

Golgi staining was performed to assess dendrite morphology of neurons in the layer II/III in the neocortex following TBI. (A–D) After TBI, tissue cavities can be observed in the cortex of injured groups; (E, F) 20 × images of pyramidal neurons in SHAM group; (G, H) 20× images of pyramidal neurons in the PBS-TBI group; (I, J) 20× images of pyramidal neurons in the DHF-TBI group; (K, L) 20× images of pyramidal neurons in the DMSO-TBI group. (e, f) 63× images of dendrites in SHAM group; (g, h) 63× images of dendrites in the PBS-TBI group; (i, j) 63× images of dendrites in the DHF-TBI group; (k, l) 63 × images of dendrites in the DMSO-TBI group. (M, N, O, P) Fragments of dendrites in 4 groups show the dendritic beadings. (Q) Quantification results of dendritic beading. (n=4, *, p<0.05).
To quantify the dendritic swellings, we counted the number of beadings on the dendrites. All of the pyramidal neurons in layer II/III within the 2-mm area from edge of cavity were imaged and beadings on those dendrites were counted. Quantification data showed that the dendritic beadings in the SHAM group were few; only 20±4 beadings were observed per 1-mm length of dendrite. In the TBI injury group, the dendritic beading increased dramatically to 38±3/1 mm, a 2-fold increase as compared to SHAM (p< 0.05). In the DHF treatment group, the number of dendritic beadings was significantly reduced compared to the injury group (20±2/1 mm vs 38±3/1 mm, p<0.05); while Vehicle treatment showed no effect on reducing the dendritic beading (43±4/1 mm) (Fig. 2P). These results indicated that DHF treatment can prevent dendritic swellings caused by TBI.
Dendritic beading may result in loss of dendrite branches after TBI. To assess if DHF treatment could prevent the loss of dendrite branches after TBI, we reconstructed the dendritic morphology of every pyramidal neuron from layer II/III in the ipsilateral cortex within a 2-mm area from the edge of the cavity, and we measured the dendrite complexity (Fig. 3A).
Figure 3. DHF treatment protected dendritic degeneration after moderate traumatic brain injury.

(A) Reconstruction of Golgi-stained neurons of 4 groups. (B) Branch number, total length, and average length were used to compare between 4 groups, DHF treatment showed advantages in branch number and total length compared to the other TBI groups. (n=4, **, SHAM group vs. TBI group, p<0.01, ##, DHF vs. PBS or DMSO, p<0.01 ) (C) Sholl analysis was done to determine how dendrite complexity changed after TBI and DHF treatment. (D) DHF group showed a dramatic increase of dendrite complexity compared to the other TBI group. (n=4, * p≪0.05, * DHF group vs.PBS or DMSO group, p<0.05, # SHAM vs. TBI, p<0.05).
In apical dendrites, the number of dendrite branches in the SHAM group was 6±0.4; total length of dendrite branches was 684.4±39.3 μm; and average length of dendrite branches was 111.3±4.1 μm. After TBI, injured mice with PBS treatment exhibited a significantly decreased in number of branches (from 6±0.4 [SHAM] to 5±0.5 [TBI], p<0.01) and total length of dendrite branches decreased (from 684.4±39.3μm [SHAM] to 475.7±32.3μm [TBI], p<0.01). With DHF treatment, both the number of dendrite branches (7±0.6) and the total length of dendrite branches were increased (794.5±58.8 μm) compared to the TBI-injured group without treatment (p<0.01). The Vehicle group did not show any protection; it had a similar branch number (4±0.3) and total length (557.9±29.2) compared to the PBS group.
In basal dendrites, the TBI group with PBS treatment also showed a decreased branch number (6±0.4) and total length (373.3±31.2 μm) compared to the SHAM group (branch number 8±0.4; total length 539.7±36.6 μm, p<0.05). While in the DHF treatment group, both branch number (8±0.6) and total length (637.5±52.8μm) were increased compared to the PBS group; both measures were back to SHAM level. The Vehicle group was close to the PBS group in branch number (5±0.3) and total length (387.0±26.2μm) (Fig. 3B). We then further performed a Sholl analysis to determine the branching characteristics of individual neurons (Fig. 3C). Sholl analysis showed that the complexity of both apical and basal dendrites in the TBI group without DHF treatment decreased dramatically. In apical dendrites, Sholl analysis showed that the injury group with PBS treatment had fewer intersections (2±0.2) than the SHAM group (3±0.2) (Fig. 3D). In contrast, the DHF treatment group had more intersections (3±0.2) than the PBS group, and was even better than the SHAM group. The dendrite complexity in the DMSO-treated Vehicle group (2±0.2) was similar to the PBS group (Fig. 3E). In basal dendrites, the PBS group had fewer intersections (4±0.3) than the SHAM group (5±0.3). The DHF treatment group had more intersections (5±0.5) compared to the PBS group, while the Vehicle group had a similar intersection (4±0.2) to the PBS group. Results indicated that DHF protected dendrite branches from loss caused by dendritic beading. Altogether, DHF treatment significantly reduced the dendrite deformation after moderate TBI and may be able to prevent degeneration.
DHF treatment can prevent dendritic spine degeneration after moderate TBI
Dendrites provide a surface for forming dendritic spines. To investigate whether dendrite protection by DHF can further preserve dendritic spines after moderate TBI, we assessed the dendritic spines within a 2-mm area from the edge of the cavity (Fig. 4A). High resolution images (63x) were taken for each single pyramidal neuron from layer II/III, the primary apical dendrite was chosen and the spines on it were counted. Representive images showed that after TBI, injury group mice underwent spine loss. But the loss of spine was self-limiting; the difference between injury group and SHAM group extended up to a 1.5-mm area from the edge of the cavity (Fig. 4B). So we separated the 2-mm area into 4 parts, with each part occupying 0.5 mm distance (Fig. 4A). In the 0-0.5 mm area, all TBI groups showed decreasing dendritic spines (PBS group: 45±2/100 μm; DHF group: 45±2/100 μm; and DMSO group: 45±4/100 μm) compared to SHAM (55±3/100 μm) (p<0.05). In the 0.5–1 mm area, the PBS group (47±3/100 μm) and the DMSO group (40±3/100 μm) retained the same level as in the 0–0.5 mm area, while spines of the DHF treatment group began to increase dramatically (from 45±2/100 μm to 56±7/100 μm, p<0.05). In the 1–1.5 mm area, all groups did not show obvious change compared with 0.5–1.0 mm area (SHAM 54±0.4/100 μm; PBS 44±3/100 μm; DHF 60±5/100 μm and DMSO 45±4/100 μm). In the 1.5–2 mm area, spine density in the PBS group began to increase significantly (from 44±3/100 μm to 53±3/100 μm) and got close to the density in the SHAM group (59±4/100 μm). The DMSO group (52±4/100 μm) exhibited the same trend as the PBS group, while the spine density in the DHF treatment group maintained a high level (67±6/100 μm) (Fig. 4C). These results indicated that DHF treatment can prevent dendritic spine loss after TBI.
Figure 4. Protective effect of DHF treatment on spine reduction after moderate traumatic brain injury.
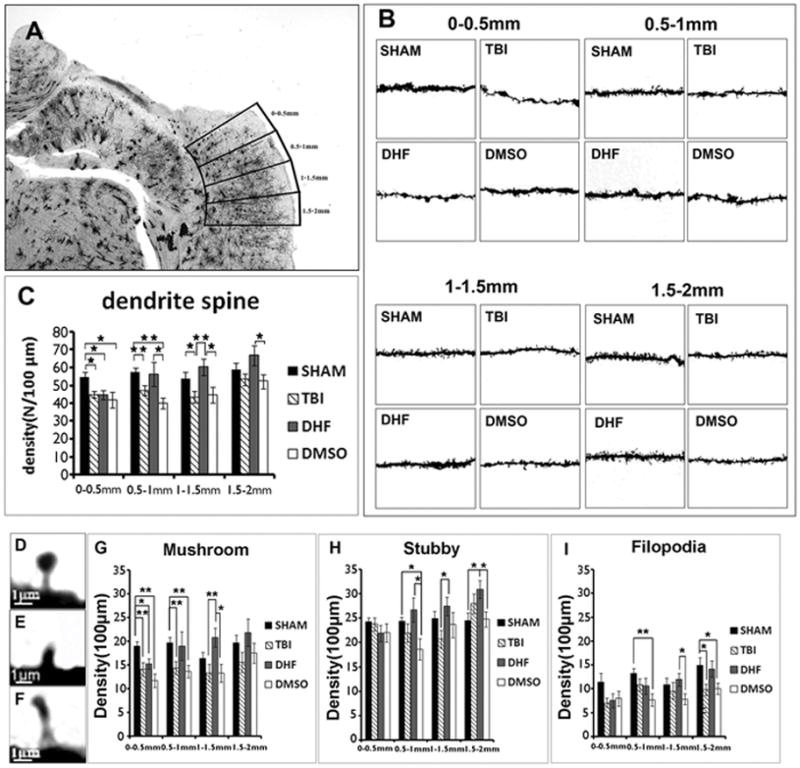
(A) Different areas from epicenter. (B) Single dendrite of 4 groups in each area. (C) Quantification results of dendritic spines in different area. (D, E, F) Different types of spines. (G, H, I) Quantification results of 3 types of dendritic spines. (n=4, *, p<0.05, **, p<0.01).
DHF affected different sub-types of dendritic spines in different ways
There are 3 different types of dendritic spines [43,44]. The first type was a mushroom-shaped spine, which has a big head with a small neck and is usually seen in mature synapses (Fig. 4D). The second one is a stubby spine, usually shorter than 2 μm and is seen in a transitional stage between an early to mature neuron (Fig. 4E). The third one is a filopodia-shaped spine, which is long and thin and is usually seen at the early stage of spine formation (Fig. 4F). When the density of these 3 types of spines were analyzed, we found that after TBI, the mushroom spines presented the same pattern change as whole spines. In all injury groups, the density of spines decreased dramatically in the area closest to the injury site (0–0.5 mm, PBS: 14±1/100 μm, DHF: 15±1/100 μm and DMSO: 12±1/100 μm vs. SHAM: 19±1/100 μm p<0.05). In the PBS group, the decrease of mushroom spines happened through the adjacent 1-mm area (0.5–1.5 mm) and was not observed until far away from the epicenter (>1.5 mm). In the DHF treatment group, the density of mushroom-shaped spines was recovered beginning from the 0.5–1 mm area (19±3/100 μm) and significantly increased with distance compared to control TBI group (1–1.5 mm, 21±2/100 μm, p<0.05; 1.5–2.0 mm, 22±3/100 μm). The change of mushroom-shaped spines in the Vehicle group was similar to the PBS group. The pattern of stubby-shaped spines was different. Compared to the SHAM group, the density of stubby spines in the PBS and the DMSO group had not significantly changed across the whole study area, while it increased steadily in the DHF-treated group. The filopodia-shaped spines were more like mushroom spines with less density. These results demonstrated that although DHF treatment could not prevent the spine loss in areas very close to the cavity, it could protect spines from degeneration in areas farther from the cavity.
DHF improved behavior outcomes
Motor function (Rotarod test)
Golgi staining had shown that DHF can prevent dendrite damage and dendritic spine degeneration. We then wanted to know whether behavior outcomes would be improved after DHF treatment. We performed the rotarod test to determine motor function. Mice completed the rotarod test for baseline before surgery and the latency in all groups was at the same level (SHAM 51.90±2.74 seconds; PBS 46.07±2.93 seconds; DHF 49.14±2.32 seconds; and DMSO 45.09±1.89 seconds). After surgery, the rotarod test was given 3 times at 3 days, 7 days, and 14 days, respectively (Fig. 5A). Latency on the stage was recorded and analyzed. Data showed that, at the third day after surgery, the latency of the SHAM group was slightly decreased compared to baseline (from 51.90±2.74 seconds to 47.4±2.34 seconds); this decrease may be due to the surgery. Meanwhile, the latency of the TBI group with PBS treatment showed a dramatic decrease (from 46.07±2.93 seconds to 37.22±2.22 seconds, p<0.05). DHF treatment significantly attenuated the dropping of latency after TBI at this time point (42.30±2.20 seconds compared to PBS treatment 37.22±2.22 seconds, p<0.05), although it was still lower than the SHAM group. DMSO did not exhibit any protection; the latency also decreased dramatically (from 45.09±1.89 seconds to 34.62±2.98 seconds, p<0.05). Seven days after TBI, latency in all groups increased because of spontaneous recovery. However, all injury groups still had less latency than the SHAM group, which was already back to baseline level (SHAM 54.67±3.81 seconds, PBS 46.47±3.10 seconds; DHF 46.26±2.82 seconds and DMSO 41.08±3.59 seconds). Fourteen days after injury, there were no significant differences between the groups, and they all returned back to the baseline level, and were even better in the DHF treatment group (Fig. 5B). This result demonstrated that DHF prevented the damage of motor function caused by moderate TBI, and thus improved the behavior outcome.
Figure 5. Post-injury treatment of DHF improved behavior outcomes after moderate traumatic brain injury.
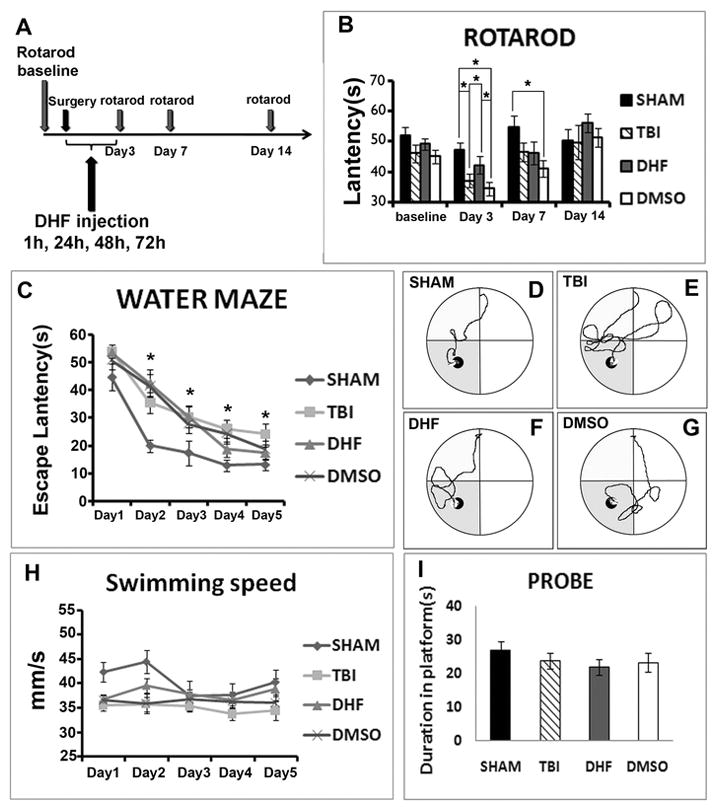
(A) Rotarod test was given to mice in 4 groups. (B) On the third day after surgery, latency of all groups decreased. TBI group and DMSO group decreased more dramatically than DHF group and SHAM group (n=16,*, p<0.05). (C) Training trials of Morris water maze. SHAM group learned much faster than other groups. The curve indicated that the latency of DHF group dropped consistently over time, but did not show a significant difference compared to the TBI group. (D, E, F, G) Swimming patterns of 4 groups on the last day of learning trial. (H) Swimming speed of 4 groups. (I) Probe trial of 4 groups.
Learning and memory function (Morris Water Maze test)
Learning and memory function were also tested by Morris Water Maze. Water maze was conducted at 2 weeks after TBI. Each mouse accepted 4 trials of training for each day from different quadrants. The learning trial was conducted for 5 days to observe learning processes of the mice. On the sixth day, the platform was removed to observe whether mice could remember the location of the platform to test their memory function. Latency of escaping, swimming speed, and latency in the platform quadrant were analyzed after the test. In the learning trial, the SHAM group demonstrated the typical learning curve with a sharp reduction of escaping latency on the second training day (from 44.82±4.79 seconds to 20.01±2.29 seconds, p<0.05). Then the escaping latency decreased steadily until the mouse reached the platform on day 4 (day 3: 17.40±4.48 seconds; day 4: 12.87±1.99 seconds; and day 5: 13.08±2.10 seconds). Mice in the injury group with PBS treatment spent more time finding the platform since the first day (54.16±2.32 seconds) compared to the SHAM group. Although the escaping latency shortened day by day, it was very slow and always siginificantly longer than the SHAM group (day 2: 35.65±3.35 seconds; day 3: 30.35±5.48 and day 4: 26.07±5.45 seconds, p<0.05). On the last training day, the latency of the PBS group mice was still 2-fold greater than the SHAM group (24.22±3.73 seconds vs. 13.08±2.10 seconds, p<0.05). The escaping latency of the DHF treatment group was at the PBS group level on the first training day and the latency reduced slowly during the first 3 days (day 1: 53.76±2.11 seconds; day 2: 42.65±4.83 seconds; and day 3: 30.44±3.92 seconds). However, latency dropped faster on the fourth day (18.61±2.75 seconds) and reached almost the SHAM level on the final day (17.38±2.47 seconds) (Fig. 5C).
The Vehicle group showed a similar trend to the DHF treatment group (day 1: 50.92±3.31 seconds; day 2: 41.72±4.07 seconds; day 3: 27.93±3.39 seconds; day 4: 24.48±3.66 seconds; and day 5: 18.81±2.86 seconds) (Fig. 5C). The swimming patterns showed this difference more visually (Fig. 5D, E, F, and G). We also analyzed the swimming speed and results showed no significant differences among the 4 groups (Fig. 5H). This result indicated that the motor function did not significantly affect the latency. In the memory trial, the latency of mice in the injury group with PBS treatment (23.84±2.32 seconds) was slightly shorter than in the SHAM group (26.82±2.74 seconds) in the target quadrant, but neither the DHF treatment group (22.00±2.80 seconds) nor the Vehicle group (23.31±2.32 seconds) showed a significant difference compared to the PBS group (Fig. 5I).
Discussion
Moderate CCI-TBI causes not only direct damage on the brain, but also induces secondary damage. Among types of secondary damage, massive cell death is just one aspect. Light microscopic studies of MAP2 staining revealed a prominent loss of MAP2 immunofluorescence in apical dendrites of pyramidal neurons in the injured cortex [45,46]. The spine density in the forebrain regions [47,3] and numner of synapses in the hippocampus [48,4] significantly reduced following TBI as well. Golgi staining further demonstrated that even in the cortex of a mildly injured brain, in which no remarkable cell death was found, there was extensive dendrite degeneration, which was not limited to the primary injury site but spread out to a very large area [3]. It suggests that even if many neurons are spared after TBI, many of them were suffering from injury. Therefore, protecting the spared neurons from injury or helping them recover gives us another opportunity to reduce the damage. BDNF, one of the important neurotrophic factors was implicated as a means to help neurons survive and prevent neural degeneration after TBI [49][32]. However, the clinical application of BDNF is limited because of the delivery problem, an immune response problem caused by huge molecular weight, a short half-life, and so on. We need a alternative, which can mimic BDNF without those limitations.
One of the promising alternatives is 7,8-dihydroxyflavone, a BDNF receptor TrkB angonist which can activate BDNF signal pathway. A small molecular weight makes it possible for 7,8-dihydroxyflavone to go through the BBB without causing an immune response. It is proven that 7,8-dihydroxyflavone showed a protective effect on other models, such as an ischemia and reperfusion model [29] and an Alzheimer’s model [30]. Previous research in our laboratory revealed that pretreatment of 7,8-dihydroxyflavone could prevent neuron death in the cortex and hippocampus caused by moderate TBI (seperated manucript, revising). In this study we used posttreatment of 7,8-dihydroxyflavone to explore whether it was able to prevent the dendrite degeneration and, in turn, to improve behavior outcomes of TBI mice. Results showed that although posttreated TBI with 7,8-dihydroxyflavone did not significantly reeuce cortical cavity after TBI, it dramatically reduced dendrite damage. The number of beadings, a hallmark of degenerated dendrites, was significantly decreased. The dendrite complexity was well preserved, as branch number and total length of both apical and basal dendrites in the DHF treatment group were remarkably increased compared to control TBI groups.
High resolution images and quantification data showed that post-injury treatment with DHF reduced tissue lesion volume dendrtie spine degeneration in the cortex in the TBI group. When we further assessed the three types of spines—mushroom, stubby, and filopodia—this protective effect was seen mainly in mushroom and stubby spines, of which mushroom spines are usually seen in mature synapses and stubby spines are seen in transitional stages of spine formation. However, this spine protection was regionally limited. Data showed that only in an area 0.5–1.5 mm from the edge of cavity, the DHF treatment group showed a significant preventive effect compared to TBI control groups. But, in an area of 0–0.5 mm, which was adjacent to the edge of cavity in the epicenter, DHF did not show an advantage for saving spine loss. This may be due to the following reasons: After TBI, in the impact site and surrounding area, the blood vessel system was totally disrupted, so that the DHF may not be able to be delivered there. Further, the damage in the close area of the cavity happened quickly and was severe and irreversible. If DHF cannot be delivered in time, then its protection may be too mild to reverse the initated damage.
DHF is also reported to prevent synaptical loss [30] and to promote axon regeneration [31] in other disease models. The neuroprotective effect of DHF also led to posttraumatic functional recovery in this study. We found that the DHF treatment group performed better in motor function than the TBI group using the rotarod test. However, the DHF group did not perform significantly better in learning and memory function than the TBI group using the water maze test. The TBI-induced functional deficits may be due to multiple reasons, including cell death, cell stress, axonal and dendritic degenerations, inflammation, and so forth. The post-treatment with DHF showed a protective effect to dendrite degeneration and cell death, which is reflected in better motor function following TBI. But there is no significant improvement in learning and memory. These data suggest that we still need to optimize the parameters of DHF treatment, such as extending the duration, increasing the dose, or combining with other therapies.
BDNF, through interacting with its receptor TrkB, activates a downstream pathway, such as PI3K and MAPK pathway. Because 7,8-dihydroxyflavone is a robust agonist of TrkB receptor, it perhaps mimics BDNF function in the same manner. Our previous study of DHF function on preventing neuronal death in TBI proved that DHF did go through the TrkB-mediated PI3K/Akt signal pathway to play the critial role in helping neurons survive after injury. However, the downstream pathway involved in dendritic protection by DHF is still unclear. Future work should be done to examine whether it also uses the TrkB-mediated signal pathway to perform its function on preventing dendrite degeneration.
Acknowledgments
This work was supported through funding from the Indiana Spinal Cord & Brain Injury Research Grants, the Ralph W. and Grace M. Showalter Research Award, Indiana University Biological Research Grant, and NIH grants RR025761 and 1R21NS072631-01A.
Footnotes
Ethnic statement: I have read and have abided by the statement of ethical standards for manuscripts submitted to Molecular Neurobiology.
Conflict of interest The authors declare that there is no conflict of interest.
References
- 1.Faul MXL, Wald MM, Coronado VG. Traumatic brain injury in the United States: emergency department visits, hospitalization, and deaths. Altanta (GA): Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; 2010. [Google Scholar]
- 2.Finkelstein ECP, Miller T, et al. The Incidence and Economic Burden of Injuries in the United States. New York (NY): Oxford University Press; 2006. [Google Scholar]
- 3.Gao X, Chen J. Mild traumatic brain injury results in extensive neuronal degeneration in the cerebral cortex. Journal of neuropathology and experimental neurology. 2011;70(3):183–191. doi: 10.1097/NEN.0b013e31820c6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gao X, Deng P, Xu ZC, Chen J. Moderate traumatic brain injury causes acute dendritic and synaptic degeneration in the hippocampal dentate gyrus. PLoS One. 2011;6(9):e24566. doi: 10.1371/journal.pone.0024566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adelson PD, Dixon CE, Robichaud P, Kochanek PM. Motor and cognitive functional deficits following diffuse traumatic brain injury in the immature rat. Journal of neurotrauma. 1997;14(2):99–108. doi: 10.1089/neu.1997.14.99. [DOI] [PubMed] [Google Scholar]
- 6.Srivastava DP, Woolfrey KM, Evans PD. Mechanisms underlying the interactions between rapid estrogenic and BDNF control of synaptic connectivity. Neuroscience. 2013;239:17–33. doi: 10.1016/j.neuroscience.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 7.Zoladz JA, Pilc A. The effect of physical activity on the brain derived neurotrophic factor: from animal to human studies. Journal of physiology and pharmacology : an official journal of the Polish Physiological Society. 2010;61(5):533–541. [PubMed] [Google Scholar]
- 8.Wu H, Lu D, Jiang H, Xiong Y, Qu C, Li B, Mahmood A, Zhou D, Chopp M. Simvastatin-mediated upregulation of VEGF and BDNF, activation of the PI3K/Akt pathway, and increase of neurogenesis are associated with therapeutic improvement after traumatic brain injury. Journal of neurotrauma. 2008;25(2):130–139. doi: 10.1089/neu.2007.0369. [DOI] [PubMed] [Google Scholar]
- 9.Griesbach GS, Hovda DA, Gomez-Pinilla F. Exercise-induced improvement in cognitive performance after traumatic brain injury in rats is dependent on BDNF activation. Brain research. 2009;1288:105–115. doi: 10.1016/j.brainres.2009.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu A, Ying Z, Gomez-Pinilla F. Dietary omega-3 fatty acids normalize BDNF levels, reduce oxidative damage, and counteract learning disability after traumatic brain injury in rats. Journal of neurotrauma. 2004;21(10):1457–1467. doi: 10.1089/neu.2004.21.1457. [DOI] [PubMed] [Google Scholar]
- 11.Wu A, Ying Z, Gomez-Pinilla F. Dietary curcumin counteracts the outcome of traumatic brain injury on oxidative stress, synaptic plasticity, and cognition. Experimental neurology. 2006;197(2):309–317. doi: 10.1016/j.expneurol.2005.09.004. S0014-4886(05)00327-4 [pii] [DOI] [PubMed] [Google Scholar]
- 12.Mahmood A, Lu D, Chopp M. Intravenous administration of marrow stromal cells (MSCs) increases the expression of growth factors in rat brain after traumatic brain injury. Journal of neurotrauma. 2004;21(1):33–39. doi: 10.1089/089771504772695922. [DOI] [PubMed] [Google Scholar]
- 13.Mahmood A, Lu D, Wang L, Chopp M. Intracerebral transplantation of marrow stromal cells cultured with neurotrophic factors promotes functional recovery in adult rats subjected to traumatic brain injury. Journal of neurotrauma. 2002;19(12):1609–1617. doi: 10.1089/089771502762300265. [DOI] [PubMed] [Google Scholar]
- 14.Wu A, Molteni R, Ying Z, Gomez-Pinilla F. A saturated-fat diet aggravates the outcome of traumatic brain injury on hippocampal plasticity and cognitive function by reducing brain-derived neurotrophic factor. Neuroscience. 2003;119(2):365–375. doi: 10.1016/s0306-4522(03)00154-4. [DOI] [PubMed] [Google Scholar]
- 15.Gao X, Arlotta P, Macklis JD, Chen J. Conditional knock-out of beta-catenin in postnatal-born dentate gyrus granule neurons results in dendritic malformation. J Neurosci. 2007;27(52):14317–14325. doi: 10.1523/JNEUROSCI.3206-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao X, Chen J. Conditional knockout of brain-derived neurotrophic factor in the hippocampus increases death of adult-born immature neurons following traumatic brain injury. Journal of neurotrauma. 2009 doi: 10.1089/neu.2008-0744. [DOI] [PubMed] [Google Scholar]
- 17.Harborne JB, Williams CA. Advances in flavonoid research since 1992. Phytochemistry. 2000;55(6):481–504. doi: 10.1016/s0031-9422(00)00235-1. [DOI] [PubMed] [Google Scholar]
- 18.Spencer JP. Food for thought: the role of dietary flavonoids in enhancing human memory, learning and neuro-cognitive performance. The Proceedings of the Nutrition Society. 2008;67(2):238–252. doi: 10.1017/S0029665108007088. [DOI] [PubMed] [Google Scholar]
- 19.Jang SW, Liu X, Yepes M, Shepherd KR, Miller GW, Liu Y, Wilson WD, Xiao G, Blanchi B, Sun YE, Ye K. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(6):2687–2692. doi: 10.1073/pnas.0913572107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andero R, Daviu N, Escorihuela RM, Nadal R, Armario A. 7, 8 dihydroxyflavone, a TrkB receptor agonist, blocks long term spatial memory impairment caused by immobilization stress in rats. Hippocampus. 2010 doi: 10.1002/hipo.20906. [DOI] [PubMed] [Google Scholar]
- 21.Andero R, Heldt SA, Ye K, Liu X, Armario A, Ressler KJ. Effect of 7,8-dihydroxyflavone, a small-molecule TrkB agonist, on emotional learning. Am J Psychiatry. 2011;168(2):163–172. doi: 10.1176/appi.ajp.2010.10030326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen J, Chua KW, Chua CC, Yu H, Pei A, Chua BH, Hamdy RC, Xu X, Liu CF. Antioxidant activity of 7,8-dihydroxyflavone provides neuroprotection against glutamate-induced toxicity. Neuroscience letters. 2011;499(3):181–185. doi: 10.1016/j.neulet.2011.05.054. [DOI] [PubMed] [Google Scholar]
- 23.Devi L, Ohno M. 7,8-dihydroxyflavone, a small-molecule TrkB agonist, reverses memory deficits and BACE1 elevation in a mouse model of Alzheimer's disease. Neuropsychopharmacology. 2012;37(2):434–444. doi: 10.1038/npp.2011.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson RA, Lam M, Punzo AM, Li H, Lin BR, Ye K, Mitchell GS, Chang Q. 7,8-dihydroxyflavone exhibits therapeutic efficacy in a mouse model of Rett syndrome. J Appl Physiol. 2012;112(5):704–710. doi: 10.1152/japplphysiol.01361.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu X, Chan CB, Jang SW, Pradoldej S, Huang J, He K, Phun LH, France S, Xiao G, Jia Y, Luo HR, Ye K. A Synthetic 7,8-Dihydroxyflavone Derivative Promotes Neurogenesis and Exhibits Potent Antidepressant Effect. J Med Chem. 2010 doi: 10.1021/jm101206p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park HY, Kim GY, Hyun JW, Hwang HJ, Kim ND, Kim BW, Choi YH. 7,8-Dihydroxyflavone exhibits anti-inflammatory properties by downregulating the NF-kappaB and MAPK signaling pathways in lipopolysaccharide-treated RAW264.7 cells. Int J Mol Med. 2012;29(6):1146–1152. doi: 10.3892/ijmm.2012.935. [DOI] [PubMed] [Google Scholar]
- 27.Park HY, Kim GY, Hyun JW, Kim ND, Kim CG, Kim WJ, Yoo YH, Choi YH. 7,8-dihydroxyflavone induces G1 arrest of the cell cycle in U937 human monocytic leukemia cells via induction of the Cdk inhibitor p27 and downregulation of pRB phosphorylation. Oncology reports. 2012;28(1):353–357. doi: 10.3892/or.2012.1773. [DOI] [PubMed] [Google Scholar]
- 28.Zeng Y, Liu Y, Wu M, Liu J, Hu Q. Activation of TrkB by 7,8-Dihydroxyflavone Prevents Fear Memory Defects and Facilitates Amygdalar Synaptic Plasticity in Aging. J Alzheimers Dis. 2012 doi: 10.3233/JAD-2012-120886. [DOI] [PubMed] [Google Scholar]
- 29.Zeng Y, Lv F, Li L, Yu H, Dong M, Fu Q. 7,8-dihydroxyflavone rescues spatial memory and synaptic plasticity in cognitively impaired aged rats. J Neurochem. 2012;122(4):800–811. doi: 10.1111/j.1471-4159.2012.07830.x. [DOI] [PubMed] [Google Scholar]
- 30.Zhang R, Kang KA, Piao MJ, Ko DO, Wang ZH, Chang WY, You HJ, Lee IK, Kim BJ, Kang SS, Hyun JW. Preventive effect of 7,8-dihydroxyflavone against oxidative stress induced genotoxicity. Biol Pharm Bull. 2009;32(2):166–171. doi: 10.1248/bpb.32.166. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Z, Liu X, Schroeder JP, Chan CB, Song M, Yu SP, Weinshenker D, Ye K. 7,8- dihydroxyflavone prevents synaptic loss and memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacology. 2014;39(3):638–650. doi: 10.1038/npp.2013.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andero R, Ressler KJ. Fear extinction and BDNF: translating animal models of PTSD to the clinic. Genes Brain Behav. 2012;11(5):503–512. doi: 10.1111/j.1601-183X.2012.00801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao X, Enikolopov G, Chen J. Moderate traumatic brain injury promotes proliferation of quiescent neural progenitors in the adult hippocampus. Experimental neurology. 2009;219(2):516–523. doi: 10.1016/j.expneurol.2009.07.007. S0014-4886(09)00270-2 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao X, Deng Bryant Y, Cho W, Carrico KM, Hall ED, Chen J. Selective death of newborn neurons in hippocampal dentate gyrus following moderate experimental traumatic brain injury. Journal of neuroscience research. 2008;86(10):2258–2270. doi: 10.1002/jnr.21677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao X, Enikolopov G, Chen J. Direct isolation of neural stem cells in the adult hippocampus after traumatic brain injury. Journal of neurotrauma. 2008;25(8):985–995. doi: 10.1089/neu.2008.0460. [DOI] [PubMed] [Google Scholar]
- 36.Brody DL, Mac Donald C, Kessens CC, Yuede C, Parsadanian M, Spinner M, Kim E, Schwetye KE, Holtzman DM, Bayly PV. Electromagnetic controlled cortical impact device for precise, graded experimental traumatic brain injury. Journal of neurotrauma. 2007;24(4):657–673. doi: 10.1089/neu.2006.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou H, Chen L, Gao X, Luo B, Chen J. Moderate traumatic brain injury triggers rapid necrotic death of immature neurons in the hippocampus. J Neuropathol Exp Neurol. 2012;71(4):348–359. doi: 10.1097/NEN.0b013e31824ea078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sholl DA. The measurable parameters of the cerebral cortex and their significance in its organization. Prog Neurobiol. 1956;(2):324–333. [PubMed] [Google Scholar]
- 39.Uylings HB, Ruiz-Marcos A, van Pelt J. The metric analysis of three-dimensional dendritic tree patterns: a methodological review. J Neurosci Methods. 1986;18(1–2):127–151. doi: 10.1016/0165-0270(86)90116-0. [DOI] [PubMed] [Google Scholar]
- 40.Risher WC, Lee MR, Fomitcheva IV, Hess DC, Kirov SA. Dibucaine mitigates spreading depolarization in human neocortical slices and prevents acute dendritic injury in the ischemic rodent neocortex. PLoS One. 2011;6(7):e22351. doi: 10.1371/journal.pone.0022351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao X, Chen J. Moderate traumatic brain injury promotes neural precursor proliferation without increasing neurogenesis in the adult hippocampus. Experimental neurology. 2012 doi: 10.1016/j.expneurol.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saatman KE, Duhaime AC, Bullock R, Maas AI, Valadka A, Manley GT. Classification of traumatic brain injury for targeted therapies. Journal of neurotrauma. 2008;25(7):719–738. doi: 10.1089/neu.2008.0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sorra KE, Harris KM. Overview on the structure, composition, function, development, and plasticity of hippocampal dendritic spines. Hippocampus. 2000;10(5):501–511. doi: 10.1002/1098-1063(2000)10:5<501::AID-HIPO1>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 44.Harris KM. Structure, development, and plasticity of dendritic spines. Curr Opin Neurobiol. 1999;9(3):343–348. doi: 10.1016/s0959-4388(99)80050-6. [DOI] [PubMed] [Google Scholar]
- 45.Posmantur RM, Kampfl A, Taft WC, Bhattacharjee M, Dixon CE, Bao J, Hayes RL. Diminished microtubule-associated protein 2 (MAP2) immunoreactivity following cortical impact brain injury. Journal of neurotrauma. 1996;13(3):125–137. doi: 10.1089/neu.1996.13.125. [DOI] [PubMed] [Google Scholar]
- 46.Chen LJ, Wang YJ, Tseng GF. Compression alters kinase and phosphatase activity and tau and MAP2 phosphorylation transiently while inducing the fast adaptive dendritic remodeling of underlying cortical neurons. Journal of neurotrauma. 2010;27(9):1657–1669. doi: 10.1089/neu.2010.1308. [DOI] [PubMed] [Google Scholar]
- 47.Campbell JN, Register D, Churn SB. Traumatic brain injury causes an FK506-sensitive loss and an overgrowth of dendritic spines in rat forebrain. Journal of neurotrauma. 2012;29(2):201–217. doi: 10.1089/neu.2011.1761. [DOI] [PubMed] [Google Scholar]
- 48.Scheff SW, Price DA, Hicks RR, Baldwin SA, Robinson S, Brackney C. Synaptogenesis in the hippocampal CA1 field following traumatic brain injury. Journal of neurotrauma. 2005;22(7):719–732. doi: 10.1089/neu.2005.22.719. [DOI] [PubMed] [Google Scholar]
- 49.Kishino A, Ishige Y, Tatsuno T, Nakayama C, Noguchi H. BDNF prevents and reverses adult rat motor neuron degeneration and induces axonal outgrowth. Experimental neurology. 1997;144(2):273–286. doi: 10.1006/exnr.1996.6367. [DOI] [PubMed] [Google Scholar]